

Abstract
Startle disease or hereditary hyperekplexia has been shown to result from mutations in the α1-subunit gene of the inhibitory glycine receptor (GlyR). In hyperekplexia patients, neuromotor symptoms generally become apparent at birth, improve with age, and often disappear in adulthood. Loss-of-function mutations of GlyR α or β-subunits in mice show rather severe neuromotor phenotypes. Here, we generated mutant mice with a transient neuromotor deficiency by introducing a GlyR β transgene into the spastic mouse (spa/spa), a recessive mutant carrying a transposon insertion within the GlyR β-subunit gene. In spa/spa TG456 mice, one of three strains generated with this construct, which expressed very low levels of GlyR β transgene-dependent mRNA and protein, the spastic phenotype was found to depend upon the transgene copy number. Notably, mice carrying two copies of the transgene showed an age-dependent sensitivity to tremor induction, which peaked at ∼ 3–4 weeks postnatally. This closely resembles the development of symptoms in human hyperekplexia patients, where motor coordination significantly improves after adolescence. The spa/spa TG456 line thus may serve as an animal model of human startle disease.
Keywords: glycine receptor, hereditary hyperekplexia, spa/spa TG456 mice, startle syndrome
Introduction
Distinct hereditary neuromotor disorders have been shown to be due to disinhibition of motoneurons resulting from mutations in the receptor for the inhibitory amino acid neurotransmitter glycine (GlyR). Amino acid substitutions in the ligand binding α1-subunit of the GlyR have been identified in both genetically recessive and dominant forms of hereditary hyperekplexia or startle syndrome (Shiang et al. 1993;Rees et al. 1994). This disease is characterized by startle-induced, generalized muscle contractions, which are often accompanied with hypertonia. Severe forms can be detected already at birth and are known as stiff baby syndrome (Lingam et al. 1981). The disease phenotype correlates well with the functional role and the anatomical localization of the GlyR, the major inhibitory ligand-gated chloride channel in the spinal cord and brain stem. In the adult spinal cord, the GlyR is composed of three copies of the ligand-binding α1-subunit and two copies of the structural β-subunit (Langosch et al. 1988;Kuhse et al. 1993); the latter targets the GlyR to the postsynaptic membrane by interaction with the cytoskeletal-anchoring protein gephyrin (Kirsch et al. 1995;Meyer et al. 1995). During development and in other brain regions, additional differentially expressed α-subunit isoforms (α2–α4) create GlyR diversity (Grenningloh et al. 1990a;Kuhse et al. 1990;Malosio et al. 1991;Betz 1992;Matzenbach et al. 1994).
In the mouse, several mutations of GlyR-subunit genes with complete or partial loss of GlyR function have been described. A null mutation in the α1-subunit in the mouse oscillator (spdot) produces a strong neuromotor phenotype that causes death at ∼ 3 weeks postnatally (Buckwalter et al. 1994). A partial loss of function is observed in the recessive mutant spasmodic (spd), which carries a single base pair substitution within the GlyR α1 gene that decreases the agonist affinity of the GlyR (Ryan et al. 1994;Saul et al. 1994). In contrast, in the recessive mouse mutant spastic (spa), GlyR levels are reduced to ∼ 10–20% of the wild-type level, but the receptor is pharmacologically normal (White & Heller 1982;Becker et al. 1986). This phenotype is due to an insertion of a repetitive LINE 1 element in the GlyR β gene, leading to deficient mRNA splicing with functional β mRNA levels being decreased to ∼ 10% (Kingsmore et al. 1994;Mülhardt et al. 1994). Because the β-subunit is required for the synaptic localization of the receptor (Kirsch et al. 1995;Meyer et al. 1995), drastically lowered receptor levels are found in postsynaptic membrane specializations of spastic mice (White & Heller 1982;Becker et al. 1986;Mülhardt et al. 1994). Homozygous spa/spa and spd/spd mice display complex neuromotor phenotypes characterized by an exaggerated startle response, an impaired righting reflex, the development of characteristic tremors and reduced male fertility. These disease symptoms become manifest at ∼ 2 weeks of age, i.e. the time when neonatal α2 GlyRs are lost from the spinal cord and brain stem of normal mice (Becker et al. 1988,1992).
In an earlier study, we have reported that the spa/spa phenotype can be rescued by transgenic expression of an exogenous rat GlyR β minigene (Hartenstein et al. 1996). This experiment proved the causal relationship between the GlyR β gene mutation and the spastic phenotype, and showed that mRNA expression levels corresponding to ∼ 25% of that seen in wt mice suffice to completely rescue the disease symptoms. Here we describe a novel mouse strain expressing even lower levels of the rat β transgene that displays a transient phenotype, which closely resembles the symptoms characterizing the most common forms of hereditary hyperekplexia in humans (Suhren et al. 1966;Andermann et al. 1980;Kurczynski 1983). These mice might provide an animal model that may help to develop novel strategies for therapeutic intervention in hyperekplexia and spasticity.
Materials and methods
Generation of the transgenic line TG456
The fragment used for microinjection containing the rat GlyR-subunit cDNA under the control of the rat NSE-promotor ( Forss-Petter et al. 1990) and the poly(A) site of the SV40-small T antigen including its splice-site (Gorman et al. 1982) has been described earlier (Hartenstein et al. 1996). Microinjection into pronuclei of zygotes generated from F1 hybrid female mice (C57bl/6 × DBA/2) mated to DBA/2 males was performed according to published procedures (Hogan et al. 1986). The transgenic founder TG456 was identified by Southern blot analysis and used to breed mice of the TG456/+ genotype.
Generation of TG456 animals on spa/spa background
Homozygous spa/spa mice were bred by intercrossing heterozygous spa/+ breeders (B6C3Fe-a/a-spa/+) purchased from Jackson Laboratory (Bar Harbor, ME, USA). For the rescue experiments, homozygous spa/spa females were crossed with TG456/+ males. Transgenic littermates of the F1 generation were then intercrossed to obtain transgenic spa/spa mice. These were intercrossed further to obtain animals of all relevant genotypes: spa/spaTG456/+, spa/spaTG456/TG456, and spa/spa non-transgenic littermates.
Genotyping and mRNA analysis
The isolations of DNA from tail biopsies and mRNA from tissue as well as PCR, Southern, Northern and dot blot techniques were performed as described in Hartenstein et al. (1996). For genotyping, the 32P-labelled Dra II fragment of the rat GlyR β-subunit cDNA (Grenningloh et al. 1990b) was used as a hybridization probe. Northern blotting was performed with a 264-bp SspI–NcoI fragment representing exons 4 and 5 of the GlyR β cDNA. The spa allele was followed by allele-specific PCR using the primers described inMülhardt et al. (1994).
Western blot and strychnine binding analysis
The preparation of membranes from the spinal cord and brain stem, and the solubilization, extraction and precipitation of membrane proteins were performed as described in Becker et al. (1986). For sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE), membrane pellets were suspended in Laemmli sample buffer and heated to 95 °C for 5 min.; 10–15 μg of total protein per lane was then electrophoresed on a 10% SDS–polyacrylamide gel, blotted onto Immobilon-P membrane (Millipore, Eschborn, Germany), and reacted with the monoclonal antibodies GlyR mAb4a (Becker et al. 1993) and anti-synaptophysin (anti-SVP, S-5768, Sigma, München, Germany). Bound immunoglobulins were visualized with horseradish peroxidase-conjugated anti-mouse IgG using the ECL system (Amersham, Braunschweig, Germany).
Binding of 3[H]-strychnine (NEN Du Pont, Köln, Germany) to membrane fractions from the brain was performed essentially as described (Pfeiffer & Betz 1981;Becker et al. 1986).
Phenotype characterization by handling
Animals were examined over a time period of 2–4 weeks two to three times a week by several handling tests. Inducible tremor and hind feet clasping behaviour was monitored after lifting the animals by their tails. The righting response was quantitated as follows. Animals were brought into a supine position by twitching the tail; the time that elapsed after release of the tail until resuming an upright position was measured by a second investigator; and each test was repeated in three consecutive trials.
Electromechanical tremor recording
For tremor recording, the mice were fixed by their tail to a F30 force transducer (Type 372) connected to a bridge amplifier (Type 336, both from Hugo Sachs Elektronik, 79229 March-Hugstetten, Germany) using a 5-cm thread of sewing silk. Electric signals were recorded by a conventional recorder ( Hartenstein 1996). All procedures were approved by the Regierungspräsidium Karlsruhe.
Results
Generation of the mouse strain spa/spa-TG456
Transgenic mice expressing the rat GlyR β gene thoughout the central nervous system were generated using an expression construct described earlier ( Hartenstein et al. 1996), in which the rat β cDNA was joined to the neuron-specific enolase (NSE) promoter from rat and flanked by the SV40 small T antigen polyadenylation signal. The NSE promoter has been shown to provide brain-specific expression of transgenes, however, large variations in expression efficiency have been reported (Forss-Petter et al. 1990;Hartenstein et al. 1996). Founder animals carrying this transgene were crossed into the spa/spa background. One of these founder strains, TG456, displayed a particularly interesting phenotype; in contrast to other transgenic strains carrying the same construct (Hartenstein et al. 1996), a haploid copy of the TG456 transgene did not rescue the phenotype. However, when spa/+-TG456/+ animals were intercrossed, several animals homozygous for the spa allele showed intermediate phenotypes displaying noticeably alleviated symptoms. This suggested that their phenotype might depend on gene dosage and prompted us to study the correlation between transgene copy number and phenotype in such mice in more detail.
Transgene expression in spa/spa-TG 456 mice
Animals resulting from double heterozygous crosses (see above) were first analysed for their transgene status by dot blot hybridization on genomic DNA with a rat GlyR β cDNA fragment (data not shown). Adult animals displaying a transgene hybridization signal twofold higher than those obtained with TG456/+ mice consistently showed a less severe spastic phenotype than non-transgenic spa/spa littermates (see below for detailed analysis). Thus, a gene dosage effect on the spa phenotype expression was clearly present. To monitor the corresponding transgene expression levels, we performed Northern analyses on brain mRNA of spa/spa-TG 456/+ and spa/spa-TG456/TG456 transgenic mice, respectively. Because the GlyR β RNA transcribed from the endogenous spa allele is < 10% functional and > 90% aberrantly spliced (Mülhardt et al. 1994), we used a probe covering exons 4 and 5 which does not detect the aberrantly spliced endogenous GlyR β RNA (Hartenstein et al. 1996). The transgene-specific band should comigrate with the endogenous 3.5-kb mature functional mRNA. Five independent Northern analyses on the four genotypes wt, spa/spa, spa/spa-TG456/+, and spa/spa-TG456/TG456 were performed, one of which is depicted inFig. 1(A). When the hybridization signal intensities were analysed, of all of them only very subtle differences between spa/spa and the transgenic lines were detected. We therefore concluded that the transgene-specific GlyR β mRNA expression was very low.
FIG. 1.
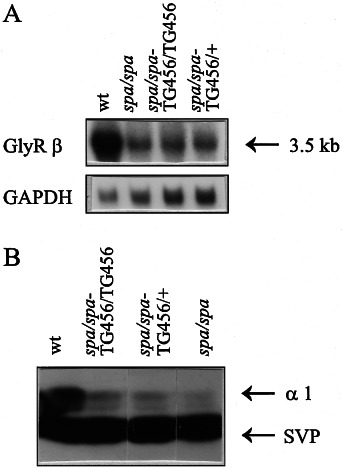
Expression of the transgene in spa/spa mice hetero- and homozygous for the TG 456 insertion. (A) Northern analysis. Total brain RNA was probed with a 264-bp SspI–NcoI fragment encompassing exons 4 and 5 of the GlyR β cDNA. Control hybridizations of the same blot were performed using the GAPDH gene probe. wt, wild-type control mice. (B) Western analysis. Membrane proteins from spinal cord and brain stem were separated on SDS–PAGE gels, transferred to nitrocellulose, and specific bands were detected with the antibodies mAb 4a and anti-SVP against synaptophysin as a control. Immunoglobulins were visualized using horseradish peroxidase-coupled second antibodies and the ECL system (Amersham).
In order to investigate the GlyR protein levels in the transgenics, we performed Western blot analyses and ligand-binding assays. While suitable anti β-subunit antibodies did not exist, in Western blots the antibody mAb 4a was used, which recognizes mainly the 48-kDa α1-subunit within the adult receptor (Pfeiffer et al. 1984;Kirsch et al. 1995). Under conditions of limiting β-subunit expression, as in adult spa/spa mice, membrane-bound mAb 4a immune reactivity is a valid measure of β-subunit surface expression and GlyR complex formation, because stable expression of the adult α1-subunit on the neuronal surface requires β-subunit expression in animals (Becker et al. 1986; 1992;Hartenstein et al. 1996).Figure 1(B) shows that consistent with the mRNA analysis, spa/spa-TG456/+ as well as spa/spa-TG456/TG456 animals express membrane-bound 48 α1-subunit at about the same levels as non-transgenic spa/spa mice. To enhance the sensitivity of the detection and at the same time assess the GlyR function we performed binding assays with the competitive GlyR antagonist strychnine.Figure 2(A) shows the isotherm for binding of 3[H]-strychnine to membrane preparations of wt, spa/spa homozygotes and spa/spa-TG 456/TG456 mice. Compared with wt mice, the spa/spa and spa/spa-TG456/TG456 animals both showed very low levels of binding. However, in triplicate measurements the latter genotype appeared to show consistently higher binding than spa/spa animals. Scatchard analysis of these data (Fig. 2B) indicated that in all three genotypes tested the affinity of the GlyR is very similar. The KD values for wt, spa/spa-TG456/456, and non-transgenic spa/spa animals were 10.4, 9.5 and 10.5 nm, respectively. The corresponding Bmax values were 1271 (wt), 173 (spa/spa-TG456/456) and 166 (spa/spa) fmoles/mg protein. In conclusion, the expression of the transgene in TG456 mice is very low.
FIG. 2.
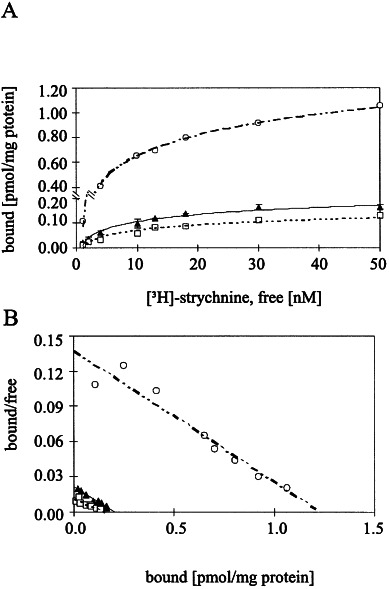
Binding of 3[H] strychnine to spinal cord membranes. Genotypes: (○), wt; (□), spa/spa; and (▴), spa/spa-TG456/TG456. Data represent the means from two (wt, spa/spa) or three (spa/spa-TG456/TG456) animals, respectively. (A) Saturation binding curves and (B) Scatchard analyses of specific 3[H]-strychnine binding assays were performed in triplicate as described inBecker et al. (1993).
Correlation of transgene dosage with motor phenotype
To unravel whether the phenotypic variations observed in the transgenic animals could be correlated to transgene dosage, we quantified the phenotypic properties of spa/spa animals hetero- and homozygous for the TG456 transgene more systematically. To this end, we performed a few simple handling assays. Firstly, we screened animals of different genotypes daily for the development of tremors. As depicted inFig. 3, in non-transgenic spa/spa mice the onset of tremor inducibility started at ∼ 14–17 days old and lasted throughout adulthood. This time of onset of handling-induced tremor proneness coincides with the replacement of α2-subunit-containing neonatal by α1-subunit-containing adult GlyRs (Becker et al. 1988; 1992). In spa/spa-TG456/+ mice carrying one copy of the transgene 456, tremor inducibility was not significantly altered. However, animals homozygous for the transgene became tremor prone only after 20–35 days. Notably, tremor inducibility was only transient; strong tremor was seen only over a few (2–10) days. Thereafter, the spa/spa-TG456/TG456 animals displayed tremor development upon handling only rarely.
FIG. 3.
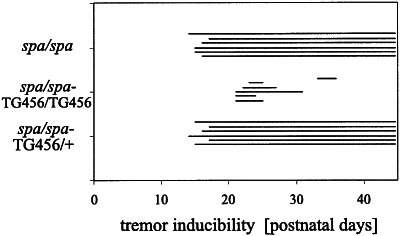
Tremor inducibility in spa/spa, spa/spa-TG456 and spa/spa-TG456/TG456 animals. Animals of each genotype were tested at several time points for tremor inducibility by handling. Periods of tremor inducibility are indicated by solid lines. Note delayed and transient appearance of tremor symptoms in spa/spa-TG456/TG456 mice. Of the non-transgenic spa/spa animals, four were non-transgenic littermates from doubly heterozygous crosses, and three animals were derived from the original stock obtained from Jackson Laboratories.
Tremor induction can be easily sensed when spastic animals are picked up by the tail. To monitor the tremor we used an electromechanical transducer onto which the mouse was fixed by the tail. Vibration resulting from tremor could then be recorded over time as apparent weight changes, with the amplitudes of the tracings reflecting the intensity of the movements. Figure 4 shows that two copies of the TG456 transgene strongly reduced the tremor-specific amplitudes recorded from spa/spa mice.
FIG. 4.
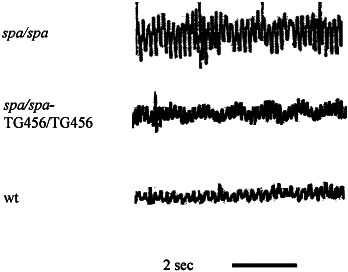
Electromechanical tracings of tremor-derived movement recorded from spa/spa, spa/spa-TG456/TG456 and wt animals. The amplitude is proportional to the strength of the movement recorded over time. For experimental details see text and Materials and methods.
As a second parameter to monitor motor performance, we measured the mean righting time by turning the animals on their back and determining the time required to regain an upright position. With this assay, gradual differences in the righting response can be observed ( Hartenstein et al. 1996). Here, we only quantified whether the animals regained their normal position in < 1 s. The presence of only a single copy of the transgene markedly influenced this parameter (Tables 1, 58% of measurements at different times on seven animals). In mice homozygous for the transgene, rescue was observed in the vast majority of these animals (86% of measurements on 28 individuals). In contrast, in non-transgenic spa/spa animals we never observed righting in < 5 s; most spa/spa mice required > 30 s to perform this task (see alsoHartenstein et al. 1996).
Table 1.
Phenotype characteristics of the different mouse lines
| spa/spa | spa/spa-TG456/+ | spa/spa-TG456/TG456 | wt | |
|---|---|---|---|---|
| Animals tested (n) | 10 | 7 | 28 | 10 |
| Hind feet clasping (%) | 100 | 71 | 32 | 0 |
| Righting time ≤ 1 s (%) | 0 | 58 | 86 | 100 |
Of the non-transgenic spa/spa animals, four were non-transgenic littermates from doubly heterozygous crosses, and six animals were derived from the original stock obtained from Jackson Laboratories.
A third criterion to evaluate neuromotor performance was the so-called ‘hind feet clasping’ phenomenon which represents an abnormal behaviour of spa/spa homozygotes which clasp their hind feet when picked up by the tail (Buckwalter et al. 1994). This phenotype was rescued in two-thirds of the spa/spa-TG456/TG456 double homozygotes and in one-third of the spa/spa-TG456/+ heterozygous transgenics (Table 1). Notably, hind feet clasping was still observed in several of the spa/spa-TG456/TG456 animals that no longer showed induced tremor (seeFig. 3). Thus, the hind feet clasping behaviour appears to be a more sensitive parameter to monitor reduced GlyR levels than tremor development.
Finally, we observed profound differences in male fertility correlating with transgene dosage. Spa/spa males are known to perform very poorly in siring offspring. Although we did not precisely quantify reproductive performance, we also experienced this in our mouse colony. By contrast, spa/spa-TG456/TG456 males displayed fertility indistinguishable from wt studs, while heterozygous transgene carriers showed an intermediate phenotype in this respect.
The behavioural and motor performance of mutant phenotypes may not only depend on the expression of the gene in question but also on the genetic background ( Logue et al. 1997;Kelly et al. 1998). Because the breeding of the transgene into the spa/spa genetic background produced genetic hybrid strains, we had to establish whether this by itself could possibly alleviate the spa/spa phenotype. Therefore, we included both the original spa/spa strain from Jackson Laboratories and non-transgenic spa/spa littermates from our transgene breedings into our experiments. In particular, for the analyses summarized inTable 1 four out of 10 spa/spa mice were non-transgenic littermates. Into the tremor analysis depicted inFig. 3, also four non-transgenic littermates were included. We found that with respect to the behavioural assays performed here, non-transgenic spa/spa mice on both genetic backgrounds behaved indistinguishably, thus allowing us to treat them as one experimental group (seeFig. 3 andTable 1). These data corroborate our earlier results derived from analogous breedings of two other GlyR β-transgenic spa/spa lines, in which we never observed a phenotypic modification in non-transgenic littermates (Hartenstein et al. 1996;Hartenstein 1996). In summary, we therefore conclude that the behavioural modifications observed here were dependent upon the presence and copy number of the low-expression transgene TG456. In addition, the different disease symptoms were differentially affected by the transgene dosage.
Discussion
In this paper, we describe a mouse strain transgenic for the rat GlyR β-subunit gene expressed at very low levels. This transgene can modify by partial rescue the phenotype of the spastic mutation in a gene dosage-dependent manner. It revealed that the rescue of the different behavioural and motor deficits showed distinct transgene dosage requirements. For example, in spa/spa-TG456/+ heterozygotes, no rescue was seen with respect to tremor inducibility, whereas male fertility, hind feet clasping and righting response were clearly improved. Furthermore, in TG456 homozygotes male fertility and righting response were completely rescued, whereas tremor induction and hind feet clasping behaviour were only partially improved. Our data also show that the different deficiencies appeared independently of each other, e.g. hind feet clasping did not require a defective righting response or tremor proneness. Thus, the different symptoms may originate from deficiencies at distinct sites of GlyR action.
The age dependence of tremor inducibility in spa/spa mice correlates with the appearance of the adult and the disappearance of the neonatal GlyR during the postnatal life of rodents (Becker et al. 1988). In spa/spa-TG456/TG456 mice, the onset of tremor inducibility was not only delayed but also of a transient nature. This is particularly interesting because alleviation of hyperexcitability with age is also seen in human hereditary hyperekplexia. A remarkable parallel thus appears to exist between mouse and man in the developmental dependence on efficient GlyR function.
The relatively subtle phenotype of spa/spa-TG456/TG456 animals constitutes a much better animal model of human GlyR disease than that seen in the spontaneous spastic mutant. Because of the high sensitivity of the disease phenotype to changes in GlyR levels, these mice might be of value for pharmacological studies. For example, although clonazepam and valproate have been successfully implemented in the therapy of human hyperekplexia (Dooley & Andermann 1989;Ryan et al. 1992), the exact mechanisms of action of these drugs on the organism level are still not clear.
In startle disease patients, characteristic symptoms, e.g. touch- or sound-induced tremor formation are often detectable already at birth, resembling the so-called ‘stiff baby syndrome’. In spastic and spasmodic mutant mice, however, motor deficiencies appear only between 2 and 3 weeks of postnatal life, i.e. a period corresponding to adolescence. This relative delay in mice may be attributed to the postnatal expression of the neonatal α2-subunit, which can form functional homo-oligomeric GlyRs (Hoch et al. 1989;Schmieden et al. 1992). In humans, a homologue of the α2-subunit exists (Grenningloh et al. 1990a), but nothing is known about its temporal expression pattern in early childhood.
Another difference between the human GlyR disease and the phenotype of spa/spa mice concerns the strongly reduced fertility of male mice. Consistent with the motor deficiencies seen, this could reflect an inability of spa/spa males to mount females. An alternative explanation is offered by the recent identification of GlyR expression in porcine sperm (Melendrez & Meizel 1996). Also, strychnine has been shown to block in vitro the acrosome reaction of porcine and human sperm (Melendrez & Meizel 1995). Whatever the underlying mechanisms might be, in spa/spa-TG456/TG456 animals the male fertility is normal. Again, this particular genotype resembles human startle disease in that no fertility problems have been reported for such patients.
There are dominant and recessive forms of hereditary hyper-ekplexia in man. At the molecular level, the spa/spa-TG456 mice represent a partial loss of function model and therefore may be regarded as correlates of the recessively inherited form of the human disease. The more frequent forms of hyperekplexia are dominantly inherited. The respective mutant GlyR α1 subunits display dominant negative effects upon heterologous expression in mammalian cell lines and Xenopus oocytes (Rajendra et al. 1994;Langosch et al. 1994). The consequences of expression of such mutations may be explored further by introducing such human hyperekplexia antimorphs as transgenes into mice.
Acknowledgments
This work was supported in part by the Deutsche Forschungsgemeinschaft (H.W.), The Bundesministerium für Forschung und Bildung (GF KV 01 411698, H.W., H.B.), and the Fond der Chemischen Industrie (H.B.).
Glossary
- GlyR
glycine receptor
- NSE
neuron-specific enolase
- SDS–PAGE
sodium dodecyl sulphate–polyacrylamide gel electrophoresis
References
- Andermann F, Keene DL, Andermann E, Qeesney L, Qeesney F. Startle disease or hyperekplexia; further delineation of the syndrome. Brain. 1980;103:985–997. doi: 10.1093/brain/103.4.985. [DOI] [PubMed] [Google Scholar]
- Becker C-M, Hermans-Borgmeyer I, Schmitt B, Betz H. The glycine receptor deficiency of the mutant mouse spastic: evidence for normal glycine receptor structure and localization. J. Neurosci. 1986;5:1358–1364. doi: 10.1523/JNEUROSCI.06-05-01358.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C-M, Hoch W, Betz H. Glycine receptor heterogeneity in rat spinal cord during postnatal development. EMBO J. 1988;7:3717–3726. doi: 10.1002/j.1460-2075.1988.tb03255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C-M, Schmieden V, Tarroni P, Strasser U, Betz H. Isoform-selective deficit of glycine receptors in the mouse mutant. Spastic. Neuron. 1992;8:283–289. doi: 10.1016/0896-6273(92)90295-o. [DOI] [PubMed] [Google Scholar]
- Becker C-M, Betz H, Schröder H. Expression of inhibitory glycine receptors in postnatal rat cerebral cortex. Brain Res. 1993;26:220–226. doi: 10.1016/0006-8993(93)90988-y. [DOI] [PubMed] [Google Scholar]
- Betz H. Structure and function of inhibitory glycine receptors. Q. Rev. Biophys. 1992;25:283–289. doi: 10.1017/s0033583500004340. [DOI] [PubMed] [Google Scholar]
- Buckwalter MS, Cook SA, Davisson MT, White WF, Camper SA. A frameshift mutation in the mouse α1 glycine receptor gene (Glra1) results in progressive neurological symptoms and juvenile death. Hum. Mol. Genet. 1994;3:2025–2030. doi: 10.1093/hmg/3.11.2025. [DOI] [PubMed] [Google Scholar]
- Dooley JM, Andermann F. Startle disease or hyperekplexia: adolescent onset and response to valproate. Pediatr. Neurol. 1989;2:126–127. doi: 10.1016/0887-8994(89)90041-6. [DOI] [PubMed] [Google Scholar]
- Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe JG. Transgenic mice expressing β-galactosidase in mature neurons under neuron-specific enolase promotor control. Neuron. 1990;5:187–197. doi: 10.1016/0896-6273(90)90308-3. [DOI] [PubMed] [Google Scholar]
- Gorman CM, Moffat LF, Horward BH. Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol. Cell. Biol. 1982;2:1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenningloh G, Schmieden V, Schofield PR, Seeburg PH, Siddique T, Mohandas TK, Becker C-M, Betz H. Alpha subunit variants of the human glycine receptor: primary structures, functional expression and chromosomal localization of the corresponding genes. EMBO J. 1990a;9:771–776. doi: 10.1002/j.1460-2075.1990.tb08172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenningloh G, Pribilla I, Prior P, Multhaup G, Beyreuther K, Taleb O, Betz H. Cloning and expression of the 58 kd β subunit of the inhibitory glycine receptor. Neuron. 1990b;4:963–970. doi: 10.1016/0896-6273(90)90149-a. [DOI] [PubMed] [Google Scholar]
- Hartenstein B. 1996. Entwicklung und Analyse von Mausmodellen zur Untersuchung von Glyzinrezeptordefekten in vivo PhD Thesis, Johann Wolfgang Goethe-Universität, Frankfurt am Main.
- Hartenstein B, Schenkel J, Kuhse J, Besenbeck B, Kling C, Becker C-M, Betz H, Weiher H. Low level expression of glycine receptor β subunit transgene is sufficient for phenotype correction in spastic mice. EMBO J. 1996;15:1275–1282. [PMC free article] [PubMed] [Google Scholar]
- Hoch W, Betz H, Becker C-M. Primary cultures of mouse spinal cord express the neonatal isoform of the inhibitory glycine receptor. Neuron. 1989;3:339–348. doi: 10.1016/0896-6273(89)90258-4. [DOI] [PubMed] [Google Scholar]
- Hogan B, Costantini F, Lacy E. Manipulating the Mouse Embryo. A Laboratory Manual. Cold Spring Harbor, NY, USA: Cold Spring Laboratory; 1986. [Google Scholar]
- Kelly MA, Rubinstein M, Phillips TJ, Lessov CN, Burkhart-Kasch S, Zhang G, Bunzow JR, Fang Y, Gerhardt GA, Grandy DK, Low MJ. Locomotor activity in D2 dopamine receptor-deficient mice is determined by gene dosage, genetic background, and developmental adaptations. J. Neurosci. 1998;18:3470–3479. doi: 10.1523/JNEUROSCI.18-09-03470.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsmore SF, Giros B, Suh D, Bieniarz M, Caron MG, Seldin MF. Glycine receptor β-subunit gene mutation in spastic mouse associated with LINE-1 element insertion. Nature Genet. 1994;7:136–141. doi: 10.1038/ng0694-136. [DOI] [PubMed] [Google Scholar]
- Kirsch J, Kuhse J, Betz H. Targeting of glycine receptor subunits to gephyrin-rich domains in transfected human embryonic kidney cells. Mol. Cell. Neurosci. 1995;6:450–461. doi: 10.1006/mcne.1995.1033. [DOI] [PubMed] [Google Scholar]
- Kuhse J, Schmieden V, Betz H. Identification and functional expression of a novel ligand binding subunit of the inhibitory glycine receptor. J. Biol. Chem. 1990;265:22 317–22. 320. [PubMed] [Google Scholar]
- Kuhse J, Laube B, Magalei D, Betz H. Assembly of the inhibitory glycine receptor: identification of amino acid sequence motifs governing subunit stoichiometry. Neuron. 1993;11:1049–1056. doi: 10.1016/0896-6273(93)90218-g. [DOI] [PubMed] [Google Scholar]
- Kurczynski TW. Hyperekplexia. Arch. Neurol. 1983;40:246–248. doi: 10.1001/archneur.1983.04050040076015. [DOI] [PubMed] [Google Scholar]
- Langosch D, Thomas L, Betz H. Conserved quaternary structure of ligand gated ion channels: the postsynaptic glycine receptor is a pentamer. Proc. Natl Acad. Sci. USA. 1988;85:7394–7398. doi: 10.1073/pnas.85.19.7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langosch D, Laube B, Rundström N, Schmieden V, Bormann J, Betz H. Decreased agonist affinity and chloride conductance of mutant glycine receptors associated with human hereditary hyperekplexia. EMBO J. 1994;13:4223–4228. doi: 10.1002/j.1460-2075.1994.tb06742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingam S, Wilson J, Hart EW. Hereditary stiff-baby syndrome. Am. J. Dis. Child. 1981;135:909–911. doi: 10.1001/archpedi.1981.02130340021008. [DOI] [PubMed] [Google Scholar]
- Logue SF, Owen EH, Rasmussen DL, Wehner JM. Assessment of locomotor activity, acoustic and tactile startle in inbred mouse strains and F1 hybrids: implications of genetic background for single gene and quantitative trait loci analyses. Neuroscience. 1997;80:1075–1086. doi: 10.1016/s0306-4522(97)00164-4. [DOI] [PubMed] [Google Scholar]
- Malosio M-L, Marqèze-Pouey B, Kuhse J, Betz H. Widespread expression of glycine receptor subunit mRNAs in the adult and developing rat brain. EMBO J. 1991;10:2401–2409. doi: 10.1002/j.1460-2075.1991.tb07779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzenbach B, Maulet I, Sefton L, Courtier B, Avner P, Guenet JL, Betz H. Structural analysis of mouse glycine receptor a subunit genes. J. Biol. Chem. 1994;269:2607–2612. [PubMed] [Google Scholar]
- Melendrez CS, Meizel S. Studies of porcine and human sperm suggesting a role for a sperm glycine receptor/Cl channel in the zona pelluzida-initiated acrosome reaction. Biol. Reprod. 1995;53:676–683. doi: 10.1095/biolreprod53.3.676. [DOI] [PubMed] [Google Scholar]
- Melendrez CS, Meizel S. Immunochemical identification of the glycine receptor/Cl-channel in porcine sperm. Biochem. Biophys. Res. Commun. 1996;223:675–678. doi: 10.1006/bbrc.1996.0954. [DOI] [PubMed] [Google Scholar]
- Meyer G, Kirsch J, Betz H, Langosch D. Identification of a gephyrin binding motif on the glycine receptor β subunit. Neuron. 1995;15:563–572. doi: 10.1016/0896-6273(95)90145-0. [DOI] [PubMed] [Google Scholar]
- Mülhardt C, Fischer M, Gass P, Simon-Chazottes D, Guenet J-L, Kuhse J, Betz H, Becker C-M. The spastic mouse: aberrant splicing of glycine receptor β subunit mRNA caused by intronic insertion of L1 element. Neuron. 1994;13:1003–1015. doi: 10.1016/0896-6273(94)90265-8. [DOI] [PubMed] [Google Scholar]
- Pfeiffer F, Betz H. Solubilization of the glycine receptor from rat spinal cord. Brain Res. 1981;226:273–279. doi: 10.1016/0006-8993(81)91099-4. [DOI] [PubMed] [Google Scholar]
- Pfeiffer F, Simler R, Grenningloh G, Betz H. Monoclonal antibodies and peptide mapping reveal structural similarities between the subunits of the glycine receptor of rat spinal cord. Proc. Natl Acad. Sci. USA. 1984;81:7224–7227. doi: 10.1073/pnas.81.22.7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra S, Lynch JW, Pierce KD, French CR, Barry HB, Schofield PR. Startle disease mutations reduce the agonist sensitivity of the human inhibitory glycine receptor. J. Biol. Chem. 1994;269:18 739–18. 742. [PubMed] [Google Scholar]
- Rees MI, Andrew M, Jawad S, Owen MJ. Evidence for recessive as well as dominant forms of startle disease (hyperekplexia) caused by mutations in the α1 subunit of the inhibitory glycine receptor. Hum. Mol. Genet. 1994;3:2175–2179. doi: 10.1093/hmg/3.12.2175. [DOI] [PubMed] [Google Scholar]
- Ryan SG, Sherman SL, Terry JC, Sparkes RS, Torres MC, Mackey RW. Startle disease, or hyperekplexia: response to clonazepam and assignment of the gene (STHE) to chromosome 5q by linkage analysis. Ann. Neurol. 1992;31:663–668. doi: 10.1002/ana.410310615. [DOI] [PubMed] [Google Scholar]
- Ryan SG, Buckwalter MS, Lynch JW, Handford CA, Segura L, Shiang R, Wasmuth JJ, Camper SA, Schofield P, O’Connell P. A missense mutation in the gene encoding the α1 subunit of the inhibitory glycine receptor in the spasmodic mouse. Nature Genet. 1994;7:131–135. doi: 10.1038/ng0694-131. [DOI] [PubMed] [Google Scholar]
- Saul B, Schmieden V, Kling C, Mülhardt C, Gass P, Kuhse J, Becker C-M. Point mutation of glycine receptor α1 subunit in the spasmodic mouse affects agonist response. FEBS Lett. 1994;350:71–76. doi: 10.1016/0014-5793(94)00736-5. [DOI] [PubMed] [Google Scholar]
- Schmieden V, Kuhse J, Betz H. Agonist pharmacology of neonatal and adult glycine receptor alpha subunits: identification of amino acid residues involved in taurine activation. EMBO J. 1992;11:2025–2032. doi: 10.1002/j.1460-2075.1992.tb05259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiang R, Ryan SG, Zhu Y-Z, Hahn AF, O’Connell P, Wasmuth JJ. Mutations in the α1 subunit of the inhibitory glycine receptor cause the dominant neurological disorder, hyperekplexia. Nature Genet. 1993;5:351–358. doi: 10.1038/ng1293-351. [DOI] [PubMed] [Google Scholar]
- Suhren O, Bruyn GW, Tuynman JA. Hyperekplexia: a hereditary startle syndrome. J. Neurol. Sci. 1966;3:577–605. [Google Scholar]
- White WF, Heller AH. Glycine receptor alteration in the mutant mouse. Spastic. Nature. 1982;298:655–657. doi: 10.1038/298655a0. [DOI] [PubMed] [Google Scholar]