

Abstract
Histone Deacetylase Inhibitors (HDIs) have shown promise as candidate radiosensitizers for many types of cancers, including prostate cancer (CaP). However, the mechanisms of action are not well understood. In this study, we demonstrate in CaP cells that valproic acid (VPA) at low concentrations has minimal cytotoxic effects, yet can significantly increase radiation induced apoptosis. VPA appears to stabilize a specific acetyl modification (lysine 120) of the p53 tumor suppressor protein, resulting in an increase of its pro-apoptotic function at the mitochondrial membrane. These effects of VPA are independent of any action of the p53 protein as a transcription factor in the nucleus, since these effects were also observed in native and engineered CaP cells containing mutant forms of p53 protein having no transcription factor activity. Transcription levels of p53-related or Bcl-2 family member proapoptotic proteins were not affected by VPA exposure. The results of this study suggest that, in addition to nuclear-based pathways previously reported, HDIs may also result in radiosensitization at lower concentrations via a specific p53 acetylation and its mitochondrial-based pathway(s).
Keywords: histone deacetylation inhibitors, apoptosis, prostate cancer, p53 acetylation, radiation therapy
Introduction
Radiation therapy is a key modality in the treatment of cancer patients. The predominant mechanism by which therapeutic irradiation kills most tumor cells is through clonogenic death. DNA double-stranded breaks (DSB) are regarded as the specific lesions that initiate this lethal response. In some cell types, ionizing radiation (IR) also triggers apoptosis, although this appears to be less significant than clonogenic cell death for many tumors. The pleiotropic nature of death pathways induced by radiation suggests that radiation resistance is likely regulated by a variety of mechanisms, each of which is associated with a specific death pathway. The molecular basis of radiation response of human tumors is complex and is likely to be multi-factorial (1).
Radiation therapy has been an effective form of treatment for prostate cancer (CaP) for decades, and the utilization of new technologies has increased the radiation doses that can be safely administered for this purpose and which has resulted in improved control rates. However, local relapses after IR can still occur(2-4). Therefore, new approaches to sensitize CaP to radiotherapy are needed if clinical outcomes are to be further improved.
It is known that some drugs can act as radiosensitizers and can enhance the tumor cytotoxic effects of radiotherapy. Recently, an emerging class of drugs known as histone deacetylase inhibitors (HDI) has shown promise in laboratory studies as candidate radiosensitizers for CaP, as well as many other types of tumors (5-11). The mechanism through which HDI seem to work in this context involves blockage of histone deacetylase (HDAC) activity resulting in hyperacetylation of core histones, and leading to a more relaxed chromatin structure (12). DNA in decondensed chromatin is relatively sensitive to damage induced by IR (13).In humans, 18 HDAC enzymes have been identified and classified, based on homology to yeast HDACs. Class I HDACs include HDAC 1, 2, 3 and 8, which are related to yeast RPD3 deacetylase and have high homology in their catalytic sites. Class II HDACs are related to yeast Hda1 (histone deacetylase 1) and include HDAC 4, -5, -6, -7, -9 and -10. All class I and II HDACs are zinc-dependent enzymes. Members of a third class, sirtuins, require NAD+ for their enzymatic activity. Class IV HDAC is represented by HDAC11, which, like yeast Hda 1 similar 3, has conserved residues in the catalytic core region shared by both class I and II enzymes (14). The potential effect of HDI as radiosensitizer has been supported by recent studies which have demonstrated in a variety of cancer cell types that HDIs, such as trichostatin A (TSA), sodium butyrate (NaB), FK228 and MS-275 enhance radiation-induced cell death, and increase IR-induced DNA double-strand breaks(9, 15-17). However, the molecular mechanisms involved in HDI mediated radiosensitization are not clear and a better understanding of these mechanisms is needed to identify those CaP patients who may benefit most from this treatment strategy.
This study investigates in detail a non-nuclear p53 based mechanism involved in the radiosensitization of CaP cells exposed to the HDI, valproic acid (VPA). VPA is an 8-carbon branched-chained fatty acid, and has been identified to be an effective inhibitor to HDACs class I and II (14). We found that VPA at low concentrations stabilizes a specific acetyl modification of the p53 tumor suppressor protein, thereby increasing p53's direct, pro-apoptotic functions at the mitochondrial membrane. Results from this study also demonstrate that p53-dependent radiosensitization with low doses of VPA is independent of any action of p53 protein in the nucleus as a transcription factor, as they take place in CaP cells containing mutant forms of p53 protein having no such activity.
Methods And Materials
Reagents and antibodies
VPA was obtained from Sigma Aldrich Co., Ltd. (St. Louis, MO). Suberoylanilide hydroxamic acid (SAHA) was obtained from Regulatory Affairs Branch, CTEP, NCI, NIH (Rockville, MD). Monoclonal antibody with specificity for phospho-histone H2A.X (γ-H2A.X, ser-139), Caspase 3 (4-1-18), and anti-acetyl-histone H4 serum were purchased from Upstate (Charlottesville, VA). Polyclonal antibodies against GST (1-109), Bcl-2 (100), Bcl-xL (H-62), Bax (N-20), and a monoclonal antibody against p53 (DO-1), PCNA were from Santa Cruz Biotech. Inc. (Santa Cruz, CA). Monoclonal antibody against mitochondria Hsp-70 (MA3-028) was from Affinity Bioreagents (Golden, CO). Monoclonal antibody against cytochrome c (SA-226) was obtained from BIOMOL (Plymouth Meeting, PA). Monoclonal antibody against betaactin was obtained from Abcam Inc. (Cambridge, MA). Polyclonal antibody specific for the acetylated p53 at K120 (anti-Ack120-p53) was a kind gift from Dr. Wei Gu (Columbia University, New York, NY). z-VAD-fmk was purchased from Promega (Madison WI).
Cell culture and treatment
Human LnCaP, DU145 and PC-3 cells were obtained from the American Type Culture Collection. Human colon cancer HCT 116 cell lines (p53+/+, and p53−/−) were kindly provided by Dr. Bert Vogelstein (John, Hopkins University, Baltimore, MD). LnCaP cells were maintained in RPMI-1640 medium with 2 mM L-Glutamine, 4.5 g/L glucose, 10 mM HEPES, 1.0 mM sodium pyruvate, 1.5 g/L sodium bicarbonate and 10% fetal bovine serum; DU145 cells were maintained as adherent monolayer cultures in DMEM medium containing 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 1.5 g/L sodium bicarbonate and 10% heat-inactivated fetal bovine serum; PC-3 cells were maintained in Ham's F12K medium with 2 mM L-glutamine, 1.5 g/L sodium bicarbonate and 10% heat-inactivated fetal bovine serum; HCT-116 and 293 cells were maintained in McCoy's 5A medium containing 10% heat-inactivated fetal bovine serum. Cells growing as monolayers in standard 6 well plates or 60 mm tissue culture plates were irradiated using a Mark I Cs-137 Irradiator (J.L. Shepherd Association, San Fernando, CA) at a dose rate between 1.43 to 1.49 Gy/min. Administered doses were validated using commercially available nanodot optically stimulated luminescence dosimeters (Landauer, Inc. Glenwood, Illinois).
Clonogenic assay
To evaluate radiosensitivity, cells in log phase were plated for 8 hours, and treated with VPA at the indicated concentrations or PBS as a control; IR was then delivered 12 hours later. Irradiated cells were maintained in VPA-containing medium for 14-20 days until colony counting. Colonies greater than 50 cells were counted as surviving colonies and the number of colonies was normalized to that observed for unirradiated controls. Mean inactivation doses were determined by the method of Fertil et al (18), and the sensitizer enhancement ratio (SER) for HDAC inhibitor treatment was calculated as the ratio of mean inactivation dosecontrol/mean inactivation doseHDAC inhibitor-treated.
Transfections
Plasmid encoding wt-tip60 (pCDNA3.1-HA-wt-Tip60 was a gift from Dr. Ediwge Col (Université Joseph Fourier, La Tronche Cedex, France). Plasmid encoding wild-type p53 (pCMV-Neo-Bam-p53wt) was kindly provided by Dr. Bert Vogelstein; Plasmids encoding mutant p53 at codon 223 (pLPC-p53-223) and codon 274 (pPSH-p53-274) were kindly provided by Dr. Andrei V. Gudkov. Wild-type or mutant p53 fragments were re-amplified by PCR and inserted into pcDNA3.1-C at BamHI and EcoR V sites. Plasmids encoding other mutations of p53 (p53K120R, p53223Leu+K120R and p53274Phe+K120R) were generated by mutagenesis PCR. All p53 plasmid constructs were then confirmed by sequencing. Electroporation were performed with Multiporator® Electroporation Systems (Cole-Parmer, 625 East Bunker Court, Vernon Hills, Illinois) as per manufacturer's instructions. Stable transfectants were then selected by G418 in vitro.
Validated SiRNAs targeting human p53 (target sequence: 5′- GGAAATTTGCGTGTGGAGT-3′) was obtained from Ambion (Austin, TX); Annealed SiRNA oligos at final concentrations of 25 nM were transiently transfected into LnCaP cells by using siPORT™ XP-1 reagent per manufacturer's instructions.
Assessment of apoptosis
Cells were incubated with or without VPA for 12 hours, followed by irradiation of 10 Gy. After incubation in VPA-containing medium for additional 48 hours, cells were collected and stained with annexin V-FITC as per manufacturer's instructions (Annexin V-FITC Apoptosis Detection Kit, BD Biosciences, San Diego, CA). The extent of apoptosis was quantified using a Becton Dickinson flow cytometer. Apoptosis was also quantified by determining the enrichment of mono- and oligonucleosomes in the cytoplasm of the apoptotic cells as per manufacturer's instruction (Cell Death Detection ELISAPLUS, Roche Applied Science, Indianapolis, IN). Briefly, 1×105 cells were collected for analysis. After cell lysis, cytoplasmic fractions were subjected to an ELISA assay that detects apoptosis-released histones and DNA in the cytoplasm. The net absorbance of samples at 405-nm (reference wavelength at 490-nm) was then normalized to the corresponding negative controls (cells without irradiation, valproic acid and/or z-vad-fmk treatments) as the changes of apoptotic cells.
Assay of mitochondrial membrane potential (MMP)
Changes in the mitochondrial membrane potential were evaluated by staining cells with the fluorescent cationic dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzamidazolocarbocyanin iodide (JC-1) by using Mīt-E-Psi™ Apoptosis & Mitochondria Permeability Detection Kit (Biomol, Plymouth Meeting, PA). Briefly, JC-1 was added directly to the cell suspension in complete medium and incubated for 15 min; after washing twice with pre-warmed culture medium, cells were suspended in fresh medium. Fluorescence was monitored in a flow cytometry by measuring both the monomer (527-nm emission; green) and J-aggregate (590-nm emission; red) forms of JC-1 following 488-nm excitation. The percentage of monomeric form or fluorescence green was then quantified as the changes of MMP.
Preparation of mitochondrial and cytosolic protein fractions and whole cell lysate for immunoblotting
After the indicated treatments, approximately 2×107 cells were collected. The mitochondria and cytosolic protein fractions were separated and prepared from cell pellets utilizing a reagent-based method per manufacturer's instruction (Mitochondria Isolation Kit, PIERCE, Rockford, IL). The mitochondria were then lysed with RIPA buffer (150mM NaCl, 50mM Tris-HCl, 1% Triton X-100, 1% Deoxycholate, 0.1% SDS, 2mM EDTA, 2mM PMSF, 10μg/ml aprotinin, 10μg/ml leupeptin, 50mM NaF, 1mM Na2VO4, 50μg/ml soybean trypsin inhibitor, and 20mM iodoacetamide). 10 μg of mitochondrial protein or 25 μg of the cytosolic fraction were then directly loaded on the SDS-PAGE gel for immunoblotting analysis.
For whole cell lysates, cells were washed with cold PBS twice, and then lysed in RIPA buffer with mild sonication. To determine the acetyl-histone H4 and phosphohistone H2A.X, whole cells were lysed in RIPA buffer containing 1 mM TSA and 5 mM nicotinamide.
RNase Protection Assay
Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA). The RNase Protection Assay was performed as per manufacturer's instruction (BD Biosciences, San Diego, CA). Briefly, 2μg of RNA was incubated with α-32P-UTP labeled single-stranded RNA probes overnight at 56°C and treated with RNAse for 45 min at 30°C. The RNA-RNA complexes were resolved by electrophoresis in 6% denaturing polyacrylamide gel and analyzed by autoradiography. Template set for hApo-2c, and custom-designed template sets for Double-Strand Break-related and p53-related genes were purchased from BD Biosciences.
In Vitro cytochrome c release assay
Mitochondria were purified using a differential centrifugation method from human HCT-116/p53−/− cells (19). Briefly, cells were harvested, pelleted and resuspended in fractionation buffer A (10 mM Hepes-KOH at pH. 7.4, 0.1 mM EDTA, 1 mM EGTA and 250 mM sucrose) supplemented with protease inhibitors cocktail (Sigma, St. Louis, MO). Cell disruption was performed by passing the cells through a 23-gauge needle 3-5 times. The homogenates were spun at 700 g for 10 min at 4 °C. The supernatants were removed and spun at 3000 g for 15 min at 4 °C. The mitochondrial pellets were washed with fraction buffer B (10 mM Hepes-KOH at pH 7.4, 5 mM KH2PH4, 5 mM succinate and 250 mM sucrose) and resuspended in fraction buffer B to a final protein concentration of 2-3 mg ml−1.
For in vitro cytochrome c release assay, engineered PC-3 cells with wild-type or mutant p53 were treated with VPA, irradiation or the combination as described above. 5 hours after the delivery of irradiation, 1×107 cells were harvested, washed with cold PBS, and resuspended in 200 μl of modified KCl buffer (15 mM Hepes/NaOH, 125 mM KCl, 4 mM Mg2Cl, 5 mM Na2HPO4, 0.5 mM EGTA, 5 μM Rotenon, and 5 mM Succinate, 250 mM sucrose at pH.7.4). Cells were swallowed on ice for 2 min, and disrupted by passing the cells through a 23-gauge needle 5 times. The homogenates were spun at 12000 g for 15 min at 4 °C. The supernatants were collected as cytosolic fractions. After the determination of protein concentrations using the Bradford Reagent (Bio-Rad Laboratories, Inc. Hercules, CA), cytosolic fractions with 150 μg protein each were incubated with 20 μg of purified mitochondria for 40 min at 30 °C in 100 μl KCl buffer (15 mM Hepes/NaOH, 125 mM KCl, 4 mM Mg2Cl, 5 mM Na2HPO4, 0.5 mM EGTA, 5 μM Rotenon, and 5 mM Succinate at pH.7.4), then centrifuged at 12000 g for 10 min at 4 °C. The mitochondrial pellets and corresponding supernatants were immunoblotted for cytochrome c, and actin antibodies to verify equal loading.
Purification of Recombinant Proteins
Bac-Pak-Tip60 was generously provided by Dr. Edwige Col (Université Joseph Fourier, La Tronche Cedex, France). pGEX-5X1-GST-p53wt was a gift from Dr. Jean Dorsey (The Wistar Institute, Philadelphia, PA). The recombinant GST-tagged proteins for mutant p53 were generated by re-insert of corresponding p53 fragments from plasmids encoding mutations of p53 in pcDNA3.1-C vector into pGEX-5X1-p53wt at PshAI/Stul sites.
The Flag/HA-Tip60 was purified from the infected insect SF-9 cells (obtained from Dr. Binghui Shen, City of Hope, Duarte, CA) to homogeneity under native conditions (Ni-NTA Spin kit, Qiagen, Hilden, German). The GST-p53wt, GST-p53K120R, GST-p53223L and GST-p53223L+K120R proteins were purified from bacteria cells BL-21 (obtained from Dr. Binghui Shen, City of Hope, Duarte, CA), and dialyzed with KCl buffer.
In Vitro Acetylation Assay
1 μg of purified Tip60 protein was rebound to 20 μl Ni-NTA agarose beads (Qiagen, Hilden, Germany) in modified Ni-NTA lysis buffer (50 mM NaH2PH4, 300 mM NaCl, 10 mM imidazole), and washed once with HAT buffer (50 mM Tris (pH 7.9), 10% Glycerol, 1 mM DTT, 10 mM sodium butyates). As a negative control, 1 μg of BSA was also incubated with Ni-NTA agarose beads at 4 °C for 2 hours. After preincubation of beads with 20 μM of acetyl-CoA in HAT buffer at RT for 30 min, 10 μg of p53 proteins were mixed with beads in 100 μl HAT buffer containing 20 μM acetyl-CoA, and incubated at 30 °C for one hour. The mixtures were then centrifuged at 1000 g for 3 min, and the supernatant were collected for immunoblot against anti-ac-K120-p53. To investigate the role of p53 protein acetylation on cytochrome c release, 40 μl of corresponding supernatants were incubated with 30 μg purified mitochondria at 30 °C for 40 min in 100 μl KCl buffer, then centrifuged at 12000 g for 10 min at 4 °C. The mitochondrial pellets and corresponding supernatants were immunoblotted for cytochrome c, and GST antibodies to verify equal loading.
Tumor Growth Assay
DU145 cells (3×106/0.2ml HBSS 1x + 1% HSA) were inoculated s.c. into the right thigh of male athymic nude mice 4-6 weeks old. When tumor volumes reached a size of 50-100 mm3 (approx. day 7 after inoculation), mice were randomly grouped into 4 groups (n=5-7) that received the following treatments: (a) Saline, 0.2ml (b) Valproic Acid (300mg/kg) (c) IR 10 Gy (d) VPA (300mg/kg) + IR 10 Gy. Mice were treated with i.p. injections of VPA (300mg/kg) every 12 hours for 3 days. After the third VPA injection. The mice were irradiated locally on the right thigh with a Mark I Cs-137 Irradiator using a collimator with a 30mm opening at a dose rate between 5.29 and 5.25 Gy/minute. Doses were validated using commercially available nanodot optically stimulated luminescence dosimeters (Landauer, Inc., Glenwood, Illinois). Tumors were measured biweekly and tumor volumes were determined from caliper measurements of tumor length (L) and width (W) according to the formula (L×W2)/2.
Results
VPA induced changes in histone acetylation, and survival after Ionizing Radiation (IR)
Dose response data for alteration of histone H4 acetylation levels by VPA in CaP lines LnCaP, DU145 and PC-3 were obtained prior to investigating potential radiosensitizing effects of the drug (Supplementary figure 1); a minimum of 50 μM VPA markedly increased histone H4 acetylation in all three lines. We next examined the effects of VPA on cell response to IR by clonogenic survival assay. When exposed to relatively high concentrations of VPA (500 μM and 1 mM), marked radiosensitization was detected in LnCaP and DU145 cells (Figure 1A), with observed SER0.1 of 1.43 and 1.62 for LnCaP and of 1.41 and 1.58 for DU145, respectively. The VPA exposures using these concentrations also caused appreciable cytotoxicity in all three CaP lines (Supplementary table 1). Importantly, using a 10-fold lower concentrations of VPA (50 μM) did not lead to any direct toxicity of the drug, but significant radiosensitization of LnCaP and DU145 cells was still detected; the SER0.1 were 1.41 for LnCaP and 1.46 for DU145. Although VPA induced acetylation of histone protein H4 in PC-3 cells comparable to the other two lines, significant radiosensitization was not observed for any drug concentration tested (SER0.1 were 1.17 for 50 μM, 1.25 for 500 μM and 1.16 for 1000 μM, respectively),
Figure 1. VPA differentially induced radiosensitization and apoptosis in CaP cells.

(A) Clonogenic survival assays of CaP cells pretreated with indicated doses of VPA. Log phase cells were trypsinized and plated as single cells. After 8 hours incubation to allow for cell attachment, cells were pretreated with different concentrations of VPA for 12 hours and then exposed to IR. Colony survival was determined 14-20 days later. (B) VPA increased apoptotic response to irradiation. Cells were first exposed to 50 μM VPA or left untreated for 12 hours before 10 Gy of IR. Cells were then collected 48 hours later, and apoptosis was evaluated by Annexin V-FITC apoptosis assay. Data represent the average of three to four experiments. Error bars indicate standard deviation. * indicates significant difference (ρ < 0.01).
IR produces DNA double-strand breaks (DSBs) in chromosomal DNA that can initiate signaling cascades and lead to cell cycle arrest or apoptosis. One of the earliest known responses to radiation-induced DSB formation is phosphorylation of the C-terminal tails of variant H2AX (γ-H2AX) in chromatin on either side of the break (20). With prior exposure to 50 μM of VPA, radiation-induced γ-H2AX formation was greater in all tested cells lines, however, the persistence of γ-H2AX at 24 hours postirradiation, which was described as corresponding well to radiosensitivity (21), were only observed in LnCaP and DU145 cells, but not in PC-3 cells, (Supplementary figure 2), suggesting VPA exposure potentially inhibits the repair of radiation-induced DSBs in LnCaP and DU145 cells.
VPA increases apoptotic response to irradiation in CaP cells
It has been reported that apoptosis is not a prominent mechanism of IR-induced cell death in CaP cells in culture (22). Consistently, only ∼5% of cells undergoing apoptosis 48 hours after exposure to 10 Gy were detected in these three cell lines (Figure 1 B). Control exposures to 50 μM of VPA alone did not induce apoptosis. However, when cells were exposed to 50 μM VPA prior to 10 Gy IR, a significant increase in the percentage of cells undergoing apoptosis was observed in LnCaP (∼ 19%) and DU145 (∼ 17%), but not in PC-3 (Figure. 1B). These augmented apoptotic responses in LnCaP and DU145 cell were sufficient to account for the radiosensitization measured by clonogenic assay (Figure 1A).
Previous studies have shown that HDI treatments can induce cell growth arrest at G1/G0 or G2/M phase in a number of tumor cell lines, including CaP. These effects were attributed to dysregulation of cell cycle regulators such as p21CIP/WAF1 and cyclin B1(23, 24). It has also been reported that treatment at lower concentrations of HDI had no effect on cell cycle distribution of CaP cells (25). Results from this study showed that exposure to VPA at 50 μM had no significant influence on cell cycle distribution compared to IR alone in all cell lines tested (data not shown), indicating that the observed VPA-enhanced radioresponse was not due to reassortment of cells into the more radiosensitive phases of the cell cycle.
VPA enhancement of apoptosis after IR involves mitochondria and caspase pathways
Increased mitochondrial membrane permeability and unleashing of caspase protease activity have each been shown to accompany cellular commitment to apoptosis (26, 27). To further elucidate the mechanism of low dose VPA enhancement of apoptosis after IR, we examined the effects of VPA on mitochondrial membrane potential (MMP) and caspase activation. Significant changes in MMP (Figure 2A) and cytochrome c release into the cytosolic fraction (Figure 2B) were detected with VPA exposure prior to 10 Gy IR in LnCaP and DU145 cells, but these were not observed with VPA alone or IR alone in those cells. In contrast, VPA plus IR did not produce comparable effects in PC-3 cells. HDI treatment has been reported to enhance the IR-induced triggering of caspase activity in DU145 cells (28). Activation of caspase-3, signaled by detection of its cleaved 17kDa subunit, was observed in LnCaP and DU145 cells exposed to VPA + IR, while IR alone or VPA alone resulted in no cleavage products (Figure 2C). Furthermore, the presence of the caspase inhibitor, z-VAD-fmk, completely abolished VPA-enhanced apoptotic response to IR in both LnCaP and DU145 (Figure 2D). These results suggest that low dose VPA exposure increase the susceptibility of cells to undergo apoptosis in response to IR, in association with stereotypical losses of mitochondrial membrane integrity, and alteration of caspase-3 activity.
Figure 2. Changes in mitochondrial membrane potential and caspase cascade activation.
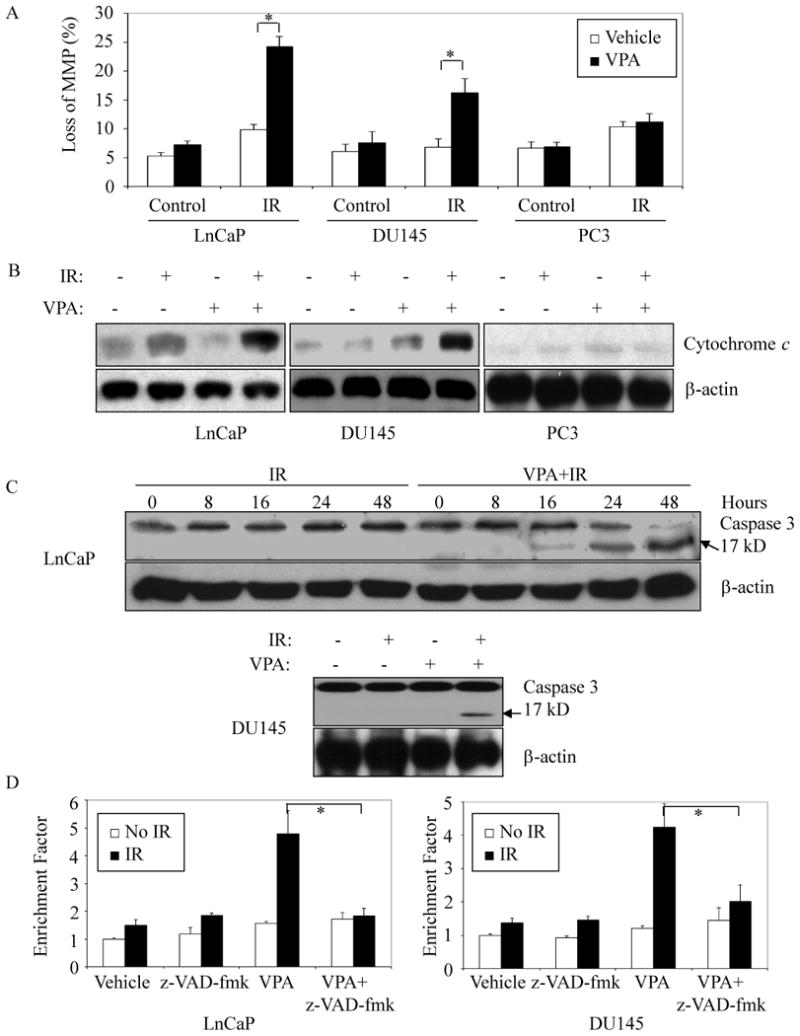
(A and B) VPA prior to irradiation increased radiation-induced mitochondrial membrane potential and cytochrome c release. Cells were pretreated with 50 μM VPA or left untreated for 12 hours followed by 10 Gy IR. Cells were then trypsinized and collected 24 hours later. Mitochondrial membrane potential was quantified by measurement of JC-1 fluorescence intensity (A); cytosolic protein was prepared as described in Materials and Methods, and analyzed by immunoblot to determine the release of cytochrome c from mitochondria. β-actin was included to confirm equivalent protein loadings (B). (C) VPA induced activation of caspase 3 in response to irradiation. Whole cell lysates were prepared 24 hours later, or at different time points as indicated after radiation. Immunoblots were performed to determine the activation of caspase-3. Cleaved caspase-3 presents product at 17kD. (D) Inhibition of VPA-effects on IR-induced apoptosis by the caspase inhibitor z-VAD-fmk. LnCaP or DU145 cells were pretreated with or without 50 μM VPA for 12 hours followed by 10 Gy IR. z-VAD-fmk (10 μM) was added to cells 2 hours before IR. Cells were maintained in VPA-containing medium for 48 hours. 1×105 cells were then collected and lysed, and cell lysates were subjected to ELISA analysis as per manufacturer's instructions (Cell Death Detection ELISAPLUS). Error bars indicate one standard deviation from three to four individual experiments. * indicates significant difference (ρ < 0.01).
Localization of p53 and Bcl-2 family proteins in mitochondria following IR is modified by VPA treatment
Previous studies have provided evidence that p53 protein is involved in apoptosis induction through rapid, “direct” mechanisms that entail the physical interactions with regulators such as Bax, Bcl-2 and Bcl-XL at the mitochondria (29, 30). We investigated p53 protein association with the mitochondrial subcellular fraction following treatments with IR, VPA, or both. Figure 3A shows that IR alone or combined with VPA caused p53 protein to accumulate in mitochondrial fractions in LnCaP cells as early as 1-3 hours after IR. With IR alone the p53 mitochondrial content had decreased by 8-12 hours, while with VPA treatment prior to IR the p53 in mitochondrial fractions had not returned to baseline by 24 hours. We also examined the effects of VPA on mitochondrial localization of Bcl-2 family proteins in the three cell lines with different p53 genotypes. As shown in Figure 3B, no changes in Bcl-2 mitochondrial accumulation were seen after IR alone or after VPA followed by IR. Treatment with combination of VPA and IR did lead to a redistribution of Bcl-xL out of the mitochondria compared to IR alone in LnCaP and DU145 cells. In contrast, Bax mitochondrial concentrations increased with the addition of VPA to IR in these cells, The lower observed level of Bax in DU145 extracts is likely due to a monoallelic Bax frameshift mutation and a second silent Bax allele in this line (31). Comparable changes for Bcl-xL and Bax with these treatments were not observed in p53 null-mutant PC-3. Furthermore, our results suggested that the observed changes of mitochondrial Bax and Bcl-Xl seemed to be correlated to the changes of mitochondrial p53, rather than the changes of protein expressions in corresponding cells.
Figure 3. Mitochondrial accumulations of p53 and Bcl-2 family membrane proteins.

(A) LnCaP cells were treated with IR (10 Gy) alone or with 50 μM VPA for 12 hours prior to IR. Mitochondrial fractions were prepared at indicated times and subjected to immunoblot using anti-p53 antibody (DO-1). Anti β-actin, or mt-HSP70 antibodies were included to confirm equivalent protein loading, respectively. (B) LnCaP, DU145 and PC-3 cells were pretreated with or without 50 μM VPA for 12 hours before 10 Gy of IR was delivered. Mitochondrial fractions (top) and whole cell lysates (bottom) were then prepared 24 hours later. p53, Bcl-2, Bcl-xL and Bax levels were analyzed by immunoblot. mt-Hsp70 was included to show equivalent protein loading. The purity of mitochondrial fractions was assessed by immunoblot with anti-cytochrome c, HSP70 and PCNA, a cytosolic/nuclear protein proliferating cell nuclear antigen (right panel). CF: cytosolic fraction; MF: mitochondrial fraction. The data are representative of three independent experiments.
VPA enhancement of radiation-induced apoptosis is p53 dependent
The ability of low dose VPA to enhance apoptosis after IR seems to be correlated with the presence of p53 protein in the lines—LnCaP and DU145—that exhibit this response, but not with the ability of that protein to function as a nuclear transcription factor (Supplementary figure 3). To establish that p53 protein is a requirement of this apoptotic response, we used specific siRNA expression to transiently down-regulate the p53 protein level in LnCaP cells. As shown in Figures 4, knock-down of p53 significantly reduced the enhancement of IR-induced apoptosis mediated by VPA pre-treatment. Enhancement of Bax mitochondrial accumulation and cytochrome c release by VPA after IR were similarly impaired by the p53 knock-down (Fig 4C).
Figure 4. Requirement of p53 in VPA-enhanced apoptotic response to IR.

LnCaP cells were transiently transfected with p53 SiRNA oligos to reduce protein level of p53 (A), and pretreated with or without 50 μM VPA for 12 hours before IR (10 Gy). Cells were collected 48 hours after irradiation, and annexin V-FITC apoptosis assay was performed to analyze apoptosis (B). Mitochondrial and cytosolic fractions were prepared and analyzed by Immunoblot using anti-Bax, Bcl-xL and cytochrome c antibodies. β-actin and mt-HSP70 were included to confirm equivalent protein loading (C). Data represent the average of three experiments. Error bars indicate one standard deviation. * indicates significant difference (ρ < 0.01).
Requirement of p53 for VPA-enhanced apoptotic response after irradiation was further confirmed by restoring wild type p53 protein expression in engineered PC-3 cells (Figure 5). Two different mutant alleles, p53223Leu and p53274Phe from DU145 cells (32), were also separately expressed in PC-3 cells. Stable transfectant clones were derived and propagated, and then examined for apoptotic response after IR alone or 50 μM VPA pretreatment followed by IR. Compared to empty vector controls, engineered expression of either p53wt or p53223Leu restored the ability of VPA to enhance IR-induced apoptosis (Figure 5B) and to sensitize cells to IR with an SER0.1>1.3 (Supplementary table 2 and Supplementary figure 4); these two proteins also significantly increased Bax protein levels in mitochondrial, and promoted cytochrome c release, with combined VPA and IR treatments (Figure 5C). In contrast, expression of p53274Phe in PC-3 cells did not result in similar changes. These results indicate that p53 plays an essential role in VPA-mediated enhancement of apoptosis and radiosensitization, and that these properties are retained by some p53 mutant alleles that are deficient in nuclear function as a transcriptional activator of target genes. Judging by relative protein levels and the degree to which apoptosis is enhanced (Figure 5A), it appears that, molecule-for-molecule, wild-type p53 may be more potent than p53223Leu in mediating these effects, however.
Figure 5. Engineering expression of wild-type p53 or mutant p53223Leu, but not mutant p53274Phe, restores the ability of VPA to enhance radiation-induced apoptotic response in p53 null PC-3 cells.

PC-3 cells were stably transfected with wild-type p53, mutant p53223Leu, mutant p53274Phe or empty vector as control (A), and pretreated with or without 50 μM VPA for 12 hours before IR (10 Gy). For apoptosis analysis, cells were collected 48 hours later, and analyzed by annexin V-FITC apoptosis assay (B). Whole cell lysates or mitochondrial fraction were also prepared 24 hours after IR, and analyzed by immunoblot. β-actin and mt-HSP70 were included to confirm equivalent protein loading (C). Data represent the average of three experiments. Error bars indicate one standard deviation from three individual experiments. * and ** indicate significant differences (ρ < 0.01 or 0.05).
Loss of K120 acetylation diminishes p53-mediated radiosensitization by VPA
p53 is known to be a substrate for acetylation at multiple lysine residues, and for de-acetylation by HDACs (33). Acetylation of p53 at lysine residue 120 specifically has been shown to be important for regulation of apoptosis through its nucleus function as transcriptional factor following IR and other genotoxic cellular insults (34, 35). We reasoned that this post-translational modification could have similar regulatory effects at the mitochondria, and so hypothesized that increasing mitochondrial levels of acetyl-K120 p53 with VPA + IR treatments would amplify p53-dependent apoptotic effects there and so produce the observed radiosensitization. We investigated the presence of K120 acetylation in mitochondrial p53 after exposure to VPA and IR by using a specific antibody (34). As shown in Figure 6A, acetyl-K120 p53 was indeed present in mitochondrial fractions following IR. Using Alphaview software (CellBiosciences, Santa Clara, CA) to quantify protein levels demonstrated that the ratio of acetylated p53 at K120 in the mitochondrial fractions between cells treated with IR alone, and IR in combination with VPA was 1: 2.48. In contrast, the ratio of total p53 in the mitochondrial fraction was 1:1.97 between cells treated with these two treatments, suggesting that the increases in K120 acetylation observed after VPA and IRwas not solely due to an increase in total amount of p53.
Figure 6. Loss of p53 acetylation at K120 reduced VPA-enhanced apoptosis response and radiosensitization.

(A) VPA induced acetylation of mitochondrial p53 at K120 in response to irradiation. LnCaP cells were pretreated with 50 μM VPA for 12 hours and then irradiated (10Gy). The mitochondrial fraction was isolated 8 hours later, and subjected to immunoprecipitation with ac-K120-p53 antiserum. The resultant proteins were analyzed by immunoblot with p53 antibody (DO-1). Total p53 and mt-HSP70 were included to confirm equivalent protein loading; (B) Mutation at K120 of p53 (K120R) diminished VPA-enhanced loss of mitochondrial membrane potential (MMP) in response to irradiation in PC-3 cells engineered to express wild-type p53 or mutant p53223Leu. Stable transfectants of PC-3 cells with empty vector, wild-type p53, and mutant p53 (p53K120R, p53223Leu, p53274Phe, p53223Leu+K120R, and p53274Phe+K120R) were pretreated with VPA for 12 hours, and then irradiated (10Gy). Cells were collected by trypsinization 48 hours later, and stained with JC-1. Loss of MMP was analyzed by flow cytometry assay. Data represent the average of three experiments. Error bars indicate one SD from three individual experiments.* indicates significant difference (ρ < 0.01).
In keeping with this hypothesis, p53K120R expression in PC-3 cells did not lead to increased apoptosis or loss of MMP following IR when cells were also first treated with VPA (Figure 6B and supplementary figure 5). To genetically dissect apart any nuclear and non-nuclear regulatory effects of p53 lysine 120 acetylation, we constructed the double mutant p53223Leu+K120R that is defective both for nuclear transactivation and, by the model, for enhanced pro-apoptotic effects at the mitochondria as well. When expressed in PC-3 cells, and in contrast to the p53223Leu single mutant, the double mutant did not support increased apoptosis with VPA + IR. Correspondingly, substitutions of lysine 120 with arginine in wild-type p53 or p53223L eliminated radiosensitization by VPA as assessed by clonogenic assays in engineered PC-3 cells (Supplementary figure 4).
Taken together these data indicate that acetylation of the K120 residue of p53 plays an important regulatory role for the pro-apoptotic effects that p53 protein mediates at the mitochondria, as has been shown to be the case for the corresponding nuclear effects, and that these mitochondrial effects are modulated by HDI.
The role of acetylated K120 was confirmed further by incubating purified mitochondria (isolated from human colon cancer HCT116/p53−/− cells) with cytoplasmic extracts from engineered PC-3 cells treated with VPA, irradiation or the combination. As shown in figure 7A, incubating the mitochondria with cytoplasmic extracts from empty vector–transfected (neo) PC-3 cells, that had been treated with VPA + IR, or from p53wt expressing PC-3 cells that had not been exposed to IR, produced little cytochrome c release from mitochondria into the supernatant fraction. Extracts from irradiated PC-3/p53wt cells resulted in modest cytochrome c release, and this markedly increased with VPA treatment of the cells prior to IR. Mitochondria incubation with extracts prepared from p53223Leu-expressing PC-3 cells supported increased cytochrome c release after VPA +IR treatment, while those from p53K120R and p53223L+K120R did not (Figure 7B).
Figure 7. In Vitro cytochrome c release assay and acetylation assay.

(A and B) Engineered PC-3 cells with empty vector, p53wt, p53K120R, p53223L or p53274P were treated with 50 μM VPA, irradiation or the combination as described above. Five hours later after the delivery of 10 Gy of IR, cytoplasmic extracts were prepared, and incubated with purified mitochondrial fraction from HCT116/p53−/− cells at 30°C for 40 min. The supernatant fraction and the pellets, after centrifugation at 12000 g for 10 min, were analyzed by immunoblots. (C) Purified p53 protein was incubated with Tip60, or BSA as control at 30°C for one hour, and the supernatant was directly analyzed for acetylation of K120 or p53, or incubated with purified mitochondria for cytochrome c release. (D) DU145 cells were engineered for wt-Tip60 expression, and pretreated with 50 μM VPA or 200 nM SAHA, followed by 10 Gy of irradiation. Cells were analyzed for apoptosis 48 hours later by determining the enrichment of mono- and oligonucleosomes in the cytoplasm using ELISA assay. Error bars indicate one SD from three individual experiments.
Tip60, a MYST family acetyltransferase, acetylates K120 and regulates HDI-mediated apoptotic response to irradiation
Lysine 120 of p53 has been identified recently to be a substrate of Tip60, a histone acetyltransferase belonging to the MYST family (34, 36). We therefore manipulated K120 acetylation levels by incubating purified p53 protein with recombinantTip60 in vitro. As seen in figure 7C, Tip60 induced K120 acetylation of p53wt and p53223Leu proteins, and enhanced corresponding p53-dependent cytochrome c release from mitochondria. No changes in acetylation or cytochrome c release were observed with p53 proteins with K120 mutations.
In addition, manipulating levels of Tip60 in irradiated cancer cells modulated apoptotic response to HDI drugs. Figure 7D shows that engineered DU145 cells with expression of wt-Tip60 demonstrated a modest, but reproducible, increase in apoptosis after HDI + IR compared to parental cells. DU145 cells were selected for these experiments to avoid any effects on p53 target gene transcription by Tip60 expression.
VPA induces radiosensitization of prostate cancer cells in vivo
Studies were extended to an in vivo model. We investigated the effects of VPA, IR or VA combined with IR on tumor growth of DU145 xenografts. VPA increased tumor growth suppression compared to IR alone (Figure 8). These results are consistent with results seen in vitro.
Figure 8. VPA induces radiosensitization in prostate cancer in vivo.

Athymic nude mice bearing isogenic DU145 xenograft tumors were treated with VPA (300mg/kg × 6) administered intraperitoneally every 12 hours for 3 days and/or 10 Gy IR. In the combined treatment group, IR was delivered after the third injection of VPA. The growth curves represent the average value in each group of 5-8 mice. Error bars represent one standard error (S.E.).
Discussion
Advances in molecular radiobiology have led to a better understanding of mechanisms involved in radioresponse and has helped to identify potential strategies to utilizing certain agents as radiosensitizer increase tumor radiosensitivity. HDI have shown promise as radiosensitizers in laboratory studies and are being actively evaluated in clinical trials. Possible mechanisms may involve cell cycle arrest, gene expression regulation or activation of apoptosis. Our previous study (37), as well as others (38), have also suggested that HDI induced differential radiosensitivity in cancer cells may be influenced by p53 expression. In this study, we demonstrated the role of a specific acetyl-modification of p53 and its mitochondrial accumulation involved in HDI-mediated apoptotic response to IR in CaP cells.
The tumor-suppressor protein p53 plays a pivotal role in governing cellular responses to genotoxic damage. p53 appears to selectively activate, at the transcriptional level, networks of genes whose products support growth arrest or apoptosis (39, 40). In cells with functional p53 protein, a post-translational increase in levels of p53 occurs in response to irradiation, which appears to be mediated through the ATM proteins (41) and ultimately results in an increase in the level of the cyclindependent kinase inhibitory protein p21Waf1/cip1 (42). The role of p53 in stress induced apoptosis was thought initially to center on its role as a transcription factor modulating gene expression. Upon activation by DNA-damage signaling pathways, p53 promotes the expression of a number of genes that promote apoptosis. In cells with mutated or nonfunctional p53, apoptotic function is often compromised or abrogated (43). In this study, we also investigated the effects of VPA on p53 transcriptional activities in response to IR. Our data clearly showed that VPA up-regulated p21 protein in p53 positive LnCaP cells was dose-dependent (Supplementary figure 3B). Interestingly, while low dose of VPA enhanced p53-dependent apoptotic response and radiosensitization in CaP cells, no significant effects on expression of genes known to be p53-related were observed in radiosensitized CaP cells, suggesting an exclusion of p53 as transcriptional factor in this radiosensitization process. Indeed, recent studies have provided strong evidence that p53 promotes apoptosis not only through target gene activation in the cell nucleus but also through the initiation of extra-nuclear proapoptotic mechanisms that involve direct interaction of p53 on the mitochondria. Although, the mechanisms involved remain unclear, direct physical interaction of p53 with antiapoptotic proteins such as Bcl-2 and Bcl-xL has been demonstrated. These interactions allow Bax to oligomerize and then mediate mitochondrial membrane permeability leading to release of cytochrome c (44, 45). Cytochrome c release then triggers activity of a family of caspases, including caspases 9 and 3, that have proteolytic function and appear to be downstream effectors of apoptotic cell death (26, 27). Our current data also demonstrated that the radiosensitizing effects of low dose VPA in CaP cells were p53-dependent, and involved a non-nuclear mitochondrial pathway. With VPA exposure prior to IR, p53 mitochondrial protein activation and mitochondrial caspase-mediated apoptosis increased. Moreover, particular p53 mutants, such as p53223Leu with a missense mutation located in DNA binding domain, also demonstrated a capability to mediate biological effects of VPA on IR-induced MMP, apoptosis and radiosensitization in CaP cells, further supporting the involvement of a p53-mediated extra-nuclear proapoptotic pathway.
Of interest is the observed difference between PC-3 cells expressing p53223Leu versus p53274Phe. Although these two mutations are located very close in the same functional domain, increased levels of apoptosis after VPA and IR were only observed with the p53223Leu mutation. The reasons why some p53 DNA binding domain mutants can trigger apoptosis while others fail remains unexplained, but may involve a critical conformational requirement for p53 activation after VPA exposure. This hypothesis is consistent with reports demonstrating that some small molecular compounds, including SAHA, have the ability to restore the proper conformation of mutant p53, by posttranslational modification, such as acetylation, of p53, and trigger p53-dependent, transcription-independent apoptosis (46, 47). The possibility of using low dose HDI to restore wild-type p53 activity to some p53 mutants to enhance IR induced apoptosis is intriguing, particularly since it has been reported that local failures after radiation therapy are more common in CaP cells with abnormal p53 (48-50). Identifying which p53 mutants are most amenable to this strategy will be clinically important.
The precise mechanism(s) by which p53 is activated in response to cellular stress is not completely understood. However, it is generally thought to primarily involve posttranslational modifications of p53, resulting in conformational changes that affect its protein levels, cellular localization, and binding specificity for both protein partners and DNA target sequences. p53 protein in vivo undergoes extensive posttranslational modifications including phosphorylation of specific serine and threonine residues and acetylation of lysine in response to irradiation. Early studies indicated that p53 is specifically acetylated at multiple lysine residues of the C-terminal regulatory domain by the HATs CBP/p300 and the HAT cofactor PCAF (51-54). Acetylated p53 can dramatically stimulate its sequence-specific DNA binding activity in vitro to modulate transcriptional pro-apoptotic responses in cancer cells (55-58). However, C-terminal acetylation of p53 also blocks the ε -amino group of the acetylated lysine residues for monoubiquitination (59-61). The latter promotes its nuclear export (62). Thus, C-terminal acetylation of p53 is expected to inhibit apoptotic direct p53 functions at the mitochondria following nuclear export (61). However, studies do not rule out the possibility that acetylation of other lysine residue(s) of p53 protein also regulate its mitochondrial localization and/or its interactions with Bcl-2 family proteins in response to DNA damage.
Recently, several groups have reported on the involvement of acetylated p53 in modulation of transcription activity and in its direct impact on the mitochondria as a cell undergoes apoptosis. These posttranslational modifications include acetylation of p53 at lysine 120 and lysine 3 (34-36, 47). Studies have also demonstrated that lysine 120 on p53 was a substrate for acetylation after DNA damage via the MYST family acetyltransferases, Tip60 and hMOF (36, 63), and that acetylated p53 regulate transcription-independent apoptosis (35, 47). In contrast to other p53 acetylation sites at the C terminus referred to above, K120 is within the core DNA binding domain of the protein. Conservative, non-acetylatable mutations (arginine for lysine) of these two residues are specifically defective for either apoptosis induction, or its interaction with other proteins such as mdm2. Results from our study also demonstrated that Tip60 acetylated wild-type p53 and mutant p53223Leu at K120, and resulted in subsequent enhancement of p53 proteins on release of cytochrome c in vitro. However, other p53 mutants having peptide sequence changes located elsewhere within the DNA binding domain were not substrates for K120 acetylation by Tip60, suggesting the critical role of conformational structure for this modification. In addition, manipulating levels of Tip60 in irradiated DU-145 cells increased the effects of HDI drugs to IR induced apoptosis. This was seen not only for VPA but also for SAHA (data not present), suggesting other HDI may work through the same pathways. Of note, a very recent study also demonstrated that deacetylation of p53 K120 was predominantly regulated by histone deacetylase I (HDAC 1), a potential target for both VPA and SAHA (14, 64). Whether HDAC1, or other HDACs are involved in the VPA enhancement of apoptotic response to IR requires further investigation.
Recurrence of radiation-resistant tumors is a common problem in clinical oncology including with the treatment of CaP (65, 66). An attractive solution to this problem of CaP cell radioresistance would be to endow them with an increased susceptibility to undergo apoptosis in response to the DNA damage IR inflicts. Reducing the threshold to undergo apoptosis by interference with apoptosis resistance pathways would be expected to sensitize tumor cells to IR by affording them an additional mechanism for induction of cell death. The fact that this may be clinically achievable with low and possibly less toxic doses of VPA, or other HDI, is theoretically attractive. Although the precise molecular mechanism(s) by which HDI-potentiation of radiation-induced apoptosis remains unclear, our data demonstrate that exposure to low concentrations of VPA and possibly other HDI prior to ionizing radiation in prostate cancers with select p53 genotypes could be a novel strategy to enhance the radiosensitivity of prostate cancers in the clinic.
Supplementary Material
Supplementary figure 1: VPA induced dose-dependent histone protein acetylation in vitro. Prostate cancer LnCaP, DU145 and PC-3 cells were exposed to different concentrations of VPA for 12 hours. Total cell lysates were analyzed by immunoblots. The anti-acetyl-histone-H4 rabbit antiserum (Upstate, Charlottesville, VA) was used to detect acetylation of histone protein. Anti-β-actin antibody was utilized to determine the equal loading of proteins.
Supplementary figure 2: VPA enhanced and prolonged γ-H2A.X levels after IR. LnCaP, DU145 and PC-3 cells were exposed to 50 μM VPA for 12 hours prior to 10 Gy of IR. Total cell lysates collected at indicated times were analyzed by immunoblot using the anti-phospho-histone H2A.X (Ser139) monoclonal antibody, and anti-β-actin antibody, respectively.
Supplementary figure 3: Effects of VPA on pro-apoptotic gene expression in response to irradiation. Cells were treated with or without 50 μM of VPA for 12 hours, followed by 10 Gy IR. (A) Total RNA were prepared 24 hours later, and subjected to RNase protection assay with p53-related, double strand break-related or hApo2c probe sets. (BD Biosciences, San Diego, CA). (B) Total RNA or cell lysates were prepared 24 hours later after IR. RNase protection assay or immunoblot analyses were performed.
Supplementary figure 4: Role of K120 acetylation of p53 in VPA-mediated radiosensitization and clonogenic survival. PC-3 cells were engineered to express either wild-type or mutant p53 as indicated. Clonogenic survival assays were performed as described in Figure 1B. Empty-vector (EV) transfected PC-3 were included as a control. Error bars indicate one SD from three individual experiments.
Supplementary figure 5: Role of K120 acetylation of p53 in VPA-mediated apoptosis. PC-3 cells engineered to express either wild-type or mutant p53 (K120R) were treated with VPA and IR. Apoptosis was analyzed by annexin V-FITC assay 48 hours after IR. Error bars indicate one SD from three individual experiments. * indicates significant difference (ρ < 0.01).
Supplementary table 1: Effects of different concentrations of VPA on plating efficiencies of prostate cancer cells in vitro
Supplementary table 2: Effects of p53 acetylation on VPA-induced radiosensitizations of engineered PC-3 cells in vitro
References
- 1.Howell SB. Resistance to apoptosis in prostate cancer cells. Mol Urol. 2000;4:225–229. discussion 231. [PubMed] [Google Scholar]
- 2.Zietman AL, DeSilvio ML, Slater JD, Rossi CJ, Jr, Miller DW, Adams JA, Shipley WU. Comparison of conventional-dose vs high-dose conformal radiation therapy in clinically localized adenocarcinoma of the prostate: a randomized controlled trial. Jama. 2005;294:1233–1239. doi: 10.1001/jama.294.10.1233. [DOI] [PubMed] [Google Scholar]
- 3.Pollack A, Zagars GK, Starkschall G, Antolak JA, Lee JJ, Huang E, von Eschenbach AC, Kuban DA, Rosen I. Prostate cancer radiation dose response: results of the M. D. Anderson phase III randomized trial. Int J Radiat Oncol Biol Phys. 2002;53:1097–1105. doi: 10.1016/s0360-3016(02)02829-8. [DOI] [PubMed] [Google Scholar]
- 4.Zelefsky MJ, Fuks Z, Hunt M, Lee HJ, Lombardi D, Ling CC, Reuter VE, Venkatraman ES, Leibel SA. High dose radiation delivered by intensity modulated conformal radiotherapy improves the outcome of localized prostate cancer. J Urol. 2001;166:876–881. [PubMed] [Google Scholar]
- 5.Camphausen K, Tofilon PJ. Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J Clin Oncol. 2007;25:4051–4056. doi: 10.1200/JCO.2007.11.6202. [DOI] [PubMed] [Google Scholar]
- 6.Baschnagel A, Russo A, Burgan WE, Carter D, Beam K, Palmieri D, Steeg PS, Tofilon P, Camphausen K. Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol Cancer Ther. 2009;8:1589–1595. doi: 10.1158/1535-7163.MCT-09-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camphausen K, Burgan W, Cerra M, Oswald KA, Trepel JB, Lee MJ, Tofilon PJ. Enhanced radiation-induced cell killing and prolongation of gammaH2AX foci expression by the histone deacetylase inhibitor MS-275. Cancer Res. 2004;64:316–321. doi: 10.1158/0008-5472.can-03-2630. [DOI] [PubMed] [Google Scholar]
- 8.Camphausen K, Cerna D, Scott T, Sproull M, Burgan WE, Cerra MA, Fine H, Tofilon PJ. Enhancement of in vitro and in vivo tumor cell radiosensitivity by valproic acid. Int J Cancer. 2005;114:380–386. doi: 10.1002/ijc.20774. [DOI] [PubMed] [Google Scholar]
- 9.Camphausen K, Scott T, Sproull M, Tofilon PJ. Enhancement of xenograft tumor radiosensitivity by the histone deacetylase inhibitor MS-275 and correlation with histone hyperacetylation. Clin Cancer Res. 2004;10:6066–6071. doi: 10.1158/1078-0432.CCR-04-0537. [DOI] [PubMed] [Google Scholar]
- 10.Chinnaiyan P, Cerna D, Burgan WE, Beam K, Williams ES, Camphausen K, Tofilon PJ. Postradiation sensitization of the histone deacetylase inhibitor valproic acid. Clin Cancer Res. 2008;14:5410–5415. doi: 10.1158/1078-0432.CCR-08-0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Jung M, Dritschilo A. Enhancement of radiation sensitivity of human squamous carcinoma cells by histone deacetylase inhibitors. Radiat Res. 2004;161:667–674. doi: 10.1667/rr3192. [DOI] [PubMed] [Google Scholar]
- 12.Florenes VA, Skrede M, Jorgensen K, Nesland JM. Deacetylase inhibition in malignant melanomas: impact on cell cycle regulation and survival. Melanoma Res. 2004;14:173–181. doi: 10.1097/01.cmr.0000129576.49313.26. [DOI] [PubMed] [Google Scholar]
- 13.Bedford JS, Dewey WC. Radiation Research Society. 1952-2002. Historical and current highlights in radiation biology: has anything important been learned by irradiating cells? Radiat Res. 2002;158:251–291. doi: 10.1667/0033-7587(2002)158[0251:hachir]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 14.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 15.Zhang F, Zhang T, Teng ZH, Zhang R, Wang JB, Mei QB. Sensitization to gamma-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biol Ther. 2009;8:823–831. doi: 10.4161/cbt.8.9.8143. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Adachi M, Zhao X, Kawamura R, Imai K. Histone deacetylase inhibitors FK228, N-(2-aminophenyl)-4-[N-(pyridin-3-yl-methoxycarbonyl)amino- methyl]benzamide and m-carboxycinnamic acid bishydroxamide augment radiation-induced cell death in gastrointestinal adenocarcinoma cells. Int J Cancer. 2004;110:301–308. doi: 10.1002/ijc.20117. [DOI] [PubMed] [Google Scholar]
- 17.Munshi A, Kurland JF, Nishikawa T, Tanaka T, Hobbs ML, Tucker SL, Ismail S, Stevens C, Meyn RE. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin Cancer Res. 2005;11:4912–4922. doi: 10.1158/1078-0432.CCR-04-2088. [DOI] [PubMed] [Google Scholar]
- 18.Fertil B, Dertinger H, Courdi A, Malaise EP. Mean inactivation dose: a useful concept for intercomparison of human cell survival curves. Radiat Res. 1984;99:73–84. [PubMed] [Google Scholar]
- 19.Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6:443–450. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- 20.Sedelnikova OA, Pilch DR, Redon C, Bonner WM. Histone H2AX in DNA damage and repair. Cancer Biol Ther. 2003;2:233–235. doi: 10.4161/cbt.2.3.373. [DOI] [PubMed] [Google Scholar]
- 21.Taneja N, Davis M, Choy JS, Beckett MA, Singh R, Kron SJ, Weichselbaum RR. Histone H2AX phosphorylation as a predictor of radiosensitivity and target for radiotherapy. J Biol Chem. 2004;279:2273–2280. doi: 10.1074/jbc.M310030200. [DOI] [PubMed] [Google Scholar]
- 22.Bromfield GP, Meng A, Warde P, Bristow RG. Cell death in irradiated prostate epithelial cells: role of apoptotic and clonogenic cell kill. Prostate Cancer Prostatic Dis. 2003;6:73–85. doi: 10.1038/sj.pcan.4500628. [DOI] [PubMed] [Google Scholar]
- 23.Noh EJ, Lee JS. Functional interplay between modulation of histone deacetylase activity and its regulatory role in G2-M transition. Biochem Biophys Res Commun. 2003;310:267–273. doi: 10.1016/j.bbrc.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 24.Rosato RR, Almenara JA, Yu C, Grant S. Evidence of a functional role for p21WAF1/CIP1 down-regulation in synergistic antileukemic interactions between the histone deacetylase inhibitor sodium butyrate and flavopiridol. Mol Pharmacol. 2004;65:571–581. doi: 10.1124/mol.65.3.571. [DOI] [PubMed] [Google Scholar]
- 25.Rashid SF, Moore JS, Walker E, Driver PM, Engel J, Edwards CE, Brown G, Uskokovic MR, Campbell MJ. Synergistic growth inhibition of prostate cancer cells by 1 alpha,25 Dihydroxyvitamin D(3) and its 19-nor-hexafluoride analogs in combination with either sodium butyrate or trichostatin A. Oncogene. 2001;20:1860–1872. doi: 10.1038/sj.onc.1204269. [DOI] [PubMed] [Google Scholar]
- 26.Orrenius S. Mitochondrial regulation of apoptotic cell death. Toxicol Lett. 2004;149:19–23. doi: 10.1016/j.toxlet.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 27.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 28.Chinnaiyan P, Vallabhaneni G, Armstrong E, Huang SM, Harari PM. Modulation of radiation response by histone deacetylase inhibition. Int J Radiat Oncol Biol Phys. 2005;62:223–229. doi: 10.1016/j.ijrobp.2004.12.088. [DOI] [PubMed] [Google Scholar]
- 29.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 30.Mihara M, Moll UM. Detection of mitochondrial localization of p53. Methods Mol Biol. 2003;234:203–209. doi: 10.1385/1-59259-408-5:203. [DOI] [PubMed] [Google Scholar]
- 31.Wendt J, von Haefen C, Hemmati P, Belka C, Dorken B, Daniel PT. TRAIL sensitizes for ionizing irradiation-induced apoptosis through an entirely Bax-dependent mitochondrial cell death pathway. Oncogene. 2005;24:4052–4064. doi: 10.1038/sj.onc.1208580. [DOI] [PubMed] [Google Scholar]
- 32.Isaacs WB, Carter BS, Ewing CM. Wild-type p53 suppresses growth of human prostate cancer cells containing mutant p53 alleles. Cancer Res. 1991;51:4716–4720. [PubMed] [Google Scholar]
- 33.Roy S, Tenniswood M. Site-specific acetylation of p53 directs selective transcription complex assembly. J Biol Chem. 2007;282:4765–4771. doi: 10.1074/jbc.M609588200. [DOI] [PubMed] [Google Scholar]
- 34.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 35.Sykes SM, Stanek TJ, Frank A, Murphy ME, McMahon SB. Acetylation of the DNA binding domain regulates transcription-independent apoptosis by p53. J Biol Chem. 2009;284:20197–20205. doi: 10.1074/jbc.M109.026096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Wong P, Radany E, Wong JY. HDAC inhibitor, valproic acid, induces p53-dependent radiosensitization of colon cancer cells. Cancer Biother Radiopharm. 2009;24:689–699. doi: 10.1089/cbr.2009.0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim IA, Shin JH, Kim IH, Kim JH, Kim JS, Wu HG, Chie EK, Ha SW, Park CI, Kao GD. Histone deacetylase inhibitor-mediated radiosensitization of human cancer cells: class differences and the potential influence of p53. Clin Cancer Res. 2006;12:940–949. doi: 10.1158/1078-0432.CCR-05-1230. [DOI] [PubMed] [Google Scholar]
- 39.Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. J Cell Sci. 2003;116:4077–4085. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- 40.Norbury CJ, Zhivotovsky B. DNA damage-induced apoptosis. Oncogene. 2004;23:2797–2808. doi: 10.1038/sj.onc.1207532. [DOI] [PubMed] [Google Scholar]
- 41.Sarkaria JN, Eshleman JS. ATM as a target for novel radiosensitizers. Semin Radiat Oncol. 2001;11:316–327. doi: 10.1053/srao.2001.26030. [DOI] [PubMed] [Google Scholar]
- 42.Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59:928–942. doi: 10.1016/j.ijrobp.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 43.Hostanska K, Vuong V, Rocha S, Soengas MS, Glanzmann C, Saller R, Bodis S, Pruschy M. Recombinant mistletoe lectin induces p53-independent apoptosis in tumour cells and cooperates with ionising radiation. Br J Cancer. 2003;88:1785–1792. doi: 10.1038/sj.bjc.6600982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manfredi JJ. p53 and apoptosis: it's not just in the nucleus anymore. Mol Cell. 2003;11:552–554. doi: 10.1016/s1097-2765(03)00106-0. [DOI] [PubMed] [Google Scholar]
- 45.Chipuk JE, Green DR. Cytoplasmic p53: bax and forward. Cell Cycle. 2004;3:429–431. [PubMed] [Google Scholar]
- 46.Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 47.Yamaguchi H, Woods NT, Piluso LG, Lee HH, Chen J, Bhalla KN, Monteiro A, Liu X, Hung MC, Wang HG. p53 acetylation is crucial for its transcription-independent proapoptotic functions. J Biol Chem. 2009;284:11171–11183. doi: 10.1074/jbc.M809268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rakozy C, Grignon DJ, Li Y, Gheiler E, Gururajanna B, Pontes JE, Sakr W, Wood DP, Jr, Sarkar FH. p53 gene alterations in prostate cancer after radiation failure and their association with clinical outcome: a molecular and immunohistochemical analysis. Pathol Res Pract. 1999;195:129–135. doi: 10.1016/S0344-0338(99)80024-7. [DOI] [PubMed] [Google Scholar]
- 49.MacGrogan D, Bookstein R. Tumour suppressor genes in prostate cancer. Semin Cancer Biol. 1997;8:11–19. doi: 10.1006/scbi.1997.0048. [DOI] [PubMed] [Google Scholar]
- 50.Prendergast NJ, Atkins MR, Schatte EC, Paulson DF, Walther PJ. p53 immunohistochemical and genetic alterations are associated at high incidence with post-irradiated locally persistent prostate carcinoma. J Urol. 1996;155:1685–1692. [PubMed] [Google Scholar]
- 51.Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci U S A. 2004;101:2259–2264. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, Berger SL. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol. 1999;19:1202–1209. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin Y, Zeng SX, Lee H, Lu H. MDM2 mediates p300/CREB-binding protein-associated factor ubiquitination and degradation. J Biol Chem. 2004;279:20035–20043. doi: 10.1074/jbc.M309916200. [DOI] [PubMed] [Google Scholar]
- 55.Zhang S, Cao HJ, Davis FB, Tang HY, Davis PJ, Lin HY. Oestrogen inhibits resveratrol-induced post-translational modification of p53 and apoptosis in breast cancer cells. Br J Cancer. 2004;91:178–185. doi: 10.1038/sj.bjc.6601902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fu M, Wang C, Zhang X, Pestell RG. Acetylation of nuclear receptors in cellular growth and apoptosis. Biochem Pharmacol. 2004;68:1199–1208. doi: 10.1016/j.bcp.2004.05.037. [DOI] [PubMed] [Google Scholar]
- 57.Vaghefi H, Neet KE. Deacetylation of p53 after nerve growth factor treatment in PC12 cells as a post-translational modification mechanism of neurotrophin-induced tumor suppressor activation. Oncogene. 2004;23:8078–8087. doi: 10.1038/sj.onc.1207953. [DOI] [PubMed] [Google Scholar]
- 58.Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 59.Li M, Luo J, Brooks CL, Gu W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem. 2002;277:50607–50611. doi: 10.1074/jbc.C200578200. [DOI] [PubMed] [Google Scholar]
- 60.Nakamura S, Roth JA, Mukhopadhyay T. Multiple lysine mutations in the C-terminal domain of p53 interfere with MDM2-dependent protein degradation and ubiquitination. Mol Cell Biol. 2000;20:9391–9398. doi: 10.1128/mcb.20.24.9391-9398.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y, Sun J, Yu Y, Zhou W, Zheng Q, Wu M, Otterson GA, Zhu WG. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1) Mol Cell Biol. 2006;26:2782–2790. doi: 10.1128/MCB.26.7.2782-2790.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grossman SR, Deato ME, Brignone C, Chan HM, Kung AL, Tagami H, Nakatani Y, Livingston DM. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science. 2003;300:342–344. doi: 10.1126/science.1080386. [DOI] [PubMed] [Google Scholar]
- 63.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mellert HS, Stanek TJ, Sykes SM, Rauscher FJ, 3rd, Schultz DC, McMahon SB. Deacetylation of the DNA-binding domain regulates p53-mediated apoptosis. J Biol Chem. doi: 10.1074/jbc.M110.184663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Devi GR. XIAP as target for therapeutic apoptosis in prostate cancer. Drug News Perspect. 2004;17:127–134. doi: 10.1358/dnp.2004.17.2.829046. [DOI] [PubMed] [Google Scholar]
- 66.Kaufmann SH, Vaux DL. Alterations in the apoptotic machinery and their potential role in anticancer drug resistance. Oncogene. 2003;22:7414–7430. doi: 10.1038/sj.onc.1206945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1: VPA induced dose-dependent histone protein acetylation in vitro. Prostate cancer LnCaP, DU145 and PC-3 cells were exposed to different concentrations of VPA for 12 hours. Total cell lysates were analyzed by immunoblots. The anti-acetyl-histone-H4 rabbit antiserum (Upstate, Charlottesville, VA) was used to detect acetylation of histone protein. Anti-β-actin antibody was utilized to determine the equal loading of proteins.
Supplementary figure 2: VPA enhanced and prolonged γ-H2A.X levels after IR. LnCaP, DU145 and PC-3 cells were exposed to 50 μM VPA for 12 hours prior to 10 Gy of IR. Total cell lysates collected at indicated times were analyzed by immunoblot using the anti-phospho-histone H2A.X (Ser139) monoclonal antibody, and anti-β-actin antibody, respectively.
Supplementary figure 3: Effects of VPA on pro-apoptotic gene expression in response to irradiation. Cells were treated with or without 50 μM of VPA for 12 hours, followed by 10 Gy IR. (A) Total RNA were prepared 24 hours later, and subjected to RNase protection assay with p53-related, double strand break-related or hApo2c probe sets. (BD Biosciences, San Diego, CA). (B) Total RNA or cell lysates were prepared 24 hours later after IR. RNase protection assay or immunoblot analyses were performed.
Supplementary figure 4: Role of K120 acetylation of p53 in VPA-mediated radiosensitization and clonogenic survival. PC-3 cells were engineered to express either wild-type or mutant p53 as indicated. Clonogenic survival assays were performed as described in Figure 1B. Empty-vector (EV) transfected PC-3 were included as a control. Error bars indicate one SD from three individual experiments.
Supplementary figure 5: Role of K120 acetylation of p53 in VPA-mediated apoptosis. PC-3 cells engineered to express either wild-type or mutant p53 (K120R) were treated with VPA and IR. Apoptosis was analyzed by annexin V-FITC assay 48 hours after IR. Error bars indicate one SD from three individual experiments. * indicates significant difference (ρ < 0.01).
Supplementary table 1: Effects of different concentrations of VPA on plating efficiencies of prostate cancer cells in vitro
Supplementary table 2: Effects of p53 acetylation on VPA-induced radiosensitizations of engineered PC-3 cells in vitro