

Abstract
3-Hydroxymethyl-4-methyl-DCK (3, HMDCK) was discovered previously as a potent HIV non-nucleoside reverse transcriptase inhibitor (NNRTIs) (EC50: 0.004 μM, TI: 6225) with a novel mechanism of action. It exerts anti-HIV activity by inhibiting the production of HIV-1 double-stranded viral DNA from a single-stranded DNA intermediate, rather than blocking the generation of single-stranded DNA from a RNA template, which is the mechanism of action of current HIV-1 RT inhibitors. However, the insufficient metabolic stability of 3 limits its further clinical development. In the current study, a series of ester prodrugs of 3 was designed and synthesized to explore the new drug candidates as NNRTIs. The l-alanine ester prodrug 10 exhibited desirable pharmacokinetic properties in vitro and in vivo and showed improved oral bioavailability of 26% in rat, and would be a potential clinical candidate as a new anti-AIDS drug.
Keywords: 3-Hydroxymethyl-4-methyl-DCK, Synthesis, Pharmacokinetic, Prodrug
1. Introduction
With approximately 4.2 million HIV/AIDS patients world-wide, 26 current anti-HIV drugs including reverse transcriptase (RT), protease, integrase, and entry inhibitors are used alone or in combination to treat HIV infection and AIDS patients. However, HIV develops resistance to all current drugs, resulting in rapidly decreased drug efficacy. Therefore, new anti-HIV agents with novel structures or mechanism(s) of action are urgently needed to overcome this problem.
In our previous studies, 3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone (1, DCK, Fig. 1) and its analogs were identified as a novel class of potent anti-HIV agents1–4 acting by a unique mechanism. These compounds inhibit the production of HIV-1 double-stranded viral DNA from a single-stranded DNA intermediate5, in contrast to current HIV-1 RT inhibitors that block the generation of single-stranded DNA from a RNA template. Due to the novel mechanism of action, we were strongly motivated to develop potent DCK analogs as potential clinical candidates. Due to the high potency (EC50: 0.0015 μM, TI: 15,733) and easy synthesis, 4-methyl-DCK (2) was first selected as a candidate for preclinical studies. However, its high hydrophobicity resulted in poor bioavailability. Subsequent structure modification then focused on the 3-position of the coumarin ring of 2 via the addition of polar functionalities to improve water-solubility and enhance drug oral absorption and bioavailability. Consequently, 3-hydroxymethyl-4-methyl-DCK (3, HMDCK) was discovered to be a potential drug candidate with high potency (EC50: 0.004 μM, TI: 6225) and moderate bioavailabity (F: 15%) in rat. However, further studies revealed that 3 lacked sufficient metabolic stability, preventing further development of this compound class.6
Figure 1.
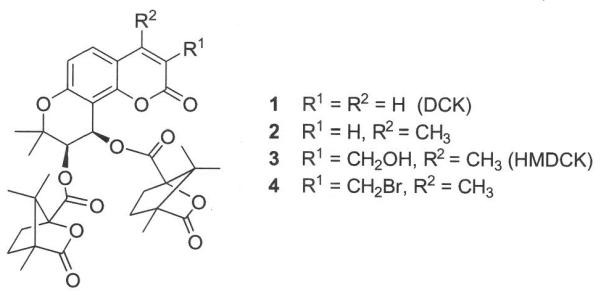
Structures of active DCK analogs.
Therefore, our current research has turned to improve DCK analogs' pharmacokinetic properties and eventually enhance oral availability. Because the hydroxy group at the 3-positon of the coumarin ring of 3 is a good “handle”, compound 3 served as the new starting point for the design of prodrugs, which has proven to be a successful approach in development of new marketed drugs, including chloramphenicol succinate, docarpamine7, valganciclovir8, valaciclovir, and others. Our prodrug design focused on masking the hydroxy group at the 3-positon of the coumarin ring of 3 by introducing polar functional groups selected to decrease the rapid metabolism of parent compound by such conjugation as well as improve molecular water-solubility and absorption9. Therefore, nine ester prodrugs (5–13) with a free carboxylic or amine group were designed and synthesized, and their pharmacokinetic properties and metablic stabilities were evaluated in vitro and in vivo.
2. Results and discussion
2.1. Chemistry
As shown in Scheme 1, the hydroxy group of 3 reacted with oxalyl chloride or succinic anhydride (molar ratio 1:1.1) in the presence of pyridine to provide carboxylic ester prodrugs 5 and 6, respectively. Next, 5 and 6 were converted to corresponding sodium carboxylates 7 and 8, respectively, by using sodium 2-ethylhexanoate. We also synthesized amino acid ester prodrugs 9–12 by using 3-bromomethyl-4-methyl-DCK (4) treated with N-Boc protected natural glycine, analine, valine, or leucine, respectively, in the presence of triethylamine in acetone, followed by hydrolysis in trifluoroacetic acid, with yields of over 50%. Furthermore, the free base of 9 was treated with hydrogen chloride in ether to form the corresponding HC1 salt 13.
Scheme 1.

Synthesis of prodrugs. (i) oxalyl chloride or suceinic anhydride, Py/CH2Cl22; (ii) sodium 2-ethylhexanoate/EtOAc; (iii) N-Boc-amino acid(s), Et3N/acetone; (iv) TFA/CH2Cl2; (v) HCl in diethyl ether.
2.2. In vitro evaluation
The in vitro metabolic stabilities of the ester prodrugs 5–13 were first investigated using Sprague-Dawley rat liver microsomes under oxidative conditions, together with the parent compound 3 and a reference compounds propranolol (PRO). The incubation mixtures of 5–13 were analyzed by analytical HPLC with MS detector to measure the release and consumption of the parent compound 3 at different time points (5, 15, 30, and 60 min). A linear relationship (R2 > 0.95) was found between the logarithm of the percentage of 3 remaining using 0 min peak height ratio as 100% versus time for all tested compounds, which was evidence of pseudo-first-order degradation kinetics. The results of the in vitro metabolic evaluation are summarized in Table 1 and Fig. 2, including the degradation rate constants (kobs), the corresponding apparent half-life (T1/2), and intrinsic clearance (CLint) of prodrugs 5–13.
Table 1.
In vitro metabolic stability profiles of prodrugs together with the parent drug in SD rat liver microsomes.
| Parameter | Compound | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | PROa | |
| k obs | 0.039 | 0.020 | 0.020 | 0.024 | 0.027 | 0.178 | 0.014 | 0.023 | 0.023 | 0.026 | 0.033 |
| T1/2 (min) | 17.92 | 34.61 | 35.27 | 29.07 | 25.93 | 3.90 | 49.26 | 30.09 | 30.77 | 26.54 | 20.71 |
| CLint (mL/min/mg) | 0.39 | 0.20 | 0.20 | 0.24 | 0.27 | 1.78 | 0.14 | 0.23 | 0.23 | 0.26 | 0.33 |
Reference compounds: propranolol (moderate metabolism in vivo with T1/2 of 3–5 h).
Figure 2.
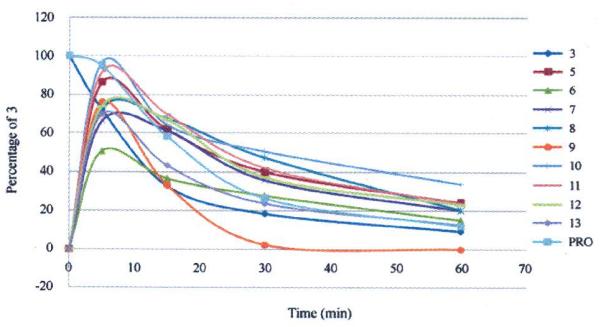
In vitro metabolic stability in Sprague-Dawley rat liver microsomes of prodrugs and parent compound (3) as well as positive control drug propranolol (PRO).
From the results, we can see that all prodrugs could release quickly and showed over 50 percent quantity of parent compound 3 at least at the 5 min time point. Except for 9, most of the ester prodrugs showed a longer half life (T1/2 25.93–49.26 min) compared with parent compound 3 (T1/2 17.92 min). The l-analine ester prodrug 10 was the best compound with the longest apparent half-time in vitro T1/2 of 49.26 min, the lowest degradation rate constant (kobs 0.014), and CLint of 0.14 mL/min/mg among this prodrug series. In contrast with 10, the glycine ester prodrug 9 was much less stable (T1/2 3.90 min) than 3, thus indicating that a methyl side chain in the amino acid must be necessary to slow down the release of the parent compound 3 from the prodrug. With T1/2 values ranging from 25.93 to 35.27 min, prodrugs 5–8 with a free carboxylic group or related sodium salt also showed longer metabolic half-lives compared with 3. Thus, our current ester prodrugs, prepared by introducing either carboxylic acids or amino acids, successfully led to significantly prolonged half-lives, decreased degradation rates, and smaller intrinsic clearance values compared with the parent compound 3. Subsequently, these new prodrugs were further evaluated in animal models.
2.3. In vivo evaluation
In the in vivo evaluation, prodrugs 5–13 were administered in 10% poly (ethylene glycol)-400 (PEG400) to adult male SD rats (180–200 g) via an oral route (i.g. suspension, 20 mg/kg), and 3 was administrated via oral and intravenous routes with i.g. 20 mg/kg and i.v. 2 mg/kg, respectively, as a reference. Plasma samples for each tested compound were collected at 6–8 time points over a 12-h period, immediately centrifuged, and stored at −20 °C, successively. All samples were analyzed with LC-MS/MS by measuring the concentration of parent compound 3, which was released from each prodrug. The pharmacokinetic parameters of prodrugs were calculated by non-compartmental analysis using DAS Version 2.0. The data are shown in Table 2 and Fig. 3.
Table 2.
Pharmacokinetic parameters of prodrugs and parent 3 in male Sprague-Dawley rats (180–200 g) by i.g. (20 mg/kg) dosing and 3 by i.v. (2 mg/kg) in 10% PEG400.
| Parameter | Compound | 3(iv) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | 12 | 13 | 3 | ||
| AUC(0-t) ng·L/mL | 521.7 ± 133.6 | 157.9 ± 42.94 | 354.2 ± 105.6 | 588.9 ± 72.06 | 384.1 ± 6.21 | 1296.5 ± 437.6 | 775.6 ± 162.9 | 477.8 ± 128.7 | 966.3 ± 219.7 | 558.4 ± 8.71 |
| CL (L/h/kg) | 0.033 ± 0.006 | 0.102 ± 0.020 | 0.046 ± 0.013 | 0.034 ± 0.004 | 0.037 ± 0.001 | 0.016 ± 0.007 | 0.026 ± 0.005 | 0.040 ± 0.010 | 0.020 ± 0.005 | 0.003 ± 0.0001 |
| Vd (L/kg) | 0.252 ± 0.130 | 0.669 ± 0.422 | 0.373 ± 0.193 | 0.102 ± 0.011 | 0.467 ± 0.063 | 0.045 ± 0.017 | 0.049 ± 0.020 | 0.123 ± 0.058 | 0.079 ± 0.039 | 0.008 ± 0.002 |
| Cmax (ng/mL) | 284.1 ± 74.73 | 80.9 ± 18.80 | 254.6 ± 32.45 | 398.5 ± 29.00 | 163.6 ± 13.56 | 621.5 ± 79.67 | 529.0 ± 192.9 | 191.1 ± 2.48 | 530.7 ± 120.8 | − |
| Tmax (h) | 0.50 | 0.38 | 0.25 | 0.50 | 0.25 | 0.75 | 0.50 | 0.75 | 0.58 | − |
| MRT(0-t) (h) | 3.092 ± 0.454 | 2.658 ± 0.681 | 3.769 ± 0.063 | 1.517 ± 0.207 | 3.581 ± 0.307 | 1.793 ± 0.181 | 1.286 ± 0.085 | 2.136 ± 0.123 | 1.983 ± 0.478 | 1.235 ± 0.168 |
| F(%) | 10.3 | 3.3 | 7.2 | 12.4 | 7.5 | 25.7 | 15.8 | 9.8 | 17.3 | − |
Figure 3.

Plasma concentration-versus-time curves of prodrugs and parent compound. (A) 3 with PEG400, i.v. administration at 2 mg/kg, and (B) prodrugs and 3 with PEG400, i.g. administration at 20 mg/kg. Results are averages of three independent assays.
The in vivo data indicated that l-analine ester prodrug 10 exhibited a better pharmacokinetic profile than 3, with a higher oral bioavailability (F: 25.7%), lower systemic clearance (CL: 0.016 L/min/kg), smaller volume of distribution (Vd: 0.045 L/kg), and moderate MRT (1.793 h). Meanwhile, the high Cmax (621.5 ng/mL) of 10 demonstrated that the presence of the l-alanine moiety greatly benefited drug absorption in vivo. With a long Tmax (0.75 h), prodrug 10 slowly and smoothly released the parent drug, allowing reduced metabolic opportunities for 3, and subsequently enhancing oral bioavailability. On the other hand, the prodrug 8, a sodium carboxylate salt of prodrug 6, exhibited obviously improved oral bioavailability (F: 12.4%) comparing to 6 (F: 3.3%). The obvious differences of 8 and 6 in AUC (588.9 and 157.9 ng·h/mL, respectively) and Cmax (398.5 and 80.9 ng/mL, respectively) values indicated that the increased dissolution rate of a salt form improved absorption. Among these prodrugs, the most promising prodrug was 10. It showed higher plasma maximal concentration (Cmax: 10 > 12 ≅ 3 > remaining compounds), lower clearance and volume of distribution (CL and Vd: 10 > 12 ≅ 3 < remaining compounds) than the other prodrugs, subsequently resulting in greatly increased drug absorption and oral bioavailability (F%: 10 > 3 > remaining compounds). These in vivo results are consistent with the in vitro data. Thus, we postulate that the presence of an amino acid moiety with a bulky side chain might benefit “drug-like” molecular physicochemical properties, resulting in improved prodrug absorption and bioavailability as shown in both in vitro and in vivo assays.
2.4. Water solubility (WS) analysis
Because water solubility is a crucial physicochemical property that affects drug absorption, the designed prodrugs contained a polar structural moiety, either amino acid or carboxylic acid, introduced to improve water solubility. We performed a WS analysis to compare the most promising prodrug 10 and parent compound 3. We first prepared a solution of each compound in acetonitrile at room temerature and diluted this stock solution into five required different concentrations to establish a standard curve. The water-solubility of each compound was determined by HPLC analysis from the correlating area of its saturated concentration in water. As shown in Table 3, prodrug 10 had better aqueous solubility (18mg/L) than 3 (8.4mg/L). Furthermore, prodrug 10 also had a lower predicted octanol-water partition coefficient (log P) and distribution coefficient (log D) values (3.84 and 3.38) than those of 3 (4.44 and 4.43, respectively). Based on its improved pharmacokinetic properties, prodrug 10 could merit further development as a drug candidate.
Table 3.
Predicted log P and log D values and water solubilities of 3 and 10*.
| Parameter | Compound |
|
|---|---|---|
| 3 | 10 | |
| Water solubility (mg/L, pH 7.4) | 8.4 | 18 |
| log P | 4.44 | 3.84 |
| log D (pH 7.4) | 4.43 | 3.38 |
The octanol-water partition coefficient log P and apparent distribution coefficient log D values were calculated by using ACD programs.
Conclusions
Ester prodrugs of 3, either carboxylic or amino acid esters, were synthesized and evaluated in the current study, leading to the discovery of the promising prodrug 10. It exhibited greatly improved oral bioavailability and other desirable pharmacokinetic properties, which were better than those of the parent compound 3. These results suggested that prodrug 10 could be further investigated as a potential new anti-AIDS NNRTI drug candidate.
4. Experimental
4.1. Chemistry
Melting points were measured with an RY-1 melting apparatus without correction. The proton nuclear magnetic resonance (1H NMR) spectra were measured on a JNM-ECA-400 (400 MHz) spectrometer using tetramethylsilane (TMS) as the internal standard. The solvent used was CDCl3. unless otherwise indicated. Mass spectra (MS) were measured on an ABI Perkin–Elmer Sciex API-150 mass spectrometer with electrospray ionization, and the relative intensity of each ion peak is presented as percent (%). The purities of target compounds were ≥95%, measured by HPLC analyses, which were performed on an Agilent 1100 HPLC system with a UV detector. HPLC analyses for purity were performed using an Agilent SC-C18 column (150 mm × 4.6 mm, 5 μm) eluting with a mixture of solvents A and B (Condition: acetonitrile/water 70:30, flow rate 1.0 mL/min), detected under UV wavelength at 320 nm and an injection volume of 40 μL. Thin-layer chromatography (TLC) and preparative TLC were performed on silica gel GF254 plates. Silica gel (200–300 mesh) from Qingdao Haiyang Chemical Company was used for column chromatography. All chemicals were obtained from Beijing Chemical Works or Sigma-Aldrich, Inc.
4.2. 2-Oxo-2-[(3′, 4′-di-O-(S)-camphanoyl-4-methyl-(+)-cis-kellactone-3-yl)methoxy] acetic acid (5) and sodium salt (7)
Oxalyl chloride (0.1 mL) in anhydrous CH2Cl2 was added slowly into a solution of 3 (133 mg, 0.2 mmol) in CH2Cl2 (20 mL, anhydrous) at ice-bath temperature, and then at room temperature with stirring for 1 h. The progress was monitored by TLC (acetone/cyclohexame 2:1) until the reaction was finished. The mixture was poured into ice-water, and extracted with EtOAc three times. The combined organic phase was washed with water, aq. NaHCO3, water, and brine successively, and dried over anhydrous Na2SO4. After removal of solvent in vacuo, the residue was purified by PTLC (eluent: acetone/cyclohexame 3:1) to obtain 108 mg of 5, yield 73%; light yellow solid, mp 208–210 °C; 1H NMR δ 0.98–1.12 (18H, m.s., 6 × CH3 in camphanoyl group), 1.45 and 1.49 (each 3H, s, CH3-2′), 1.68, 1.93, 2.23, and 2.51 (each 2H, m, CH2 in camphanoyl group), 2.49 (3H, s, CH3-4), 5.17 (2H, s, CH2-3), 5.33 (1H, d, j = 4.4 Hz, H-3′), 6.56 (1H, d, J = 4.4 Hz, H-4′), 6.87 (1H, d, j = 9.2 Hz, H-6), 7.64 (1H, d, j = 9.2 Hz, H-5). MS m/z (%) 739.3 (M + 1, 4), 649.5 (M-89, 100); HPLC purity 96.14%. Compound 5 (77 mg, 0.1 mmol) in 2 mL of EtOAc with 18 mg of sodium 2-ethylhexanoate was stirred at room temperature for 12 h. The precipitated solid was collected and dried to obtain the sodium salt 7, mp > 230 °C; HPLC purity 95.36%.
4.3. 4-Oxo-4-[(3′,4′-di-0-(S)-camphanoyl-4-methyl-(+)-cis-kellactone-3-yl)methoxy]butanok acid (6) and sodium salt (8)
To a solution of 3 (133 mg, 0.2 mmol) in CH2C12 (20 mL, anhydrous) was added 0.5 mL of pyridine and 20 mg of succinic anhydride successively. The mixture was stirred at room temperature for 12 h and monitored by TLC (acetone/cyclohexane 1:1) to the reaction completion. The mixture was added to 10 mL of aq. HCl (5%), and extracted with EtOAc three times. The combined organic phase was washed with water, aq. NaHCO3, water, and brine successively, and dried over anhydrous Na2SO4. After removal of solvent in vacuo, the residue was purified by PTLC (eluent: acetone/cyclohexame 1:1) to obtain 86 mg of 6, yield 56%; light yellow solid, mp 132–134 °C; 1H NMR δ 0.98–1.12 (18H, m.s., 6 × CH3 in camphanoyl group), 1.45 and 1.49 (each 3H, s, CH3-2′), 1.68, 1.93, 2.23, and 2.51 (each 2H, m, CH2 in camphanoyl group), 2.48 (3H, s, CH3-4), 2.64 (4H, m, 2 × CH2 in side chain), 5.17 (2H, s, CH2-3), 5.40 (1H, d, j = 4.8 Hz, H-3′), 6.63 (1H, d, j = 4.8 Hz, H-4′), 6.86 (1H, d, j = 9.2 Hz, H-6), 7.63 (1H, d, j = 9.2 Hz, H-5). MS m/z (%) 767.4 (M + 1, 27), 649.5 (M - 117, 100); HPLC purity 97.12%. Compound 6 (77 mg, 0.1 mmol) in 2 mL of EtOAc with 18 mg of sodium 2-ethylhexanoate was stirred at room temperature for 12 h. The precipitated solid was collected and dried to obtain the sodium salt 8, mp > 230 °C; HPLC purity 100.0%.
4.4. General procedure for the preparation of N-Boc-amino acids
To a solution of an amino acid (i.e., glycine, alanine, valine, or leucine) (10 mmol) in 25 mL of water was added 0.5 g of Na2CO3 with stirring until a complete solution was reached. BOC anhydride (15 mmol) in 1,4-dioxane (10 mL) was added to the above solution at 0 °C with stirring for 1 h, and then stirring continued at room temperature for an additional 12 h. The mixture was poured into water (25 mL) and washed with EtOAc to remove unreacted anhydride. The water phase was adjusted to pH 1 with aq. HC1 (10%) and extracted with EtOAc again three times. Next, the combined organic phase was washed with water and brine successively, and dried with anhydrous Na2SO4. After solvent was removed under reduced pressure, the protected amino acid was used for the next reaction without purification.
4.5. General procedure for the preparation of ester prodrugs 9–12
A mixture of 3-bromomethyl-4-methyl-DCK (4, 0.21 mmol) in acetone (10 mL), Et3N (1 mL), and Boc-protected amino acid (0.4 mmol) was stirred at room temperature for 1 h, and monitored by TLC eluting with a mixture of petroleum ether and EtOAc (3:1) until the reaction had finished. The mixture was poured into ice-water and extracted with EtOAc three times. The combined organic phase was washed with aq. HCl (5%), water, aq. NaHCO3, water, and brine successively, and dried over anhydrous Na2SO4. After removing solvent in vacuo, solid crude product was purified by column chromatograph (gradient of EtOAc and petroleum ether 0–40%) to provide corresponding ester products. The BOC group was removed by treatment with CF3COOH (0.5 mL) in CH2Cl2 (2 mL) at room temperature with stirring for 1 h. The mixture was poured into water, pH adjusted to 10 with Na2CO3, and extraction performed with EtOAc three times. After removal of solvent in vacuo, the crude product was purified by preparative TLC (eluent: acetone/cyclohexane 2:1) to provide corresponding pure ester prodrugs 9–12.
4.5.1. [3′,4′-Di-O-(S)-camphanoyl-4-methyl-(+)-cis-kellactone-3-yl]methyl 2-aminoacetate (9)
Starting with 148 mg (0.21 mmol) of 4 and 70 mg of N-Boc glycine to provide 76 mg of pure 9 as light yellow solid in 53% yield, mp 192–194 °C; 1H NMR δ 0.98–1.12 (18H, m.s., 6 × CH3 in camphanoyl group), 1.45 and 1.50 (each 3H, s, CH3-2′), 1.69, 1.92, 2.20, and 2.51 (each 2H, m, CH2 in camphanoyl group), 2.50 (3H, s, CH3-4), 3.45 (2H, s, CH2CO-3), 5.20 (2H, s, CH2O-3), 5.40 (1H, d, J=4.8 Hz, H-3′), 6. 63 (1H, d, J=4.8 Hz, H-4′), 6.86 (1H, d, J=8.8 Hz, H-6), 7.62 (1H, d, J=8.8 Hz, H-5). MS m/z (%) 724.6 (M+1, 100); HPLC purity 95.45%.
4.5.2. [3′,4′-Di-O-(S)-camphanoyl-4-methyl-(+)-cis-kellactone-3-yl]methyl 2-aminopropanoate (10)
Starting with 148 mg (0.21 mmol) of 4 and 76 mg of N-Boc alanine to provide 85 mg of pure 10 as light yellow solid in 58% yield, mp 178–180 °C; 1H NMR δ 0.98–1.12 (18H, m.s., 6 × CH3 in camphanoyl group), 1.32 (3H, d, J=7.2 Hz, alanine CH3), 1.45 and 1.49 (each 3H, s, CH3-2′), 1.68, 1.93, 2.21, and 2.51 (each 2H, m, CH2 in camphanoyl group), 2.49 (3H, s, CH3-4), 3.56 (1H, m, CHCO-3), 5.18 (2H, s, CH2-3), 5.40 (1H, d, J=4.8 Hz, H-3′), 6.63 (1H, d, J=4.8 Hz, H-4′), 6.86 (1H, d, J=8.8 Hz, H-6), 7.62 (1H, d, J=8.8 Hz, H-5). MS m/z (%) 738.1 (M+1, 100); HPLC purity 100.0%.
4.5.3. [3′,4′-Di-O-(S)-camphanoyl-4-methyl-(+)-cis-kellactone-3-yl]methyl 2-amino-3-methyl-butanoate (11)
Starting with 148 mg (0.21 mmol) of 4 and 86 mg of N-Boc valine to provide 83 mg of pure 11 as light yellow solid in 54% yield, mp 150–152 °C; 1H NMR δ 0.84–0.86 (6H, m, valine 2 × CH3), 0.98–1.12 (18H, m.s., 6 × CH3 in camphanoyl group), 1.24 (1H, m, valine CH), 1.43 and 1.49 (each 3H, s, CH3-2′), 1.68, 1.93, 2.20, and 2.49 (each 2H, m, CH2 in camphanoyl group), 2.49 (3H, s, CH3-4), 3.28 (1H, d, J=4.8 Hz, NCHCO), 5.15 (2H, s, CH2-3), 5.38 (1H, d, J=4.8 Hz, H-3′), 6.62 (1H, d, J=4.8 Hz, H-4′), 6.84 (1H, d, J=8.8 Hz, H-6), 7.60 (1H, d, J=8.8 Hz, H-5). MS m/z (%) 766.6 (M+1, 100), 649.5 (M-116, 86); HPLC purity 100.0%.
4.5.4. [3′,4′-Di-O-(S) -camphanoyl-4-methyl-(+)-cis-kellactone-3-yl]methyl 2-amino-4-methyl-pentanoate (12)
Starting with 148 mg of 4 and 92 mg of N-Boc leucine to provide 102 mg of pure 12 as light yellow solid in 65% yield, mp 158–160 °C; 1H NMR δ 0.85 (6H, m, leucine 2 × CH3), 0.98–1.12 (18H, m.s., 6 × CH3 in camphanoyl), 1.26 (3H, m, CH2CH of leucine side chain), 1.46 and 1.49 (each 3H, s, CH3-2′), 1.68, 1.93, 2.23, and 2.51 (each 2H, m, CH2 in camphanoyl group), 2.49 (3H, s, CH3-4), 3.47 (1H, m, leucine NCHCO), 5.16 (2H, s, CH2-3), 5.45 (1H, d, J=4.8 Hz, H-3′), 6.64 (1H, d, J=4.8 Hz, H-4′), 6.87 (1H, d, J=8.8 Hz, H-6), 7.63 (1H, d, J=8.8 Hz, H-5). MS m/z (%) 780.7 (M+1, 100), 649.5 (M-130, 21); HPLC purity 100.0%.
4.6. [3′,4′-Di-O-(S)-camphanoyl-4-methyl-(+)-cis-kellactone-3-yl]methyl 2-aminoacetate hydrochloride salt (13)
Compound 9 (72 mg, 0.1 mmol) in 2 mL of HCl-ether was stirred at room temperature for 30 min. After removal of solvent in vacuo, 13, the hydrochloride salt of 9, was obtained with mp 216–218 °C; HPLC purity 100.0%.
4.7. In vitro metabolic assay
4.7.1. Materials
Compounds 3 and 5–13 were synthesized and characterized in our study. NADPH, MgCl2, KH2PO4, K2HPO4 were purchased from Sigma-Aldrich. Reference compounds [fast-metabolized: terfenadine (TER) and moderate-metabolized: propranolol (PRO)] were also purchased from Sigma-Aldrich. HPLC-grade acetonitrile and water were purchased from VWR. Sprague-Dawley rat liver microsomes (Lot No 38290) were purchased from BD Biosciences (Woburn, MA).
4.7.2. Sample preparation
Stock solutions of the test compounds in DMSO (2 mM) were dissolved to 0.1 mM with acetonitrile and stored at 4 °C. For measurement of metabolic stability, the reference compound PRO and the test compounds 3 and 5–13 were brought to a final concentration of 1 μM with 0.1 M potassium phosphate buffer at pH 7.4, which contained 0.1 mg/mL SD rat liver microsomes and 5 mM MgCl2. The final incubation volumes were 300 μL. Reactions were started by adding 60 μL of NADPH (final concentration of 1 mM) and quenched with 600 μL of ice-cold acetonitrile at 5, 15, 30, 60 min time points. The mixture was then centrifuged at 12,000 rpm for 5 min at 0 °C. Samples at 0 min time point were prepared by adding 600 μL cold acetonitrile first, followed by NADPH and centrifure. The supernatant was collected and 10 μL of the supernatant was injected directly to LC-MS. The following controls were also conducted: 1) positive control: liver microsomes, NADPH and two mentioned reference compounds (fast/moderate metabolized clinical drugs); 2) negative control: omit NADPH (replaced by phosphate buffer).
4.7.3. HP LC-MS conditions
Analysis was carried out on a Shimadzu LC-20AT HPLC with auto-sampler and PDA detector, and LCMS-20 mass spectrometer (single-quadrupole with Q-array technology) in electrospray ionization (ESI) mode. An Alltima C18 (5 μm, 150 mm × 2.1 mm) column was used with a flow rate of 0.2 mL/min. The isocratic elution was conducted using acetonitrile (B) in water (A) at 60% or 75%. The MS conditions were optimized to detector voltage: +1.6 kV, acquisition mode: SIM of the appropriate molecular weights of the testing compounds. The CDL temperature was 200 °C, heat block was 230 °C and neutralizing gas flow was 1.5 L/min. The electrospray ionization was operated in the positive ion mode. Full-scan spectra were also monitored over the range of m/z 180–1000.
4.7.4. Calculations
As the degradation of parent compounds 5–13 fit pseudo-first-order kinetics (R2>0.95), linear regression was used to calculate the apparent in vitro half-life (T1/2) and intrinsic clearance (CLint)10. The measured compound 3 peak height ratios were converted to percentage drug remaining using the initial time (0 min) peak height ratio values as 100%. The slope of the linear regression from logarithm percentage remaining vs. time relationships (kobs) was determined. In vitro T1/2=−0.693/kobs. The in vitro CLint (in units of mL/min/mg protein) was done using the following formula: in vitro CLint=(0.693/in vitro T1/2) × (mL incubation/mg microsomes).
4.8. Pharmacokinetic assay in rat
Male SD rats (180–200 g) were used and triplicates were performed for each sample. Rats were dosed with a tested prodrug or parent compound 3 at 20 mg/kg for i.g. administration in 10% PEG400. Blood samples were collected at 0, 15, 30, 60, 120, 240, and 360 min and were immediately centrifuged (3000 rpm, 20 min) to separate the plasma fractions. When compound 3 was administrated by intravenous (i.v.) with a dose of 2 mg/kg, the blood samples were collected at 0, 5, 10, 15, 30, 60, 120, 240, and 360 min (Fig. 3). All obtained plasma samples were stored at −20 °C until analysis. Concentration-versus-time profiles were obtained for each analyte, and standard non-compartmental analysis was performed on the data using DAS Version 2.0 to recover the area under the curve (AUC) and other non-compartmental parameters. Bioavailability was estimated by dividing the dose-normalized AUC(0-t) resulting from oral administration by the AUC(0-t) resulting from intravenous administration (where t is the last time point with measurable drug concentrations in the study). Calibration curves for testing compounds in plasma were linear in the concentration range of 10–2000 ng/mL with correlation coefficients ≥ 0.990 for all curves.
4.9. Water solubility analysis assay
Each tested compound was added in excess to a 1.5 mL tube containing 1 mL of HPLC grade water at pH 7.4 (phosphate buffer of K2HPO4), and the mixture stirred with ultrasonic assistance at room temperature for 1 h. The excess solid was removed by filtration, and the supernatant was dispensed into glass HPLC vials. The concentration of the samples was determined by HPLC on an Agilent SB-C18 column (150 mm × 4.6 mm, 5 μm), with a flow rate of 1.0 mL/min and detection at 320 nm UV wavelength. The samples (10 μL) were injected and run with a solution of 30% water and 70% MeCN. For each compound, a standard curve consisting of five concentrations (five-fold stepwise) in MeCN was established initially.
Acknowledgments
This investigation was supported by grants from the Ministry of Science and Technology of the People's Republic of China (2009ZX09102–008) and Beijing Municipal Science & Technology Commission (D0206001040191) awarded to Lan Xie, and by NIH grant number AI 33066 awarded to K. H. Lee
References
- 1.Huang L, Kashiwada Y, Cosentino LM, Fan S, Chen CH, McPhail AT, et al. Anti-AIDS agents 15. Synthesis and anti-HIV activity of dihydroseselins and related analogs. J Med Chem. 1994;37:3947–55. doi: 10.1021/jm00049a014. [DOI] [PubMed] [Google Scholar]
- 2.Xie L, Takeuchi Y, Cosentino LM, Lee KH. Anti-AIDS agents 37. Synthesis andstructure-activity relationships of (3′R, 4′R)-(+)-cis-khellactone derivatives as novel poten anti-HIV agents. J Med Chem. 1999;42:2662–72. doi: 10.1021/jm9900624. [DOI] [PubMed] [Google Scholar]
- 3.Xie L, Takeuchi Y, Cosentino LM, McPhail AT, Lee KH. Anti-AIDS agents 42. Synthesis and anti-HIV activity of disubstituted (3′R, 4′R)-3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone analogues. J Med Chem. 2001;44:664–71. doi: 10.1021/jm000070g. [DOI] [PubMed] [Google Scholar]
- 4.Xie L, Yu D, Wild C, Allaway G, Turpin J, Smith PC, et al. Anti-AIDS agents 52. Synthesis and anti-HIV activity of hydroxymethyl (3′R, 4′R)-3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone derivatives. J Med Chem. 2004;47:756–60. doi: 10.1021/jm030416y. [DOI] [PubMed] [Google Scholar]
- 5.Huang L, Yuan X, Yu D, Lcc KH, Chen CH. Mechanism of action and resistant profile of anti-HIV-1 coumarin derivatives. Virology. 2005;332:623–8. doi: 10.1016/j.virol.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki M, Li Y, Smith PC, Swenberg JA, Martin DE, Morrisnatschke SL, et al. Anti-AIDS agents 65: investigation of the in vitro oxidative metabolism of 3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone derivatives as potent anti-HIV agents. Drug Metab Dispos. 2005;33:1588–92. doi: 10.1124/dmd.105.004218. [DOI] [PubMed] [Google Scholar]
- 7.Yoshikawa M, Mishiyama S, Takaiti O. Metabolism of dopamine prodrug, docarpamine. Hypertens Res. 1995;18:211–3. doi: 10.1291/hypres.18.supplementi_s211. [DOI] [PubMed] [Google Scholar]
- 8.Sugawara M, Huang W, Fei YJ. Transport of valganciclovir, a ganciclovir prodrug, via peptidc transporters PEPT1 and PEPT2. J Pharm Sci. 2000;89:781–9. doi: 10.1002/(SICI)1520-6017(200006)89:6<781::AID-JPS10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 9.Yang YH, Aloysius H, Inoyama D, Chen Y, Hu LQ. Enzyme-mediated hydrolyticactivation of prodrugs. Acta Pharm Sin B. 2011;1:143–59. [Google Scholar]
- 10.Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, Maclntyre F, et al. The prediction of human pharmacokinetic parameters from prcclinical and in vitro metabolism data. J Pharmacol Exp Ther. 1997;283:46–58. [PubMed] [Google Scholar]