

Abstract
Objective
Report of a 16q24.1 deletion in a premature newborn, demonstrating the usefulness of array-CGH in persistent pulmonary hypertension of the newborn (PPHN) and multiple congenital malformations.
Design
Descriptive case report
Setting
Genetic department and neonatal intensive care unit of a tertiary care children’s hospital
Patient
We report the case of a preterm male infant, born at 26 weeks of gestation. A cardiac malformation and bilateral hydronephrosis were diagnosed at 19 weeks of gestation. Karyotype analysis was normal and a 22q11.2 microdeletion was excluded by FISH analysis. A Caesarean section was performed due to fetal distress. The patient developed persistent pulmonary hypertension unresponsive to mechanical ventilation and nitric oxide treatment and expired at 16 hours of life. An autopsy revealed partial atrio-ventricular canal malformation and showed bilateral dilatation of the renal pelvocaliceal system with bilateral ureteral stenosis, and annular pancreas. Array-CGH analysis (Agilent oligoNT 44K) showed an interstitial microdeletion encompassing the FOX gene cluster in 16q24.1. Review of the pulmonary microscopic examination showed the characteristic features of alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV). Some features were less prominent due to the gestational age.
Conclusions
Our review of the literature shows that ACD/MPV is rare, but probably underreported. Prematurity is not a usual presentation and histological features are difficult to interpret. In our case, array-CGH revealed a 16q24.1 deletion leading to the final diagnosis of ACD/MPV. It emphasises the usefulness of array-CGH analysis as a diagnostic tool with implications for both prognosis and management decisions in newborns with refractory PPHN and multiple congenital malformations.
Keywords: persistent pulmonary hypertension of the newborn, alveolar capillary dysplasia with misalignment of pulmonary veins, ACD/MPV, 16q24.1, neonate, array-CGH
INTRODUCTION
Persistent pulmonary hypertension of the newborn (PPHN) is characterized by failure to achieve or sustain the normal decrease of pulmonary vascular resistance at birth. It is associated with right to left shunting of blood across the patent ductus arteriosus and/or the foramen ovale, leading to hypoxemia [1]. Its incidence is about 1: 1000 live births [2]. PPHN can be primary or secondary to a variety of disorders including fetal or perinatal hypoxia, sepsis, pneumonia, meconium aspiration, congenital diaphragmatic hernia, pulmonary hypoplasia, premature closure of the ductus arteriosus, veno-occlusive disease, capillary hemangiomatosis and congenital heart disease, including pulmonary vein stenosis [1]. Alveolar capillary dysplasia with misalignment of the pulmonary veins (ACD/MPV) is a rare interstitial lung disease affecting primarily the alveolar components [3]. It is recognized as a cause of PPHN [4]. About 200 cases of this lethal developmental disorder of the lung have been reported in the literature [5-7]. The histological features supporting the diagnosis of ACD/MPV were described first in 1981 by Janney [8]. They are characterized by a marked paucity of capillaries adjacent to the alveolar epithelium, anomalous small veins within the bronchioarterial bundles, and medial thickening of small pulmonary arteries. Lymphangiectasis is commonly observed, but is not universal feature of ACD/MPV [9]. ACD/MPV exact incidence is unknown and the diagnosis may be missed even on histological examination as some expertise on the part of the pathologist is required. In the majority of cases, affected infants are full-term newborns. Apgar scores are usually normal and respiratory deterioration occurs in the first hours to days of life, with the development of respiratory distress and severe pulmonary hypertension. The patients fail to respond to various supportive strategies of management including conventional or high-frequency ventilation, inhaled nitric oxide and extracorporeal membrane oxygenation (ECMO) [4, 7, 10-12]. The majority expire within 4 to 6 weeks of birth, although late presentation has been reported [11, 13-15]. Up to 80% of reported patients have additional malformations, affecting predominantly the gastro-intestinal, cardiac and genito-urinary systems [7, 10, 16, 17]. Although familial cases occur, most cases are sporadic and no genetic etiology supported the diagnosis until recently. FOX gene cluster deletions in 16q24.1 and FOXF1 point mutations have been identified in several cases of ACD/MPV associated with other congenital anomalies [18, 19]. We report the clinical course and cytogenetic data of a new case of 16q24.1 deletion and discuss recent insights into the etiology of ACD/MVP that illustrate the diagnostic and prognostic value of array-CGH in clinical practice.
SUBJECT AND METHODS
A preterm male infant was born at 26 weeks of gestation by emergency caesarean section due to fetal distress. He was the first child of a nonconsanguineous Caucasian couple. During pregnancy, the 2nd trimester fetal ultrasound revealed a cardiac malformation (partial atrio-ventricular canal defect) and bilateral pelvocaliceal dilatation. Cultured amniotic cells showed a normal 46,XY karyotype and FISH analysis excluded a 22q11.2 microdeletion. Apgar score was 5 at 1′ and 8 at 5′. The patient was intubated due to inadequate respiratory efforts. He remained hypoxemic (PaO2 30 mmHg) despite surfactant treatment and mechanical ventilation with 100% oxygen. Echocardiography confirmed the partial atrioventricular canal and showed suprasystemic pulmonary hypertension with a right to left shunt across the ductus arteriosus. Treatment with inhaled nitric oxide, dopamine and volume failed to improve oxygenation and the patient expired at 16 hours of life from hypoxemic respiratory failure. After informed consent an autopsy and complementary genetic analyses were performed.Growth parameters include weight of 592.4 g (appropriate for 24 weeks of gestation), crown-heel length 32.5 cm (appropriate for 26 weeks of gestation), head circumference 21.5 cm, without evidence for symmetric intrauterine growth retardation.
On internal examination the organs were normally located. The heart showed partial AV canal malformation with dysplastic tricuspid valve and two mitral valve leaflets. The pulmonary artery appeared somewhat small. Both kidneys showed dilatation of the pelvocaliceal system with proximal ureteral stenosis on the left and distal stenosis on the right. There was no tracheoesophageal fistula, but there was annular pancreas with duodenal dilatation proximal to the pancreas. The remainder of the small intestine and colon were well positioned and no stenosis, atresia, or malrotation was identified.
Histology of the placenta showed evidence of intrauterine infection with funisitis, chorioamnionitis, subchorionic fibrin deposits, and vasculitis and thrombosis at the insertion of the umbilical vessels. The umbilical cord had the usual three vessels. There were no other abnormal findings.
The lungs showed the characteristic features of ACD/MPV (Figure 1). Microscopic examination was similar in all five lobes. Some features were less prominent than usual due to the gestational age. Capillaries were reduced in normal locations, beyond that typically seen at this gestational age. Small pulmonary arteries were not as prominent as often seen in ACD/MPV, but showed mildly increased amounts of smooth muscle within their walls and typically adjacent markedly dilated thin-walled venous channels. There were also signs of congenital pneumonia.
Figure 1. Lung histopathology of ACD/MPV.

HE staining, × 100. The lobular architecture is altered with enlarged simplified and poorly subdivided alveoli. Small pulmonary arteries adjacent to bronchioles show mildly increased amounts of smooth muscle within their walls; there are abnormally positioned markedly dilated thin-walled veins adjacent to these small pulmonary arteries. Capillaries cannot be clearly delineated at this magnification, but they are deficient in numbers in the thickened alveolar walls, where there are larger centrally located dilated thin-walled vascular channels. Clusters of neutrophils are evident in some airspaces (perinatal pneumonia).
Genomic DNA from the patient was isolated from uncultured skin fibroblasts collected after death. Array CGH analysis was performed using Agilent oligoNT 44K [20]. Array CGH demonstrated a deletion in 16q24.1 encompassing the FOX gene cluster (Figure 2). The size of the deletion was estimated at 1.57 Mb. This region contained five annotated genes (IRF8, FOXF1, MTHFSD, FOXC2, and FOXL1). Metaphase FISH could not be achieved because of fibroblast culture failure, probably due to a delay between death and specimen collection. Interphase FISH was performed on paraffin slides of pulmonary, hepatic and duodenal sections, using a 16q24-specific BAC clone probe, RP11-542M13. The results were consistent with a heterozygote deletion in all three tissues (Figure 3). Parental samplings are still pending.
Figure 2. Array-CGH (Agilent 44K) showing the 16q24.1 deletion.
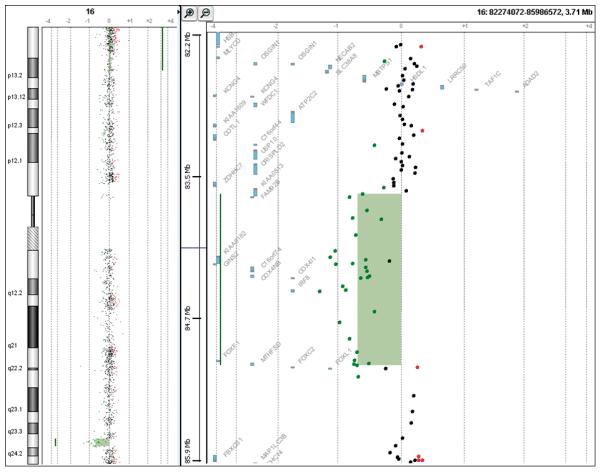
Figure 3. Interphase FISH on paraffin sections of lung biopsy.
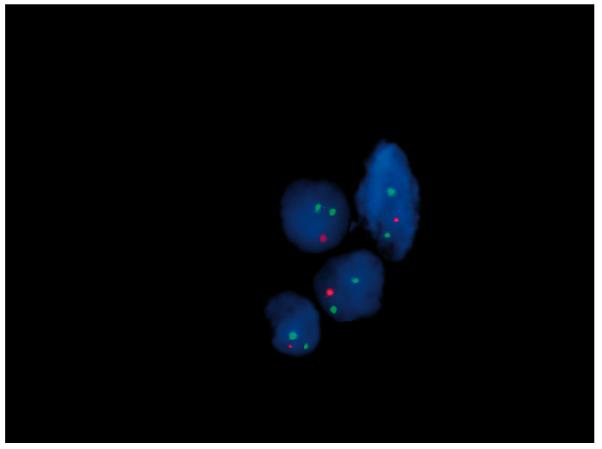
Using a 16q24-specific BAC clone, RP11-542M13 (red), and a control chromosome 16 heterochromatic probe pHUR195 (green), more than 90% of analysed nuclei showed a single red signal in 16q24.1 region, consistent with a heterozygous deletion.
A high-resolution genome wide 2.1 M Nimblegen array (Roche NimbleGen, Madison, WI, USA) and long-range PCR was used to determine the breakpoints [18]. An 8kb PCR product spanning the deletion junction was generated and DNA sequence analysis enabled breakpoint sequencing. The deletion was 1,611,503 bp in size and extended between ch16:83,666,210-85,277,713 (build NCBI 36). At the junction, there was a 25 bp AT-rich insertion TTTATTTTATTTTATTTTATTTTTA. The distal breakpoint mapped in AluYa5 and the proximal within this unique AT-rich sequence, suggesting that the event arose as a microhomology-mediated replication error repair, concordant with the previous published cases [18].
DISCUSSION
We report a new case of a 16q24.1 deletion encompassing the FOX gene cluster. Array-CGH allowed the diagnosis of this new syndrome associated with ACD/MPV. Six cases of ACD/MPV associated with FOXF1 gene deletion or mutation were included in the initial study and only one supplementary case report has been reported until now [18, 19]. All cases were full term newborns. As in the present case, malformations associated with ACD/MPV are multiple, variable in severity, and may resemble VACTERL spectrum anomalies [21]. Intestinal atresia and malrotation are the two more commonly reported gastro-intestinal malformations. Others included annular pancreas, imperforate anus and congenital short bowel. Cardiac anomalies include hypoplastic left heart syndrome, hypoplasia or coarctation of the aortic arch, tetralogy of Fallot, patent ductus arteriosus and atrial septal defect. Hydronephrosis and pelviectasis are the more frequent urinary tract anomalies.
Prior to identification of the associated genetic abnormalities in FOXF1, ACD/MVP was considered to be a rare syndromic entity, but was likely underdiagnosed, depending on i) clinical suspicion, ii) biopsy and/or autopsy rate and iii) possible misinterpretation of pathologic findings [11, 16]. Neonatologist awareness is essential in considering ACD/MPV among causes of PPHN. In the recent literature, open lung biopsy has been proposed as a means for early diagnosis of fatal lung dysplasia [17, 22-25]. In practice, it is performed only in tertiary centres on a limited number of cases and diagnosis is more often made at post mortem examination. Despite the usefulness of autopsy in establishing cause of death and associated comorbidities and complications of therapy, autopsy rates have been declining worldwide over the last decades. Table 1 shows the results of 7 retrospective studies of paediatric ECMO and/or diagnostic lung biopsy. Even with these data, it remains impossible to assess the incidence of ACD/MVP. Nevertheless, available data clearly indicate that ACD/MPV diagnosis represents an important proportion of lethal cases among neonates with PPHN [10, 22-23, 25-29]. Tibbals and Chow [26] retrospectively diagnosed ACD/MPV in 6 newborns while reviewing lung tissue from 13 cases of “idiopathic” PPHN. Congenital heart disease was an exclusion criterion in this study, inappropriately as it is now understood that congenital cardiac abnormalities are often present in ACD/MPV. The histological criteria of ACD/MPV are well defined but pathologists may have limited experience with this rare condition and special expertise may be necessary for definitive diagnosis as circumstances such as prematurity in this case may subtly alter diagnostic features.
Table 1. Retrospective studies on paediatric ECMO and open lung biopsies.
| Series | Sebire, 2005 [27] | Eulmesekian, 2005 [25] | Inwald, 2004 [23] | Tibbals, 2002 [26] | Cassidy, 2002 [22] | Ryan, 1998 [28] | Bond, 1996 [29] |
|---|---|---|---|---|---|---|---|
| Method | Retrospective study, UK |
Retrospective study, single institution, UK |
Retrospective study, single institution, UK |
Retrospective study, single institution, Australia |
Retrospective study, 4 ECMO centers, UK |
Extracorporeal Life Organization Registry (ELSO) |
Retrospective study, single institution, Kentucky |
| Period of time | 7 years (1996-2003) | 6 years (1997-2002) | 6 years (1996-2002) | 18 years (1982-2000) | 3 years (1997-2000) 2,132,525 infants born in the UK |
8 years (1986-1994) | 6 years (1989-1995) |
| Ascertainment | children receiving ECMO in whom open lung biopsy was carried out |
neonates with histological proven ACD/MVP and review of all ECMO and/or open lung biopsy cases |
children referred for and placed on ECMO who also underwent open lung biopsy (before or during ECMO) |
infants admitted to a paediatric intensive care with a diagnosis of PPHN |
full term neonates with acute hypoxemic respiratory failure treated with ECMO |
newborns who had open lung biopsies while receiving ECMO |
all neonatal and paediatric patients who had an open lung biopsy while on ECMO |
| ECMO | 22 children | 9 neonates | 506 referred and 310 patients placed on ECMO, of whom 136 neonates |
13 neonates | 173 neonates | 6000 neonatal ECMO | 213 patients |
| Open lung biopsy | 12 neonates | 10 neonates | 15/506 patients 8/136 neonates |
N/A | 5/173 neonates | 18/6000 neonates | 8/213, of whom 4 neonates |
|
Number of alveolar capillary dyplasia (ACD) |
3/12 ACD | 7 ACD | 3/8 ACD | 6/13 ACD | 5/173 ACD | 2/18 ACD | 0/4 ACD |
| ACD mortality | N/A | 7/7 | 3/3 | 6/6 | 5/5 | 2/2 | N/A |
|
Global mortality in the series |
N/A | 7/7 | N/A | 7/13 (54%) | 36/173 (20%) | N/A | N/A |
|
Premortem diagnosis of ACD |
N/A | 6/7 | N/A | 2/6 | 3/5 | N/A | N/A |
| Autopsy rate | N/A | N/A | N/A | 100% (7/7) | 56% | N/A | N/A |
PPHN= persistent pulmonary hypertension of the newborn, ECMO= extracorporeal membrane oxygenation, N/A= not available or not applicable
In our opinion, genetic testing should now be offered as a first line diagnostic tool for suspected idiopathic PPHN. As congenital cardiac and/or intestinal malformations are present in the vast majority of cases, their association with treatment refractory primary PPHN should prompt array-CGH analysis. Screening for FOXF1 mutations should also be considered. As the outcome of ACD/MPV is uniformly lethal, the identification of 16q24.1 deletion could avoid further invasive diagnostic or therapeutic procedures. Definitive diagnosis is paramount in the case of lethal neonatal disorders as it permits accurate genetic counselling. ACD/MPV is genetically heterogeneous. Familial cases of ACD/MPV account for about 10% of reported cases. Multiple affected siblings born to both consanguinous and non-consanguinous parents initially oriented towards an autosomal recessive model [5, 7, 30, 31]. FOXF1 haploinsufficiency might be responsible for about 30-40 % of sporadic ACD/MPV cases (Stankiewicz, Langston, unpublished data). In Stankiewicz’s series [18], 16q24.1 microdeletions arose de novo except one sparing FOXF1 gene, which was maternally inherited. Based on these data, the risk for recurrence should be low although the possibility of germline mosaicism cannot be completely excluded. Prenatal diagnosis might be recommended for subsequent pregnancies using array-CGH or FISH analyses.
This case supports the usefulness of systematic genetic investigation in multiple congenital abnormalities [32]. The application of array-CGH in the setting of congenital malformations has brought significant insights, currently mostly in defined cohorts [33]. Its clinical diagnosis validity has been proven and new microdeletion/duplication syndromes are continually being delineated. Increased awareness of its value in neonatology and paediatric departments could provide important new information in this area.
CONCLUSIONS
In conclusion, we identified a 16q24.1 deletion in a premature newborn with ACD/MPV, cardiac, urinary and intestinal malformations. ACD/MPV is a probably underdiagnosed developmental lung disorder. Its diagnosis remains challenging and needs pathological expert advice. The diagnosis should be considered in neonates presenting with refractory primary PPHN and multiple congenital malformations. The use of array-CGH as a diagnostic tool in the management of such patients will permit accurate clinical decision making and genetic counselling. By extension, this case highlights the importance and the new perspectives brought by the use of this technique in patients with congenital malformations.
Acknowledgements
The authors wish to thank the parents for their collaboration and the team of CHUV cytogenetic laboratory for excellent technical assistance. PS was supported by NIH grant 1 R01 HL101975-01 and by grant R13-0005-04/2008 from the Polish Ministry of Science and Higher Education.
Footnotes
Consent
Written informed consent was obtained from the parents for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interest
The authors declare that they have no competing interests.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abman SH. Recent advances in the pathogenesis and treatment of persistent pulmonary hypertension of the newborn. Neonatology. 2007;91(4):283–290. doi: 10.1159/000101343. [DOI] [PubMed] [Google Scholar]
- 2.Hageman JR, Adams MA, Gardner TH. Persistent pulmonary hypertension of the newborn. Trends in. 1984;138:592–595. doi: 10.1001/archpedi.1984.02140440076021. [DOI] [PubMed] [Google Scholar]
- 3.Clement A, Nathan N, Epaud R, Fauroux B, Corvol H. Interstitial lung diseases in children. Orphanet J Rare Dis. 2010;5:22. doi: 10.1186/1750-1172-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chelliah BP, Brown D, Cohen M, Talleyrand AJ, Shen-Schwarz S. Alveolar capillary dysplasia--a cause of persistent pulmonary hypertension unresponsive to a second course of extracorporeal membrane oxygenation. Pediatrics. 1995;96:1159–1161. [PubMed] [Google Scholar]
- 5.Gutierrez C, Rodriguez A, Palenzuela S, Forteza C, Rossello JL. Congenital misalignment of pulmonary veins with alveolar capillary dysplasia causing persistent neonatal pulmonary hypertension: report of two affected siblings. Pediatr Dev Pathol. 2000;3:271–276. doi: 10.1007/s100249910035. [DOI] [PubMed] [Google Scholar]
- 6.Antao B, Samuel M, Kiely E, Spitz L, Malone M. Congenital alveolar capillary dysplasia and associated gastrointestinal anomalies. Fetal Pediatr Pathol. 2006;25:137–145. doi: 10.1080/15513810600908230. [DOI] [PubMed] [Google Scholar]
- 7.Sen P, Thakur N, Stockton DW, Langston C, Bejjani BA. Expanding the phenotype of alveolar capillary dysplasia (ACD) J Pediatr. 2004;145:646–651. doi: 10.1016/j.jpeds.2004.06.081. [DOI] [PubMed] [Google Scholar]
- 8.Janney CG, Askin FB, Kuhn C., 3rd Congenital alveolar capillary dysplasia--an unusual cause of respiratory distress in the newborn. Am J Clin Pathol. 1981;76:722–727. doi: 10.1093/ajcp/76.5.722. [DOI] [PubMed] [Google Scholar]
- 9.Langston C. Misalignment of pulmonary veins and alveolar capillary dysplasia. Pediatr Pathol. 1991;11:163–170. doi: 10.3109/15513819109064753. [DOI] [PubMed] [Google Scholar]
- 10.Al-Hathlol K, Phillips S, Seshia MK, Casiro O, Alvaro RE, Rigatto H. Alveolar capillary dysplasia. Report of a case of prolonged life without extracorporeal membrane oxygenation (ECMO) and review of the literature. Early Hum Dev. 2000;57:85–94. doi: 10.1016/s0378-3782(99)00065-1. [DOI] [PubMed] [Google Scholar]
- 11.Michalsky MP, Arca MJ, Groenman F, Hammond S, Tibboel D, Caniano DA. Alveolar capillary dysplasia: a logical approach to a fatal disease. J Pediatr Surg. 2005;40:1100–1105. doi: 10.1016/j.jpedsurg.2005.03.067. [DOI] [PubMed] [Google Scholar]
- 12.Lally KP, Breaux CW., Jr. A second course of extracorporeal membrane oxygenation in the neonate--is there a benefit? Surgery. 1995;117:175–178. doi: 10.1016/s0039-6060(05)80082-0. [DOI] [PubMed] [Google Scholar]
- 13.Shankar V, Haque A, Johnson J, Pietsch J. Late presentation of alveolar capillary dysplasia in an infant. Pediatr Crit Care Med. 2006;7:177–179. doi: 10.1097/01.PCC.0000202570.58016.67. [DOI] [PubMed] [Google Scholar]
- 14.Abdallah HI, Karmazin N, Marks LA. Late presentation of misalignment of lung vessels with alveolar capillary dysplasia. Crit Care Med. 1993;21:628–630. doi: 10.1097/00003246-199304000-00026. [DOI] [PubMed] [Google Scholar]
- 15.Ahmed S, Ackerman V, Faught P, Langston C. Profound hypoxemia and pulmonary hypertension in a 7-month-old infant: late presentation of alveolar capillary dysplasia. Pediatr Crit Care Med. 2008;9:e43–46. doi: 10.1097/PCC.0b013e31818e383e. [DOI] [PubMed] [Google Scholar]
- 16.Rabah R, Poulik JM. Congenital alveolar capillary dysplasia with misalignment of pulmonary veins associated with hypoplastic left heart syndrome. Pediatr Dev Pathol. 2001;4:167–174. doi: 10.1007/s100240010125. [DOI] [PubMed] [Google Scholar]
- 17.Alameh J, Bachiri A, Devisme L, Truffert P, Rakza T, Riou Y, Manouvrier S, Lequien P, Storme L. Alveolar capillary dysplasia: a cause of persistent pulmonary hypertension of the newborn. Eur J Pediatr. 2002;161:262–266. doi: 10.1007/s00431-002-0927-7. [DOI] [PubMed] [Google Scholar]
- 18.Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, Ou Z, Wiszniewska J, Driscoll DJ, Maisenbacher MK, et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. 2009;84:780–791. doi: 10.1016/j.ajhg.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu S, Shao L, Kilbride H, Zwick DL. Haploinsufficiencies of FOXF1 and FOXC2 genes associated with lethal alveolar capillary dysplasia and congenital heart disease. Am J Med Genet A. 2010;152A:1257–1262. doi: 10.1002/ajmg.a.33378. [DOI] [PubMed] [Google Scholar]
- 20.Martinet D, Filges I, Besuchet Schmutz N, et al. Subtelomeric 6p deletion: clinical and array-CGH characterization in two patients. Am J Med Genet A. 2008;146A:2094–102. doi: 10.1002/ajmg.a.32414. [DOI] [PubMed] [Google Scholar]
- 21.Shaw-Smith C. Genetic factors in esophageal atresia, tracheo-esophageal fistula and the VACTERL association: roles for FOXF1 and the 16q24.1 FOX transcription factor gene cluster, and review of the literature. Eur J Med Genet. 2010;53:6–13. doi: 10.1016/j.ejmg.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cassidy J, Smith J, Goldman A, Haynes S, Smith E, Wright C, Haworth S, Davis P, Firmin R, Kasem K, Davis C. The incidence and characteristics of neonatal irreversible lung dysplasia. J Pediatr. 2002;141:426–428. doi: 10.1067/mpd.2002.126602. [DOI] [PubMed] [Google Scholar]
- 23.Inwald D, Brown K, Gensini F, Malone M, Goldman A. Open lung biopsy in neonatal and paediatric patients referred for extracorporeal membrane oxygenation (ECMO) Thorax. 2004;59:328–333. doi: 10.1136/thx.2003.010793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaklitsch MT, Linden BC, Braunlin EA, Bolman RM, 3rd, Foker JE. Open-lung biopsy guides therapy in children. Ann Thorac Surg. 2001;71:1779–1785. doi: 10.1016/s0003-4975(01)02516-4. [DOI] [PubMed] [Google Scholar]
- 25.Eulmesekian P, Cutz E, Parvez B, Bohn D, Adatia I. Alveolar capillary dysplasia: a six-year single center experience. J Perinat Med. 2005;33:347–352. doi: 10.1515/JPM.2005.067. [DOI] [PubMed] [Google Scholar]
- 26.Tibballs J, Chow CW. Incidence of alveolar capillary dysplasia in severe idiopathic persistent pulmonary hypertension of the newborn. J Paediatr Child Health. 2002;38:397–400. doi: 10.1046/j.1440-1754.2002.00014.x. [DOI] [PubMed] [Google Scholar]
- 27.Sebire NJ, Ramsay AD, Malone M. Histopathological features of open lung biopsies in children treated with extracorporeal membrane oxygenation (ECMO) Early Hum Dev. 2005;81:455–460. doi: 10.1016/j.earlhumdev.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Ryan CA, Finer NN. Open lung biopsies in neonates on ECMO: additional cases. Extracorporeal membrane oxygenation. J Pediatr Surg. 1998;33:1327–1328. doi: 10.1016/s0022-3468(98)90183-x. [DOI] [PubMed] [Google Scholar]
- 29.Bond SJ, Lee DJ, Stewart DL, Buchino JJ. Open lung biopsy in pediatric patients on extracorporeal membrane oxygenation. J Pediatr Surg. 1996;31:1376–1378. doi: 10.1016/s0022-3468(96)90832-5. [DOI] [PubMed] [Google Scholar]
- 30.Manouvrier-Hanu S, Devisme L, Farre I, Hue V, Storme L, Kacet N, Boute-Benejean O, Farriaux JP. Pulmonary hypertension of the newborn and urogenital anomalies in two male siblings: a new family with misalignment of pulmonary vessels. Genet Couns. 1996;7:249–255. [PubMed] [Google Scholar]
- 31.Vassal HB, Malone M, Petros AJ, Winter RM. Familial persistent pulmonary hypertension of the newborn resulting from misalignment of the pulmonary vessels (congenital alveolar capillary dysplasia) J Med Genet. 1998;35:58–60. doi: 10.1136/jmg.35.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]