

Abstract
Diabetic nephropathy (DN) is the leading cause of end-stage kidney disease worldwide. However, current treatments remain suboptimal. Many factors, such as genetic and nongenetic promoters, hypertension, hyperglycemia, the accumulation of advanced glycation end products (AGEs), dyslipidemia, and albuminuria/proteinuria itself, influence the progression of this disease. It is important to determine the molecular mechanisms and treatment of this disease. The development of diabetes results in the formation of AGEs, oxidative stress, and the activation of the renin-angiotensin-aldosterone system (RAAS) within the kidney, which promotes progressive inflammation and fibrosis, leading to DN and declining renal function. A number of novel therapies have also been tested in the experimental diabetic model, including exercise, inhibitors of the RAAS (angiotensin type 1 receptor blockers (ARB), angiotensin-converting enzyme (ACE) inhibitors), inhibitors of AGE (pyridoxamine), peroxisome proliferator-activated receptor (PPAR) γ agonists (pioglitazone), inhibitors of lipid accumulation (statins and eicosapentaenoic acid (EPA)), and the vitamin D analogues. This review summarizes the advances in knowledge gained from our studies and therapeutic interventions that may prevent this disease.
1. Introduction
Diabetes mellitus is a major cause of chronic kidney disease (CKD) worldwide [1] and is associated with enhanced morbidity and mortality, particularly with accelerated cardiovascular disease [2].
Current approaches to the prevention of diabetic nephropathy (DN) include the strict control of blood glucose and blood pressure. The strict control of blood glucose, as quickly as possible, was shown to be effective in major clinical trials [3, 4]. Blood pressure control has also been shown to be of major importance in many studies [5–7]. In addition, in patients who develop increased urinary albumin-creatinine ratio (ACR) levels, one or more of the medications that inhibit the renin-angiotensin-aldosterone system (RAAS) axis should be used to lower ACR levels [8, 9]. These treatments remain suboptimal, however, so much more research is needed to determine other specific pathophysiologic mechanisms in order to develop more treatments that are targeted specifically to identified mechanisms.
The pathogenesis of DN appears to be multifactorial. Several genetic and environmental factors likely contribute to its development and progression [10]. Diabetes induces the formation of advanced glycation end products (AGEs), which can alter the function of proteins and stimulate pathological cellular responses via AGE receptors. Increasing levels of AGEs, and their deposition in diabetic kidneys, correlate with the development of DN [11]. Of the pathophysiologic mechanisms that have been identified in the development and progression of DN, oxidative stress (more accurately described as increased levels of reactive oxygen species; ROS) is of major importance [12]. Recent studies have shown that kidney inflammation is crucial in promoting the development and progression of DN. Inflammation, which is activated by the metabolic, biochemical, and hemodynamic derangements known to exist in the diabetic kidney, may be a key factor [13]. DN progresses in stages, starting with the thickening of the glomerular basement membrane, mesangial cell expansion, and then gradually progressing into glomerulosclerosis and interstitial fibrosis eventually resulting in renal failure [14]. It has been postulated that the relationship between AGE effects, oxidative stress, RAAS activation, inflammation, and fibrosis pathways plays an important role in the development and progression of DN (Figure 1).
Figure 1.
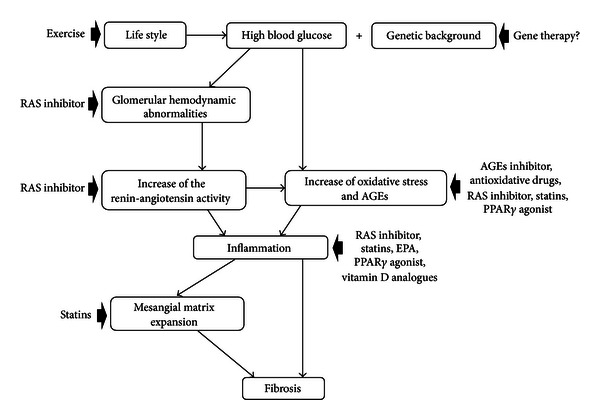
Major mechanistic pathways of diabetic renal injury identified. Various factors contribute to the progression of diabetic nephropathy, as shown in this slide, and there is crosstalk between these factors.
This review focuses on potential targets for new renoprotective therapies from our data in addition to the inhibition of the RAAS in DN.
2. Characteristics of KK and KK-Ay Mouse
The KK mouse is an inbred mouse strain established from Japanese native mice. This mouse spontaneously exhibits type 2 diabetes, associated with mild hyperglycemia, mild glucose intolerance, mild hyperinsulinemia, mild obesity, and mild microalbuminuria. Renal lesions in KK mice closely resemble those in human diabetic nephropathy. Young KK mice are considered to be suitable model for the study of the early stage of type 2 DN in humans [15].
The KK-Ay mouse line was established in 1969, and these mice are widely used as an experimental model for type 2 diabetes mellitus. KK-Ay mice spontaneously exhibit type 2 diabetes mellitus signs, including hyperglycemia, glucose intolerance, hyperinsulinemia, obesity, and microalbuminuria. The mice also develop renal lesions that show diffuse hyperplasia of the mesangial area with mesangial cell (MC) proliferation, segmental sclerosis, overexpression of TGF-β1, and the accumulation of AGEs and ROS products [16–18].
3. Treatment of Diabetic Nephropathy
3.1. The Effect of Renin-Angiotensin-Aldosterone System Inhibitors
Angiotensin II (Ang II) exerts both hemodynamic effects, leading to increased glomerular capillary pressure, and nonhemodynamic effects such as cellular hypertrophy stimulation and extracellular matrix (ECM) accumulation. These effects are mediated through the interaction of Ang II with its angiotensin type 1 (AT1) receptor. Angiotensin-converting enzyme (ACE) inhibitors and AT1 receptor blockers (ARB) have been demonstrated to improve glomerular hemodynamics and structures in both human and experimental DN [19–21]. Our study demonstrated that treatment with candesartan, an ARB, reduced blood pressure and mesangial ECM accumulation and reduced ACR and type IV collagen excretion without altering glucose metabolism [22]. Many studies have reported that high glucose and Ang II stimulate collagen production by TGF-β activation [23, 24]. TGF-β is an important mediator of fibrosis in the repair tissues. Smad7 is generally considered to be a TGF-β signaling inhibitor in mature T cells. In our study, TGF-β expression by immunohistochemistry in glomeruli was markedly increased in the mild diabetic model of KK mice. Candesartan administration significantly reduced TGF-β expression. Our data also demonstrated that candesartan treatment led to an increase in glomerular Smad7 expression [22]. It appears that these protective effects of candesartan were associated with lower glomerular hydraulic pressure, reduced TGF-β expression, and increased Smad7 expression. These data supported the previous data that the TGF-β/Smad signal system could play an important role in the development and progression of DN in KK mice [15].
Oxidative and nitrosative stresses are widely recognized as key factors in the development of DN. We demonstrated that nitrooxidative stress and AGE production are enhanced in the kidneys of KK mice. Candesartan decreased nitrooxidative stress by downregulating NAD(P)H oxidase p37phox and iNOS expression, and modified interaction between AGEs and RAGE by attenuating RAGE expression, contributing to the reduction of AGE accumulation and subsequent albuminuria [15, 25].
Evidence has accumulated over the past few years indicating that adenosine monophosphate activated protein kinase (AMPK) may be a useful target for the pharmacologic treatment of type 2 diabetes. The actions of AMPK were initially defined as the regulation of fatty acid and cholesterol synthesis pathways [26]. In parallel with their activation of AMPK, antidiabetic adipokines, that is, adiponectin, stimulate phosphorylation of acetyl CoA carboxylase (ACC), fatty acid oxidation, glucose uptake, and lactate production. Our data demonstrated that enalapril and/or losartan improved the urinary ACR levels through the activation of adiponectin and AMPK in the kidneys of KK-Ay mice [27]. These results suggested that the RAS inhibitors activated renal AMPK through its phosphorylation. Therefore, the effects of ACE inhibitors and/or ARBs, especially in combination treatment, might be associated with tissue-specific adiponectin-AMPK activity.
3.2. Effect of Angiotensin-(1–7)
There is emerging evidence that in DN, the generation of ROS is a major factor in the development of diabetes and its associated complications [28, 29]. NAD(P)H oxidase is an enzymatic complex that is responsible for ROS production. Ang II-mediated ROS is an important second messenger for the transcriptional effects of Ang II, and NAD(P)H oxidase is the central enzyme complex of Ang II-induced ROS [30, 31]. The recent discovery of the renal RAS [28, 29], ACE carboxypeptidase (ACE2), and Ang-(1–7) has changed the way the RAS is viewed. Ang-(1–7) is a biologically active heptapeptide component of the RAS and is generated in the kidney at relatively high levels via enzymatic pathways that include ACE2. The biological effects of Ang-(1–7) in the kidney are primarily mediated by their interaction with the G-protein-coupled receptor Mas [32]. We found that mice coinfused with Ang II + Ang-(1–7) had a lower increase in urinary ACR than mice infused with Ang II alone. In this animal model, Ang-(1–7) attenuated Ang II-mediated NAD(P)H oxidase activation and ROS production in diabetic glomeruli and MCs. These findings were related to improved mesangial expansion and the production of fibronectin and TGF-β1 in the diabetic kidney and cultured MCs as opposed to Ang II. We also found that Ang II-induced NF-κB and MAPK activation was attenuated by Ang-(1–7) in the MCs [18]. Our findings suggest that Ang-(1–7) may attenuate Ang II-stimulated NAD(P)H dependent ROS-mediated renal injury in diabetes. The ACE2-Ang-(1–7)-Mas receptor axis should be investigated further as a novel target of treatment of DN.
3.3. Effect of Pyridoxamine
Chronic hyperglycemia promotes the generation of AGEs as a result of sequential biochemical reactions involving the nonenzymatic glycation of protein and lipids [33]. The formation of AGEs occurs in normal aging, but it is accelerated in the diabetic state. AGEs increase the expression of growth factors and cytokines, including TGF-β, connective tissue growth factor (CTGF), and vascular endothelial growth factor (VEGF) [34]. AGEs can induce the expression of the monocyte chemoattractant protein-1 (MCP-1) in podocytes through the activation of the AGE receptor (RAGE) and the generation of intracellular ROS [35]. Eventually, these products cause glomerular and tubulointerstitial injury. In therapeutic interventions for reducing AGEs, many compounds have been reported as AGE inhibitors, such as aminoguanidine, phenacylthiazolium bromide, 2-isopropylidenehydrazono-4-oxo-thiazolidine-5-yl-acetanilide (OPB-9195), 2,3-diaminophenazine, vitamin C, vitamin E, Ang II receptor inhibitor, and pyridoxamine [25, 36–38]. Pyridoxamine was introduced by Khalifah et al. [39, 40] as an inhibitor of AGE formation from Amadori products [41]. The effects of pyridoxamine include (a) the inhibition of AGE formation by blocking the oxidative degradation of the Amadori intermediate of the Millard reaction, (b) the scavenging of toxic carbonyl products of glucose and lipid degradation, and (c) the trapping of ROS [42]. We demonstrated that pyridoxamine (K-163) ameliorates the levels of urinary ACR and serum 3-deoxyglucosone (3DG) in KK-Ay mice without changing systemic blood pressure. Furthermore, pyridoxamine prevented accumulations of Nq-(carboxymethyl)lysine (CML), nitrotyrosine, TGF-β1, and laminin-β1 in the kidney tissues [41]. AGEs and oxidative stress might activate autocrine Ang II signaling and subsequently induce TGF-β1-Smad signaling in mesangial cells [43, 44]. Our findings suggested that the amelioration of urinary ACR was related to the improvement of TGF-β1 and laminin-β1 expressions in the kidney because CML and nitrotyrosine accumulations were improved and the levels of serum 3DG were reduced by anti-AGE and/or the antioxidant effects of pyridoxamine.
3.4. Effect of Pioglitazone
Thiazolidinediones (TZDs), PPARγ agonists, such as troglitazone, pioglitazone, and rosiglitazone, are insulin-sensitizing agents [45]. It is generally considered that these drugs have preventive effects on impaired glucose tolerance (IGT) and urinary ACR excretion in diabetics [46–48]. In the kidney, PPARγ messages were localized predominantly in the inner medullary collecting ducts and renal medullary interstitial cells but not in the cortex [49]. However, previous reports have shown that TZDs ameliorate renal microcirculation, glomerular hyperfiltration, and mesangial expansion in DN [50–52]. The increase of renal perfusion and glomerular filtration rate (GFR) occurs early in the course of DN. Nitric oxide (NO) might be one of the causes of glomerular hyperfiltration [53]. Veelken et al. [54] reported that early glomerular hyperfiltration was dependent on increased NO generation due to greater expression and activity of endothelial constitutive NOS (ecNOS) in glomeruli and afferent arterioles in untreated hyperfiltrating diabetic rats. Therefore, ecNOS might be a more important hemodynamic factor in the early stage of DN. We demonstrated that pioglitazone, one of the TZDs, ameliorates urinary ACR and IGT in diabetic KK mice without changing systemic blood pressure and blood glucose levels. We localized ecNOS protein in the endothelium of preglomerular arteries, arterioles, and glomerular tufts. Moreover, this positive staining in KK mice treated with pioglitazone was less than that in control mice. It appears that the decrease of urinary ACR excretion might be related to the improvement of glomerular enlargement, including hyperfiltration, since the levels of ecNOS protein were reduced by pioglitazone in the glomerular vessels [55].
3.5. Effect of Eicosapentaenoic Acid
Eicosapentaenoic acid (EPA) is one of the n-3 polyunsaturated fatty acids (PUFA) which are contained in fish oil. It was shown that EPA has many effects, such as antithrombotic, hypolipidemic, antiatherogenic, anti-inflammatory, and antimitogenic actions. The feeding of fish oil rich in n-3 PUFA reduces the ex vivo production of interleukin-1 (IL-1), IL-6, tumour necrosis factor (TNF), and IL-2 by peripheral blood mononuclear cells and reduces the response to endotoxin and to proinflammatory cytokines, resulting in increased survival. MCP-1 is the strongest known chemokine, which has the function of recruiting and activating monocytes/macrophages from the circulation to inflammatory sites. Macrophages and its products play an important pathogenic role in the tubulointerstitial inflammatory and noninflammatory conditions and have been implicated as effector cells of tubulointerstitial damage and mesangial matrix accumulation in DN [56]. We demonstrated that EPA, one of the n-3PUFA, ameliorates urinary ACR and MCP-1 levels and attenuates mesangial matrix accumulation and tubulointerstitial fibrosis in KK-Ay mice without changing systemic blood pressure and fasting blood glucose levels. Moreover, EPA ameliorates IGT and hypertriglyceridemia and lowers leptin levels in KK-Ay mice. Because MCP-1 induces monocyte immigration and differentiation to macrophages, which augment extracellular matrix production and tubulointerstitial fibrosis, and also because it directly induces tubulointerstitial inflammation and vascular damage in the kidney, we propose that the observed downregulation of MCP-1 is critically involved in the beneficial effect of EPA, probably in concert with the improvement of other clinical parameters. The potential of EPA in the treatment of DN might be of particular relevance to patients with comorbidities such as dyslipidaemia and obesity [57].
3.6. Effect of Statins
The 3-hydroxy-3-methylglutaryl-coenzyme A (HMGCoA) reductase inhibitors (statins) have pleiotropic effects on cardiovascular, cerebrovascular, and microvascular diseases independent of their cholesterol-lowering effect [58, 59]. Statins also have beneficial effects on kidney disease, including DN. Previous reports have shown the pleiotropic effects of statins, such as their anti-inflammatory effects and antioxidative stress effects in vitro and in vivo [58, 60]. Mechanisms of improvement of urinary ACR by statin treatment have been proposed in some reports [59, 61, 62]. These reports have stated that statins improved the urinary ACR of diabetic rats through an anti-inflammatory effect and/or through the inhibition of macrophage recruitment and activation and also by the inhibition of TGF-β overexpression. We suggested that oxidative stress and nitrotyrosine are related to the progression of DN [41]. Oxidative stress is defined as a tissue injury induced by an increase of ROS, such as the hydroxyl radical, superoxide anion, or hydrogen peroxide. Thus, oxidative stress is considered to be one of the factors involved in the development of diabetic complications [63, 64]. Isoprenoids, such as farnesyl pyrophosphate or geranylgeranyl pyrophosphate, are generated from HMG-CoA through mevalonate depletion. Isoprenoids inhibit the generation of eNOS and GTPCH-1, and they also increase NAD(P)H oxidase through the inhibition of the Rho pathway and activation of Rac-1. Since pitavastatin inhibits HMGCoA reductase and blocks synthesis of the isoprenoids, the generation of NAD(P)H oxidase is inhibited and signals to generate eNOS are upregulated. Furthermore, statins activate GTPCH-1 and lead to the upregulation of BH4, which is essential for eNOS to form a dimer. As a result, pitavastatin activates eNOS dimerization and enforces their stability through this cascade. As the monomerization of eNOS, which involves NO and ROS imbalance, is decreased, oxidative stress is decreased. Moreover, upregulated dimeric eNOS acts as an NO generator and may work against shear stress in the early stage of DN [17]. We demonstrated that pitavastatin improves the levels of urinary ACR, urinary 8-OHdG, and insulin resistance in KK-Ay mice independent of cholesterol-lowering effect. Furthermore, pitavastatin prevented the accumulation of monomeric eNOS, nitrotyrosine, and p47 phox in kidney tissues. It appears that pitavastatin improved not only urinary ACR but also HbA1c and impaired glucose tolerance in KK-Ay mice, which might be because of the suppression of eNOS uncoupling and its antioxidant effects on DN.
3.7. Effect of Vitamin D Analogues
The natural activator of the vitamin D receptor, calcitriol, is produced by the kidney, but plasma concentrations decline as estimated GFR (eGFR) reduces [65]. In a multivariable analysis of patients with chronic kidney disease (CKD), lower calcitriol concentrations strongly correlated with higher risk of diabetes, higher urinary ACR, and lower eGFR [65]. Calcitriol, 1,25-dihydroxyvitamin D 3 (1,25(OH) 2 D 3), and its analogs have been shown to attenuate renal diseases [66–68]. For example, the knockout of the vitamin D receptor in diabetic mice was associated with severe albuminuria and glomerulosclerosis from increased thickening of the glomerular basement membrane (GBM) and podocyte effacement [69]. 1,25(OH) 2 D 3 is a negative endocrine regulator of RAS and suppresses renin biosynthesis [70, 71]. These studies provide the molecular basis for exploring the potential of 1,25(OH) 2 D 3 to regulate the RAS by inhibition of renin [70]. We investigated the effect of therapy with 1,25(OH) 2 D 3 upon DN in KK-Ay mice. It appears that therapy with 1,25(OH) 2 D 3 reduced the urinary ACR level by suppressing the compensatory renin increase in type 2 DN. These beneficial effects might be related to suppressed renal expression of renin, ERK1/2, and TGF-β which may or may not be Ang II dependent [72]. Moreover, a recent trial reported antialbuminuric effects of another analogue, paricalcitol, further strengthening the evidence for vitamin D analogues as renoprotective agents [73]. Paricalcitol lowers residual albuminuria in patients with DN and could be a novel approach to lower residual renal risk in diabetes.
3.8. Effect of Exercise
Lifestyle modification, especially appropriate exercise, is recommended for the management of type 2 DN through improvements of metabolic risk factors such as blood pressure, blood glucose, plasma lipids, and oxidative stress markers. On the other hand, appropriate exercise also consumes considerable amounts of oxygen, leading to the production of high levels of ROS. There is also evidence that ROS and high glucose exposure contribute to podocyte apoptosis in experimental DN [74]. There are several mechanisms for the renoprotective effects of exercise in DN. In general, exercise training ameliorates renal function by improving metabolic factors such as plasma lipids, blood glucose, blood pressure, and body weight. It is also known to improve renal histology without altering metabolic factors. Boor et al. [75] demonstrated that exercise training reduced AGEs in both serum and kidney tissues of obese Zucker rats, an animal model of type 2 diabetes, without altering inflammatory biomarkers or metabolic factors. In contrast, our study clearly showed that the exercised mice showed attenuated renal expression of MCP-1 and infiltration of macrophage in the kidneys. We demonstrated that the exercise training improved urinary NAG levels as well as the change rate of urinary ACR, independent of body weight and glycemic status in the kidneys of KK-Ay mice, although moderate-intensity exercise increased expression of HIF-1α in the kidneys. In our study, no significant changes were observed in the levels of Ccr between sedentary KK-Ay and exercised KK-Ay mice. Therefore, it is indicated that the decrease of urinary ACR was not due to the reduction of renal blood flow/glomerular filtration rate, but more likely to the effect of exercise. It is thought that appropriate exercise increases antioxidant enzymes, although excessive exercise causes inflammation, increases oxidative stress associated with ROS, and decreases the renal blood flow and glomerular filtration rate. In our study, both exercises decreased urinary 8-OHdG levels, an oxidative stress marker. However, contrary to our expectation, low-intensity exercise was more effective than moderate-intensity exercise in terms of renal function. Further investigation is required to determine appropriate exercise intensity. It appears that low-intensity exercise attenuates the progression of early DN without affecting marked renal ischemia. Thus, attention should be paid to renal ischemia even though albuminuria has improved. Reductions in the rate of urinary ACR change, urinary NAG, and maintained podocyte numbers, with parallel improvements in oxidative damage and chronic inflammation, might be related to beneficial effects of exercise in DN [76].
4. Conclusion
Despite the successful use of lifestyle changes, metabolic control, and blood pressure control, including ACE inhibitors and ARB therapy, residual renal risk remains very high, leaving the diabetic population with a clear unmet need for novel treatment options. As outlined in this review, various drugs are in development. It is anticipated that some of the newer agents that are currently the focus of clinical trials will ultimately lead to improvements in slowing the progression and eventually improving the prognosis of this devastating disease.
Conflict of Interests
The authors declare no conflict of interests.
Acknowledgments
The authors thank S. Horikoshi, Ph.D., K. Funabiki, Ph.D., Y. Makita, Ph.D., T. Ito, Ph.D., S. Hagiwara, Ph.D., T. Yamazaki, Ph.D., I. Ohara, Ph.D., M. Murakoshi, Ph.D., M. Matsumoto, Ph.D., T. Aoki, Ph.D., Y. Ishikawa, Ph.D., and J. Y. Moon, Ph.D., for their support.
References
- 1.Stenvinkel P. Chronic kidney disease: a public health priority and harbinger of premature cardiovascular disease. Journal of Internal Medicine. 2010;268(5):456–467. doi: 10.1111/j.1365-2796.2010.02269.x. [DOI] [PubMed] [Google Scholar]
- 2.Sarnak MJ, Levey AS, Schoolwerth AC, et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation. 2003;108(17):2154–2169. doi: 10.1161/01.CIR.0000095676.90936.80. [DOI] [PubMed] [Google Scholar]
- 3.Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. Journal of the American Medical Association. 2003;290(16):2159–2167. doi: 10.1001/jama.290.16.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA, Holman RR. Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64) Kidney International. 2003;63(1):225–232. doi: 10.1046/j.1523-1755.2003.00712.x. [DOI] [PubMed] [Google Scholar]
- 5.Bakris GL, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working Group. American Journal of Kidney Diseases. 2000;36(3):646–661. doi: 10.1053/ajkd.2000.16225. [DOI] [PubMed] [Google Scholar]
- 6.Schrier RW, Estacio RO, Esler A, Mehler P. Effects of aggressive blood pressure control in normotensive type 2 diabetic patients on albuminuria, retinopathy and strokes. Kidney International. 2002;61(3):1086–1097. doi: 10.1046/j.1523-1755.2002.00213.x. [DOI] [PubMed] [Google Scholar]
- 7.Shankar A, Klein R, Klein BEK, Nieto FJ, Moss SE. Relationship between low-normal blood pressure and kidney disease in type 1 diabetes. Hypertension. 2007;49(1):48–54. doi: 10.1161/01.HYP.0000252431.75154.3a. [DOI] [PubMed] [Google Scholar]
- 8.Remuzzi G, Ruggenenti P, Perna A, et al. Continuum of renoprotection with losartan at all stages of type 2 diabetic nephropathy: a post hoc analysis of the RENAAL trial results. Journal of the American Society of Nephrology. 2004;15(12):3117–3125. doi: 10.1097/01.ASN.0000146423.71226.0C. [DOI] [PubMed] [Google Scholar]
- 9.Ritz E, Orth SR. Nephropathy in patients with type 2 diabetes mellitus. The New England Journal of Medicine. 1999;341(15):1127–1133. doi: 10.1056/NEJM199910073411506. [DOI] [PubMed] [Google Scholar]
- 10.Aoki T, Kaneko S, Tanimoto M, et al. Identification of quantitative trait loci for diabetic nephropathy in KK-Ay/Ta mice. Journal of Nephrology. 2012;25(1):127–136. doi: 10.5301/JN.2011.8433. [DOI] [PubMed] [Google Scholar]
- 11.Tesch GH, Lim AKH. Recent insights into diabetic renal injury from the db/db mouse model of type 2 diabetic nephropathy. American Journal of Physiology. 2011;300(2):F301–F310. doi: 10.1152/ajprenal.00607.2010. [DOI] [PubMed] [Google Scholar]
- 12.Stanton RC. Oxidative stress and diabetic kidney disease. Current Diabetes Reports. 2011;11(4):330–336. doi: 10.1007/s11892-011-0196-9. [DOI] [PubMed] [Google Scholar]
- 13.Lim AK, Tesch GH. Inflammation in diabetic nephropathy. Mediators of Inflammation. 2012;2012:12 pages. doi: 10.1155/2012/146154.146154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karihaloo A. Anti-fibrosis therapy and diabetic nephropathy. Current Diabetes Reports. 2012;12(4):414–422. doi: 10.1007/s11892-012-0290-7. [DOI] [PubMed] [Google Scholar]
- 15.Tomino Y, Tanimoto M, Shike T, et al. Pathogenesis and treatment of type 2 diabetic nephropathy: lessons from the spontaneous KK/Ta mouse model. Current Diabetes Reviews. 2005;1(3):281–286. doi: 10.2174/157339905774574374. [DOI] [PubMed] [Google Scholar]
- 16.Ito T, Tanimoto M, Yamada K, et al. Glomerular changes in the KK-Ay/Ta mouse: a possible model for human type 2 diabetic nephropathy. Nephrology. 2006;11(1):29–35. doi: 10.1111/j.1440-1797.2006.00543.x. [DOI] [PubMed] [Google Scholar]
- 17.Matsumoto M, Tanimoto M, Gohda T, et al. Effect of pitavastatin on type 2 diabetes mellitus nephropathy in KK-Ay/Ta mice. Metabolism. 2008;57(5):691–697. doi: 10.1016/j.metabol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 18.Moon JY, Tanimoto M, Gohda T, et al. Attenuating effect of angiotensin-(1-7) on angiotensin II-mediated NAD(P)H oxidase activation in type 2 diabetic nephropathy of KK-Ay/Ta mice. American Journal of Physiology. 2011;300(6):F1271–F1282. doi: 10.1152/ajprenal.00065.2010. [DOI] [PubMed] [Google Scholar]
- 19.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. The New England Journal of Medicine. 1993;329(20):1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 20.Andersen S, Tarnow L, Rossing P, Hansen BV, Parving HH. Renoprotective effects of angiotensin II receptor blockade in type 1 diabetic patients with diabetic nephropathy. Kidney International. 2000;57(2):601–606. doi: 10.1046/j.1523-1755.2000.00880.x. [DOI] [PubMed] [Google Scholar]
- 21.Hong SW, Isono M, Chen S, Iglesias-De La Cruz MC, Han DC, Ziyadeh FN. Increased glomerular and tubular expression of transforming growth factor-β1, its type II receptor, and activation of the smad signaling pathway in the db/db mouse. American Journal of Pathology. 2001;158(5):1653–1663. doi: 10.1016/s0002-9440(10)64121-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lioa J, Kobayashi M, Kanamuru Y, et al. Effects of candesartan, an angiotensin II type 1 receptor blocker, on diabetic nephropathy in KK/Ta mice. Journal of Nephrology. 2003;16(6):841–849. [PubMed] [Google Scholar]
- 23.Isono M, Iglesias-De La Cruz MC, Chen S, Hong SW, Ziyadeh FN. Extracellular signal-regulated kinase mediates stimulation of TGF-β1 and matrix by high glucose in mesangial cells. Journal of the American Society of Nephrology. 2000;11(12):2222–2230. doi: 10.1681/ASN.V11122222. [DOI] [PubMed] [Google Scholar]
- 24.Kagami S, Border WA, Miller DE, Noble NA. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-β expression in rat glomerular mesangial cells. Journal of Clinical Investigation. 1994;93(6):2431–2437. doi: 10.1172/JCI117251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan Q, Liao J, Kobayashi M, et al. Candesartan reduced advanced glycation end-products accumulation and diminished nitro-oxidative stress in type 2 diabetic KK/Ta mice. Nephrology Dialysis Transplantation. 2004;19(12):3012–3020. doi: 10.1093/ndt/gfh499. [DOI] [PubMed] [Google Scholar]
- 26.Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Letters. 1987;223(2):217–222. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- 27.Yamazaki T, Tanimoto M, Gohda T, et al. Combination effects of enalapril and losartan on lipid peroxidation in the kidneys of KK-Ay/Ta mice. Nephron. 2009;113(2):e66–e76. doi: 10.1159/000228714. [DOI] [PubMed] [Google Scholar]
- 28.Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40(4):405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- 29.Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999;48(1):1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- 30.Ritz E, Haxsen V. Angiotensin II and oxidative stress: an unholy alliance. Journal of the American Society of Nephrology. 2003;14(11):2985–2987. doi: 10.1097/01.asn.0000096784.86791.21. [DOI] [PubMed] [Google Scholar]
- 31.Wolf G. Free radical production and angiotensin. Current Hypertension Reports. 2000;2(2):167–173. doi: 10.1007/s11906-000-0078-z. [DOI] [PubMed] [Google Scholar]
- 32.Zimmerman D, Burns KD. Angiotensin-(1–7) in kidney disease: a review of the controversies. Clinical Science. 2012;123:333–346. doi: 10.1042/CS20120111. [DOI] [PubMed] [Google Scholar]
- 33.Cooper ME. Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia. 2001;44(11):1957–1972. doi: 10.1007/s001250100000. [DOI] [PubMed] [Google Scholar]
- 34.Wendt TM, Tanji N, Guo J, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. American Journal of Pathology. 2003;162(4):1123–1137. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu L, Hagiwara S, Fan Q, et al. Role of receptor for advanced glycation end-products and signalling events in advanced glycation end-product-induced monocyte chemoattractant protein-1 expression in differentiated mouse podocytes. Nephrology Dialysis Transplantation. 2006;21(2):299–313. doi: 10.1093/ndt/gfi210. [DOI] [PubMed] [Google Scholar]
- 36.Singh R, Barden A, Mori T, Beilin L. Advanced glycation end-products: a review. Diabetologia. 2001;44(2):129–146. doi: 10.1007/s001250051591. [DOI] [PubMed] [Google Scholar]
- 37.Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science. 1986;232(4758):1629–1632. doi: 10.1126/science.3487117. [DOI] [PubMed] [Google Scholar]
- 38.Nakamura S, Makita Z, Ishikawa S, et al. Progression of nephropathy in spontaneous diabetic rats is prevented by OPB-9195, a novel inhibitor of advanced glycation. Diabetes. 1997;46(5):895–899. doi: 10.2337/diab.46.5.895. [DOI] [PubMed] [Google Scholar]
- 39.Khalifah RG, Todd P, Booth AA, Yang SX, Mott JD, Hudson BG. Kinetics of nonenzymatic glycation of ribonuclease a leading to advanced glycation end products. Paradoxical inhibition by ribose leads to facile isolation of protein intermediate for rapid post-Amadori studies. Biochemistry. 1996;35(15):4645–4654. doi: 10.1021/bi9525942. [DOI] [PubMed] [Google Scholar]
- 40.Khalifah RG, Baynes JW, Hudson BG. Breakthroughs and views. Amadorins: novel post-Amadori inhibitors of advanced glycation reactions. Biochemical and Biophysical Research Communications. 1999;257(2):251–258. doi: 10.1006/bbrc.1999.0371. [DOI] [PubMed] [Google Scholar]
- 41.Tanimoto M, Gohda T, Kaneko S, et al. Effect of pyridoxamine (K-163), an inhibitor of advanced glycation end products, on type 2 diabetic nephropathy in KK-Ay/Ta mice. Metabolism. 2007;56(2):160–167. doi: 10.1016/j.metabol.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 42.Voziyan PA, Hudson BG. Pyridoxamine as a multifunctional pharmaceutical: targeting pathogenic glycation and oxidative damage. Cellular and Molecular Life Sciences. 2005;62(15):1671–1681. doi: 10.1007/s00018-005-5082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsuchida K, Makita Z, Yamagishi S, et al. Suppression of transforming growth factor beta and vascular endothelial growth factor in diabetic nephropathy in rats by a novel advanced glycation end product inhibitor, OPB-9195. Diabetologia. 1999;42(5):579–588. doi: 10.1007/s001250051198. [DOI] [PubMed] [Google Scholar]
- 44.Fukami K, Ueda S, Yamagishi SI, et al. AGEs activate mesangial TGF-β-Smad signaling via an angiotensin II type I receptor interaction. Kidney International. 2004;66(6):2137–2147. doi: 10.1111/j.1523-1755.2004.66004.x. [DOI] [PubMed] [Google Scholar]
- 45.Day C. Thiazolidinediones: a new class of antidiabetic drugs. Diabetic Medicine. 1999;16(3):179–192. doi: 10.1046/j.1464-5491.1999.00023.x. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz S, Raskin P, Fonseca V, Graveline JF. Effect of troglitazone in insulin-treated patients with type II diabetes mellitus. Troglitazone and Exogenous Insulin Study Group. The New England Journal of Medicine. 1998;338(13):861–866. doi: 10.1056/NEJM199803263381302. [DOI] [PubMed] [Google Scholar]
- 47.Imano E, Kanda T, Nakatani Y, et al. Effect of troglitazone on microalbuminuria in patients with incipient diabetic nephropathy. Diabetes Care. 1998;21(12):2135–2139. doi: 10.2337/diacare.21.12.2135. [DOI] [PubMed] [Google Scholar]
- 48.Fujii M, Takemura R, Yamaguchi M, et al. Troglitazone (CS-045) ameliorates albuminuria in streptozotocin-induced diabetic rats. Metabolism. 1997;46(9):981–983. doi: 10.1016/s0026-0495(97)90264-x. [DOI] [PubMed] [Google Scholar]
- 49.Yang T, Michele DE, Park J, et al. Expression of peroxisomal proliferator-activated receptors and retinoid X receptors in the kidney. American Journal of Physiology. 1999;277(6):F966–F973. doi: 10.1152/ajprenal.1999.277.6.F966. [DOI] [PubMed] [Google Scholar]
- 50.Arima S, Kohagura K, Takeuchi K, et al. Biphasic vasodilator action of troglitazone on the renal microcirculation. Journal of the American Society of Nephrology. 2002;13(2):342–349. doi: 10.1681/ASN.V132342. [DOI] [PubMed] [Google Scholar]
- 51.Isshiki K, Haneda M, Koya D, Maeda S, Sugimoto T, Kikkawa R. Thiazolidinedione compounds ameliorate glomerular dysfunction independent of their insulin-sensitizing action in diabetic rats. Diabetes. 2000;49(6):1022–1032. doi: 10.2337/diabetes.49.6.1022. [DOI] [PubMed] [Google Scholar]
- 52.McCarthy KJ, Routh RE, Shaw W, Walsh K, Welbourne TC, Johnson JH. Troglitazone halts diabetic glomerulosclerosis by blockade of mesangial expansion. Kidney International. 2000;58(6):2341–2350. doi: 10.1046/j.1523-1755.2000.00418.x. [DOI] [PubMed] [Google Scholar]
- 53.Bank N, Aynedjian HS. Role of EDRF (nitric oxide) in diabetic renal hyperfiltration. Kidney International. 1993;43(6):1306–1312. doi: 10.1038/ki.1993.183. [DOI] [PubMed] [Google Scholar]
- 54.Veelken R, Hilgers KF, Hartner A, Haas A, Böhmer KP, Sterzel RB. Nitric oxide synthase isoforms and glomerular hyperfiltration in early diabetic nephropathy. Journal of the American Society of Nephrology. 2000;11(1):71–79. doi: 10.1681/ASN.V11171. [DOI] [PubMed] [Google Scholar]
- 55.Tanimoto M, Fan Q, Gohda T, Shike T, Makita Y, Tomino Y. Effect of pioglitazone on the early stage of type 2 diabetic nephropathy in KK/Ta mice. Metabolism. 2004;53(11):1473–1479. doi: 10.1016/j.metabol.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 56.Furuta T, Saito T, Ootaka T, et al. The role of macrophages in diabetic glomerulosclerosis. American Journal of Kidney Diseases. 1993;21(5):480–485. doi: 10.1016/s0272-6386(12)80393-3. [DOI] [PubMed] [Google Scholar]
- 57.Hagiwara S, Makita Y, Gu L, et al. Eicosapentaenoic acid ameliorates diabetic nephropathy of type 2 diabetic KKAy/Ta mice: involvement of MCP-1 suppression and decreased ERK1/2 and p38 phosphorylation. Nephrology Dialysis Transplantation. 2006;21(3):605–615. doi: 10.1093/ndt/gfi208. [DOI] [PubMed] [Google Scholar]
- 58.McFarlane SI, Muniyappa R, Francisco R, Sowers JR. Clinical review 145: pleiotropic effects of statins: lipid reduction and beyond. Journal of Clinical Endocrinology and Metabolism. 2002;87(4):1451–1458. doi: 10.1210/jcem.87.4.8412. [DOI] [PubMed] [Google Scholar]
- 59.Endres M, Laufs U. Effects of statins on endothelium and signaling mechanisms. Stroke. 2004;35(11):2708–2711. doi: 10.1161/01.STR.0000143319.73503.38. [DOI] [PubMed] [Google Scholar]
- 60.Epstein M, Campese VM. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors on renal function. American Journal of Kidney Diseases. 2005;45(1):2–14. doi: 10.1053/j.ajkd.2004.08.040. [DOI] [PubMed] [Google Scholar]
- 61.Usui H, Shikata K, Matsuda M, et al. HMG-CoA reductase inhibitor ameliorates diabetic nephropathy by its pleiotropic effects in rats. Nephrology, Dialysis, Transplantation. 2003;18(2):265–272. doi: 10.1093/ndt/18.2.265. [DOI] [PubMed] [Google Scholar]
- 62.Danesh FR, Kanwar YS. Modulatory effects of HMG-CoA reductase inhibitors in diabetic microangiopathy. FASEB Journal. 2004;18(7):805–815. doi: 10.1096/fj.03-0839rev. [DOI] [PubMed] [Google Scholar]
- 63.Booth AA, Khalifah RG, Todd P, Hudson BG. In vitro kinetic studies of formation of antigenic advanced glycation end products (AGEs). Novel inhibition of post-Amadori glycation pathways. Journal of Biological Chemistry. 1997;272(9):5430–5437. doi: 10.1074/jbc.272.9.5430. [DOI] [PubMed] [Google Scholar]
- 64.Miyoshi H, Taguchi T, Sugiura M, et al. Aminoguanidine pyridoxal adduct is superior to aminoguanidine for preventing diabetic nephropathy in mice. Hormone and Metabolic Research. 2002;34(7):371–377. doi: 10.1055/s-2002-33478. [DOI] [PubMed] [Google Scholar]
- 65.Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney International. 2007;71(1):31–38. doi: 10.1038/sj.ki.5002009. [DOI] [PubMed] [Google Scholar]
- 66.Makibayashi K, Tatematsu M, Hirata M, et al. A vitamin D analog ameliorates glomerular injury on rat glomerulonephritis. American Journal of Pathology. 2001;158(5):1733–1741. doi: 10.1016/S0002-9440(10)64129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Panichi V, Migliori M, Taccola D, et al. Effects of 1,25(OH)2D3 in experimental mesangial proliferative nephritis in rats. Kidney International. 2001;60(1):87–95. doi: 10.1046/j.1523-1755.2001.00775.x. [DOI] [PubMed] [Google Scholar]
- 68.Schwarz U, Amann K, Orth SR, Simonaviciene A, Wessels S, Ritz E. Effect of 1,25(OH)2 vitamin D3 on glomerulosclerosis in subtotally nephrectomized rats. Kidney International. 1998;53(6):1696–1705. doi: 10.1046/j.1523-1755.1998.00951.x. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Z, Sun L, Wang Y, et al. Renoprotective role of the vitamin D receptor in diabetic nephropathy. Kidney International. 2008;73(2):163–171. doi: 10.1038/sj.ki.5002572. [DOI] [PubMed] [Google Scholar]
- 70.Li YC, Qiao G, Uskokovic M, Xiang W, Zheng W, Kong J. Vitamin D: a negative endocrine regulator of the renin-angiotensin system and blood pressure. Journal of Steroid Biochemistry and Molecular Biology. 2004;89-90:387–392. doi: 10.1016/j.jsbmb.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 71.Yuan W, Pan W, Kong J, et al. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. Journal of Biological Chemistry. 2007;282(41):29821–29830. doi: 10.1074/jbc.M705495200. [DOI] [PubMed] [Google Scholar]
- 72.Ohara I, Tanimoto M, Gohda T, et al. Effect of combination therapy with angiotensin receptor blocker and 1,25-dihydroxyvitamin D3 in type 2 diabetic nephropathy in KK-A y/Ta mice. Nephron. 2011;117(4):e124–e132. doi: 10.1159/000320284. [DOI] [PubMed] [Google Scholar]
- 73.de Zeeuw D, Agarwal R, Amdahl M, et al. Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. The Lancet. 2010;376(9752):1543–1551. doi: 10.1016/S0140-6736(10)61032-X. [DOI] [PubMed] [Google Scholar]
- 74.Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225–233. [PubMed] [Google Scholar]
- 75.Boor P, Celec P, Behuliak M, et al. Regular moderate exercise reduces advanced glycation and ameliorates early diabetic nephropathy in obese Zucker rats. Metabolism. 2009;58(11):1669–1677. doi: 10.1016/j.metabol.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 76.Ishikawa Y, Gohda T, Tanimoto M, et al. Effect of exercise on kidney function, oxidative stress, and inflammation in type 2 diabetic KK-A(y) mice. Experimental Diabetes Research. 2012;2012:10 pages. doi: 10.1155/2012/702948.702948 [DOI] [PMC free article] [PubMed] [Google Scholar]