

Abstract
Response regulators (RRs) comprise a major family of signaling proteins in prokaryotes. A modular architecture which consists of a conserved receiver domain and a variable effector domain allows RRs to function as phosphorylation-regulated switches that couple a wide variety of cellular behaviors to environmental cues. Recently, advances have been made in understanding RR functions both at genome-wide and molecular levels. Global techniques have been developed to analyze RR input and output, expanding the scope of characterization of these versatile components. Meanwhile, structural studies have revealed that despite common structures and mechanisms of function within individual domains, a range of interactions between receiver and effector domains confer great diversity in regulatory strategies, optimizing individual RRs for the specific regulatory needs of different signaling systems.
Two-component signal transduction
Bacteria frequently encounter significant changes in their living conditions. Consequently, most characterized species contain numerous signaling systems that allow the coupling of a diverse array of adaptive responses to specific environmental stimuli. The abundance of signal transduction proteins in bacteria has often been underestimated because the major families of signaling proteins identified in eukaryotes play only a minor role in bacteria. Instead, bacteria contain their own distinct repertoire of signaling components [1]. Prevalent among these are the two-component systems (TCSs) that are based on a conserved phosphotransfer pathway between a histidine protein kinase (HPK) and a response regulator (RR) protein (Box 1). HPKs catalyze autophosphorylation at a conserved histidine residue and often possess phosphatase activity toward cognate phosphorylated RRs. RRs catalyze phosphoryl transfer from phosphorylated HPKs and possess autodephosphorylation activity that limits the lifetime of their phosphorylation. The RR is the fundamental control element within these pathways, functioning as a phosphorylation-activated switch that mediates the output response.
Box 1. Two-component signal transduction phosphotransfer pathways.
In the simplest cases, two-component systems (TCSs) consist of just two proteins: a histidine protein kinase (HPK) and a response regulator (RR) protein that function together in a phosphotransfer pathway (Box 1, Figure I) (for reviews see [21, 22, 66]). HPKs are typically transmembrane proteins with variable extracytoplasmic sensing domains that detect specific stimului and conserved cytoplasmic regions containing an ATP-binding kinase domain and a His-containing dimerization domain that contains the site of phosphorylation (for reviews see [67]). HPKs autophosphorylate in trans between monomers of the dimeric kinase, creating a high energy N~P bond between the phosphoryl group and the imidazole ring of histidine. Unlike the phosphoesters of eukaryotic Ser, Thr, and Tyr protein kinases, the high-energy phosphoHis precludes stoichiometric phosphorylation. The phosphorylated HPK, thought to exist as <1% of the total HPK population, serves as an intermediate for phosphotransfer.
RRs are typically multidomain proteins, consisting of a conserved N-terminal regulatory (or receiver) domain and a variable C-terminal effector domain that elicits the specific output response of the system, most commonly transcriptional regulation. The conserved regulatory domain catalyzes transfer of a phosphoryl group from the phosphoHis of the HPK to one of its own aspartic acid residues, stabilizing a conformation capable of promoting activity of the effector domain. An intrinsic autophosphatase activity regulates the lifetime of the phosphorylated RR, yielding half-lives ranging from seconds to hours.
The level of RR phosphorylation ultimately determines the output response of the system and all regulatory features in TCSs are consequently directed at influencing the level of RR phosphorylation. Stimuli regulate one or both opposing activities of the HPK: the autophosphorylation activity of the HPK which determines the pool of phosphoryl groups available for phosphotransfer and/or a specific RR phosphatase activity of the HPK which achieves more rapid adjustment of the level of RR phosphorylation [68]. Many systems contain auxiliary proteins that influence the activities of the HPK or the lifetime of the phosphorylated RR.
Additional complexity and a greater number of loci for regulation are achieved by expansion of the basic phosphotransfer pathway into a phosphorelay. These systems contain multiple His- and Asp-containing domains that enable multiple phosphotransfer steps. Such systems typically contain hybrid HPKs that incorporate a RR receiver domain as well as His-phosphotransfer (HPt) proteins, versions of His-containing dimerization domains that exist independently of HPKs.
Most sequenced bacterial genomes encode numerous TCSs, with the number of systems increasing with the genome size and the complexity of the lifestyle of the organism (see refs. [1, 2], http://www.ncbi.nlm.nih.gov/Complete_Genomes/SignalCensus.html and http://genomics.ornl.gov/mist/ for comprehensive lists). While a few bacteria contain no TCSs (e.g. Mycoplasma, Candidatus Blochmannia floridanus), many contain several dozen (e.g. Bacillus anthracis, Escherichia coli), and a few contain over 100 (e.g. Myxococcus xanthus, cyanobacteria [3]). Consistent with their great prevalence, TCSs are involved in most aspects of bacterial regulation, coupling diverse physical and chemical stimuli to an equally diverse array of outputs. Responses range from basic metabolic regulation such as carbon/nitrogen utilization, phosphate assimilation, and aerobic/anaerobic growth to very specialized and complex developmental responses such as formation of spores, biofilms, and fruiting bodies.
Understanding and manipulating TCS pathways holds enormous potential promise for beneficial environmental applications such as bioremediation and nitrogen fixation for agricultural purposes. However, most research has emphasized TCSs of pathogens [4]. Because of their complete absence from animals (though present in other eukaryotes such as yeasts [5] and plants [6]), two-component proteins have been targeted for the development of antibacterial drugs [7]. However, little progress has been made in developing drugs that inhibit two-component proteins, which for several different reasons have proven to be difficult targets. It is hoped that a more focused approach aimed at specific RR proteins may yet be successful.
The field of TCS research has expanded and evolved significantly since its inception approximately two decades ago. In the early years, studies were focused on establishing the enzymatic activities of the two conserved proteins and on characterizing their roles within a relatively small number of model pathways. A significant effort is still focused on characterizing individual systems but the studied systems represent only a small percentage of the thousands within the rapidly expanding database of TCSs identified by genome sequencing.
Outside of system-specific investigations, recent advances in TCS research have been made at the extremes of the spectrum, by global genome-wide analyses and by atomic resolution characterization of specific protein components. Though seemingly disparate, these two approaches together are beginning to provide an overview of TCS signaling that could be useful in guiding detailed studies of individual systems. This review will focus on these two broad areas from the perspective of the RR protein, the central control element of the signaling pathway, and a more tractable experimental subject than the transmembrane HPK. Global approaches have been aimed at characterizing the repertoire of two-component proteins within an organism with the ultimate goal of identifying the specific physiological responses they control. Structural and functional studies of RR proteins have provided detailed descriptions of the mechanisms of phosphorylation-mediated regulation in individual proteins. These studies have defined similarities and differences among RRs and have begun to address the extent to which similar sequence is predictive of similar mechanisms of function.
Global approaches to identifying RR input and output
In recent years, functional genomic approaches have become increasingly important to the understanding of RR functions at a system level. Complete genome sequences have presented a vast number of RR genes with undefined regulatory roles. Global analyses are helping delineate these unknown signaling pathways as well as to define the comprehensive regulon of well-studied systems. An image of regulatory networks with interacting signaling pathways has begun to emerge.
Phosphotransfer between specific HPKs and RRs is central to regulation of the output responses. However, the cognate HPK and RR are not always encoded in close proximity to each other, complicating identification. For example, 5 out of 32 RRs in E. coli [8] and 19 of 44 RRs in Caulobacter crescentus [9] do not have an adjacent cognate HPK gene. Clues to pairing components have come from a systematic biochemical analysis of phosphotransfer from each HPK to different RRs in E. coli which revealed that HPKs have a kinetic preference for phosphorylation of their cognate RRs [9]. Such phosphotransfer profiling in C. crescentus successfully led to the pairing of an orphan HPK and RR, CenK-CenR [9]. Besides the typical one-to-one HPK/RR pair, TCS pathways can be branched, with multiple HPKs or RRs involved in phosphotransfer. A comprehensive investigation of in vitro phosphorylation for all possible HPK-RR combinations in E. coli showed transphosphorylation between a small number (3%) of non-cognate pairs [10], implicating potential cross-talk between specific TCS pathways. One of such transphosphorylation pairs, ArcB-RssB, has been shown by genetic and biochemical studies to be physiologically important for RssB functions [11].
Phenotypes of RR deletion mutants contain important traits to identify the physiological function of individual TCSs. A high-throughput method, Phenotype MicroArrays, has been applied to all the HPK/RR deletion mutants of E. coli to screen for the growth-related phenotypes in the presence of various nutrients or inhibitors [12]. This technology could be valuable for drug discovery against TCS targets and it also allows a global perspective on the regulatory networks although the phenotypes screened are far from complete and additional methods are required for identification of the specific genes that are regulated.
Transcriptional regulation is the most common output of TCS pathways [13]. Thus transcriptome analysis using DNA microarrays becomes increasingly attractive to characterize the regulons of RRs. The differentially expressed genes identified by this method represent the genes potentially regulated by the RR, either directly or indirectly. The number of regulated genes varies greatly from system to system, ranging from a few genes for CreB in E. coli [14] to 15% of all genes for CovR in group A Streptococcus [15].
Traditionally, studies of RR-mediated regulation have been focused on a small number of representative genes. Transcription profiling broadens the view of gene regulation on a genome-wide scale for known pathways and provides important insights for unveiling the functions of uncharacterized systems. Moreover, the transcriptome approach has been used at a system level on all of the TCS mutants in an organism [14, 16]. Statistical analyses of the correlation between expression profiles of distinct systems help to predict and identify the co-regulation or interaction between different regulatory pathways [14]. However, RRs are known to regulate other TCSs or transcription factors [17, 18] and transcription analysis cannot distinguish direct regulation from these indirect or cascade regulatory events. Genome-wide location analysis has been performed to map the direct binding sites of the master regulator CtrA in C. crescentus [19]. In combination with transcription profiling, a detailed hierarchy of regulatory events can be derived for a better understanding of signaling networks.
Another challenging problem of regulon characterization is to identify those differentially regulated genes that cannot be readily recognized by traditional genetic or genomic tools. These genes might have promoters with low affinity for RRs, weak consensus sequences that are elusive to sequence searches, or transcription levels that change insufficiently to be distinguished by microarray analyses under experimental growth conditions. A computational method, termed Gene Promoter Scan, was recently developed to uncover these promoters in the E. coli and Salmonella PhoP regulatory network [20]. Gene expression patterns from genome-wide transcription profiling and sequence features of known PhoP-regulated promoters were used to predict new PhoP binding sites. Subsequent biochemical analyses verified the predictions, suggesting that this approach can lead to a more comprehensive characterization of RR-regulated genes.
RR diversity
The large number of complete bacterial genome sequences has allowed a broad assessment of the structural and functional diversity of RRs. Recent surveys of ~400 sequenced bacterial and archaeal genomes have provided databases of ~9000 RRs [2, 13]. Approximately 17% of RRs consist of an isolated receiver domain and presumably either regulate target effectors through intermolecular interactions, such as the regulation of the flagellar motor by the chemotaxis protein CheY, or function as phosphoryl shuttle proteins within phosphorelays, such as the sporulation protein Spo0F. The remainder can be classified into subfamilies based on sequence similarity of their effector domains (Figure 1, Table 1). DNA-binding domains of several different folds dominate the subfamilies, accounting for ~60% of RRs. The remaining subfamilies include a variety of enzymatic, protein-protein interaction, and RNA-binding domains.
Figure 1.

Classification of bacterial RRs. RRs are categorized by their effector domains and are further divided into subfamilies based on functions or structures. The distribution of respective types are indicated as percentage values in the set of ~9000 bacterial RRs analyzed by Galperin (http://www.ncbi.nlm.nih.gov/Complete_Genomes/SignalCensus.html) [13].
Table 1.
Functional and structural diversity in RR effector domains
| Function/Structure Classification |
Name/Representatives |
|---|---|
| DNA-binding | |
| wHTHa | OmpR/PhoB |
| HTHb | NarL/FixJ |
| AAA-FIS c,d | NtrC/DctD |
| LytTR | LytR |
| FIS | PrrA(RegA) |
| HTH_AraC | YesN |
| RNA-binding | AmiR/NasR |
| Protein-binding | CheV |
| Enzymatic | |
| Methylesterase | CheB |
| Diguanylate cyclase | GGDEF |
| c-di-GMP phosphodiesterase | EAL |
| c-di-GMP phosphodiesterase | HD-GYP |
| Protein phosphatase | PP2C |
| Histidine kinase | HisKA-HATPase |
| Stand-alone receivers | |
| Phosphotransfer component | Spo0F |
| Chemotaxis motility control | CheY |
winged Helix Turn Helix
Helix Turn Helix
AAA+ ATPase
Factor for Inversion Stimulation (belonging to the HTH family)
The great diversity of RRs raises a fundamental question. How does the common regulatory domain (20–30% sequence identity) control the activities of such structurally and functionally dissimilar effector domains? An elegantly simple answer to this question emerged about a decade ago when the inactive and active states of isolated receiver domains were first characterized. Regulatory domains were observed to exist primarily in two conformations, designated inactive and active, with the latter stabilized by phosphorylation. It was postulated that molecular surfaces that differed in the two states could be exploited for regulatory protein-protein interactions, enabling a variety of regulatory strategies.
Genetic and biochemical analyses of different RRs have provided solid evidence of both inhibitory and activating mechanisms of regulation. However, it is only recently that the nature of a significant number of these regulatory interactions has been described. As these descriptions accumulate, it becomes possible to ascertain the structural and functional conservation among RRs and to delineate regulatory features that are shared from those that differ.
Common mechanisms of receiver domain activation
The defining feature of RRs is the presence of a structurally conserved α/β domain referred to as a regulatory or receiver domain (Figure 2a). This consists of a five-stranded parallel β sheet surrounded by five amphipathic helices. A small number of highly conserved residues in the receiver domain of RRs play important roles in signal propagation and in catalysis of phosphotransfer and auto-dephosphorylation (see reviews [21, 22]). The active site contains a cluster of conserved acidic residues including the aspartic acid at the C-terminal end of β3 that is the site of phosphorylation. Two additional acidic residues in the β1–α1 loop position a divalent metal ion, commonly Mg+2 (Figure 2b), required for both phosphotransfer and phosphate hydrolysis [23, 24].
Figure 2.

Conserved features of RR receiver domains. (a) A ribbon diagram of E. coli CheY (1FQW) displays the classic (βα)5 fold of receiver domains with the site of phosphorylation (Asp57) shown in ball-and-stick mode. The α4-β5-α5 signaling face, the region that shows the largest structural perturbations upon phosphorylation, is colored green. (b) A stereo diagram of the active site of CheY (1FWQ) illustrates the roles of highly conserved residues in coordinating the phosphate and divalent metal ion. Due to the lability of the acyl phosphate, structural analyses of active RRs have often been carried out in the presence of the non-covalent phosphoryl analog beryllofluoride [65]. The BeF3− complex (beryllium, orange; fluorides, purple), which coordinates to Asp57, is stabilized by interactions with the side chains of Lys109 and Thr87, and a divalent metal ion (metallic blue). The metal ion (Mn2+ in this structure) is required for catalysis of both phosphotransfer and autodephosphorylation and is positioned by octahedral coordination to the side chains of Asp12 (water-mediated), Asp13, and Asp57, the backbone carbonyl of Asn59, a fluoride of BeF3−, and an additional water molecule (green). (c) RRs utilize a common mechanism to couple phosphorylation to surface changes. This mechanism involves the reorientation of two highly conserved residues, a hydroxyl-containing residue (Ser/Thr) and an aromatic residue (Phe/Tyr). The relative orientations of these side chains (Thr87 and Tyr106 in CheY) and that of the phosphorylated aspartate (Asp57) in their inactive (yellow) and active (blue) conformations are shown in ball-and-stick mode following superpositioning of inactive and active CheY structures (2CHE and 1FWQ). In the inactive conformation, the Ser/Thr and Phe/Tyr are oriented away from the active site with the aromatic side chain in a surface exposed position on the α4-β5-α5 face. In the active conformation, both side chains are oriented towards the active site with the Phe/Tyr side chain buried and the hydroxyl group of the Ser/Thr forming a hydrogen bond with a phosphate oxygen (in this structure, a fluoride of BeF3−). The view represents a rotation of ~90° about the x-axis relative to the view in (a) and a Cα trace of active CheY with colors as in (a) is shown for reference. In all panels, oxygen atoms are colored red; nitrogen atoms, blue; and carbon atoms, black, unless noted otherwise.
The other highly conserved residues in receiver domains are involved in propagation of long-range conformational changes that accompany RR phosphorylation. Phosphorylation does not result in substantial changes in secondary structure; rather, it usually involves subtle displacements of the backbone (typically ~1 Å) and perturbations of the molecular surface localized primarily to the α4-β5-α5 face. Structural characterizations of RRs in inactive and active states have suggested a molecular mechanism for signal propagation that involves a highly, although not exclusively, conserved repositioning of a Ser/Thr residue at the C terminus of β4 and a Phe/Tyr residue in β5 that are positioned between the active site and the α4-β5-α5 surface (see reviews [21, 25]) (Figure 2c). As shown (Figure 2c), activation leads to reorientation of these Phe/Tyr and Ser/Thr residues so that they are directed toward the active site. Although the different rotamer conformations of these two residues are the most readily distinguishable feature, packing interactions of many residues contribute to stabilizing each state. The importance of all interactions is underscored by the fact that many different single-residue substitutions have been identified that activate individual RRs, but corresponding substitutions rarely confer similar phenotypes to other RRs [26, 27]. Furthermore, the specific spatial extent and magnitudes of the conformational alterations between the two states vary among RRs.
Regulatory domain dynamics
The receiver domains of RRs are conformationally dynamic. The most dynamic regions of the unphosphorylated form of Spo0F and the receiver domain of NtrC are the functional surfaces, regions that are known to undergo conformational changes upon phosphorylation and that are used for interactions with other protein partners [28–30]. In phosphorylated NtrC, these millisecond molecular motions are quenched.
Further reflecting the dynamic nature of RRs, the active conformation of the receiver domain is not restricted to phosphorylated proteins but accessible to unphosphorylated forms as well. Direct evidence of an equilibrium between inactive and active conformations has come from NMR and X-ray diffraction analyses of several RRs [28, 30, 31]. These and other data led to a “two-state” activation model in which two sub-populations of inactive and active states coexist and phosphorylation or binding of targets shifts the distribution from primarily inactive to primarily active conformations.
The two-state equilibrium model is also supported by in vivo and in vitro biochemical data. For example, in vitro, the rates of phosphorylation of RRs are enhanced by binding to targets, as has been observed for the transcription factor OmpR in the presence of specific DNA recognition elements [32] and the chemotaxis protein CheY in the presence of peptides of the flagellar motor protein FliM or its phosphatase CheZ [33]. However, the physiological significance of target-induced enhancement of phosphorylation in vivo is unknown. The molecular basis of these effects can be explained by analogy with recent dynamics studies of other enzymes, in which it was observed that catalysis occurred only in a preexisting subpopulation of molecules with a conformation compatible with substrate binding [34]. Similarly, it might be expected that phosphorylation of receiver domains occurs only in the subpopulation that exists in an active conformation and that phosphorylation rates might correlate directly with the two-state equilibrium distribution.
Additional physiological significance of the two-state equilibrium is suggested by RRs, such as UhpA, PhoP and HrpY, for which overexpression of an unphosphorylatable mutant is sufficient to complement the RR-deletion phenotype and allow target gene transcription [35–37]. The observation that a physiologically significant subpopulation of active RR can be obtained by increased levels of unphosphorylated RRs poses an interesting problem for RRs, such as PhoB from E. coli, that autoregulate their own expression [38]. Induction of these systems results in higher levels of RR protein, in addition to RR phosphorylation. Whereas the latter can be readily reversed by phosphatase activities, elevated protein levels are typically longer lived. In such systems, additional mechanisms might be required to ensure inactivity of RRs at high concentrations.
The examples described above are consistent with a two-state model of activation. Structurally, these two states have been distinguished by the orientation of the conserved Ser/Thr and Phe/Tyr pair, either being “inward” and active or “outward” and inactive. However, there is emerging evidence that a two-state model might be an over-simplification of a more complex multi-state equilibrium [39]. Two recent structural studies of CheY bound to peptides of its targets CheZ and FliM suggest the existence of physiologically relevant intermediates that possess features that are associated with both the active and inactive conformations [40, 41]. Thus, although it appears that activation involves the redistribution of a population the number of physiologically relevant states within the population could vary from RR to RR.
Diverse regulatory strategies via different domain interactions
Despite common mechanisms shared by conserved receiver domains within the RR superfamily, interdomain communication between the receiver and effector domains appears highly diversified, partly due to a wide variety of effector domains with distinct structures and functions. It has long been noticed that removal of the regulatory domain results in completely inactive proteins in some RRs and constitutively active or partially active ones in others [42–44]. Thus different mechanisms have been proposed to account for the positive, negative or a combination of both regulatory roles of the receiver domain in individual RRs.
Initial insight into how receiver domains negatively regulate the activity of effector domains came from structural analyses of two full-length inactive RRs, the methylesterase CheB and the transcription factor NarL [45, 46]. In both proteins, the receiver domains make extensive contacts with the functional regions of the effector domains, sterically blocking access to the methylesterase active site of CheB and the DNA recognition helix of NarL. As for positive regulation seen in some RRs, such as FixJ [47] and NtrC [48], phosphorylation of the receiver domain promotes the dimerization or oligomerization of RRs to regulate transcription. A single RR can employ both negative and positive regulation as demonstrated for CheB [44] and FixJ [47].
Even with such diverse regulatory strategies, it was initially perceived that diversity perhaps arose from the different structures of effector domains and that regulatory mechanisms might be conserved within RR subfamilies. It was attractive to assume that sequence and structural similarity of the effector domains in the same RR subfamily might imply a similar regulatory strategy, facilitating the design of studies on new RRs as well as global analyses. However, as structural details of multiple full-length RRs of several subfamilies have become available, the lack of conserved interdomain interactions and corresponding mechanistic diversity within individual subfamilies have been revealed.
Distinct inhibitory and stimulatory regulation by receiver domains has been observed within the NtrC/DctD subfamily. The NtrC/DctD subfamily of RRs is characterized by the presence of a central AAA+ ATPase domain located between the receiver domain and the helix-turn-helix DNA-binding motif (Figure 3). The role of the AAA+ ATPase domain is to induce open complex formation in σ54-containing RNA polymerases in an ATP-dependent manner and the DNA-binding domain directs this activity to a particular promoter. The ATPase activity requires oligomerization of the AAA+ ATPase domain into a ring structure that is regulated by the receiver domain. High sequence similarity (40% sequence identity) and conserved structural folds of individual domains are shared by the three most extensively characterized members of the NtrC/DctD subfamily, NtrC1 from Aquifex aeolicus, DctD from Sinorhizobium meliloti, and NtrC from Salmonella enterica. However, structural and biochemical analyses have indicated that ring assembly of the ATPase domains is regulated by two different mechanisms involving inhibition or activation by the receiver domains (see Figure 3) [49–51]. Distinctive features in protein sequences, especially the helical structure in linker regions between the domains, have been correlated with these two mechanisms [52].
Figure 3.

Activation mechanisms of NtrC subfamily RRs. NtrC subfamily members share a similar domain composition of receiver (R), ATPase (C) and DNA-binding (D) domains and are all dependent on the ring assembly of the central ATPase domains (alternating blue and green) to interact with σ54 for transcription regulation. However, despite these similarities, two distinct assembly mechanisms have been discovered within the subfamily so far. In DctD and NtrC1 (top), the central ATPase domain is intrinsically competent for ring assembly but the inactive receiver domain negatively regulates assembly by holding the ATPase (C) domains in a front-to-front dimer, unfavorable for a front-to-back assembly [50]. Phosphorylation or removal of the receiver domain exposes the surface that is buried in the inactive dimer, relieving the inhibition. In NtrC (bottom), the isolated ATPase domain lacks significant ATPase activity. Phosphorylation causes conformational changes that enable the receiver domain from one subunit to be in contact with the ATPase domain of a second subunit, stabilizing the ring structure and promoting ATPase activity [49].
The wide range of mechanistic diversity is especially apparent within the OmpR/PhoB subfamily that is defined by the presence of a winged helix-turn-helix DNA-binding domain that typically binds in tandem to direct repeat half-sites. The OmpR/PhoB subfamily represents about one third of all RRs and displays a high degree of conservation in the α4-β5-α5 face of the receiver domain not seen in other subfamilies [53, 54]. A common active state is believed to be conserved within the subfamily and it features a head-to-head dimer of the receiver domains, utilizing the conserved α4-β5-α5 face for intermolecular interactions [53, 55], paired with a head-to-tail dimer of the winged helix-turn-helix motifs that bind to tandem DNA repeats (Figure 4). Notably, this model implies the presence of flexible linkers connecting the domains, because no unique intramolecular interface in the two monomers can accommodate the different symmetries of the domain dimers.
Figure 4.

Inactive and active domain arrangements in OmpR/PhoB subfamily members. OmpR/PhoB RRs have different domain orientations in the inactive state, yet all assume a common active state. Structures are available for three inactive full-length multidomain RRs: (a) T. maritima DrrB (1P2F), (b) M. tuberculosis PrrA (1YS6) and (c) T. maritima DrrD (1KGS). Domain arrangements in a fourth RR, (d) E. coli PhoB, can be modeled from structures of the isolated receiver domain dimer (1B00) and the isolated DNA-binding domain (1GXQ). The orientation of the DNA-binding domains (bracketed) relative to the receiver domains in PhoB is unknown, but the short linkers that connect the domains (depicted as dotted lines) restrict placement of the DNA-binding domains to diagonal positions across the receiver domain dimer. Although no structures of active multidomain OmpR/PhoB RRs have been determined, an active state can be readily envisioned (e). The common α4-β5-α5 dimer observed for all active OmpR/PhoB receiver domains paired with a tandem dimer of DNA-binding domains bound to direct repeat half-sites is compatible with only a single active state. The different symmetries of the N- and C-terminal domain dimers preclude a unique intramolecular interface between the domains. Thus flexible linkers (depicted as dotted lines) are proposed to tether the domain dimers, with domain orientations restricted only by the linker length. The depicted model is constructed from structures of the isolated active receiver domain dimer of PhoB (1ZES) and the complex of PhoB DNA-binding domains bound to target DNA (1GXP). Receiver domains are shown in teal with α4-β5-α5 faces highlighted in green; DNA-binding domains are shown in gold with recognition helices highlighted in red.
The regulatory diversity is manifested in a variety of interactions between the receiver and effector domains in their inactive states. Three full-length structures of inactive RRs in the OmpR/PhoB subfamily have been solved [56–58] and each displays significantly different interdomain interactions (Figure 4). DrrB and PrrA both form significant contacts between the DNA-binding domain and the α4-β5-α5 face of the receiver domain, though different surfaces of the DNA-binding domain are involved (Figure 4a and b). In DrrB the so-called β-platform contributes to the interface, whereas in PrrA, the positioning and recognition helices mediate this interaction. Therefore the regulation of PrrA seems to involve steric inhibition of the recognition helix from binding to DNA but this does not occur in DrrB which has an exposed recognition helix. An additional feature of the extensive domain interfaces is their stabilization of the inactive conformation of the receiver domains, which, as previously discussed, might pose a barrier to activation by reducing their propensity for phosphorylation. In contrast, another OmpR/PhoB subfamily member, DrrD, displays no significant interface and it is not clear how or if the receiver domain influences the DNA-binding domain in the inactive state (Figure 4c). Furthermore, two independent structural characterizations of the inactive PhoB receiver domain identified a similar dimer, which is different from the active dimer that associates through an α4-β5-α5 interface [55, 59] (Figure 4d). Yet another regulatory mechanism is potentially provided by the alternative inactive dimer that orients the effector domains in a position incompatible with DNA binding. It is evident that different interdomain interactions have been exploited by the OmpR/PhoB subfamily RRs for distinct regulatory strategies to maintain the inactive state. Phosphorylation alters these interactions and promotes a common active dimer.
Different interactions between receiver and effector domains have also been observed in the NarL/FixJ subfamily. For example, the individual domains of Pseudomonas fluorescens StyR [60] are structurally similar to those of NarL [46], but their spatial arrangements differ. Transcription regulation by the NarL/FixJ subfamily is not as well understood as the NtrC/DctD and OmpR/PhoB subfamilies. The sequence and orientation of DNA recognition sites, the oligomerization states and the intermolecular surface within the subfamily all display great flexibility, consistent with diverse regulatory mechanisms.
The majority of research in past years has focused on a rather small subset of RRs, especially those well-represented RR subfamilies with DNA-binding domains. A wide variety of regulatory schemes have already been discovered in these structurally similar proteins and there are probably more in the RR superfamily. Perhaps there are recognizable relationships between protein sequences and regulatory strategies, but few correlations have been discerned to date. Another fundamental question is how RRs have evolved to accommodate such diverse effectors and employ different regulatory strategies. A recent analysis of HPKs reveals clues to the evolution history of TCSs [61]. Both horizontal gene transfer (HGT) and gene duplication through lineage-specific expansion (LSE) contribute to the evolution of new HPKs in individual genomes. Strikingly, domain shuffling or incorporation of novel domains often accompanies gene duplication events whereas HGT is more likely to retain its original signaling domains. RRs might follow a similar evolutionary pathway to generate diversity.
Concluding remarks – The predictive limits of homology
With the increasing pace of bacterial genome sequencing, the discovery of TCSs far exceeds the rate at which they can be characterized on an individual basis. Thus it seems likely that genomic approaches will play an increasingly important role in identifying TCSs that are of particular interest and warrant detailed study. Such investigations will require both in vitro and in vivo analyses. The level of RR phosphorylation is tightly regulated in vivo and such control has been shown to be essential for virulence in M. tuberculosis [62] and S. enterica [63]. The complex interplay between HPK autokinase and phosphatase activities, levels of RR protein, levels of RR phosphorylation, promoter occupancy, and transcription of target genes is just beginning to be explored in vivo [63]. Defining the intrinsic and extrinsic mechanisms that regulate RR activation is fundamental to understanding the complex regulation that exists within TCS pathways.
As detailed studies are pursued, it will be valuable to know the extent of similarity that can be expected between different TCSs. A sufficiently large body of knowledge has been gathered for RRs that the issue of similarity can begin to be addressed. Activities and mechanisms within domains are largely conserved, as are the structures of individual domains. In contrast, the ways in which the domains interact, both their structural orientations and the regulatory consequences of these interactions are remarkably different among RRs, even among proteins within the same subfamily, with the only exception to date being the common active state adopted by OmpR/PhoB subfamily members. Differences in domain interactions provide extraordinary versatility, allowing for a variety of inhibition and activation strategies in the unphosphorylated and phosphorylated states. Such great diversity of regulatory strategies within the same protein superfamily, as seen in RRs as well as in eukaryotic signaling proteins such as Ras family GTPases [64], might reflect the divergent or convergent evolutionary histories of signaling pathways. These different mechanisms appear to allow RRs to be optimized for the specific signaling pathways in which they function. It is clear that much of what we learn about one RR will not be applicable to all, and that a reasonable understanding of any particular TCS is likely to require individual characterization.
Box 1, Figure I.
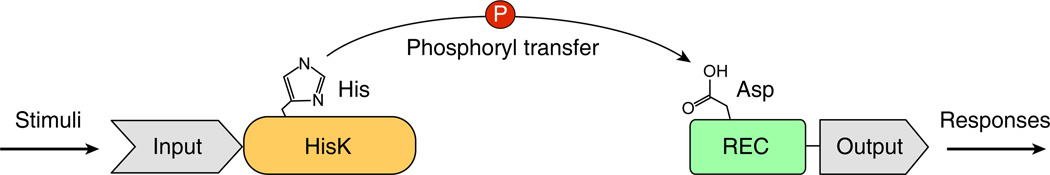
Schematic diagram of a typical two-component system. The conserved His-containing kinase domain of the HPK is shown in orange and the conserved Asp-containing receiver domain of the RR is in green. The variable domains that confer specificity of input (sensing domain of the HPK) and output (effector domain of the RR) to each TCS are shown in grey.
Box 2. Glossary.
Regulon: the set of genes regulated either directly or indirectly by a specific transcription factor.
Phosphotransfer profiling: a biochemical technique used to systematically compare phosphotransfer efficiency between different combinations of HPKs and RRs in vitro [9]. In this approach the cytoplasmic kinase domain of a HPK is autophosphorylated and then mixed with different full-length RRs to allow the phosphotransfer to proceed. Phosphotransfer profiling has demonstrated that there is a kinetic preference for phosphotransfer between cognate HPK/RR pairs that is ~1000-fold greater than between non-cognate pairs. Thus, this method is a valuable tool for mapping HPK/RR connectivity.
Phenotype MicroArrays: a high-throughput method in which a large number of phenotypes are analyzed simultaneously [12, 69]. Cells are grown under a wide variety of conditions while their physiological state is monitored in real time by measuring cell respiration. Comparing various mutant or deletion strains to the wild-type allows genotype-phenotype relationships to be evaluated.
Transcriptome analysis: an examination of the relative expression levels of all mRNAs in a given organism using DNA microarray technology. Gene transcription profiles between wild type and RR-deletion strains or profiles from the same strain grown under different conditions are compared. Identification of differentially expressed genes contributes to the comprehensive characterization of genes regulated by specific RRs.
Genome-wide location analysis: a modified chromatin-immunoprecipitation procedure intended to map the genomic binding sites of transcriptional regulatory proteins [19, 70]. Proteins are crosslinked to DNA targets in vivo, followed by fragmentation of the genomic DNA and finally immunoprecipitation of the proteins. The co-precipitated DNA fragments are amplified, labeled and analyzed by DNA microarrays.
Gene Promoter Scan: a computational technique used to identify uncharacterized promoter sites of specific transcription factors. This technique was initially applied to the RR PhoP in both E. coli and Salmonella enterica [20]. Profiles of PhoP-regulated promoters were built and classified based on common subsets of features, such as gene expression levels, PhoP box submotifs, the orientation and position of the PhoP box, etc. These promoter profiles were searched against the intergenic sequences of the genome to uncover putative PhoP promoter sites. It differs from other prediction methods as it uses both gene expression patterns and sequence features of specific known promoters. Furthermore, different modes of regulation are taken into account by categorizing these features into subgroups.
Acknowledgement
We thank Dr. Michael Galperin for providing the updated RR database prior to public release.
References
- 1.Galperin MY. A census of membrane-bound and intracellular signal transduction proteins in bacteria: bacterial IQ, extroverts and introverts. BMC Microbiol. 2005;5:35. doi: 10.1186/1471-2180-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ulrich LE, Zhulin IB. MiST: a microbial signal transduction database. Nucl. Acids Res. 2007;35:D386–D390. doi: 10.1093/nar/gkl932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashby MK, Houmard J. Cyanobacterial two-component proteins: structure, diversity, distribution, and evolution. Microbiol. Mol. Biol. Rev. 2006;70:472–509. doi: 10.1128/MMBR.00046-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beier D, Gross R. Regulation of bacterial virulence by two-component systems. Curr. Opin. Microbiol. 2006;9:143–152. doi: 10.1016/j.mib.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Kruppa M, Calderone R. Two-component signal transduction in human fungal pathogens. FEMS Yeast Res. 2006;6:149–159. doi: 10.1111/j.1567-1364.2006.00024.x. [DOI] [PubMed] [Google Scholar]
- 6.Mizuno T. Two-component phosphorelay signal transduction systems in plants: from hormone responses to circadian rhythms. Biosci. Biotechnol. Biochem. 2005;69:2263–2276. doi: 10.1271/bbb.69.2263. [DOI] [PubMed] [Google Scholar]
- 7.Stephenson K, Hoch JA. Developing inhibitors to selectively target two-component and phosphorelay signal transduction systems of pathogenic microorganisms. Curr. Med. Chem. 2004;11:765–773. doi: 10.2174/0929867043455765. [DOI] [PubMed] [Google Scholar]
- 8.Mizuno T. Compilation of all genes encoding two-component phosphotransfer signal transducers in the genome of Escherichia coli. DNA Res. 1997;4:161–168. doi: 10.1093/dnares/4.2.161. [DOI] [PubMed] [Google Scholar]
- 9.Skerker JM, et al. Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: A system-level analysis. PLoS Biology. 2005;3:e334. doi: 10.1371/journal.pbio.0030334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto K, et al. Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J. Biol. Chem. 2005;280:1448–1456. doi: 10.1074/jbc.M410104200. [DOI] [PubMed] [Google Scholar]
- 11.Mika F, Hengge R. A two-component phosphotransfer network involving ArcB, ArcA, and RssB coordinates synthesis and proteolysis of {sigma}S (RpoS) in E. coli. Genes Dev. 2005;19:2770–2781. doi: 10.1101/gad.353705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou L, et al. Phenotype MicroArray analysis of Escherichia coli K-12 mutants with deletions of all two-component systems. J. Bacteriol. 2003;185:4956–4972. doi: 10.1128/JB.185.16.4956-4972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galperin MY. Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J. Bacteriol. 2006;188:4169–4182. doi: 10.1128/JB.01887-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oshima T, et al. Transcriptome analysis of all two-component regulatory system mutants of Escherichia coli K-12. Mol. Microbiol. 2002;46:281–291. doi: 10.1046/j.1365-2958.2002.03170.x. [DOI] [PubMed] [Google Scholar]
- 15.Graham MR, et al. Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc. Natl. Acad. Sci. USA. 2002;99:13855–13860. doi: 10.1073/pnas.202353699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi K, et al. Comprehensive DNA microarray analysis of Bacillus subtilis two-component regulatory systems. J. Bacteriol. 2001;183:7365–7370. doi: 10.1128/JB.183.24.7365-7370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McAdams HH, Shapiro L. A bacterial cell-cycle regulatory network operating in time and space. Science. 2003;301:1874–1877. doi: 10.1126/science.1087694. [DOI] [PubMed] [Google Scholar]
- 18.Kato A, Groisman EA. Connecting two-component regulatory systems by a protein that protects a response regulator from dephosphorylation by its cognate sensor. Genes Dev. 2004;18:2302–2313. doi: 10.1101/gad.1230804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laub MT, et al. Genes directly controlled by CtrA, a master regulator of the Caulobacter cell cycle. Proc. Natl. Acad. Sci. USA. 2002;99:4632–4637. doi: 10.1073/pnas.062065699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zwir I, et al. Dissecting the PhoP regulatory network of Escherichia coli and Salmonella enterica. Proc. Natl. Acad. Sci. USA. 2005;102:2862–2867. doi: 10.1073/pnas.0408238102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stock AM, et al. Two-component signal transduction. Annu. Rev. Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 22.West AH, Stock AM. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem. Sci. 2001;26:369–376. doi: 10.1016/s0968-0004(01)01852-7. [DOI] [PubMed] [Google Scholar]
- 23.Lukat GS, et al. Divalent metal ion binding to the CheY protein and its significance to phosphotransfer in bacterial chemotaxis. Biochemistry. 1990;29:5436–5442. doi: 10.1021/bi00475a004. [DOI] [PubMed] [Google Scholar]
- 24.Stock AM, et al. Structure of the Mg2+-bound form of CheY and mechanism of phosphoryl transfer in bacterial chemotaxis. Biochemistry. 1993;32:13375–13380. doi: 10.1021/bi00212a001. [DOI] [PubMed] [Google Scholar]
- 25.Robinson VL, et al. A tale of two components: a novel kinase and a regulatory switch. Nat. Struct. Biol. 2000;7:628–633. doi: 10.1038/77915. [DOI] [PubMed] [Google Scholar]
- 26.Smith JG, et al. A search for amino acid substitutions that universally activate response regulators. Molecular Microbiology. 2004;51:887–901. doi: 10.1046/j.1365-2958.2003.03882.x. [DOI] [PubMed] [Google Scholar]
- 27.Porter SL, et al. The CheYs of Rhodobacter sphaeroides. J. Biol. Chem. 2006;281:32694–32704. doi: 10.1074/jbc.M606016200. [DOI] [PubMed] [Google Scholar]
- 28.Kern D, et al. Structure of a transiently phosphorylated switch in bacterial signal transduction. Nature. 1999;40:894–898. doi: 10.1038/47273. [DOI] [PubMed] [Google Scholar]
- 29.Feher VA, Cavanagh J. Millisecond-timescale motions contribute to the function of the bacterial response regulator protein Spo0F. Nature. 1999;400:289–293. doi: 10.1038/22357. [DOI] [PubMed] [Google Scholar]
- 30.Volkman BF, et al. Two-state allosteric behavior in a single domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 31.Simonovic M, Volz K. A distinct meta-active conformation in the 1.1-Å resolution structure of wild-type apoCheY. J. Biol. Chem. 2001;276:28637–28640. doi: 10.1074/jbc.C100295200. [DOI] [PubMed] [Google Scholar]
- 32.Ames SK, et al. C-terminal DNA binding stimulates N-terminal phosphorylation of the outer membrane protein regulator OmpR from Escherichia coli. Proc. Natl. Acad. Sci. USA. 1999;96:11792–11797. doi: 10.1073/pnas.96.21.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuster M, et al. Conformational coupling in the chemotaxis response regulator CheY. Proc. Natl. Acad. Sci. USA. 2001;98:6003–6008. doi: 10.1073/pnas.101571298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eisenmesser EZ, et al. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 35.Lejona S, et al. PhoP can activate its target genes in a PhoQ-independent manner. J. Bacteriol. 2004;186:2476–2480. doi: 10.1128/JB.186.8.2476-2480.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webber CA, Kadner RJ. Involvement of the amino-terminal phosphorylation module of UhpA in activation of uhpT transcription in Escherichia coli. Mol. Microbiol. 1997;24:1039–1048. doi: 10.1046/j.1365-2958.1997.4021765.x. [DOI] [PubMed] [Google Scholar]
- 37.Merighi M, et al. The HrpX/HrpY two-component system activates hrpS expression, the first step in the regulatory cascade controlling the Hrp regulon in Pantoea stewartii subsp. stewartii. Mol. Plant-Microbe. Interact. 2003;16:238–248. doi: 10.1094/MPMI.2003.16.3.238. [DOI] [PubMed] [Google Scholar]
- 38.Hoffer SM, et al. Autoamplification of a two-component regulatory system results in "learning" behavior. J. Bacteriol. 2001;183:4914–4917. doi: 10.1128/JB.183.16.4914-4917.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Formaneck MS, et al. Reconciling the "old" and "new" views of protein allostery: a molecular simulation study of chemotaxis Y protein (CheY) Proteins. 2006;63:846–867. doi: 10.1002/prot.20893. [DOI] [PubMed] [Google Scholar]
- 40.Dyer CM, Dahlquist FW. Switched or not? The structure of unphosphorylated CheY bound to the N terminus of FliM. J. Bacteriol. 2006;188:7354–7363. doi: 10.1128/JB.00637-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guhaniyogi J, et al. Crystal structures of beryllium fluoride-free and beryllium fluoride-bound CheY in complex with the conserved C-terminal peptide of CheZ reveal dual binding modes specific to CheY conformation. J. Mol. Biol. 2006;359:624–645. doi: 10.1016/j.jmb.2006.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drummond MH, et al. The function of isolated domains and chimaeric proteins constucted from the transcriptional activators NifA and NtrC of Klebsiella pneumoniae. Mol. Microbiol. 1990;4:29–37. doi: 10.1111/j.1365-2958.1990.tb02012.x. [DOI] [PubMed] [Google Scholar]
- 43.Huala E, et al. The central domain of Rhizobium leguminosarum DctD functions independently to activate transcription. J. Bacteriol. 1992;174:1428–1431. doi: 10.1128/jb.174.4.1428-1431.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anand GS, et al. Activation of methylesterase CheB: evidence of a dual role for the regulatory domain. Biochemistry. 1998;37:14038–14047. doi: 10.1021/bi980865d. [DOI] [PubMed] [Google Scholar]
- 45.Djordjevic S, et al. Structural basis for methylesterase CheB regulation by a phosphorylation-activated domain. Proc. Natl. Acad. Sci. USA. 1998;95:1381–1386. doi: 10.1073/pnas.95.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baikalov I, et al. Structure of the Escherichia coli response regulator NarL. Biochemistry. 1996;35:11053–11061. doi: 10.1021/bi960919o. [DOI] [PubMed] [Google Scholar]
- 47.Da Re S, et al. Phosphorylation-induced dimerization of the FixJ receiver domain. Mol. Microbiol. 1999;34:504–511. doi: 10.1046/j.1365-2958.1999.01614.x. [DOI] [PubMed] [Google Scholar]
- 48.Wyman C, et al. Unusual oligomerization required for activity of NtrC, a bacterial enhancer-binding protein. Science. 1997;275:1658–1661. doi: 10.1126/science.275.5306.1658. [DOI] [PubMed] [Google Scholar]
- 49.De Carlo S, et al. The structural basis for regulated assembly and function of the transcriptional activator NtrC. Genes Dev. 2006;20:1485–1495. doi: 10.1101/gad.1418306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee SY, et al. Regulation of the transcriptional activator NtrC1: structural studies of the regulatory and AAA+ ATPase domains. Genes Dev. 2003;17:2552–2563. doi: 10.1101/gad.1125603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park S, et al. Two-component signaling in the AAA+ ATPase DctD: binding Mg2+ and BeF3− selects between alternate dimeric states of the receiver domain. FASEB J. 2002;16:1964–1966. doi: 10.1096/fj.02-0395fje. [DOI] [PubMed] [Google Scholar]
- 52.Doucleff M, et al. Negative regulation of AAA+ ATPase assembly by two component receiver domains: a transcription activation mechanism that is conserved in mesophilic and extremely hyperthermophilic bacteria. J. Mol. Biol. 2005;353:242–255. doi: 10.1016/j.jmb.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 53.Toro-Roman A, et al. Structural analysis and solution studies of the activated regulatory domain of the response regulator ArcA: a symmetric dimer mediated by the α4-β5-α5 face. J. Mol. Biol. 2005;349:11–26. doi: 10.1016/j.jmb.2005.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toro-Roman A, et al. A common dimerization interface in bacterial response regulators KdpE and TorR. Protein Sci. 2005;14:3077–3388. doi: 10.1110/ps.051722805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bachhawat P, et al. Mechanism of activation for transcription factor PhoB suggested by different modes of dimerization in the inactive and active states. Structure. 2005;13:1353–1363. doi: 10.1016/j.str.2005.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson VL, et al. Structural analysis of the domain interface in DrrB, a response regulator of the OmpR/PhoB subfamily. J. Bacteriol. 2003;185:4186–4194. doi: 10.1128/JB.185.14.4186-4194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buckler DR, et al. Evidence of intradomain and interdomain flexibility in an OmpR/PhoB homolog from Thermotoga maritima. Structure. 2002;10:153–164. doi: 10.1016/s0969-2126(01)00706-7. [DOI] [PubMed] [Google Scholar]
- 58.Nowak E, et al. The structural basis of signal transduction for the response regulator PrrA from Mycobacterium tuberculosis. J. Biol. Chem. 2006;281:9659–9666. doi: 10.1074/jbc.M512004200. [DOI] [PubMed] [Google Scholar]
- 59.Solà M, et al. Three-dimensional crystal structure of the transcription factor PhoB receiver domain. J. Mol. Biol. 1999;285:675–687. doi: 10.1006/jmbi.1998.2326. [DOI] [PubMed] [Google Scholar]
- 60.Milani M, et al. An active-like structure in the unphosphorylated StyR response regulator suggests a phosphorylation-dependent allosteric activation mechanism. Structure. 2005;13:1289–1297. doi: 10.1016/j.str.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 61.Alm E, et al. The evolution of two-component systems in bacteria reveals different strategies for niche adaptation. PLoS Comput. Biol. 2006;2:e143. doi: 10.1371/journal.pcbi.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fol M, et al. Modulation of Mycobacterium tuberculosis proliferation by MtrA, an essential two-component response regulator. Mol. Microbiol. 2006;60:643–657. doi: 10.1111/j.1365-2958.2006.05137.x. [DOI] [PubMed] [Google Scholar]
- 63.Shin D, et al. A positive feedback loop promotes transcription surge that jump-starts Salmonella virulence circuit. Science. 2006;314:1607–1609. doi: 10.1126/science.1134930. [DOI] [PubMed] [Google Scholar]
- 64.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 65.Wemmer DE, Kern D. Beryllofluoride binding mimics phosphorylation of aspartate in response regulators. J. Bacteriol. 2005;187:8229–8230. doi: 10.1128/JB.187.24.8229-8230.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoch JA, Silhavy TJ, editors. Two-component signal transduction. American Society for Microbiology Press; 1995. [Google Scholar]
- 67.Inouye M, Dutta R, editors. Histidine kinases in signal transduction. Academic Press; 2003. [Google Scholar]
- 68.Mascher T, et al. Stimulus perception in bacterial signal-transducing histidine kinases. Microbiol. Mol. Biol. Rev. 2006;70:910–938. doi: 10.1128/MMBR.00020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bochner BR. New technologies to assess genotype-phenotype relationships. Nat. Rev. Genet. 2003;4:309–314. doi: 10.1038/nrg1046. [DOI] [PubMed] [Google Scholar]
- 70.Ren B, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]