

Abstract
Despite remarkable advances in therapy, heart failure remains a leading cause of morbidity and mortality. Although enhanced β-adrenergic receptor stimulation is part of normal physiologic adaptation to either the increase in physiologic demand or decrease in cardiac function, chronic β-adrenergic stimulation has been associated with increased mortality and morbidity in both animal models and humans. For example, overexpression of cardiac Gsα or β-adrenergic receptors in transgenic mice results in enhanced cardiac function in young animals, but with prolonged overstimulation of this pathway, cardiomyopathy develops in these mice as they age. Similarly, chronic sympathomimetic amine therapy increases morbidity and mortality in patients with heart failure. Conversely, the use of β-blockade has proven to be of benefit and is currently part of the standard of care for heart failure. It is conceivable that interrupting distal mechanisms in the β-adrenergic receptor-G protein-adenylyl cyclase pathway may also provide targets for future therapeutic modalities for heart failure. Interestingly, there are two major isoforms of adenylyl cyclase (AC) in the heart (type 5 and type 6), which may exert opposite effects on the heart, i.e., cardiac overexpression of AC6 appears to be protective, whereas disruption of type 5 AC prolongs longevity and protects against cardiac stress. The goal of this review is to summarize the paradigm shift in the treatment of heart failure over the past 50 years from administering sympathomimetic amine agonists to administering β-adrenergic receptor antagonists, and to explore the basis for a novel therapy of inhibiting type 5 AC.
Keywords: adenylyl cyclase, longevity, heart failure, β-adrenergic receptors
Introduction
Current medical management of heart failure depends upon the degree of heart failure and extent of symptoms as well as the severity of the existing co-morbidities. In general the current management of heart failure includes the use of angiotensin converting enzyme inhibitors or angiotensin receptor blockers, combined with β-adrenergic receptor (β-AR) blockers, diuretics, aldosterone antagonists, digitalis, nitrates and in cases of severe heart failure implantable cardioverter defibrillators with or without biventricular pacing. Finally, in those patients with severe refractory heart failure the options are often limited to palliation with or without chronic inotropes, cardiac transplantation, ventricular assist devices or experimental surgeries or drugs. However, this was not the case 50 years ago; at that time the treatment for heart failure consisted of bed rest, diuretics, digitalis, and the use of β-AR agonists. The latter is based on the reasoning that decreased cardiac contractility is a sine qua non in heart failure, and it was known that β-AR stimulation is one of the most potent stimulators of cardiac contractility [1–3]. It was also recognized that sympathetic stimulation via the cardiac nerves or via sympathomimetic amines was less effective in heart failure, as established by Braunwald and coworkers [4–5]. Later Bristow and coworkers identified this desensitization to sympathetic stimulation as due to β-AR down regulation [6–7]. The initial reasoning underlying therapy with sympathetic agonists was based on the simple concept that these pharmacological agents improved contractility, which was the fundamental derangement in the failing heart.
Physiologically the activation of the β-AR neurohormonal activity is a compensatory mechanism for the reduction in cardiac output and systemic blood pressure during heart failure. When the β-AR system is activated in the heart, the result is the activation of the associated adenylyl cyclase (AC) – cAMP – protein kinase A (PKA) pathway, which is predominantly mediated through β1-ARs. The resulting increase in PKA activity in turn leads to the release of Ca2+ from the sarcoplasmic reticulum with consequent increase in inotropy [8–10] (Figure 1). The activity of cAMP/PKA is kept in check by the activity of cAMP phosphodiesterases (PDE) and protein phosphatases [11–14], the activation of these enzymes leads to the breakdown of cAMP and inactivation of PKA. As noted above, with chronic heart failure one of the key phenotypic changes independent of the etiology of heart failure is the diminished cardiac response to catecholamine stimulation as indicated by the blunted contractile response to β-AR agonists [15–18]. This alteration in catecholamine response appears to be associated with both a decrease in β-AR density and subtype composition [19–20], an increase in β-AR kinase activity resulting in an increase in the dissociation of β-AR from Gs [21], leading to the impairment of cAMP – PKA signaling pathway [22–23]. Due to the decreased cardiac function seen in heart failure and the positive inotropic response seen with β-AR stimulation, it was attractive 30–50 years ago to attempt to overcome this β-AR desensitization and increase cardiac function through the stimulation of the adrenergic system in patients as a way to re-establish hemodynamic stability. However, it was not appreciated until much later that β-adrenergic desensitization in heart failure is actually a compensatory mechanism and accordingly interfering with this mechanism can be deleterious.
Fig. 1. Schematic representation of β-AR signaling pathway in cardiac myocytes.

Upon β-AR stimulation, the Gsα subunit-bound AC produces cAMP which activates target molecules, PKA and Epac. PKA regulates Ca2+ homeostasis through SR and LTCC, increases contractility. Under conditions of excessive sympathetic stimulation, PKA-induced Ca2+ leads to hypertrophy. On the other hand, activated PKA inhibits Raf, which results in the activation of the MEK/ERK signaling, and thus suppression of apoptosis. In contrast, Epac, the other target protein of cAMP than PKA, induces myocyte apoptosis via the P38MAPK pathway. Upon activation of β2-AR, the Giβγ subunits are released and activate the PI3K/Akt pathway. Activated Akt inhibits apoptosis, and accelerates hypertrophy
Adrenergic Receptors
The β-ARs serve as an interface between the adrenergic signaling originating from sympathetic nerves or circulating catecholamines and the cellular response of the end organ. The interest in how the sympatho-adrenal system regulates the heart and blood vessels dates back over a hundred years. Epinephrine was first purified in 1899 by Abel, and then in 1906 Dale first introduced the concept of sympathetic receptors based on his observation with the effects of ergot alkaloid antagonists in various organs and blood vessels. He noted ergot alkaloids had stimulatory effects in muscular organs, while having inhibitory effects in structures associated with sympathetic stimulation, and suggested the possible existence of a receptor mechanism for adrenalin [24]. Much work has been done since that time to understand the mechanisms behind this signaling pathway. Prior to 1948 adrenergic signals were thought to be mediated by two fundamentally opposing classes of chemical transmitters, sympathin E and sympathin I, with either excitatory or inhibitory effects [25]. At that time various types of sympathomimetic amines were used for the study of adrenergic systems including norepinephrine (arterenol), α-methylnorepinephrine (cobefrine), racemic-epinephrine, levo-epinephrine, α-methylepinephrine (methyl-epi), and isoproterenol (N-isopropylarterenol). Due to the fact that the then available preparations of adrenalin were fundamentally impure and displayed a wide variability in contents of epinephrine and norepinephrine, the resulting conclusions based on these studies were often difficult to interpret [26]. This was further compounded by the lack of awareness for the existence of subtypes of ARs, resulting in a lack of understanding for the receptor specificity found with the different sympathomimetic amines used. A major step forward was taken with the work of Ahlquist, who proposed the existence of different types of ARs, which when stimulated by the sympatho-adrenal system would elicit different physiological responses. In 1948 Ahlquist described and characterized these receptors based on the observation that there are two patterns of response to adrenergic stimulation and he theorized the existence of two types of adrenergic receptors and hence named them α and β-AR [27–28]. It wasn’t until the late 1960s through the early 1990s that the current classification of the existing subtypes of these two major classes of adrenergic receptors, α (α1, α2) and β (β1, β2, β3), was fully established. Each of these two major types of receptors can be further subdivided into various subtypes. Currently six subtypes of α-ARs have been described, α1-AR can be divided into α1A, α1B, α1D and the subtypes of α2-AR includes α2A, α2B, and α2C [29–30]. Although α-ARs are primarily involved in the regulation of peripheral resistance, there is now substantial evidence that they play a role in regulating cardiac contractility as well [31]. In the heart α2-ARs were found to regulate sympathetic neurotransmission, and the disruption of α2-ARs led to increased cardiac hypertrophy and decreased cardiac contractility [32]. The α1-, α1A- and α1B-ARs account for the majority of the cardiac α1-ARs [33]. Furthermore, in 2001 Fang et al. demonstrated the ability of α1A-AR overexpression to enhance cardiac contractility in transgenic mice with cardiac specific overexpression of α1A-AR [34]. There appears to be evidence pointing to the ability of α1A-AR overexpression to protect against myocardial ischemia [35]. Conversely, work in transgenic mice overexpressing α1B-AR showed decreased LV function as well as contractile response to β-AR stimulation, likely due to the activation of Gi [36–37]. Although α1D-AR accounts for a minor proportion of α1-AR in the heart; it does appear to be the predominant α1-AR within human coronary arteries [38].
Use of inotropic agents in the treatment of heart failure
The initial enthusiasm for the pharmacologic manipulation of the adrenergic system for the management of heart failure revolved largely around the use of the sympatho-adrenal hormones, epinephrine and norepinephrine, to increase inotropy through stimulation of the β-ARs. However, this initial enthusiasm waned even before the age of controlled clinical trials, since it was readily recognized that patients either became hypertensive or arrhythmic upon catecholamine therapy. This led to the search for synthetic sympathomimetic amines that retained the characteristic of β-AR stimulation to increase cardiac contractility, but are devoid of α-AR effects that lead to increased peripheral resistance, reduced renal flow and hypertension. Isoproterenol, followed by dopamine, dobutamine, selective β1-AR agonists and the PDE inhibitor milrinone; all of which turned out to exert adverse outcomes when administered chronically in heart failure [39–45].
Isoproterenol, a sympathomimetic agent structurally similar to epinephrine, functions as a non-selective β1- and β2-AR agonist with strong chronotropic and inotropic effects was first used in the treatment of heart failure in the 1960s and was found to elicit fatal arrhythmias [39] as well as increase myocardial ischemia in patients with coronary artery disease [40]. These findings led the pharmaceutical industry to develop β-adrenergic agents that selectively stimulated inotropy without increasing chronotropy or arterial pressure. Interestingly, the adverse actions of increasing chronotropy and arterial pressure were limited in these newer agents by the powerful arterial baroreceptors, which helped maintain pressure and heart rate in the face of sympathomimetic stimulation [46–47]. This was found by Vatner, et, al., using conscious dogs with arterial baroreceptor denervation, vagotomy, and muscarinic blockade, i.e., that the sympathomimetic amines stimulated the baororecpetors to increase parasympathetic tone and decrease sympathetic tone thereby buffering the pressor and chronotropic effects of the drugs, and that in the absence of reflexes, the adverse positive chronotropic and pressor effects of the β-AR agonists were unmasked [48]. Thus, the absence of the chronotropic and pressor responses, in vivo, was actually due to arterial reflex buffering of these actions.
In the search for a β-AR agonist that did not increase arterial pressure and heart rate, but did improve cardiac contractility, dopamine, a naturally occurring amine with prominent actions in the central nervous system and peripheral vascular beds, was found to have these properties. For example, dopamine, a non-selective β- and αAR agonist, at low doses (In humans ≤ 2μg/kg/min) leads to the activation of dopamine receptors in splanchnic and renal arterial beds and results in vasodilatation of these vessels [49], infusion at intermediate doses (2 to 5 μg/kg/min) results in direct stimulation of the cardiac β-ARs. However, at higher doses both α- and β-ARs are activated by dopamine and this results in the undesirable effects of increases in both heart rate [41] and vascular tone [42]. Although there have been sporadic reports of dopamine’s use in heart failure since 1977 [43, 50], clinical trials where dopamine was compared to dobutamine, a β1- selective inotropic agent, dopamine led to greater tachycardia, higher aortic blood pressure as well as it was less effective in its ability to lower left ventricular end diastolic pressure when compared to dobutamine [43], and was felt to be inferior to dobutamine for the management of heart failure. Furthermore, dopamine was found to have respiratory depressive effects, it decreased minute ventilation in patients with heart failure [44]. This respiratory depressive effect is an important consideration in hypoxic patients with heart failure. While dopamine has less chronotropic effect compared to isoproterenol, it did induce greater tachycardia when compared to dobutamine; in addition, its arrhythmogenic potential remained a concern. Furthermore, the decrease in endogenous noradrenaline stores in severe heart failure contributes to the unpredictability of dopamine’s effects in heart failure [51].
Although there was some initial success in the early 1980s with the use of the β1-selective inotropic agent, prenalterol, in its ability to improve hemodynamics acutely [52–53], studies involving long term administration proved it to be detrimental [54]. Furthermore, the observation that prenalterol reduced the maximal and submaximal heart rate responsiveness to exercise led to the suggestion that prenalterol may have β-AR blocking effects [55] and that the observed benefits may be secondary to this β-AR blocking effect. Similar results were seen in other heart failure trials involving other β1-AR selective inotropic agents such as xamoterol [56]. The randomized placebo controlled Xamoterol in Severe Heart Failure Study Group [56] found no significant benefit of oral xamoterol over placebo; however the study again showed variable agonist and antagonist effects over 24 hours on heart rate. The first clear indication that utilization of a β1-AR selective inotropic agent in heart failure was detrimental came with dobutamine, a predominately β1-AR selective inotropic agent. In heart failure, dobutamine leads to an increase in mortality, and this was demonstrated in the 1999 Flolan International Randomized Survival Trial [57], the results of which were further confirmed by a series of large scale clinical trials [58–59]. Clinical trials in the 1990s involving the β2-AR agonist, dopexamine, showed some initial clinical benefit over placebo [60–61], however much of this clinical benefit was attributed to the vasodilatory and cardiac unloading effects of this β2-AR agonist. Since many of the inotropic agents also have vasodilatory effects, it is important to review briefly the use of vasodilators in the treatment of heart failure. It has been known since the 1970s that vasodilators such as nitroprusside can lead to significant left ventricular functional improvement in the setting of heart failure [62–64]. Appropriately vasodilators were compared against inotropic agents, mostly dobutamine, for their clinical efficacy in heart failure. The use of vasodilators was associated with a lower mortality rate when compared to inotropic agents [59], however, the combined use of inotropes with vasodilators resulted in the highest mortality rate [65]. The use of downstream targets of the β-AR to improve cardiac function has also been explored. Milrinone, a PDE inhibitor that is capable of prolonging the half life of cAMP and thus increasing intracellular Ca2+ concentration and enhancing myocardial contractility, was first approved for intravenous use in the late 1980s. In the peripheral vascular system, increases in cAMP result in dilation of both the arterial and venous systems and may lead to subsequent hypotension. Clinical trials involving the use of milrinone in heart failure found that the use of milrinone led to no clear clinical benefit over placebo in non-ischemic heart failure and can increase mortality in patients with ischemic heart disease [45]. In 1986 the other PDE inhibitor, enoximone, was studied and thought to have a favorable profile for the management of heart failure [66–68]. In 1987 enoximone was compared to dobutamine and nitroprusside in the management of heart failure and found to be superior to both [69]. This clinical improvement was also demonstrated in other small placebo controlled clinical trials [70–75]. In 1993 the use of enoximone along and in combination with β-blockade was studied and this combination was found to be synergistic [76], the result was later confirmed in a 1998 study by Shakar et al. [77]. The first signs of increased mortality with the use of enoximone came in 1994, when enoximone was administered to patients with severe end stage heart failure [78]. Then in 2005 results from larger phase III, randomized, double blind placebo controlled clinical trials involving 1854 patients at 211 sites in 16 countries (ESSENTIAL-I and ESSENTIAL-II) became available, the trials found no benefit of enoximone treatment over placebo [79], and this finding lead to the termination of further development of enoximone.
It became progressively clearer that the use of inotropic agents in the treatment of heart failure resulted in an increase in arrhythmia frequency and myocardial ischemia with associated injury. Furthermore, the potential of direct myocyte toxicity due to intracellular calcium overload is of additional concern. All of these factors mediate the increased mortality observed with the use of inotropic agents in the setting of heart failure.
Animal models of cardiac receptor overexpression in the β-AR-Gs pathway
In the mammalian heart, β-ARs predominantly mediate the cardiac response to adrenergic stimulation and thus have been the focus for much of the heart failure research. In the healthy mammalian heart, β1-ARs account for the majority (70–80%) of total β-ARs, while β2-ARs account for approximately (20–30%) and β3-ARs have a minimal contribution [80–81]. Interestingly, there appears to be a species specific difference in the physiologic response to β3-AR stimulation after autonomic blockade, with a significant response observed in dogs and rodents and a near complete absence of response in primates [82], which is an important consideration for translational research involving animal models.
Paradoxically, there is down regulation of β-ARs in heart failure as a compensatory mechanism elicited to protect the heart from too much sympathetic stimulation. It was noted that in the setting of heart failure the β1-AR undergoes subtype-selective down regulation, resulting in an approximate 50:50 β1, β2-AR ratio [6, 83], with both sub-types becoming desensitize to adrenergic stimulation secondary to the uncoupling of these ARs from their downstream signaling pathways [84–86]. Again following the reasoning that β-AR signaling is desensitized in heart failure, resulting in the need for higher doses of an agonist to achieve effective increases in myocardial contractility, it was thought that this desensitization of chronic adrenergic stimulation in heart failure could be overcome with genetic overexpression of these receptors. Accordingly, transgenic mice with β1-AR, β2-AR and Gsα expression were generated, where the receptors would be increased in the heart several fold to allow greater efficacy from a given dose of norepinephrine released from the sympathetic nerves. The initial publications in this field [87–90] were quite promising and the authors suggested that overexpression of β2-ARs might be a new treatment for heart failure. It was noted that mice with up to 60-fold overexpression of β2-AR showed improvement in cardiac function without cardiac pathology [88, 91]. This was further supported by the gene transfer experiments of Koch et al., where improvement in cardiac response to β-AR stimulation was seen in failing rabbit hearts that received overexpression of the β2-AR transgene [89]. It was also noted that in the failing hearts, β-ARs are re-sensitized to adrenergic stimulation after a period of β-AR blockade, and this was characterized by the normalization of β-AR density and subtype distribution [92–94]. Consequentially, the restoration of β2-AR was theorized to be a viable therapeutic option for heart failure.
The crucial missing data in the previous animal studies that demonstrated a therapeutic benefit to β-AR overexpression, as well as many others, was that most of the transgenic mice were studied only for a few months, when they were young, and in terms of gene transfer experiments the follow up is generally only for several weeks. Importantly, diametrically opposite conclusions were derived when the transgenic animals were studied for several months to a year. Under those conditions the transgenic animals developed cardiomyopathy and heart failure. These data in mice parallel the findings in studies in patients, when comparing acute versus chronic effects of sympathomimetic amine therapy. The first evidence of this paradoxical response, i.e., enhanced response to sympathetic stimulation in young adults, but development of cardiomyopathy and heart failure as the animals age, was observed by Iwase, et al, in Gsα transgenic mice, showing a shift to the left in response to β-AR stimulation in young mice [95–96], but when the mice were studied several months later it was found that these mice developed cardiomyopathy and heart failure [95, 97–99]. Similar results were found for transgenic mice with overexpression of β1-ARs [100] and with overexpression of β2-ARs [101].
β1-AR overexpression in transgenic mice has been found to induce apoptosis, which was attributed to the associated increase in intracellular calcium and the activation of calcineurin, while β2-AR overexpression was initially thought not to lead to the promotion of apoptosis [102–104]. One possible explanation for this difference could be the differences in the downstream effectors of these two major β-ARs. Although it is well accepted that both β1- and β2-ARs are able to activate Gsα, β2-AR have been found to be associated with the inhibitory protein Gi [105–108], capable of inhibiting the main downstream effector, AC, thus raising questions about differences in the regulation of apoptosis by these two β-ARs. Interestingly, when β2-ARs are overexpressed in the heart of transgenic mice, apoptosis increases as cardiomyopathy develops, [91, 109]. This major difference between in vitro and in vivo studies again points out the importance of verifying in vitro work with in vivo models. It could be that the in vitro work was conducted in young cells, and as noted above, and the adverse effects of chronic β-AR stimulation are only observed in older mice. An additional consideration is the duration (12–24 hr) of the treatments in the in vitro studies, this short duration maybe insufficient to complete all the changes in protein expression. It is now clear that the overexpression of both β1-[100] and β2-AR [101] leads to development of cardiomyopathy and heart failure, as reflected by decreased cardiac function, increased fibrosis, cadiomyocyte apoptosis and mortality in these transgenic mice (Figure 2). Further complicating this picture is the differential compartmentation of β1-ARs and β2-ARs.
Fig. 2. Effects of cardiac overexpression of β1-AR, β2-AR or Gsα in mice on development of cardiomyopathy.

Panel a. The ages at 50% mortality in Tg mice with overexpression of β1-AR (blue bar, unpublished results), β2-AR (green bar) [101] or Gsα (red bar) [98] are shown in panel compared against wild type (WT) (FVB [204], or C57 [203]). The time to 50% mortality for the transgenic mice was less than half of that for the respective WT mice (*, p<0.05 compared to WT). Panel b. Decreased LV ejection fraction (LVEF) in the Tg animals vs WT [98,119]. Panel c. Cardiac fibrosis was increased in the Tg animals as compared to WT animals [98,119]. Panel d. Increased cardiac apoptosis in the Tg animals. The percent apoptotic cells in the transgenic mice were normalized against WT [99,119] (*, p<0.05 compared to WT). Figures used and modified with permission
Subtype specific β-AR induced cAMP compartmentation and heart failure
It was traditionally thought that in the cardiovascular system, both β1-AR and β2-AR are capable of activating the AC-cAMP-PKA pathway primarily through coupling to Gs proteins. However, there is growing evidence that β1-AR and β2-ARs elicit different physiological responses upon selective stimulation [110–111] and mediate distinct signaling pathways [112–113]. It has been suggested that β2-ARs are not only coupled to Gs, but also to Gi proteins, which results in different effects from β1-AR on activating Ca2+ handling, cardiac contractility, and PKA mediated phosphorylation of proteins that are involved in excitation and contraction coupling, such as phospholamban and myofilament proteins [107, 110, 114]. The inhibitory effect of β2-AR stimulation on contractility by coupling to Gi proteins was further confirmed using mice where β1- and β2-AR were knocked out [113]. Furthermore, previous studies have also suggested that β1-AR and β2-AR affect apoptosis and myocyte hypertrophy differently, that is, β1-AR stimulation induces cardiac myocyte hypertrophy and apoptosis [103, 115] while the activation of β2-AR significantly protects the myocardium from apoptosis [116] and hypertrophy [117–118]. As noted above, the concept that β2-AR stimulation protects against apoptosis was derived from experiments conducted in young myocytes cultured in vitro. In transgenic mice with cardiac overexpression of β2-ARs, Peter et al. found that apoptosis actually increases as they develop cardiomyopathy [119]. This apparent paradox has resulted despite β1-AR and β2-AR sharing cAMP as a common second messenger, and may be explained by the differentially regulated cardiomyocyte function through the compartmentation of cAMP signaling [120–121]. However, this spatial compartmentation of cAMP regulated by β1-AR and β2-AR was not recognized until recently with the emergence of fluorescence resonance energy transfer (FRET) microscopy technique, which was utilized to observe the dynamic changes of cAMP in living cardiomyocytes under β1-AR or β2-AR stimulation [122–123]. Furthermore, using combined nanoscale live-cell scanning ion conductance and FRET microscopy techniques, Nikolaev et al. [124] recently demonstrated that the spatial localization of β1-ARs and β2-ARs are different in normal myocytes, i.e., β2-ARs and their induced cAMP signals are localized to the transverse tubules, whereas β1-ARs are widely distributed at the cell crest. However, in the failing heart, the β2-ARs were redistributed from the transverse tubules to the cell crest, leading to a change in β2-AR associated compartmentation of cAMP. Thus β2-ARs in heart failure behave like β1-ARs. Whether the cAMP signals generated by the redistribution of β2-ARs are similar to those generated by β1-ARs in the regulation of contractility, cell death and apoptosis signaling pathways remain to be determined. In addition, if overexpression of β2-ARs induces abnormal compartmentation of cAMP in transgenic mice, this may reconcile the findings of increased apoptosis in chronically enhanced β2-AR signaling as in with β2-AR cardiomyopathy vs. protection in cultured neonatal cells [91, 101, 119]. To prove that abnormal compartmentation of β-ARs is the cause of the observed cardiomyopathy in transgenic mice overexpressing β-ARs, further work is needed for the localization of β1-ARs and β2-ARs in the mouse heart overexpressing β1-ARs or β2-ARs..
Downstream effectors of β-adrenergic receptor signaling
Further downstream of the β-AR – Gsα – AC – cAMP – PKA pathway, transgenic animals overexpressing AC (to be discussed later) and PKA have also been generated. PKA is a cAMP dependent tetrameric protein consists of two regulatory and two catalytic subunits [125], where the binding of cAMP to the regulatory subunits results in the disassociation of the regulatory subunits from the catalytic subunits and the activation of the enzyme. Within the mouse model there exist four isoforms of the regulatory subunit (RIα, RIβ, RIIα, and RIIβ) and two isoforms of the catalytic subunit (Cα and Cβ), under normal physiologic conditions the major holoenzyme consists of RIIβ and Cα [126]. In 2001 Antos et al. published the results on transgenic mice with PKA Cα overexpression, and found the activity of PKA to be elevated, and this was associated with dilated cardiomyopathy, heart failure and increased risk of sudden death, further supporting the adverse effects of chronic β-AR stimulation and PKA activation [127].
As noted above, both cardiac overexpressed β1-AR and β2-AR transgenic mice develop cardiomyopathy [91, 101, 119]. However, the molecular mechanisms mediating the cardiomyopathy in these mice could differ. Recent studies have demonstrated some common as well as different signaling pathways mediated by β1-AR and β2-AR (Figure 1). p38α MAPK was activated in both β1-AR and β2-AR transgenic hearts, but in contrast to the rescue observed when β2-AR transgenic mice were crossed with dominant negative (DN) p38α mice (bigenic β2-AR x DNp38α), when β1-AR transgenic mice were crossed with DNp38α mice (bigenic β1-AR x DNp38α) the cross did not rescue the β1-AR overexpression associated cardiomyopathy [119], suggesting other signaling pathways may be more important in ameliorating the adverse effects of chronic β1-AR stimulation. For example, mammalian sterile 20-like kinase 1 (Mst1), was found to be up regulated significantly in old β1-AR transgenic mice, but not in old β2-AR transgenic mice. Our recent studies have demonstrated that type 5 AC (AC5) could be a critical enzyme mediating cardiomyopathy in β-AR transgenic mice, and that inhibition of AC5 rescues the cardiomyopathy in these transgenic mice [128].
The results of these studies in transgenic mice with overexpressed β-ARs, Gsα or PKA have further established the pathological effects of chronic adrenergic stimulation and are akin to the studies in patients with heart failure, demonstrating improved cardiac function with short term inotropic therapy, but exacerbation of heart failure, development of lethal arrhythmias and premature mortality with more chronic therapy [57, 65, 129–130].
Paradigm Shift: Advent of β-AR blockade in heart failure
The groundwork for the advent of treating patients with heart failure with β-blockers, rather than agonists, was laid by the parallel studies noted above demonstrating adverse effects of chronic β-AR stimulation in transgenic mice with overexpressed β-ARs or Gsα or PKA along with the studies in patients with prolonged β-AR agonist therapy noted above. These studies in combination with the work of Bristow and colleagues demonstrating down regulation of β-ARs in patients with heart failure as a compensatory, protective mechanism, led to the exploration of β-AR blockers in heart failure. β-AR blockade was initially attempted as a therapy to control the tachycardia associated with heart failure, unexpectedly it was found to significantly reduce mortality in patients with heart failure [131–132] (Figure 3). The potential benefit of β-AR blockade in heart failure was first demonstrated in 1974 by Waggstein in a 7 patient series involving the use of a non-selective β-AR blocker, alprenolol, in the setting of congestive cardiomyopathy [133]. From that point on additional series of case reports and case series continued to emerge, further demonstrating the utility of β-AR blockade in the management of heart failure. However, as debate persisted over the use of β-AR blockade in the management of heart failure, discussions and case reports continued. In 1993 clinical benefits of β-AR blockade were demonstrated in a moderate sized placebo controlled clinical trial with the β1AR selective blocker, metoprolol. The trial involved 383 patients with idiopathic dilated cardiomyopathy and heart failure (MDC trial study) [134]. The MDC trial was followed by another β1 selective β-AR blocker trial, the Cardiac Insufficiency Bisoprolol Study (CIBIS) in 1994[135], then came the US Carvedilol (non-selective β/α1 blocker) Study in 1996 [131, 136]. The first truly large randomized clinical trial was released in 1999, The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II) [137–140] involving 2647 heart failure patients randomized to either bisoprolol or placebo and followed for a mean duration of 1.3 years, the study was terminated early due to clear evidence of mortality benefit with all cause mortality of 11.8% vs. 17.3% bisprolol vs. placebo, and a sudden death rate of 3.6% vs. 6.3%. In 2000 The Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERIT-heart failure) study was released. This study involved 3991 patients randomized to either metoprolol or placebo, and again a significant (19%) reduction in total mortality or all cause hospitalization led to the early termination of the study and demonstrated a clear benefit in using β-AR blockers in the management of heart failure [141]. By this time the benefit of β-blocker on mortality and use of β-AR blockers in treatment of heart failure had been well established. Current clinical decision in the use of β-AR blockers requires familiarity with the pharmacologic variability among the available β-blockers. In general β-blockers can be classified as β1-AR selective, non-selective β-AR blockers, and non-selective β/α-AR blockers. β1-AR selective β-blockers such as atenolol, while able to provide effective blockade of β1-AR, leaves vascular α-AR unopposed, and this in turn leads to peripheral vasoconstriction and a reflex increase in aortic pressure as demonstrated by the decreased ability of atenolol to reduce aortic pressure in the CAFÉ study [142]. Non-selective β/α-AR blockers such as carvedilol resulted in vasodilation secondary to α-AR blockade, thus allowing actual lowering of aortic pressure [143–144]. The use of these non-selective β/α-AR blockers have been tested in clinical trials against placebo in the COPRENICUS trial [145] where 2289 heart failure patients were randomized to either carvedilol or placebo, and were found to lead to a decrease in mortality (hazard ratio of 0.75), as well as hospitalization (hazard ratio 0.85). When compared to the β1-AR selective blocker metoprolol in the COMET trial [146], carvedilol was found to be superior to metoprolol in reduction of all causes mortality (hazard ratio 0.83), without any differences being detected in the incidence of side effects of drug withdrawals. Based on these clinical trials it is clear that β-AR blockade provides clear mortality benefit in the setting of heart failure, and this mortality benefit may be further enhanced in the setting of vasodilator therapy as in the case of combined β/α-AR blockade.
Fig. 3. β-blockade increases survival, and preserves cardiac function in heart failure patients and Gsα Tg mice.

Panel a. Kaplan-Meier survival curve showing mortality improvement in survival in heart failure patients on carvedilol (red line) vs placebo patients (blue line) [131] and Gsα Tg mice treated with propranolol (solid black line) vs vehicle (broken line) [98] indicating prolongation of lifespan with β-blockade. Panel b. β-blockade (blue bars) also resulted in improvement in LV function in heart failure patients [132] and Gsα Tg mice [98] as indicated by improved LVEF (*, p<0.05 vs placebo vs vehicle). Figures used and modified with permission
There were also parallel studies in animal models showing the effectiveness of β-AR blocking agents in heart failure [147]. In addition, the study by Asai et al. [98] demonstrated that propranolol could prevent the cardiomyopathy that develops in Gsα transgenic mice as they age, resulting in normal cardiac function and normal myocardial histology, as well as eliminating the premature mortality (Figure 3).
Adenylyl Cyclase
Adenylyl cyclase is a transmembrane protein with two hydrophobic and two cytoplasmic domains capable of converting ATP to cAMP upon the stimulation with various G-protein coupled receptors such as β-AR (Figure 1). Thus far, at least nine mammalian isoforms of AC (AC1–9) have been identified, and each isoform demonstrates a distinct tissue distribution, biological as well as pharmacological properties [148–151]. AC5 and AC6 are the major mammalian AC cardiac isoforms [152–153]. Furthermore, upon characterization, they were found to be sensitive to and inhibited by low concentration of Ca2+ [154–155]. Interestingly, prior studies suggest that these two isoforms may exert opposite effects on cardioprotection, i.e., overexpression of AC6 appears cardioprotective, whereas, AC5 disruption is also cardioprotective.
Adenylyl Cyclase Type 5
In 1999 Tepe et al. showed enhanced baseline heart rate and LV function in transgenic mice with cardiac specific overexpression of AC5 [156]. Furthermore, mating AC5 transgenic with Gαq transgenic mice, was able to rescue Gαq transgenic mice from LV dysfunction (but not hypertrophy) [157], thus establishing the down regulation of AC5 seen in the transgenic Gαq overexpressing mice as the dominant pathological mechanism leading to the observed β-AR signaling dysfunction. In direct contrast, Hanoune et al. reported that mice with AC5 overexpression (AC5 transgenic) developed cardiomyopathy with age [158]. Although the original study was not published, the review suggests that mice with AC5 overexpression behave like mice with β-AR overexpression as those seen in Gsα transgenic [97], β1-AR transgenic [100] or β2-AR transgenic, i.e., hyperfunction at a young age with the development of cardiomyopathy as they age. This finding was confirmed by our observation that AC5 transgenic mice have an increased heart rate and LV ejection fraction at baseline, however under the condition of chronic pressure overload or catecholamine stimulation, decreased LV function, increased LV hypertrophy, apoptosis and fibrosis, as well as the development of pulmonary congestion and heart failure occurs [158].
Conversely, mice with AC5 disruption, i.e., knockout (AC5KO), develop resistance to the adverse effects of chronic pressure overload, having the ability to maintain LV function, as well as resistance to the apoptotic process with chronic transverse aortic banding [159]. Furthermore, these AC5KO mice were found to have a more effective physiologic response and desensitization to chronic isoproterenol infusion and a lower number of apoptotic myocytes after chronic stimulation [160] (Figure 4). This improvement in desensitization to chronic catecholamine stress is a major defense mechanism against the pathogenesis of heart failure [161–162]. These results speak to the central role of AC5 in the regulation of the apoptosis induction and the subsequent development of heart failure. By the same token, the inhibition of AC5 could prove to be a novel therapy for heart failure.
Fig. 4. AC5KO in mice protects against the development of cardiomyopathy.

AC5KO (yellow bar) enhances protection against heart failure induced by pressure overload [159] or catecholamine stress (chronic isoproterenol) [160]. Preserved LVEF (Panel a) and reduced cardiomyocyte apoptosis (Panel b) was observed in AC5KO mice compared with WT in response to catecholamine stress (*, p<0.05 compared to no treatment, gray bar) [160]. Figures used and modified with permission
Adenylyl Cyclase Type 6
Interestingly, the other major cardiac AC isoform, AC6, most often has been reported to play a protective role in the heart. Hammond et al. demonstrated that mice with cardiac specific overexpression of AC6 exhibited normal cardiac function at baseline and an increased response to β-AR stimulation, without any signs of physiological or histological abnormality up to 21 months of age [163]. The same group examined the effects of AC6 overexpression in pigs with intracoronary injection of recombinant adenovirus encoding AC6 or LacZ. They found the treatment with AC6 led to an increase in AC6 protein and stimulated cAMP production, associated with an increase in ventricular function, as demonstrated by increased peak LV dP/dt and cardiac output under isoproterenol challenge in vivo [164]. In 1999 and 2002 Roth et al. demonstrated the ability of cardiac directed AC6 overexpression to rescue mice with cardiomyopathy secondary to Gq overexpression [165–166]. Then in 2006, the same group found enhanced survival after myocardial infarction in transgenic mice with cardiac specific AC6 overexpression [167]. Additionally, AC6 deletion led to impaired cardiac cAMP generation and calcium handling which resulted in depressed LV function [168]. Together, these studies indicate that enhancement of AC6 activity could be a potential therapeutic target for the treatment of congestive heart failure. However, a recent study from our laboratory revealed quite different conclusions. We found that AC6 overexpression in mice led to impaired LV function due to chronic aortic banding (4 weeks) compared with wild type mice, which may have been due to enhanced LV systolic wall stress [169]. It is important to note that previous studies demonstrating the salutary effects of AC6 overexpression did not examine chronic pressure overload [165–167], which was the intervention we studied in our AC6 transgenic mice [169]. Again it could be that distal mechanisms might differ with the stress of chronic pressure overload than that induced by other stresses, e.g., ischemia. However, Tang et al. recently reported chronic aortic banding in female AC6KO mice resulted in improved function [170], which is inconsistent with the previous work from that group on the ischemic interventions [171], but is consistent with our findings of the adverse effects of chronic pressure overload in AC6 overexpression [169]. However, an appendix in the study by Tang et al. [170] indicated that male AC5KO mice fared worse than wild type with chronic pressure overload.
Concept of longevity and stress resistance
It has been recognized for some time that aging and longevity are regulated by evolutionarily conserved molecular pathways [172] and the association between longevity and stress resistance rests with studies utilizing yeast, C. elegans, and Drosophila as models to study the triggers and the molecular basis for aging and longevity. By utilizing these models, several longevity regulators have been found, including Sir2, an NAD-dependent histone deacetylase, whose increased expression is associated with extended lifespan in yeast [173], C. elegans [174–175] and fruit flies [176–178].
Longevity in mice with AC5 disruption
We have recently reported a novel, genetically engineered animal model, where by AC type 5 isoform is knocked out (AC5KO). AC5KO mice exhibit increased longevity [179] and are protected against cardiac stress related to heart failure [159]. When AC5 is disrupted in this mouse model, phenotypic changes are summarized as [159–160, 179]: 1) AC5KO mice live a third longer than wild type (Figure 5). 2) AC5KO mice are protected from aging-induced cardiomyopathy, including decreased left ventricular hypertrophy, a reduction in fibrosis and decreased cardiac apoptosis compared to wild type (Figure 5). 3) AC5KO mice are protected from aging associated reduction in bone density and fracture risk. 4) AC5KO mice are protected from pressure overload and chronic catecholamine stimulation induced apoptosis and the associated impairment in cardiac function (Figure 4). 5) AC5KO mice have increased exercise capacity [180]. 6) Bigenic mice with AC5KO and β2-AR overexpression, did not develop the typical cardiomyopathy observed in β2-AR overexpressing mice, hence rescuing the mice from β2-AR overexpression associated cardiomyopathy [128]. 7) Most importantly, AC5KO have several features that resemble caloric restriction, the most widely studied model of longevity. For example, both the AC5KO and caloric restriction models appear to affect metabolism similarly. Both caloric restriction and AC5KO mice weigh less than their control littermates; the caloric restriction mice weighed less because of restricted food intake, however, the AC5KO weighed less despite augmented food intake. In both cases, a decrease in glycogen and blood glucose was observed. 8) Mice on a high fat diet and mice treated with a pharmacologic AC5 inhibitor showed a decreased amount of weight gain and improved glucose tolerance [181]. All of these phenotypic changes in AC5KO mice point to the beneficial effects of AC5 disruption not only in the prolongation of life but also an improvement of health with aging, i.e., improvement in body weight, cardiac function, bone density, glucose tolerance and exercise capacity.
Fig. 5. Disruption of AC5 in mice protects against aging cardiomyopathy.

Kaplan-Meier survival curve for AC5KO (yellow square) shows prolonged life span compared with WT (open square), accompanied by preservation of LVEF (left in inset) and a reduction in myocardial apoptosis (right in inset) in the older mice. *, p<0.05 vs WT. [179]. Figures used and modified with permission
Although the exact mechanism thorough which AC5 disruption leads to longevity is still unclear at present, resistance to oxidative stress is clearly involved. An upregulation of Manganese-Superoxide Dismutase (MnSOD) [182], a protein with a known role in resistance to oxidative stress was seen in the AC5KO mice compared to wild type littermates [179]. The current evidence also suggests that AC5 disruption leads to the activation of the MEK/ERK signaling pathway. The activation of ERK in turn activates MnSOD, which protects against oxidative stress, apoptosis, resulting in cell survival, which leads to the observed longevity in AC5KO mice [179] (Figure 6). However, this appears to be only part of the story. As noted above, one keytrait of the AC5KO mice is that they eat more but weigh less than wild type mice reflecting a favorable difference in metabolic rate compared to wild type mice. Thus, similar to caloric restriction models which are dependent on changes in metabolism, it appears that the AC5KO model of longevity may share some of these same mechanisms.
Fig. 6. Mechanism of AC5-mediated regulation of longevity.
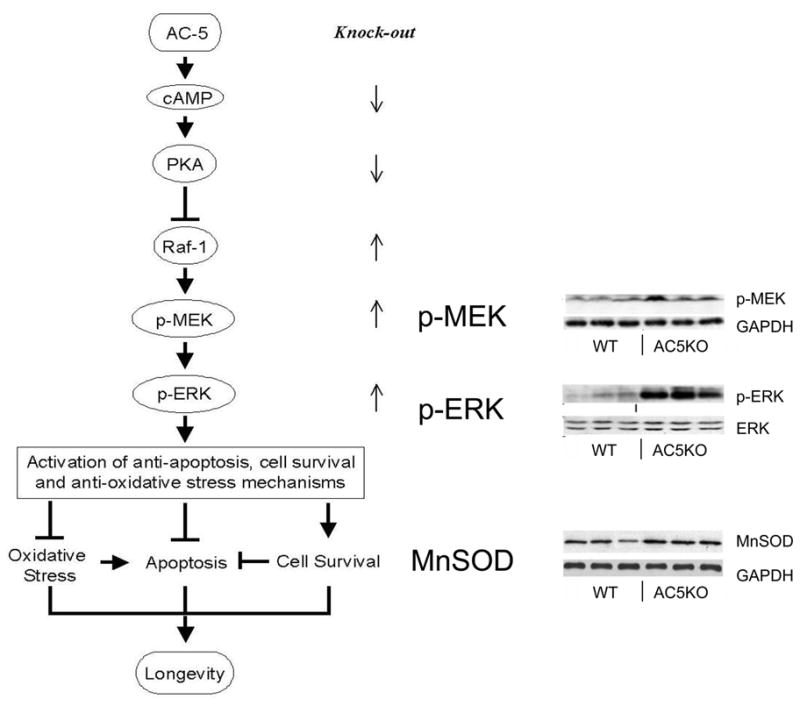
Disruption of AC5 activates the MEK/ERK signaling pathway [179]. The activation of ERK leads to increased expression of manganese superoxide dismutase (MnSOD), which results in inhibition of apoptosis. On the other hand, the ERK signaling activates the cell survival signaling (ref). Taken together, these mechanisms contribute to increased longevity in AC5KO mice. Western blots demonstrate the changes of key enzymes in the heart by disruption of AC5 [179]. Figures used and modified with permission
Future Directions
The data derived from genetically engineered mouse models suggest that pharmacologic inhibition of AC5 could be a therapeutic strategy for heart failure and longevity. Much attention has been paid to specific AC isoforms which play a major role in the β-AR pathway. The cloning of AC isoforms in 1990’s [183–192] enabled studies of specific AC inhibitors. SpecificAC5 inhibitors have been developed, based on studies of P-site inhibitors, which are known to inhibit AC [193–195], using purified AC5 in baculovirus [196–202]. However, due to high IC50, low selectivity for AC5 or membrane impermeability of those compounds, it is still premature to translate these compounds to the clinic.
Summary
This review summarized the paradigm shift that occurred over the past half century, where the treatment of heart failure changed diametrically from the use of β-AR agonists to antagonists. The development of a variety of sympathomimetic amines to stimulate cardiac contractility in the failing heart was based on syllogistic reasoning, i.e., that a heart characterized by depressed contractility would benefit from increasing its inotropic state. This concept was supported by numerous experiments demonstrating that catecholamines could restore LV function acutely in hearts with cardiac depression and by studies in transgenic mice with cardiac overexpression of β-AR, where cardiac contractility was enhanced. An important take home message from this review is that the pathophysiology of any disease state in animals or patients, e.g., heart failure, is not simple, but rather is exceedingly complex. We know now that distal signaling pathways and compartmentation of β-ARs and AC may modify dramatically the effects of β-AR agonists in heart failure. Based on studies in patients with heart failure on chronic sympathomimetic amine therapy demonstrating increased mortality and morbidity, and based on studies in transgenic mice overexpressing components of the β-AR signaling pathway, which developed cardiomyopathy as they age, a paradigm shift emerged resulting in a diametrically different direction of therapy, i.e., β-AR blockade. However, even though β-AR blockers have improved therapy in patients with heart failure, there is still significant room for further improvement. Our hypothesis is that a new direction will emerge involving distal components of the β-AR signaling pathway. Our recent work suggests that inhibition of type 5 AC is one such mechanism that could develop into a novel therapeutic approach for heart failure.
Acknowledgments
This work was supported by National Institutes of Health grants HL095888, HL102354, HL059139, HL069020, AG014121, AG028854, HL033107, AG023137, HL65183, and HL069752
References
- 1.Goldberg LI, et al. The direct effects of norepinephrine, epinephrine, and methoxamine on myocardial contractile force in man. Circulation. 1960;22:1125–32. doi: 10.1161/01.cir.22.6.1125. [DOI] [PubMed] [Google Scholar]
- 2.Gazes PC, Goldberg LI, Darby TD. Heart force effects of sympathomimetic amines as a basis for their use in shock accompanying myocardial infarction. Circulation. 1953;8(6):883–92. doi: 10.1161/01.cir.8.6.883. [DOI] [PubMed] [Google Scholar]
- 3.Cotten MD, Pincus S. Comparative effects of a wide range of doses of L-epinephrine and of L-norepinephrine on the contractile force of the heart in situ. J Pharmacol Exp Ther. 1955;114(1):110–8. [PubMed] [Google Scholar]
- 4.Chidsey CA, et al. Myocardial Norepinephrine Concentration in Man. Effects of Reserpine and of Congestive Heart Failure. N Engl J Med. 1963;269:653–8. doi: 10.1056/NEJM196309262691302. [DOI] [PubMed] [Google Scholar]
- 5.Chidsey CA, et al. Cardiac Norephinephrine Stores in Experimental Heart Failure in the Dog. J Clin Invest. 1964;43:2386–93. doi: 10.1172/JCI105113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bristow MR, et al. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ Res. 1986;59(3):297–309. doi: 10.1161/01.res.59.3.297. [DOI] [PubMed] [Google Scholar]
- 7.Bristow MR, et al. Reduced beta 1 receptor messenger RNA abundance in the failing human heart. J Clin Invest. 1993;92(6):2737–45. doi: 10.1172/JCI116891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415(6868):198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 9.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 10.Rapundalo ST. Cardiac protein phosphorylation: functional and pathophysiological correlates. Cardiovasc Res. 1998;38(3):559–88. doi: 10.1016/s0008-6363(98)00063-7. [DOI] [PubMed] [Google Scholar]
- 11.Cohen PT. Protein phosphatase 1--targeted in many directions. J Cell Sci. 2002;115(Pt 2):241–56. doi: 10.1242/jcs.115.2.241. [DOI] [PubMed] [Google Scholar]
- 12.Ceulemans H, Bollen M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol Rev. 2004;84(1):1–39. doi: 10.1152/physrev.00013.2003. [DOI] [PubMed] [Google Scholar]
- 13.Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev. 2000;80(1):173–210. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- 14.Marks AR, Marx SO, Reiken S. Regulation of ryanodine receptors via macromolecular complexes: a novel role for leucine/isoleucine zippers. Trends Cardiovasc Med. 2002;12(4):166–70. doi: 10.1016/s1050-1738(02)00156-1. [DOI] [PubMed] [Google Scholar]
- 15.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93(10):896–906. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- 16.Movsesian MA, Bristow MR. Alterations in cAMP-mediated signaling and their role in the pathophysiology of dilated cardiomyopathy. Curr Top Dev Biol. 2005;68:25–48. doi: 10.1016/S0070-2153(05)68002-7. [DOI] [PubMed] [Google Scholar]
- 17.El-Armouche A, et al. Inhibitory G-proteins and their role in desensitization of the adenylyl cyclase pathway in heart failure. Cardiovasc Res. 2003;60(3):478–87. doi: 10.1016/j.cardiores.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 18.Vatner SF, Vatner DE, Homcy CJ. beta-adrenergic receptor signaling: an acute compensatory adjustment-inappropriate for the chronic stress of heart failure? Insights from Gsalpha overexpression and other genetically engineered animal models. Circ Res. 2000;86(5):502–6. doi: 10.1161/01.res.86.5.502. [DOI] [PubMed] [Google Scholar]
- 19.Bristow MR, et al. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307(4):205–11. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 20.Engelhardt S, et al. Analysis of beta-adrenergic receptor mRNA levels in human ventricular biopsy specimens by quantitative polymerase chain reactions: progressive reduction of beta 1-adrenergic receptor mRNA in heart failure. J Am Coll Cardiol. 1996;27(1):146–54. doi: 10.1016/0735-1097(95)00425-4. [DOI] [PubMed] [Google Scholar]
- 21.Ungerer M, et al. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation. 1993;87(2):454–63. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- 22.Diviani D. Modulation of cardiac function by A-kinase anchoring proteins. Curr Opin Pharmacol. 2008;8(2):166–73. doi: 10.1016/j.coph.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Zakhary DR, Moravec CS, Bond M. Regulation of PKA binding to AKAPs in the heart: alterations in human heart failure. Circulation. 2000;101(12):1459–64. doi: 10.1161/01.cir.101.12.1459. [DOI] [PubMed] [Google Scholar]
- 24.Dale HH. On some physiological actions of ergot. J Physiol. 1906;34(3):163–206. doi: 10.1113/jphysiol.1906.sp001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cannon W, Rosenbueth A. Studies on the conditions of activity in endocrine organs: XXIX. Sympathin E and sympathin I. Am J Physiol. 1933;104:557–574. [Google Scholar]
- 26.Bylund DB. Alpha- and beta-adrenergic receptors: Ahlquist’s landmark hypothesis of a single mediator with two receptors. Am J Physiol Endocrinol Metab. 2007;293(6):E1479–81. doi: 10.1152/ajpendo.00664.2007. [DOI] [PubMed] [Google Scholar]
- 27.Ahlquist RP. Development of the concept of alpha and beta adrenotropic receptors. Ann N Y Acad Sci. 1967;139(3):549–52. doi: 10.1111/j.1749-6632.1967.tb41228.x. [DOI] [PubMed] [Google Scholar]
- 28.Ahlquist RP. A study of the adrenotropic receptors. Am J Physiol. 1948;153(3):586–600. doi: 10.1152/ajplegacy.1948.153.3.586. [DOI] [PubMed] [Google Scholar]
- 29.Bylund DB. Subtypes of alpha 1- and alpha 2-adrenergic receptors. FASEB J. 1992;6(3):832–9. doi: 10.1096/fasebj.6.3.1346768. [DOI] [PubMed] [Google Scholar]
- 30.Graham RM, et al. alpha 1-adrenergic receptor subtypes. Molecular structure, function, and signaling. Circ Res. 1996;78(5):737–49. doi: 10.1161/01.res.78.5.737. [DOI] [PubMed] [Google Scholar]
- 31.Triposkiadis F, et al. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. 2009;54(19):1747–62. doi: 10.1016/j.jacc.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 32.Hein L, Altman JD, Kobilka BK. Two functionally distinct alpha2-adrenergic receptors regulate sympathetic neurotransmission. Nature. 1999;402(6758):181–4. doi: 10.1038/46040. [DOI] [PubMed] [Google Scholar]
- 33.Wolff DW, et al. Distribution of alpha1-adrenergic receptor mRNA species in rat heart. J Cardiovasc Pharmacol. 1998;32(1):117–22. doi: 10.1097/00005344-199807000-00018. [DOI] [PubMed] [Google Scholar]
- 34.Lin F, et al. Targeted alpha(1A)-adrenergic receptor overexpression induces enhanced cardiac contractility but not hypertrophy. Circ Res. 2001;89(4):343–50. doi: 10.1161/hh1601.095912. [DOI] [PubMed] [Google Scholar]
- 35.Rorabaugh BR, et al. alpha1A- but not alpha1B-adrenergic receptors precondition the ischemic heart by a staurosporine-sensitive, chelerythrine-insensitive mechanism. Cardiovasc Res. 2005;65(2):436–45. doi: 10.1016/j.cardiores.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 36.Woodcock EA. Roles of alpha1A- and alpha1B-adrenoceptors in heart: insights from studies of genetically modified mice. Clin Exp Pharmacol Physiol. 2007;34(9):884–8. doi: 10.1111/j.1440-1681.2007.04707.x. [DOI] [PubMed] [Google Scholar]
- 37.Lemire I, et al. Cardiac-specific overexpression of alpha1BAR regulates betaAR activity via molecular crosstalk. J Mol Cell Cardiol. 1998;30(9):1827–39. doi: 10.1006/jmcc.1998.0746. [DOI] [PubMed] [Google Scholar]
- 38.Jensen BC, et al. The alpha-1D Is the predominant alpha-1-adrenergic receptor subtype in human epicardial coronary arteries. J Am Coll Cardiol. 2009;54(13):1137–45. doi: 10.1016/j.jacc.2009.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith HJ, et al. Hemodynamic studies in cardiogenic shock. Treatment with isoproterenol and metaraminol. Circulation. 1967;35(6):1084–91. doi: 10.1161/01.cir.35.6.1084. [DOI] [PubMed] [Google Scholar]
- 40.Maroko PR, et al. Factors influencing infarct size following experimental coronary artery occlusions. Circulation. 1971;43(1):67–82. doi: 10.1161/01.cir.43.1.67. [DOI] [PubMed] [Google Scholar]
- 41.Goldberg LI. Cardiovascular and renal actions of dopamine: potential clinical applications. Pharmacol Rev. 1972;24(1):1–29. [PubMed] [Google Scholar]
- 42.Vatner SF, Millard RW, Higgins CB. Coronary and myocardial effects of dopamine in the conscious dog: parasympatholytic augmentation of pressor and inotropic actions. J Pharmacol Exp Ther. 1973;187(2):280–95. [PubMed] [Google Scholar]
- 43.Stoner JD, 3rd, Bolen JL, Harrison DC. Comparison of dobutamine and dopamine in treatment of severe heart failure. Br Heart J. 1977;39(5):536–9. doi: 10.1136/hrt.39.5.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van de Borne P, Oren R, Somers VK. Dopamine depresses minute ventilation in patients with heart failure. Circulation. 1998;98(2):126–31. doi: 10.1161/01.cir.98.2.126. [DOI] [PubMed] [Google Scholar]
- 45.Felker GM, et al. Heart failure etiology and response to milrinone in decompensated heart failure: results from the OPTIME-CHF study. J Am Coll Cardiol. 2003;41(6):997–1003. doi: 10.1016/s0735-1097(02)02968-6. [DOI] [PubMed] [Google Scholar]
- 46.Vatner SF, et al. Effects of carotid sinus nerve stimulation on the coronary circulation of the conscious dog. Circ Res. 1970;27(1):11–21. doi: 10.1161/01.res.27.1.11. [DOI] [PubMed] [Google Scholar]
- 47.Vatner SF, et al. Extent of carotid sinus regulation of the myocardial contractile state in conscious dogs. J Clin Invest. 1972;51(4):995–1008. doi: 10.1172/JCI106893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vatner SF, Rutherford JD, Ochs HR. Baroreflex and vagal mechanisms modulating left ventricular contractile responses to sympathomimetic amines in conscious dogs. Circ Res. 1979;44(2):195–207. doi: 10.1161/01.res.44.2.195. [DOI] [PubMed] [Google Scholar]
- 49.Bayram M, et al. Reassessment of dobutamine, dopamine, and milrinone in the management of acute heart failure syndromes. Am J Cardiol. 2005;96(6A):47G–58G. doi: 10.1016/j.amjcard.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 50.Miller AJ. Dopamine in the treatment of heart failure. Proc R Soc Med. 1977;70(Suppl 2):16–24. doi: 10.1177/00359157770700S204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tuttle RR, Mills J. Dobutamine: development of a new catecholamine to selectively increase cardiac contractility. Circ Res. 1975;36(1):185–96. doi: 10.1161/01.res.36.1.185. [DOI] [PubMed] [Google Scholar]
- 52.Fitzpatrick D, et al. Hemodynamic, hormonal and electrolyte responses to prenalterol infusion in heart failure. Circulation. 1983;67(3):613–9. doi: 10.1161/01.cir.67.3.613. [DOI] [PubMed] [Google Scholar]
- 53.Petch MC, et al. Acute haemodynamic effects of oral prenalterol in severe heart failure. Br Heart J. 1984;52(1):49–52. doi: 10.1136/hrt.52.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lambertz H, Meyer J, Erbel R. Long-term hemodynamic effects of prenalterol in patients with severe congestive heart failure. Circulation. 1984;69(2):298–305. doi: 10.1161/01.cir.69.2.298. [DOI] [PubMed] [Google Scholar]
- 55.Glover DR, et al. Are the clinical benefits of oral prenalterol in ischaemic heart failure due to beta blockade? A six month randomised double blind comparison with placebo. Br Heart J. 1985;53(2):208–15. doi: 10.1136/hrt.53.2.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xamoterol in severe heart failure. The Xamoterol in Severe Heart Failure Study Group. Lancet. 1990;336(8706):1–6. [PubMed] [Google Scholar]
- 57.O’Connor CM, et al. Continuous intravenous dobutamine is associated with an increased risk of death in patients with advanced heart failure: insights from the Flolan International Randomized Survival Trial (FIRST) Am Heart J. 1999;138(1 Pt 1):78–86. doi: 10.1016/s0002-8703(99)70250-4. [DOI] [PubMed] [Google Scholar]
- 58.Coletta AP, et al. Clinical trials update from the European Society of Cardiology Heart Failure meeting: SHAPE, BRING-UP 2 VAS, COLA II, FOSIDIAL, BETACAR, CASINO and meta-analysis of cardiac resynchronisation therapy. Eur J Heart Fail. 2004;6(5):673–6. doi: 10.1016/j.ejheart.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 59.Abraham WT, et al. In-hospital mortality in patients with acute decompensated heart failure requiring intravenous vasoactive medications: an analysis from the Acute Decompensated Heart Failure National Registry (ADHERE) J Am Coll Cardiol. 2005;46(1):57–64. doi: 10.1016/j.jacc.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 60.Gollub SB, et al. Efficacy and safety of a short-term (6-h) intravenous infusion of dopexamine in patients with severe congestive heart failure: a randomized, double-blind, parallel, placebo-controlled multicenter study. J Am Coll Cardiol. 1991;18(2):383–90. doi: 10.1016/0735-1097(91)90590-6. [DOI] [PubMed] [Google Scholar]
- 61.Asanoi H, et al. Intravenous dopexamine in the treatment of acute congestive heart failure: results of a multicenter, double-blind, placebo-controlled withdrawal study. Cardiovasc Drugs Ther. 1995;9(6):791–7. doi: 10.1007/BF00879873. [DOI] [PubMed] [Google Scholar]
- 62.Franciosa JA, et al. Improved left ventricular function during nitroprusside infusion in acute myocardial infarction. Lancet. 1972;1(7752):650–4. doi: 10.1016/s0140-6736(72)90460-6. [DOI] [PubMed] [Google Scholar]
- 63.Guiha NH, et al. Treatment of refractory heart failure with infusion of nitroprusside. N Engl J Med. 1974;291(12):587–92. doi: 10.1056/NEJM197409192911201. [DOI] [PubMed] [Google Scholar]
- 64.Miller RR, et al. Clinical use of sodium nitroprusside in chronic ischemic heart disease. Effects on peripheral vascular resistance and venous tone and on ventricular volume, pump and mechanical performance. Circulation. 1975;51(2):328–36. doi: 10.1161/01.cir.51.2.328. [DOI] [PubMed] [Google Scholar]
- 65.Elkayam U, et al. Use and impact of inotropes and vasodilator therapy in hospitalized patients with severe heart failure. Am Heart J. 2007;153(1):98–104. doi: 10.1016/j.ahj.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 66.Weber KT, Janicki JS, Jain MC. Enoximone (MDL 17,043) for stable, chronic heart failure secondary to ischemic or idiopathic cardiomyopathy. Am J Cardiol. 1986;58(7):589–95. doi: 10.1016/0002-9149(86)90281-x. [DOI] [PubMed] [Google Scholar]
- 67.Likoff MJ, et al. Comparison of acute hemodynamic response to dobutamine and intravenous MDL 17,043 (enoximone) in severe congestive heart failure secondary to ischemic cardiomyopathy or idiopathic dilated cardiomyopathy. Am J Cardiol. 1986;57(15):1328–34. doi: 10.1016/0002-9149(86)90213-4. [DOI] [PubMed] [Google Scholar]
- 68.Gilbert EM, Bristow MR, Mason JW. Acute hemodynamic response to low dose enoximone (MDL 17,043): an oral dose-range study. Am J Cardiol. 1987;60(5):57C–62C. doi: 10.1016/0002-9149(87)90527-3. [DOI] [PubMed] [Google Scholar]
- 69.Installe E, et al. Comparative effects on hemodynamics of enoximone (MDL 17,043), dobutamine and nitroprusside in severe congestive heart failure. Am J Cardiol. 1987;60(5):46C–52C. doi: 10.1016/0002-9149(87)90525-x. [DOI] [PubMed] [Google Scholar]
- 70.Choraria SK, Taylor D, Pilcher J. Double-blind crossover comparison of enoximone and placebo in patients with congestive heart failure. Circulation. 1987;76(6):1307–11. doi: 10.1161/01.cir.76.6.1307. [DOI] [PubMed] [Google Scholar]
- 71.Treese N, et al. Long-term treatment with oral enoximone for chronic congestive heart failure: the European experience. Am J Cardiol. 1987;60(5):85C–90C. doi: 10.1016/0002-9149(87)90533-9. [DOI] [PubMed] [Google Scholar]
- 72.Zipperle G, et al. A double-blind dose response comparison of oral enoximone and placebo for congestive heart failure. Am J Cardiol. 1987;60(5):72C–74C. doi: 10.1016/0002-9149(87)90530-3. [DOI] [PubMed] [Google Scholar]
- 73.Narahara KA. Oral enoximone therapy in chronic heart failure: a placebo-controlled randomized trial. The Western Enoximone Study Group. Am Heart J. 1991;121(5):1471–9. doi: 10.1016/0002-8703(91)90154-a. [DOI] [PubMed] [Google Scholar]
- 74.Itoh H, et al. Effects of enoximone on exercise tolerance in patients with mild to moderate heart failure. Am J Cardiol. 1991;68(4):360–4. doi: 10.1016/0002-9149(91)90832-6. [DOI] [PubMed] [Google Scholar]
- 75.Dec GW, et al. Long-term outcome of enoximone therapy in patients with refractory heart failure. Am Heart J. 1993;125(2 Pt 1):423–9. doi: 10.1016/0002-8703(93)90021-z. [DOI] [PubMed] [Google Scholar]
- 76.Galie N, et al. Effect of enoximone alone and in combination with metoprolol on myocardial function and energetics in severe congestive heart failure: improvement in hemodynamic and metabolic profile. Cardiovasc Drugs Ther. 1993;7(3):337–47. doi: 10.1007/BF00880157. [DOI] [PubMed] [Google Scholar]
- 77.Shakar SF, et al. Combined oral positive inotropic and beta-blocker therapy for treatment of refractory class IV heart failure. J Am Coll Cardiol. 1998;31(6):1336–40. doi: 10.1016/s0735-1097(98)00077-1. [DOI] [PubMed] [Google Scholar]
- 78.Cowley AJ, Skene AM. Treatment of severe heart failure: quantity or quality of life? A trial of enoximone. Enoximone Investigators. Br Heart J. 1994;72(3):226–30. doi: 10.1136/hrt.72.3.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Metra M, et al. Effects of low-dose oral enoximone administration on mortality, morbidity, and exercise capacity in patients with advanced heart failure: the randomized, double-blind, placebo-controlled, parallel group ESSENTIAL trials. Eur Heart J. 2009;30(24):3015–26. doi: 10.1093/eurheartj/ehp338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gauthier C, et al. Functional beta3-adrenoceptor in the human heart. J Clin Invest. 1996;98(2):556–62. doi: 10.1172/JCI118823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alexander RW, Williams LT, Lefkowitz RJ. Identification of cardiac beta-adrenergic receptors by (minus) [3H]alprenolol binding. Proc Natl Acad Sci U S A. 1975;72(4):1564–8. doi: 10.1073/pnas.72.4.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shen YT, et al. Differences in beta 3-adrenergic receptor cardiovascular regulation in conscious primates, rats and dogs. J Pharmacol Exp Ther. 1996;278(3):1435–43. [PubMed] [Google Scholar]
- 83.Brodde OE. Pathophysiology of the beta-adrenoceptor system in chronic heart failure: consequences for treatment with agonists, partial agonists or antagonists? Eur Heart J. 1991;12(Suppl F):54–62. doi: 10.1093/eurheartj/12.suppl_f.54. [DOI] [PubMed] [Google Scholar]
- 84.Bristow MR, et al. Beta-adrenergic pathways in nonfailing and failing human ventricular myocardium. Circulation. 1990;82(2 Suppl):I12–25. [PubMed] [Google Scholar]
- 85.Bristow MR, et al. Beta 1- and beta 2-adrenergic receptor-mediated adenylate cyclase stimulation in nonfailing and failing human ventricular myocardium. Mol Pharmacol. 1989;35(3):295–303. [PubMed] [Google Scholar]
- 86.Bristow MR, et al. Differences in beta-adrenergic neuroeffector mechanisms in ischemic versus idiopathic dilated cardiomyopathy. Circulation. 1991;84(3):1024–39. doi: 10.1161/01.cir.84.3.1024. [DOI] [PubMed] [Google Scholar]
- 87.Cross HR, et al. Overexpression of the cardiac beta(2)-adrenergic receptor and expression of a beta-adrenergic receptor kinase-1 (betaARK1) inhibitor both increase myocardial contractility but have differential effects on susceptibility to ischemic injury. Circ Res. 1999;85(11):1077–84. doi: 10.1161/01.res.85.11.1077. [DOI] [PubMed] [Google Scholar]
- 88.Milano CA, et al. Enhanced myocardial function in transgenic mice overexpressing the beta 2-adrenergic receptor. Science. 1994;264(5158):582–6. doi: 10.1126/science.8160017. [DOI] [PubMed] [Google Scholar]
- 89.Tevaearai HT, et al. Myocardial gene transfer and overexpression of beta2-adrenergic receptors potentiates the functional recovery of unloaded failing hearts. Circulation. 2002;106(1):124–9. doi: 10.1161/01.cir.0000020220.79105.fd. [DOI] [PubMed] [Google Scholar]
- 90.Dorn GW, 2nd, et al. Low- and high-level transgenic expression of beta2-adrenergic receptors differentially affect cardiac hypertrophy and function in Galphaq-overexpressing mice. Proc Natl Acad Sci U S A. 1999;96(11):6400–5. doi: 10.1073/pnas.96.11.6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liggett SB, et al. Early and delayed consequences of beta(2)-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation. 2000;101(14):1707–14. doi: 10.1161/01.cir.101.14.1707. [DOI] [PubMed] [Google Scholar]
- 92.Heilbrunn SM, et al. Increased beta-receptor density and improved hemodynamic response to catecholamine stimulation during long-term metoprolol therapy in heart failure from dilated cardiomyopathy. Circulation. 1989;79(3):483–90. doi: 10.1161/01.cir.79.3.483. [DOI] [PubMed] [Google Scholar]
- 93.Kubo H, et al. Patients with end-stage congestive heart failure treated with beta-adrenergic receptor antagonists have improved ventricular myocyte calcium regulatory protein abundance. Circulation. 2001;104(9):1012–8. doi: 10.1161/hc3401.095073. [DOI] [PubMed] [Google Scholar]
- 94.Lowes BD, et al. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med. 2002;346(18):1357–65. doi: 10.1056/NEJMoa012630. [DOI] [PubMed] [Google Scholar]
- 95.Iwase M, et al. Cardiomyopathy induced by cardiac Gs alpha overexpression. Am J Physiol. 1997;272(1 Pt 2):H585–9. doi: 10.1152/ajpheart.1997.272.1.H585. [DOI] [PubMed] [Google Scholar]
- 96.Lader AS, et al. Cardiac Gsalpha overexpression enhances L-type calcium channels through an adenylyl cyclase independent pathway. Proc Natl Acad Sci U S A. 1998;95(16):9669–74. doi: 10.1073/pnas.95.16.9669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Iwase M, et al. Adverse effects of chronic endogenous sympathetic drive induced by cardiac GS alpha overexpression. Circ Res. 1996;78(4):517–24. doi: 10.1161/01.res.78.4.517. [DOI] [PubMed] [Google Scholar]
- 98.Asai K, et al. Beta-adrenergic receptor blockade arrests myocyte damage and preserves cardiac function in the transgenic G(salpha) mouse. J Clin Invest. 1999;104(5):551–8. doi: 10.1172/JCI7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Geng YJ, et al. Apoptosis of cardiac myocytes in Gsalpha transgenic mice. Circ Res. 1999;84(1):34–42. doi: 10.1161/01.res.84.1.34. [DOI] [PubMed] [Google Scholar]
- 100.Engelhardt S, et al. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999;96(12):7059–64. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Du XJ, et al. Age-dependent cardiomyopathy and heart failure phenotype in mice overexpressing beta(2)-adrenergic receptors in the heart. Cardiovasc Res. 2000;48(3):448–54. doi: 10.1016/s0008-6363(00)00187-5. [DOI] [PubMed] [Google Scholar]
- 102.Communal C, et al. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. 1998;98(13):1329–34. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- 103.Communal C, et al. Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein. Circulation. 1999;100(22):2210–2. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- 104.Zaugg M, et al. Beta-adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation. 2000;102(3):344–50. doi: 10.1161/01.cir.102.3.344. [DOI] [PubMed] [Google Scholar]
- 105.Koch WJ, et al. The binding site for the beta gamma subunits of heterotrimeric G proteins on the beta-adrenergic receptor kinase. J Biol Chem. 1993;268(11):8256–60. [PubMed] [Google Scholar]
- 106.Koch WJ, et al. Direct evidence that Gi-coupled receptor stimulation of mitogen-activated protein kinase is mediated by G beta gamma activation of p21ras. Proc Natl Acad Sci U S A. 1994;91(26):12706–10. doi: 10.1073/pnas.91.26.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xiao RP, et al. Coupling of beta2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circ Res. 1999;84(1):43–52. doi: 10.1161/01.res.84.1.43. [DOI] [PubMed] [Google Scholar]
- 108.Zou Y, et al. Both Gs and Gi proteins are critically involved in isoproterenol-induced cardiomyocyte hypertrophy. J Biol Chem. 1999;274(14):9760–70. doi: 10.1074/jbc.274.14.9760. [DOI] [PubMed] [Google Scholar]
- 109.Burniston JG, Tan LB, Goldspink DF. beta2-Adrenergic receptor stimulation in vivo induces apoptosis in the rat heart and soleus muscle. J Appl Physiol. 2005;98(4):1379–86. doi: 10.1152/japplphysiol.00642.2004. [DOI] [PubMed] [Google Scholar]
- 110.Xiao RP, et al. Beta 2-adrenergic receptor-stimulated increase in cAMP in rat heart cells is not coupled to changes in Ca2+ dynamics, contractility, or phospholamban phosphorylation. J Biol Chem. 1994;269(29):19151–6. [PubMed] [Google Scholar]
- 111.Xiao RP, Lakatta EG. Beta 1-adrenoceptor stimulation and beta 2-adrenoceptor stimulation differ in their effects on contraction, cytosolic Ca2+, and Ca2+ current in single rat ventricular cells. Circ Res. 1993;73(2):286–300. doi: 10.1161/01.res.73.2.286. [DOI] [PubMed] [Google Scholar]
- 112.Xiang Y, Kobilka B. The PDZ-binding motif of the beta2-adrenoceptor is essential for physiologic signaling and trafficking in cardiac myocytes. Proc Natl Acad Sci U S A. 2003;100(19):10776–81. doi: 10.1073/pnas.1831718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Devic E, et al. Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from beta(1) and beta(2) adrenoceptor knockout mice. Mol Pharmacol. 2001;60(3):577–83. [PubMed] [Google Scholar]
- 114.Xiao RP, Ji X, Lakatta EG. Functional coupling of the beta 2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol Pharmacol. 1995;47(2):322–9. [PubMed] [Google Scholar]
- 115.Zhu WZ, et al. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci U S A. 2001;98(4):1607–12. doi: 10.1073/pnas.98.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chesley A, et al. The beta(2)-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through G(i)-dependent coupling to phosphatidylinositol 3′-kinase. Circ Res. 2000;87(12):1172–9. doi: 10.1161/01.res.87.12.1172. [DOI] [PubMed] [Google Scholar]
- 117.Schafer M, et al. Hypertrophic effect of selective beta(1)-adrenoceptor stimulation on ventricular cardiomyocytes from adult rat. Am J Physiol Cell Physiol. 2000;279(2):C495–503. doi: 10.1152/ajpcell.2000.279.2.C495. [DOI] [PubMed] [Google Scholar]
- 118.Morisco C, et al. Beta-adrenergic cardiac hypertrophy is mediated primarily by the beta(1)-subtype in the rat heart. J Mol Cell Cardiol. 2001;33(3):561–73. doi: 10.1006/jmcc.2000.1332. [DOI] [PubMed] [Google Scholar]
- 119.Peter PS, et al. Inhibition of p38 alpha MAPK rescues cardiomyopathy induced by overexpressed beta 2-adrenergic receptor, but not beta 1-adrenergic receptor. J Clin Invest. 2007;117(5):1335–43. doi: 10.1172/JCI29576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kuschel M, et al. G(i) protein-mediated functional compartmentalization of cardiac beta(2)-adrenergic signaling. J Biol Chem. 1999;274(31):22048–52. doi: 10.1074/jbc.274.31.22048. [DOI] [PubMed] [Google Scholar]
- 121.Chen-Izu Y, et al. G(i)-dependent localization of beta(2)-adrenergic receptor signaling to L-type Ca(2+) channels. Biophys J. 2000;79(5):2547–56. doi: 10.1016/S0006-3495(00)76495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002;295(5560):1711–5. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- 123.Nikolaev VO, et al. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ Res. 2006;99(10):1084–91. doi: 10.1161/01.RES.0000250046.69918.d5. [DOI] [PubMed] [Google Scholar]
- 124.Nikolaev VO, et al. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327(5973):1653–7. doi: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 125.Krebs EG, Beavo JA. Phosphorylation-dephosphorylation of enzymes. Annu Rev Biochem. 1979;48:923–59. doi: 10.1146/annurev.bi.48.070179.004423. [DOI] [PubMed] [Google Scholar]
- 126.McKnight GS, et al. Cyclic AMP, PKA, and the physiological regulation of adiposity. Recent Prog Horm Res. 1998;53:139–59. discussion 160–1. [PubMed] [Google Scholar]
- 127.Antos CL, et al. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ Res. 2001;89(11):997–1004. doi: 10.1161/hh2301.100003. [DOI] [PubMed] [Google Scholar]
- 128.Yan L, Dillinger J, Williams JG, Iwatsubo K, Gao S, Fritzky LF, Ge H, Vatner DE, Vatner SF. Abstract. 5936: Inhibition of Type 5 Adenylyl Cyclase Rescues Cardiomyopathy Induced by Overexpressed beta2-Adrenergic Receptors in the Heart. Circulation. 2009;(120):S1178. [Google Scholar]
- 129.Oliva F, et al. Intermittent 6-month low-dose dobutamine infusion in severe heart failure: DICE multicenter trial. Am Heart J. 1999;138(2 Pt 1):247–53. doi: 10.1016/s0002-8703(99)70108-0. [DOI] [PubMed] [Google Scholar]
- 130.Packer M, et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med. 1991;325(21):1468–75. doi: 10.1056/NEJM199111213252103. [DOI] [PubMed] [Google Scholar]
- 131.Packer M, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N Engl J Med. 1996;334(21):1349–55. doi: 10.1056/NEJM199605233342101. [DOI] [PubMed] [Google Scholar]
- 132.Lechat P, et al. Clinical effects of beta-adrenergic blockade in chronic heart failure: a meta-analysis of double-blind, placebo-controlled, randomized trials. Circulation. 1998;98(12):1184–91. doi: 10.1161/01.cir.98.12.1184. [DOI] [PubMed] [Google Scholar]
- 133.Waagstein F, Hjalmarson AC, Wasir HS. Apex cardiogram and systolic time intervals in acute myocardial infarction and effects of practolol. Br Heart J. 1974;36(11):1109–21. doi: 10.1136/hrt.36.11.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Waagstein F, et al. Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy. Metoprolol in Dilated Cardiomyopathy (MDC) Trial Study Group. Lancet. 1993;342(8885):1441–6. doi: 10.1016/0140-6736(93)92930-r. [DOI] [PubMed] [Google Scholar]
- 135.A randomized trial of beta-blockade in heart failure. The Cardiac Insufficiency Bisoprolol Study (CIBIS). CIBIS Investigators and Committees. Circulation. 1994;90(4):1765–73. doi: 10.1161/01.cir.90.4.1765. [DOI] [PubMed] [Google Scholar]
- 136.Effect of carvedilol on mortality and morbidity in patients with chronic heart failure. Circulation. 1996;94(4):592. doi: 10.1161/01.cir.94.4.592. [DOI] [PubMed] [Google Scholar]
- 137.The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353(9146):9–13. [PubMed] [Google Scholar]
- 138.Segev A, Mekori YA. The Cardiac Insufficiency Bisoprolol Study II. Lancet. 1999;353(9161):1361. doi: 10.1016/S0140-6736(05)74357-9. [DOI] [PubMed] [Google Scholar]
- 139.Drummond GA, I, Squire B. The Cardiac Insufficiency Bisoprolol Study II. Lancet. 1999;353(9161):1361. doi: 10.1016/s0140-6736(05)74356-7. [DOI] [PubMed] [Google Scholar]
- 140.Poole-Wilson PA. The Cardiac Insufficiency Bisoprolol Study II. Lancet. 1999;353(9161):1360–1. doi: 10.1016/S0140-6736(05)74354-3. [DOI] [PubMed] [Google Scholar]
- 141.Hjalmarson A, et al. Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: the Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERIT-HF). MERIT-HF Study Group. JAMA. 2000;283(10):1295–302. doi: 10.1001/jama.283.10.1295. [DOI] [PubMed] [Google Scholar]
- 142.Williams B, et al. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation. 2006;113(9):1213–25. doi: 10.1161/CIRCULATIONAHA.105.595496. [DOI] [PubMed] [Google Scholar]
- 143.Pedersen ME, Cockcroft JR. The latest generation of beta-blockers: new pharmacologic properties. Curr Hypertens Rep. 2006;8(4):279–86. doi: 10.1007/s11906-006-0065-0. [DOI] [PubMed] [Google Scholar]
- 144.Pedersen ME, Cockcroft JR. The vasodilatory beta-blockers. Curr Hypertens Rep. 2007;9(4):269–77. doi: 10.1007/s11906-007-0050-2. [DOI] [PubMed] [Google Scholar]
- 145.Krum H, et al. Effects of initiating carvedilol in patients with severe chronic heart failure: results from the COPERNICUS Study. JAMA. 2003;289(6):712–8. doi: 10.1001/jama.289.6.712. [DOI] [PubMed] [Google Scholar]
- 146.Poole-Wilson PA, et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet. 2003;362(9377):7–13. doi: 10.1016/S0140-6736(03)13800-7. [DOI] [PubMed] [Google Scholar]
- 147.Reiken S, et al. beta-adrenergic receptor blockers restore cardiac calcium release channel (ryanodine receptor) structure and function in heart failure. Circulation. 2001;104(23):2843–8. doi: 10.1161/hc4701.099578. [DOI] [PubMed] [Google Scholar]
- 148.Hanoune J, et al. Adenylyl cyclases: structure, regulation and function in an enzyme superfamily. Mol Cell Endocrinol. 1997;128(1–2):179–94. doi: 10.1016/s0303-7207(97)04013-6. [DOI] [PubMed] [Google Scholar]
- 149.Iyengar R. Molecular and functional diversity of mammalian Gs-stimulated adenylyl cyclases. FASEB J. 1993;7(9):768–75. doi: 10.1096/fasebj.7.9.8330684. [DOI] [PubMed] [Google Scholar]
- 150.Simonds WF. G protein regulation of adenylate cyclase. Trends Pharmacol Sci. 1999;20(2):66–73. doi: 10.1016/s0165-6147(99)01307-3. [DOI] [PubMed] [Google Scholar]
- 151.Sunahara RK, Dessauer CW, Gilman AG. Complexity and diversity of mammalian adenylyl cyclases. Annu Rev Pharmacol Toxicol. 1996;36:461–80. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- 152.Ishikawa Y, Homcy CJ. The adenylyl cyclases as integrators of transmembrane signal transduction. Circ Res. 1997;80(3):297–304. doi: 10.1161/01.res.80.3.297. [DOI] [PubMed] [Google Scholar]
- 153.Okumura S, et al. Type 5 adenylyl cyclase disruption alters not only sympathetic but also parasympathetic and calcium-mediated cardiac regulation. Circ Res. 2003;93(4):364–71. doi: 10.1161/01.RES.0000086986.35568.63. [DOI] [PubMed] [Google Scholar]
- 154.Guillou JL, Nakata H, Cooper DM. Inhibition by calcium of mammalian adenylyl cyclases. J Biol Chem. 1999;274(50):35539–45. doi: 10.1074/jbc.274.50.35539. [DOI] [PubMed] [Google Scholar]
- 155.Cooper DM. Molecular and cellular requirements for the regulation of adenylate cyclases by calcium. Biochem Soc Trans. 2003;31(Pt 5):912–5. doi: 10.1042/bst0310912. [DOI] [PubMed] [Google Scholar]
- 156.Tepe NM, et al. Altering the receptor-effector ratio by transgenic overexpression of type V adenylyl cyclase: enhanced basal catalytic activity and function without increased cardiomyocyte beta-adrenergic signalling. Biochemistry. 1999;38(50):16706–13. doi: 10.1021/bi991619k. [DOI] [PubMed] [Google Scholar]
- 157.Tepe NM, Liggett SB. Transgenic replacement of type V adenylyl cyclase identifies a critical mechanism of beta-adrenergic receptor dysfunction in the G alpha q overexpressing mouse. FEBS Lett. 1999;458(2):236–40. doi: 10.1016/s0014-5793(99)01147-3. [DOI] [PubMed] [Google Scholar]
- 158.Lai L, Yan L, Hu CL, Gao S, Iwatsubo K, Ishiwawa Y, Sadoshima J, Vatner SF, Vatner DE. Abstract 3290: Increased Type 5 Adenylyl Cyclase Expression Mediates Chronic Catecholamine Stress via Increases in Oxidative Stress and Down-regulation of MnSOD. Circulation. 2009;120:S781. [Google Scholar]
- 159.Okumura S, et al. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci U S A. 2003;100(17):9986–90. doi: 10.1073/pnas.1733772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Okumura S, et al. Disruption of type 5 adenylyl cyclase enhances desensitization of cyclic adenosine monophosphate signal and increases Akt signal with chronic catecholamine stress. Circulation. 2007;116(16):1776–83. doi: 10.1161/CIRCULATIONAHA.107.698662. [DOI] [PubMed] [Google Scholar]
- 161.Vatner DE, et al. Beta-adrenoceptor desensitization during the development of canine pacing-induced heart failure. Clin Exp Pharmacol Physiol. 1996;23(8):688–92. doi: 10.1111/j.1440-1681.1996.tb01760.x. [DOI] [PubMed] [Google Scholar]
- 162.Vatner DE, et al. Chronic norepinephrine elicits desensitization by uncoupling the beta-receptor. J Clin Invest. 1989;84(6):1741–8. doi: 10.1172/JCI114357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gao MH, et al. Adenylylcyclase increases responsiveness to catecholamine stimulation in transgenic mice. Circulation. 1999;99(12):1618–22. doi: 10.1161/01.cir.99.12.1618. [DOI] [PubMed] [Google Scholar]
- 164.Lai NC, et al. Intracoronary delivery of adenovirus encoding adenylyl cyclase VI increases left ventricular function and cAMP-generating capacity. Circulation. 2000;102(19):2396–401. doi: 10.1161/01.cir.102.19.2396. [DOI] [PubMed] [Google Scholar]
- 165.Roth DM, et al. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation. 1999;99(24):3099–102. doi: 10.1161/01.cir.99.24.3099. [DOI] [PubMed] [Google Scholar]
- 166.Roth DM, et al. Adenylyl cyclase increases survival in cardiomyopathy. Circulation. 2002;105(16):1989–94. doi: 10.1161/01.cir.0000014968.54967.d3. [DOI] [PubMed] [Google Scholar]
- 167.Takahashi T, et al. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation. 2006;114(5):388–96. doi: 10.1161/CIRCULATIONAHA.106.632513. [DOI] [PubMed] [Google Scholar]
- 168.Tang T, et al. Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation. 2008;117(1):61–9. doi: 10.1161/CIRCULATIONAHA.107.730069. [DOI] [PubMed] [Google Scholar]
- 169.Guellich A, et al. Effects of Cardiac Overexpression of Type 6 Adenylyl Cyclase Affects on the Response to Chronic Pressure Overload. Am J Physiol Heart Circ Physiol. 2010 doi: 10.1152/ajpheart.00148.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tang T, et al. Adenylyl cyclase 6 deletion reduces left ventricular hypertrophy, dilation, dysfunction, and fibrosis in pressure-overloaded female mice. J Am Coll Cardiol. 2010;55(14):1476–86. doi: 10.1016/j.jacc.2009.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lai NC, et al. Activation of cardiac adenylyl cyclase expression increases function of the failing ischemic heart in mice. J Am Coll Cardiol. 2008;51(15):1490–7. doi: 10.1016/j.jacc.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Longo VD, Finch CE. Evolutionary medicine: from dwarf model systems to healthy centenarians? Science. 2003;299(5611):1342–6. doi: 10.1126/science.1077991. [DOI] [PubMed] [Google Scholar]
- 173.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289(5487):2126–8. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 174.Wang Y, Tissenbaum HA. Overlapping and distinct functions for a Caenorhabditis elegans SIR2 and DAF-16/FOXO. Mech Ageing Dev. 2006;127(1):48–56. doi: 10.1016/j.mad.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 175.Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410(6825):227–30. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 176.Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004;101(45):15998–6003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Guarente L. Calorie restriction and SIR2 genes--towards a mechanism. Mech Ageing Dev. 2005;126(9):923–8. doi: 10.1016/j.mad.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 178.Chen D, et al. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310(5754):1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- 179.Yan L, et al. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007;130(2):247–58. doi: 10.1016/j.cell.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 180.Yan L, Williams JG, Dillinger J, Lai L, Ge H, Sadoshima J, Vatner DE, Vatner SF. Abstract 1625: Type 5 Adenylyl Cyclase Disruption Enhances Exercise Capacity Not Due to Improved Cardiac Output, but Rather to Resistance to Oxidative Stress in Skeletal Muscle. Circulation. 2009;120:S532. [Google Scholar]
- 181.Ho DZX, Stanley WC, Gao S, Fritzky LF, Burnett EJ, Vatner DE, Meggs LG, Malhotra A, Iwatsubo K, Vatner SF. Abstract: Inhibition of Adenylyl Cyclase Type 5, as a Novel Therapeutic Approach for Obesity and Diabetes. American Diabetes Association Scientific Sessions. 2010;2010 [Google Scholar]
- 182.Li M, et al. Down-regulation of manganese-superoxide dismutase through phosphorylation of FOXO3a by Akt in explanted vascular smooth muscle cells from old rats. J Biol Chem. 2006;281(52):40429–39. doi: 10.1074/jbc.M606596200. [DOI] [PubMed] [Google Scholar]
- 183.Bakalyar HA, Reed RR. Identification of a specialized adenylyl cyclase that may mediate odorant detection. Science. 1990;250(4986):1403–6. doi: 10.1126/science.2255909. [DOI] [PubMed] [Google Scholar]
- 184.Feinstein PG, et al. Molecular cloning and characterization of a Ca2+/calmodulin-insensitive adenylyl cyclase from rat brain. Proc Natl Acad Sci U S A. 1991;88(22):10173–7. doi: 10.1073/pnas.88.22.10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gao BN, Gilman AG. Cloning and expression of a widely distributed (type IV) adenylyl cyclase. Proc Natl Acad Sci U S A. 1991;88(22):10178–82. doi: 10.1073/pnas.88.22.10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ishikawa Y, et al. Isolation and characterization of a novel cardiac adenylylcyclase cDNA. J Biol Chem. 1992;267(19):13553–7. [PubMed] [Google Scholar]
- 187.Katsushika S, et al. Cloning and characterization of a sixth adenylyl cyclase isoform: types V and VI constitute a subgroup within the mammalian adenylyl cyclase family. Proc Natl Acad Sci U S A. 1992;89(18):8774–8. doi: 10.1073/pnas.89.18.8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Premont RT, et al. Two members of a widely expressed subfamily of hormone-stimulated adenylyl cyclases. Proc Natl Acad Sci U S A. 1992;89(20):9809–13. doi: 10.1073/pnas.89.20.9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yoshimura M, Cooper DM. Cloning and expression of a Ca(2+)-inhibitable adenylyl cyclase from NCB-20 cells. Proc Natl Acad Sci U S A. 1992;89(15):6716–20. doi: 10.1073/pnas.89.15.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Cali JJ, et al. Type VIII adenylyl cyclase. A Ca2+/calmodulin-stimulated enzyme expressed in discrete regions of rat brain. J Biol Chem. 1994;269(16):12190–5. [PubMed] [Google Scholar]
- 191.Watson PA, et al. Molecular cloning and characterization of the type VII isoform of mammalian adenylyl cyclase expressed widely in mouse tissues and in S49 mouse lymphoma cells. J Biol Chem. 1994;269(46):28893–8. [PubMed] [Google Scholar]
- 192.Paterson JM, et al. Control of a novel adenylyl cyclase by calcineurin. Biochem Biophys Res Commun. 1995;214(3):1000–8. doi: 10.1006/bbrc.1995.2385. [DOI] [PubMed] [Google Scholar]
- 193.Londos C, Wolff J. Two distinct adenosine-sensitive sites on adenylate cyclase. Proc Natl Acad Sci U S A. 1977;74(12):5482–6. doi: 10.1073/pnas.74.12.5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Holgate ST, Lewis RA, Austen KF. Role of adenylate cyclase in immunologic release of mediators from rat mast cells: agonist and antagonist effects of purine- and ribose-modified adenosine analogs. Proc Natl Acad Sci U S A. 1980;77(11):6800–4. doi: 10.1073/pnas.77.11.6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Johnson RA, et al. Isozyme-dependent sensitivity of adenylyl cyclases to P-site-mediated inhibition by adenine nucleosides and nucleoside 3′-polyphosphates. J Biol Chem. 1997;272(14):8962–6. doi: 10.1074/jbc.272.14.8962. [DOI] [PubMed] [Google Scholar]
- 196.Gille A, et al. Differential inhibition of adenylyl cyclase isoforms and soluble guanylyl cyclase by purine and pyrimidine nucleotides. J Biol Chem. 2004;279(19):19955–69. doi: 10.1074/jbc.M312560200. [DOI] [PubMed] [Google Scholar]
- 197.Rottlaender D, et al. Functional adenylyl cyclase inhibition in murine cardiomyocytes by 2′(3′)-O-(N-methylanthraniloyl)-guanosine 5′-[gamma-thio]triphosphate. J Pharmacol Exp Ther. 2007;321(2):608–15. doi: 10.1124/jpet.106.118422. [DOI] [PubMed] [Google Scholar]
- 198.Gottle M, et al. Characterization of mouse heart adenylyl cyclase. J Pharmacol Exp Ther. 2009;329(3):1156–65. doi: 10.1124/jpet.109.150953. [DOI] [PubMed] [Google Scholar]
- 199.Iwatsubo K, et al. Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J Biol Chem. 2004;279(39):40938–45. doi: 10.1074/jbc.M314238200. [DOI] [PubMed] [Google Scholar]
- 200.Levy DE, et al. Metal coordination-based inhibitors of adenylyl cyclase: novel potent P-site antagonists. J Med Chem. 2003;46(11):2177–86. doi: 10.1021/jm0205604. [DOI] [PubMed] [Google Scholar]
- 201.Levy D, et al. Hydroxamate based inhibitors of adenylyl cyclase. Part 2: the effect of cyclic linkers on P-site binding. Bioorg Med Chem Lett. 2002;12(21):3089–92. doi: 10.1016/s0960-894x(02)00654-6. [DOI] [PubMed] [Google Scholar]
- 202.Levy D, et al. Hydroxamate based inhibitors of adenylyl cyclase. Part 1: the effect of acyclic linkers on P-site binding. Bioorg Med Chem Lett. 2002;12(21):3085–8. doi: 10.1016/s0960-894x(02)00653-4. [DOI] [PubMed] [Google Scholar]
- 203.Ren J, et al. Interaction between age and obesity on cardiomyocyte contractile function: role of leptin and stress signaling. PLoS One. 2010;5(4):e10085. doi: 10.1371/journal.pone.0010085. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 204.Li Q, Ren J. Influence of cardiac-specific overexpression of insulin-like growth factor 1 on lifespan and aging-associated changes in cardiac intracellular Ca2+ homeostasis, protein damage and apoptotic protein expression. Aging Cell. 2007;6(6):799–806. doi: 10.1111/j.1474-9726.2007.00343.x. [DOI] [PubMed] [Google Scholar]