

Abstract
Background
Dravet syndrome is a devastating infantile-onset epilepsy syndrome with cognitive deficits and autistic traits caused by genetic alterations in SCN1A gene encoding the α-subunit of the voltage-gated sodium channel Nav1.1. Disease modeling using patient-derived induced pluripotent stem cells (iPSCs) can be a powerful tool to reproduce this syndrome’s human pathology. However, no such effort has been reported to date. We here report a cellular model for DS that utilizes patient-derived iPSCs.
Results
We generated iPSCs from a Dravet syndrome patient with a c.4933C>T substitution in SCN1A, which is predicted to result in truncation in the fourth homologous domain of the protein (p.R1645*). Neurons derived from these iPSCs were primarily GABAergic (>50%), although glutamatergic neurons were observed as a minor population (<1%). Current-clamp analyses revealed significant impairment in action potential generation when strong depolarizing currents were injected.
Conclusions
Our results indicate a functional decline in Dravet neurons, especially in the GABAergic subtype, which supports previous findings in murine disease models, where loss-of-function in GABAergic inhibition appears to be a main driver in epileptogenesis. Our data indicate that patient-derived iPSCs may serve as a new and powerful research platform for genetic disorders, including the epilepsies.
Keywords: Induced pluripotent stem cells, Disease modeling, Dravet syndrome, SCN1A, Nav1.1, Epileptogenesis, Action potential, Gamma aminobutyric acid
Background
Dravet syndrome (DS) is an infantile-onset epileptic encephalopathy that develops in a previously normal infant [1]. Seizures are refractory to all currently available forms of treatment; severe neuropsychiatric disabilities include cognitive deficits and autism-spectrum behaviors, and approximately 10–20% of the afflicted children do not survive [2,3]. Clearly, new and improved treatment modalities are needed, but their development hinges on research platforms that faithfully reproduce the human pathology.
Defects in the SCN1A gene, which encodes the α-subunit of the voltage-gated sodium channel Nav1.1, are seen in 70–80% of patients with DS, and approximately 50% of these defects truncate the Nav1.1 protein prematurely [4,5]. Various approaches have been used to describe and characterize the condition, most notably heterologous expression of Nav1.1 mutants [6,7] and, more recently, the development of DS mouse models, which are based on heterozygotes of an Scn1a knock-out/knock-in [8,9], or cell-type specific conditional knock-out [10,11]. These efforts have revealed the pathogenic mechanism for DS likely involves Nav1.1 haploinsufficiency [11-14]. Additionally, in the rodent forebrain, Nav1.1 is predominantly expressed in GABAergic interneurons [15], especially in the axon initial segment of a parvalbumin (PV)-positive subgroup [9], where Nav1.1 has been suggested to directly influence action potential generation and thereby exert excitation control over downstream pyramidal neurons [16]. Impaired inhibition through disruption of this suppression by forebrain GABAergic neurons may be the main pathogenic mechanism underlying the seizure susceptibility of DS [8-10,17]. A recent study has indicated that autism-related behaviors in Scn1a+/- mice result from the impaired GABAergic neurotransmission [11]. The full spectrum of factors contributing to the phenotype, however, is likely more complex with additional, so far unidentified components modifying the presentation.
Various neurological disease models have been developed using patient-derived iPSCs [18-23], but to date, no such effort has been reported toward epilepsy. Due to the early onset of the disorder and the wealth of knowledge on the associated genetic defects, DS is a highly suitable candidate for iPSC technology. We here report the first successful development of an iPSC-based DS model incorporating a nonsense mutation in SCN1A, and we show how neurons of this model have abnormal electrophysiological responses.
Results
Patient demographics and history
The tissue donor was a female patient who was born in an uneventful delivery of dizygotic twins at 38 weeks of gestation. Her birth weight was 2850 g. There were no complications during the perinatal and early postnatal period. However, at 6 months of age, she experienced her first seizure with loss of consciousness. At 7 months of age, generalized tonic-clonic seizures began, often prolonged, and induced by fever or hot baths. Despite anticonvulsant therapy, she later developed facial myoclonia and focal seizures, and obtundation status [24]. Generalized polyspikes-waves were identified in interictal electroencephalography. At the age of 21, direct sequencing of blood leukocyte-extracted genomic DNA revealed an SCN1A point mutation, c.4933C>T [GenBank: NM_001165963.1] (Figure 1A) as first reported by Fukuma and co-workers [25], which is expected to prematurely truncate the Nav1.1 protein in the fourth homologous domain (p.R1645*, Additional file 1) [GenPept: NP_001159435.1]. By 29 years of age, when a skin biopsy was performed, she had developed profound intellectual disability as well as ataxia. At that time, she had 7–8 nocturnal generalized tonic-clonic seizures a month and obtundation status once every 2–3 months.
Figure 1.
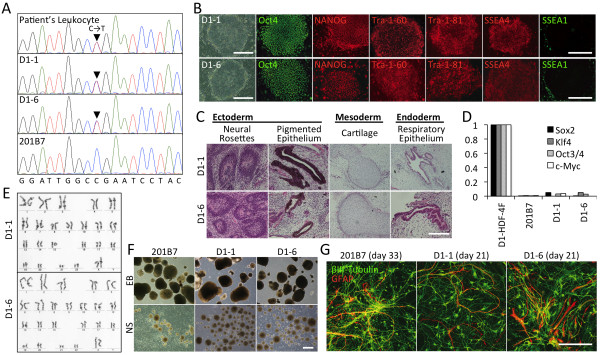
Characterization of generated iPSCs and neuronal differentiation. (A) SCN1A sequencing of the indicated cell material. Solid arrowheads point to the c.4933C>T substitution. (B) iPSC morphology and immunostaining of pluripotency markers (Oct 4, Nanog, Tra-1-60, Tra-1-81, and SSEA4 without SSEA1). Scale bar, 500 μm. (C) iPSC-derived teratomas generated in NOD-SCID mouse testes comprised tissues from all three germ layers. Scale bar, 200 μm (neural rosettes and respiratory epithelium) and 400 μm (others). (D) Real-time PCR analysis showed suppressed expression of the four reprogramming factors in both patient and control iPSCs compared to patient fibroblasts transduced with the same four factors (D1-HDF-4F). (E) G-band karyotyping showed normal chromosome numbers (46,XX) in all tested colonies (N = 20 each). (F) Representative images of embryoid bodies (EB) and neurospheres (NS). Scale bar, 500 μm. (G) Expression of βIII-tubulin, a neuronal marker (green) and GFAP, an astrocyte marker (red) in iPSC-derived neural cells. Scale bar, 200 μm. Day numbers indicate the days of differentiation in adherent culture after neurosphere formation.
Characterization of the generated iPSCs
Two lines of patient-derived iPSCs, D1-1 and D1-6, were established from skin fibroblasts obtained from the biopsy specimen. Control experiments used the iPSC line, 201B7, which was developed from the facial skin of a 36-year-old Caucasian female as reported previously [26]. All iPSC colonies had the typical human embryonic stem cell morphology with tightly-packed cells, a clear border, and a round shape (Figure 1B). Expression of pluripotency markers was confirmed (Figure 1B); additional analysis showed that the resulting teratomas consisted of tridermic tissues (Figure 1C), which supports the iPSCs’ undifferentiated state and pluripotency. Silencing of reprogramming transgenes, normal karyotype (46,XX), and the presence of the SCN1A c.4933C> T variation were confirmed (Figure 1D, E, A, respectively). Direct sequencing of additional sodium channels genes, the closely related SCN2A as well as the genes for subunits β1 and β2 (SCN1B and SCN2B) revealed wildtype status for all examined sequence regions (data not shown). During neural induction, all clones efficiently generated neurospheres (Figure 1F). In adherent cells differentiated from neurospheres, the expression of neuron and astrocyte markers was confirmed (βIII-tubulin and GFAP, respectively – Figure 1G). Staining for CNPase, an oligodendrocyte marker was negative in all cell lines.
Nav expression in iPSC-derived neurons
To determine the expression levels for those voltage-gated sodium channels, that predominate in the brain, we used real-time PCR targeting genes SCN1A, SCN2A (protein name: Nav1.2), SCN3A (Nav1.3), and SCN8A (Nav1.6) on iPSCs-derived neurons at 30 days of differentiation. In all cell lines, SCN2A expression was highest, followed by SCN1A, SCN3A, and SCN8A (Figure 2A and Additional file 2). When the expression levels were normalized to each other, we found that SCN1A tended to be expressed higher in the patient neurons than in control neurons (Figure 2B). We furthermore confirmed that, in patient neurons, SCN1A mRNA translated from the mutated allele was present (Figure 2C), which suggests that the mutated mRNA was able to escape nonsense-mediated decay, possibly owing to the mutation locating to the last coding exon [27].
Figure 2.

Nav channel expression in iPSC-derived neurons. (A) Real-time PCR addressing neuronal Nav expression at 30 days of differentiation (N = 3 in each cell line) Crossing point differences to β-actin (ΔCp = Cpβ-actin - CpNav) closer to zero indicate higher expression. PCR efficiencies were nearly identical (Additional file 2). Asterisks indicate a significant difference to SCN1A (P < 0.5, one-way ANOVA). Expression strength of the indicated Nav genes was constant across the cell lines (P = 0.92, two-way ANOVA) (B) Normalized expression levels for each Nav gene (SCN1A + SCN2A + SCN3A + SCN8A)/4 = 1. Compared to the control, SCN1A expression tended to be higher in D1-1 (P = 0.0929, one-way ANOVA), and it was significantly higher in D1-6 (*P = 0.0078). The distribution of Nav genes expression ratios in each cell line was significantly different between the control and the patient lines (P =0.0086 and <0.0001 for D1-1 and D1-6, respectively, two-way ANOVA), but identical between D1-1 and D1-6 (P = 0.11). (C) Sequencing of SCN1A reverse transcribed mRNA isolated from iPSCs-derived neurons. Patient-neurons show a double peak at mutation site (solid arrowheads), confirming the heterozygous state of the cells (D) Immunocytochemical characterization of Nav1.1 expression in control neurons: strong (solid arrows), moderate (open arrows), weak (solid arrowheads), and faint (open arrowhead). Despite weak staining in the cell body, neurite staining was often apparent (solid arrowheads). (E) Neurite co-localization of Nav1.1 and the AIS marker ankyrin G (AnkG, solid arrowheads). (F) PAN-Nav staining of SCN1A Venus-positive neurons (via anti-GFP, see Figure 3) in the AIS (arrowheads). (G) Co-localization of Nav1.1 and GAD67 staining. (H) VGlut1-positive neuron with SCN1A Venus expression. Scale bars: 100 μm (D), 30 μm (F), 200 μm (G) and 50 μm (others).
We next examined Nav1.1 expression at the protein level using a polyclonal antibody targeting the D1-D2 linker (Additional file 1). Among βIII-tubulin-positive cells, Nav1.1 immunostaining was identified in 59.0% or 105/178 in 201B7, 52.1% or 139/267 in D1-1, and 58.1% or 151/260 in D1-6 neurons. Nav1.1-immunostaining was evident in cell bodies, dendrites, and axons (Figure 2D, E); the antibody’s specificity was confirmed with epitope peptide pre-treatment (Additional file 3). Neurons with well-developed axons often displayed strong axonal Nav1.1-staining (Figure 2E). Intense expression of Nav channels (PAN-Nav) in the axon initial segment became evident after several weeks of in vitro differentiation of neurospheres (Figure 2F, Additional file 4A). This spatial and temporal expression pattern has been suggested to be critical in action potential generation [28].
Subtypes of Nav1.1-positive neurons
The majority of the Nav1.1-positive control and patient-derived neurons were GABAergic in nature as established by GAD67 staining (58.3% or 260/446 in 201B7, Figure 2G; 54.8% or 292/533 in D1-1; and 52.6% or 214/407 in D1-6, Additional file 4B). We next examined Nav1.1 expression differences among the subtypes of GABAergic neurons based on co-expression of PV, calretinin, or somatostatin. In mouse brain, strong Nav1.1 expression has been shown in PV-positive interneurons, whereas somatostatin- and calretinin-positive neurons show none [29]. This study produced several calretinin-positive 201B7 control neurons that also stained for Nav1.1 after 33 days of differentiation (44.4% or 8/18, Additional file 5A). Somatostatin-positive neurons, on the other hand, presented with either faint or negligible Nav1.1-staining in all cases (N = 10 and 7 in 201B7 and D1-6 neurons, respectively – Additional file 5B). PV expression was not detectable, even after treatment with sonic-hedgehog (shh) [30] or purmorphamine (a shh-signaling agonist) [31] for ventralization, and/or BMP4 [32] However, we did detect PV mRNA (Additional file 6) as well as mRNA for Nkx2.1, a medial ganglionic eminence neuron marker [33,34] that is elevated by the ventralizing treatments (Additional file 7) [35,36]. Thus, while PV-neuron precursors were likely present, our culture conditions may have interfered with further maturation.
Although it was technically difficult to distinguish subtypes other than GABAergic amongst the Nav1.1-positive neurons, some were positive for VGlut1, a marker of glutamatergic neurons, as a minor population (<1%), and on occasion, these neurons co-localized with SCN1A-Venus fluorescence (i.e. SCN1A expression, Figure 2H and Additional file 4C).
SCN1A-reporter for electrophysiology
To reliably identify SCN1A-expressing neurons for electrophysiological analyses, we generated a lentiviral reporter for SCN1A. The reporter contained SCN1A promoter sequence [37], some SCN1A 5′-untranslated region, as well as Venus cDNA following the ATG start codon (Figure 3A). This SCN1A-Venus construct was used to infect freshly plated cells from dissociated neurospheres. After several days of differentiation, SCN1A-Venus fluorescence developed in a few neurons, which further increased both, in the number of Venus-positive neurons and fluorescence intensity, as neuronal differentiation proceeded.
Figure 3.

Structure and characterization of the lentiviral SCN1A-reporter used in the electrophysiological analyses. (A) The reporter comprised (5′ to 3′) a 1.2-kb upstream sequence, a 5′-untranslated exon, the 5′-end of the first coding exon, and, following the ATG start codon, Venus cDNA. (B) & (C) 201B7 neurons labeled for Venus (using a GFP antibody) and Nav1.1 (B) or GABA (C). (B) Open arrowheads indicate GFP-pseudopositive neurons lacking Nav1.1 staining. (C) GFP-positive neurons with (arrowhead) and without GABA staining (open arrowhead). Scale bars: 200 μm.
To confirm co-existence of Nav1.1 and Venus protein in the same cells, we employed immunostaining and found that most of the Venus-positive neurons also expressed Nav1.1 protein (81% or 81/98 in 201B7 neurons, Figure 3B; 90.1% or 100/111 in D1-1; and 78.8% or 93/118 in D1-6, Additional file 8A). Furthermore, many SCN1A-Venus-positive neurons were also positive for GABA (79.3% or 69/87 in 201B7 neurons, Figure 3C; 83.0% or 186/224 in D1-1 neurons; and 70.3% or 152/216 in D1-6, Additional file 8B), indicating GABAergic neurons.
Neuron selection for electrophysiology
To examine the electrophysiological behavior of control and patient-derived neurons, we conducted current-clamp experiments on cells 22–50 days into neuronal differentiation; shorter differentiation times produced unreliable responses suggesting that the neurons had not fully matured. Hence, neuron selection for electrophysiological analysis was based on the following conditions: (1) clear SCN1A-Venus fluorescence; (2) mature neuronal morphology with a large and complex cell body and growth of ≥4 neurites; (3) ≥30 pF membrane capacitance; and (4) resting membrane potential at or more negative than -30 mV.
Based on these criteria, a total of 48 and 27 neurons were recruited for patient-derived cell lines D1-1 and D1-6, respectively; 33 neurons were examined for the 201B7 control cell line. We first established cell capacitance and the resting membrane potential for all cells as indicators for neuron maturity in an effort to minimize inclusion of potentially inappropriate cell responses (Additional file 9A). We found that the resting membrane potential averagely fell between -40 and -45 mV without any statistically discernible difference between the cell lines. The neurons had membrane capacitance mostly up to 70 pF. Some outliers of 100+ pF were also present, but they required excessive current injection to generate action potentials (Additional file 10), which prompted us to remove them from our analyses.
We next examined action potential generation in the current clamp configuration, using 10-ms depolarizing current injections from a holding potential of -70-mV, and we found no statistical difference between patient-derived and control neurons in terms of firing threshold and peak voltage (Additional file 9B).
We then determined the input–output relationship using sustained 500-ms injections of depolarizing current to trigger action potentials. In all cases, the number of action potentials per 500-ms stimulation period increased with the intensity of the injected current. However, as current injection intensified, amplitude attenuation became apparent (Additional file 11A). This intensified up to a certain current injection level, where action potentials not only obviously declined in amplitude but also in number, to ultimately stop completely (depolarization block, Additional file 11B). Because depolarization block was common (Additional file 12), we suspected that electrically immature neurons were abundant among the cells we had selected for analysis. For further electrophysiological characterization, we therefore admitted neurons only, if they produced 10 or more action potentials.
Action potential analysis
The numbers of neurons for electrophysiological comparisons between cell lines were 12 in D1-1, 15 in D1-6, and 16 in 201B7. Capacitance, resting membrane potential, action potential threshold, and action potential peak voltage in control and patient-derived neurons were statistically indistinguishable (Figure 4A). However, most notably in the input–output relationship, both patient-derived neuron cell lines frequently produced marked amplitude attenuation, which was not seen in control neurons (Figure 4B–C). Furthermore, both Dravet neuron cell lines showed a similar reduction in action potential firing at >50 pA, which, too, was never observed in control cells (Figure 4D). Data of individual neurons (Additional file 13) show that a higher number of D1-1 (33.3% or 4/12) and D1-6 (46.7% or 7/15) neurons reached their peak output prior to reaching a current of 100 pA compared to 201B7 (12.5% or 2/16) neurons. These differences support functional impairment in the patient-derived neurons, especially in GABAergic neurons. The essence is a reduced output capacity during intense stimulation.
Figure 4.

Electrophysiological characteristics of mature iPSC-derived neurons. (A) Capacitance, resting membrane potential (RMP), action potential (AP) firing threshold, and voltage peak were identical across all neurons analyzed (P >0.05, Kruskal-Wallis test); error bars indicate S.E.M. (B) Representative traces of AP trains triggered by a 500-ms depolarizing current at the indicated intensities. Transverse dotted lines demark 0 mV membrane potential. Scale bars: 20 mV vs. 100 ms. (C) Action potential (AP) decrement at current intensities triggering >10 APs calculated as a percentage: 10th/1st AP amplitude. Control vs. D1-1 (P = 0.078) and D1-6 (*P = 0.045, ANOVA); D1-1 vs. D1-6 (P = 0.839) (D) Total number of APs during the 500-ms stimulation period vs. current injection intensity. When exposed to strong current injections, both patient-derived cell lines produced significantly fewer APs compared to the control (the slope of AP numbers at ≥50 pA, P = 0.0102 and 0.0011 for D1-1 and D1-6, respectively, ANCOVA, *P <0.05 for D1-6 only, **P <0.05 for both D1-1 and D1-6, Wilcoxon rank-sum test).
Discussion
In this study, we report on the generation of neurons from DS patient iPSCs. Gene expression and immunocytochemical analyses demonstrated that the control and the two patient cell lines contained neurons of identical character. Electrophysiological analysis of the patient-derived cells revealed impairments in action potential generation in response to sustained current injection, especially with higher current intensities. Specifically, patient-derived iPSC neurons produced fewer action potentials with attenuated amplitudes and earlier depolarization block compared to control neurons. These results are reminiscent of the voltage responses seen in neurons isolated from rodent epilepsy models with SCN1A defects [8,9,17,38] and they are consistent with DS pathophysiology that includes an inability of neurons to adequately respond to high-intensity stimulation. Although it was technically difficult to conclusively determine whether the Nav1.1-positive neurons were GABAergic or glutamatergic (likely due to cell population heterogeneity and low marker protein expression), data from immunocytochemical analyses suggest that the Nav1.1-positive neurons were mostly GABAergic. Furthermore, the majority of SCN1A Venus-positive neurons showed GABA immunostaining, which supports that the neurons undergoing electrophysiological analysis in this study were phenotypically homogeneous. We therefore interpreted our findings in the context of a functional decline in GABAergic neuron activity – defective inhibition. Of course, we cannot exclude involvement of other neuron types. In the context of the data presented here, however, it is possible that the pathophysiology of human and mouse Dravet syndrome employs similar mechanisms.
Several differences may exist between human and rodent brains with respect to Nav1.1 expression. In rodent cerebral cortex, Nav1.1 is predominantly expressed in the axon initial segment of GABAergic interneurons. Pyramidal neurons also express Nav1.1 [8,39,40], albeit at a minor level [9,15]. Furthermore, epilepsy models with SCN1A defects have identified functional deficits in GABAergic interneurons, but not in pyramidal neurons [8,9,41]. In human brain, Nav1.1 expression differs from what is seen in rodents: Nav1.1 shows somatodendritic localization and expression in pyramidal neurons, specifically in cortical layer V and in the hippocampus [42-44]. This may be attributed to different experimental conditions and antibodies used, but, if rodent and human expression patterns indeed diverge, then it is conceivable that the associated pathophysiology differs as well. Pyramidal neurons use glutamate as their neurotransmitter, and our analyses showed that iPSCs-derived neurons expressing SCN1A-Venus were also positive for VGlut1. Unfortunately, the culture conditions in our study did not permit ready differentiation into glutamatergic neurons, which kept their number below what is usable for functional analyses. Functional characterization of non-GABAergic neurons must be addressed in the future to enhance our understanding of this DS model, and possibly unveil further pathogenic mechanisms.
With current methodology, establishing iPSC lines is labor- and time- consuming. Future research into human in vitro disease models may soon overcome these obstacles as other sources of pluripotent stem cells are considered, such as Nestin-expressing hair follicle stem cells [45,46]. They are easily accessible, they can be utilized without any genetic manipulation, and they have the potential to differentiate into neurons. If stable and efficient neural induction and maturation methods are established, for in vivo neuronal disease modeling will be possible.
Conclusions
With this study, we report the first successful generation of a human-based in vitro DS model. Our data are consistent with a functional decline in GABAergic neurons, which may contribute to DS epileptogenesis. The results are encouraging that patient-derived iPSC models can be utilized in human epilepsy research. They may, in fact, provide unparalleled insight into pathogenic mechanisms, and a uniquely suited research platform for drug development.
Materials and methods
Isolation of human skin fibroblasts and generation of iPSCs
Skin fibroblasts were isolated from a skin punch biopsy of the patient’s upper arm with the approval by the Human Ethics Committee of Fukuoka University (Approval No. 361). The parents of the patient provided signed informed consent before the study. Fibroblasts were cultured in DMEM containing 10% fetal bovine serum, 50 IU/mL penicillin, and 50 mg/mL streptomycin. The generation, maintenance, and characterization of iPSCs were performed as previously described [26,47]. Briefly, fibroblasts were lentivirus-trunsduced with Slc7a1 and plated at a density of 3.5 × 105 cells/60-mm dish. The next day, 4 reprogramming factors (Sox2, Klf4, Oct3/4, and c-Myc) were transduced using retroviruses. Seven days thereafter, the fibroblasts were re-plated at a density of 5 × 103–5 × 105 cells/100-mm dish with a mitomycin C-treated SNL feeder layer. The SNL feeder cells were obtained from the Wellcome Trust Sanger Institute (Hinxton, England). The next day, the medium was replaced with human iPS medium, which was DMEM/F12 containing 20% knockout serum replacement, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 0.1 mM 2-mercaptoethanol, 4 μg/mL basic FGF (R&D Systems, Minneapolis, MN), 50 IU/mL penicillin, and 50 mg/mL streptomycin. The medium was changed either daily or every other day until iPSC colonies were isolated, 24–28 days from the transduction of the reprogramming factors. iPSC clones were selected based on reprogramming transgene silencing (real-time PCR), colony morphology, expression of markers for undifferentiated cells (immunocytochemistry), pluripotency (teratoma formation), and neural induction efficiency. For iPSC maintenance, the medium was changed daily, and the cells passaged every 4–7 days depending on colony size. iPSCs passaged < 32 times were used for neural induction. Details on the PCR conditions for SCN1A, SCN2A, SCN1B, and SCN2B sequencing are available on request. The control iPSCs, 201B7, were provided by the RIKEN BioResource Center through the Project for Realization of Regenerative Medicine and the National Bio-Resource Project of the Ministry of Education, Culture, Sports, Science & Technology (MEXT) in Japan. iPSC production was approved by the Keio University School of Medicine Ethics Committee (Approval No. 20-16-18) and the Human Ethics Committee of Fukuoka University (Approval No. 361).
Neuronal differentiation of human iPSCs
Neuronal differentiation of human iPSCs was performed as previously described [48] with minor modifications [19,22,49]. Briefly, iPSC colonies were detached from feeder layers, and were cultured in suspension as embryoid bodies (EBs) for about 30 days in bacteriologic dishes (Kord-Valmark/Myers, Akron, OH). For neurosphere formation, EBs were enzymatically dissociated and the single cells were cultured in suspension in the serum-free neurosphere medium (media-hormone-mix) for 10 to 14 days. The resulting neurospheres could be repeatedly passaged using the dissociation procedures described above. Neurospheres passaged 1–3 times were used for analysis. For ventralization of neuronal properties in some preliminary assays, 5 or 30 nM of sonic hedgehog (R&D Systems, Minneapolis, MN) or 1 μM of purmorphamine (EMD Millipore, Billerica, MA) were added to the medium during the later phase of embryoid body and/or neurosphere formation. For terminal differentiation, neurospheres were plated onto poly-l-ornithine/fibronectin-coated coverslips in a media-hormone-mix supplemented with B27 supplement. Dissociated cells were plated at an approximate density of 1 × 105 cells/cm2. Both dissociated and undissociated cells were cultured for varying periods depending on the assay. To enhance cell viability and to promote neuronal maturation, the differentiation medium was supplemented as follows: 10 ng/mL rhBDNF and rhGDNF (R&D Systems), and 200 μg/mL L-ascorbic acid (Sigma-Aldrich, St. Louis, MO). Attempts to induce parvalbumin-positive neurons were made with 100 ng/mL of BMP4 (R&D Systems) on day 10 of neuronal differentiation and continued until the assay was conducted.
RNA isolation, reverse transcription, and real-time PCR analysis
Total cellular RNA was extracted using TRIZOL Reagent (Life Technologies, Carlsbad, CA), RNase-Free DNase Set (Qiagen, Venlo, Netherlands), and RNeasy Mini or Micro Kits (Qiagen). Complimentary DNA synthesis was performed using the SuperScript III First-Strand Synthesis System for RT-PCR (Life Technologies) with oligo-dT primers from 0.2–1.0 μg of total RNA, according to the manufacturer’s guidelines. To analyze the relative expression of different mRNAs, the amount of cDNA was normalized to β-actin mRNA expression. The mRNA expression levels in iPSC-derived neurons were determined from at least three separately cultivated samples. Real-time PCR was performed using the LightCycler 480 System II (Roche Diagnostics, Basel, Switzerland) with the SYBR Premix Ex Taq (Takara Bio, Shiga, Japan). Primer sequences for real-time PCR are listed in Table 1.
Table 1.
Primer details for real-time PCR
| Target | Forward | Reverse |
|---|---|---|
|
β-actin |
GATCAAGATCATTGCTCCTCCT |
GGGTGTAACGCAACTAAGTCA |
|
Sox2 (tg*) |
ACGGCCATTAACGGCACACTG |
CCCTTTTTCTGGAGACTAAATAAA |
|
Klf4 (tg) |
CACCTCGCCTTACACATGAAGAG |
|
|
Oct3/4 (tg) |
TCTGGGCTCTCCCATGCATTCAAAC |
|
|
c-Myc (tg) |
CTTGAACAGCTACGGAACTCTTG |
|
|
SCN1A |
AACAGAATCAGGCCACCTTG |
CACTGGGCTCTCTGGAATG |
|
SCN2A |
GCTACACGAGCTTTGACACC |
CCCAAGAAAATGACCAGCAC |
|
SCN3A |
ATGGTGTGGTTTCCTTGGTG |
TGACTTCCGTTTCTGTGGTG |
|
SCN8A |
GGACCCATGGAACTGGTTAG |
ACCCTGAAAGTGCGTAGAGC |
|
Nkx2.1 |
AGCACACGACTCCGTTCTCA |
CCCTCCATGCCCACTTTCTT |
| Parvalbumin | CTGGACAAGGACAAAAGTGG | ACAGGTCTCTGGCATCTGG |
All sequences are displayed in 5′-to-3′ direction. *tg, transgene.
Immunocytochemistry
Cells on coverslips were fixed with 4% paraformaldehyde for 10–30 min at room temperature, followed by washing 3 times with PBS. After incubating with blocking buffer (PBS containing 5% normal goat or fetal calf serum and 0.1–0.3% triton X-100) for 1 h at room temperature, the cells were incubated overnight at 4°C with primary antibodies diluted with the blocking buffer. Details of primary antibodies and the dilution conditions are presented below. The cells were then washed 3 times with PBS and incubated with secondary antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 555 (1:500, Life Technologies) and Hoechst33342 (2 μg/mL, Dojindo Laboratories, Kumamoto, Japan) for 1 h at room temperature. After washing 3 times with PBS and a single wash with distilled water, the coverslips were mounted on slides with FluorSave Reagent (EMD Millipore/Merck Group). Images were acquired using a confocal laser-scanning microscope, FV1000-D (Olympus, Tokyo, Japan). Observation through 20× objective was used to determine whether Nav1.1-positive neurons were also positive for GAD67, calretinin, or GFP (for detection of Venus).
Primary antibodies used in immunocytochemistry
Anti-Nav1.1 (rabbit IgG, 1:500, Cat No. ASC-001 – Alomone Labs, Israel). This antibody targets the peptide TASEHSREPSAAGRLSD, which corresponds to amino acids 465–481 in the internal D1–D2 linker human full-length Nav1.1 (Reference sequence: NP_001159435.1). Anti-Sodium Channel ‘PAN’ (mouse IgG1, 1:100, Cat No. S8809 – Sigma-Aldrich, St. Louis, MO) targets the peptide TEEQKKYYNAMKKLGSKK in the intracellular D3–D4 linker of Nav channels which is identical in all known vertebrate Nav channel isoforms. Anti-SSEA1 (mouse IgM, 1:500, Cat No. ab16285 – Abcam, Cambridge, MA), anti-SSEA4 (mouse IgG3, 1:500, ab16287 – Abcam), anti-TRA-1-60 (mouse IgM, 1:1000, Cat No. MAB4369 – EMD Millipore, Billerica, MA USA), anti-TRA-1-81 (mouse IgM, 1:1000, MAB4381 – EMD Millipore), anti-Oct3/4 (Rb IgG, 1:500, Cat No. sc-9081 – Santa Cruz Biotechnology, Santa Cruz, CA), anti-Nanog (rabbit IgG, 1:100, Cat No.RCAB0001P – Cosmo Bio, Carlsbad, CA), anti-βIII-tubulin (mouse IgG2b, 1:1000, Cat No. T8660 – Sigma-Aldrich, St. Louis, MO), anti-GFAP (rabbit IgG, 1:4000, Cat No. Z0334 – Dako, Denmark), anti-GFP (mouse IgG2a, 1:100, A11120 – Life Technologies, Carlsbad, CA, or rabbit IgG, 1:500, gift from Dr. Y. Misumi, Fukuoka University), anti-Ankyrin G (mouse IgG1, 1:100, 33–8800 – Life Technologies), anti-GAD67 (mouse IgG2a, 1:2000, MAB5406 – EMD Millipore), anti-Parvalbumin (mouse IgG1, 1:1000, MAB1572 – EMD Millipore), anti-Somatostatin (Rat IgG2b, 1:100, ab30788 – Abcam), anti-Calretinin (mouse, 1:1000, Cat No. 6B3 – Swant, Switzerland), anti-VGlut1 (rabbit IgG, 1:1000, Cat No. 135303 – Synaptic Systems, Germany), and anti-GABA (rabbit IgG, 1:2000, A2052– Sigma-Aldrich).
Generation of SCN1A reporter lentivirus
The upstream genomic sequence of an SCN1A 5′-untranslated exon (previously referred to as “h1b” by Martin, et al.[50], or “hB” by Nakayama, et al.[37]) was used as SCN1A promoter sequence. The 1,200-bp sequence stretch showed strong promoter activity and was obtained from the patient’s genomic DNA. Aforementioned untranslated exon connected with the 5′-end of the first coding exon, which was obtained from D1-1 iPSC-derived neuronal cDNA. These fragments were connected via PCR and transferred into the pSIN-Venus vector, which has a cloning site connected to Venus cDNA (constructed by Y. Okada, Keio University). For lentivirus production, the pSIN construct, pLP1, pLP2, and pLP/VSVG plasmids (Life Technologies) were mixed and transfected into 293FT cells using CalPhos Mammalian Transfection Kit (Clontech/Takara Bio) or Lipofectamine 2000 Reagent (Life Technologies). The medium was changed the following day. Two days thereafter, the virus-containing medium was collected, filtered, and ultracentrifuged at 25,000 rpm with an SW 28 Rotor (Beckman Coulter, Brea, CA USA), at 4°C for 90 min. The viral pellet was resuspended in 1/200 of the original medium volume with media-hormone-mix, aliquoted, and stored at -80°C until use.
Electrophysiological analysis
Electrophysiological analysis employed room temperature current-clamping of iPSC-derived neurons in the whole-cell configuration. Cell micrographs were produced with an upright microscope (BX51WI – Olympus, Melville, NY) equipped with a CMOS image sensor camera, ORCA-Flash2.8 (Hamamatsu Photonics, Japan). Reporter fluorescence Venus was detected through a 40x water-immersion objective (LUMPlanFI/IR2 – Olympus) with a U-MGFPHQ cube (excitation: 460–480 nm, dichroic mirror: 485 nm, emission: 495–540 nm – Olympus) and processed with Aquacosmos software (Hamamatsu, Japan). The extracellular solution contained 150 mM NaCl, 4 mM KCl, 2 mM CaCl2, 2 mM MgCl2·6 H2O, 10 mM HEPES, and 10 mM glucose adjusted to pH 7.4 with NaOH. Patch pipettes were made from borosilicate glass with filament (Cat No. FB150-86-0 – Sutter Instruments, Novato, CA) and pulled to resistances of 2–4 MΩ (P-97, Sutter Instruments) when filled with 0.22-μm filtered intracellular solution of the following composition: 117 mM K-methanesulfonate, 9 mM EGTA, 9 mM HEPES, 1.8 mM MgCl2·6 H2O, 29 mM sucrose, 4 mM Mg-ATP, 0.3 mM Tris-GTP, and 5 mM KCl adjusted to pH 7.3 with KOH. Whole-cell patch-clamp recordings were carried out using an Axopatch 700B amplifier (Axon Instruments, Sunnyville, CA) and pCLAMP 10 software (Axon Instruments). Signals were low-pass Bessel filtered at 10 kHz and sampled at a 50 kHz with an Axon Digidata 1440A digitizer (Axon Instruments). Cell capacitance was calculated by integrating the capacitive current evoked by a 10-mV depolarizing pulse from a holding potential of -65 mV. The resting membrane potential was determined from the mean potential during a 10-s continuous recording in zero-current clamp mode. During current-clamp experiments, cells were held at -70 mV by constant current injection, as needed. Single action potentials, operationally defined to minimally reach 0 mV, were evoked by current injection (10 ms) to determine their firing thresholds and peak voltages. The injection current amplitude was increased in 10-pA increments from sub- to supra-threshold. To investigate the input–output relationship, sustained depolarizing currents (500 ms) were injected and the current amplitude was increased from 5 to 100 pA in 5-pA increments. Final data was taken from neurons on at least 8 coverslips of at least 4 separately cultivated samples in each clone. Electrophysiological data were analyzed using pCLAMP 10 software (Axon Instruments, Sunnyvale, CA).
Statistical analysis
All of the data analyses were performed using SAS (Statistical Analysis System) Software Package (Ver. 9.2, SAS Institute Inc., Cary, NC) at Fukuoka University (Fukuoka Japan). Nav gene expression was compared with one-way ANOVA (between Nav channel genes) and two-way ANOVA (between iPSC clones). Cell capacitance, resting membrane potential, action potential firing threshold, peak voltage, action potential decrement, and area under the input–output relationship curve were compared among the clones using one-way ANOVA (with Scheffe’s post hoc test) and/or the Kruskal-Wallis test. The action potential number for each injection level in the input–output relationship was compared between the clones using the Wilcoxon rank-sum test. The slope of the number of action potentials vs. injected current in Figure 4C was compared using ANCOVA. Data are presented as mean ± standard error of the mean (S.E.M.), and P-values <0.05 were deemed significant.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NH designed, executed, and directed the study, and wrote the paper. TU performed all electrophysiological assays. CL designed the electrophysiological analyses and contributed to the writing of the manuscript. MM and SK assisted with the electrophysiological assay methods and helped with data interpretation. YM contributed to the immunocytochemical analysis. YO, YI and WA contributed to all cell culture and biologic assay procedures. KN performed the pathologic analysis of the iPSC-derived teratomas. BZ performed statistical analysis. YS contributed to clinical and genetic analysis of the patient. HO and SH coordinated the study. All authors read and approved the final manuscript.
Supplementary Material
Schematic representation of Nav1.1 topology. The typical Nav channel complex consists of one main, pore-forming α subunit (Nav1.1–Nav1.9) and one or more auxiliary β subunits. The α subunit is made up of four homologous domains (D1–D4) with six transmembrane regions each (S1–S6). Voltage sensitivity is mediated by positively charged residues in the S4 regions that move in the electrical field upon depolarization to cause a conformational change that favors opening of the channel. The antigenic regions for the Nav antibodies are shown as grey boxes; the site of the truncating mutation in D4/S4 is highlighted in red. Terminated at the R1645 residue, the Nav1.1 protein looses the faded protein portion (i.e., part of the voltage sensor, pore-lining residues and the entire C- terminus) and thereby its ability to function.
Real-time PCR efficiency, Nav genes.
Nav1.1 antibody selectivity. Representative images of Nav1.1 immunostaining (red). The image on the right was acquired after the Nav1.1 antibody had been pre-treated with epitope peptide. Scale bar, 50 μm. Nuclei are stained blue with Hoechst33342 to facilitate cell identification.
Characterization of patient-derived neurons. (A) Intense expressions of PAN-Nav in the axon initial segment (solid arrowheads) of SCN1A Venus-positive neurons. (B) Co-localization of Nav1.1 and GAD67 staining. (C) VGlut1-positive neuron co-localized with SCN1A Venus (solid arrowheads). Scale bars: 30 μm (A), 200 μm (B), and 50 μm (C).
Characterization of Nav1.1-positive neurons. (a) Calretinin-positive neurons with (arrowhead) and without Nav1.1 staining (open arrowhead). Scale bar, 50 μm. (b) Somatostatin-positive neurons are negative for Nav1.1. Scale bar, 100 μm.
RT-PCR of parvalbumin mRNA from iPSCs-derived neurons. 180-bp bands are indicated beta-actin mRNA expression. 85-bp bands demark parvalbumin (PV). When total RNA was used as template (RT-), no product was generated.
Increase in Nkx2.1 mRNA expression following treatment with sonic hedgehog (SHH) or purmorphamine. (a) During embryoid body formation (approx. 20–30 days) of cell line D1-1, the growth medium was supplemented with SHH to the indicated concentrations. This resulted in a dose-related increase in Nkx2.1 mRNA expression. Data from two different setups were averaged and normalized to the control (0 nM SHH); error bars are S.E.M. (b) Similar setup as in Panel (a), but SHH was added during neurosphere (NS) formation; cell line D1-6. This produced an increase in Nkx2.1 mRNA expression, although apparently not in dose-dependent fashion, which may relate to SHH only maintaining Nkx2.1 expression rather than inducing new ventral neuronal precursors. (c) Setup similar to Panel (a), albeit with purmorphamine treatment.
Nav1.1 and GABA expression in SCN1A Venus-positive patient neurons. Venus was detected using a GFP antibody. (A) Venus-positive neurons lacking Nav1.1 staining, open arrowheads. (B) Venus-positive neurons with (solid arrowheads) and without GABA staining (open arrowhead). Scale bars: 200 μm.
Electrophysiological characteristics of all recruited iPSC-derived neurons. (A) Capacitance & resting membrane potential (RMP) and (B) action potential (AP) firing threshold & voltage peak. No statistical differences were found in all items (P >0.05, Kruskal-Wallis test). Error bars indicate S.E.M.
Input–output relationship of large (≥100 pF) control neurons. Current clamping as in Figure 4. This produced a set number of action potentials per 500-ms stimulation period, which was plotted against the injected current amplitude. Note the size-dependent increase in the current required to trigger the same number of action potentials compared to smaller neurons (average for Figure 4C depicted in bold).
Illustration of “action potential attenuation” and “depolarization block” in current-clamped neurons. (A) The number of action potentials increased with stronger current injections but a simultaneous tapering of action potential amplitude was apparent. (B) Action potential tapering reached a state where further firing was prevented despite continued stimulation. Rectangular pulses represent current injection periods (500 ms) at the indicated intensities.
Example current-clamp traces of 201B7 control neurons with immature voltage responses. 500-ms depolarizing currents were injected at the indicated intensities. Transverse dotted lines demark 0 mV membrane potential. Scale bars, 20 mV vs. 100 ms.
Individual input–output relationship plots for control and Dravet-derived neurons. Experimental setup and plotting as in Additional file 6. Each line plot represents one cell. Current injections of <100 pA frequently maxed out the number of action potentials triggered in patient neurons (D1-1 and D1-6), but only rarely in the control neurons (201B7).
Contributor Information
Norimichi Higurashi, Email: higijh.n@gmail.com.
Taku Uchida, Email: tuchida@fukuoka-u.ac.jp.
Christoph Lossin, Email: lossinc@gmail.com.
Yoshio Misumi, Email: misumi@fukuoka-u.ac.jp.
Yohei Okada, Email: yohei@a6.keio.jp.
Wado Akamatsu, Email: akamatsu@a7.keio.jp.
Yoichi Imaizumi, Email: imaizumi@2006.jukuin.keio.ac.jp.
Bo Zhang, Email: bozhang@fukuoka-u.ac.jp.
Kazuki Nabeshima, Email: kaznabes@fukuoka-u.ac.jp.
Masayuki X Mori, Email: mxmori@fukuoka-u.ac.jp.
Shutaro Katsurabayashi, Email: shutarok@fukuoka-u.ac.jp.
Yukiyoshi Shirasaka, Email: yukisss-sc@healthcarenet.jp.
Hideyuki Okano, Email: hidokano@a2.keio.jp.
Shinichi Hirose, Email: hirose@fukuoka-u.ac.jp.
Acknowledgements
The authors thank the patient and parents for their cooperation in this study. We are indebted to Ms. Akiyo Hamachi and Ms. Minako Yonetani for excellent technical assistance. This work was supported in part by a Grants-in-Aid for Scientific Research (A) (21249062), for Challenging Exploratory Research (23659529 and 2570481), for Scientific Research on Innovative Areas, and for Bilateral Joint Research Projects from Japan Society for the Promotion of Science (JSPS) to S.H.; a Grant-in-Aid for Scientific Research on Innovative Areas “Genome Science” to S.H.; a Grant-in-Aid for Young Scientists (B) (24791095) from JSPS to N.H.; a Grant-in-Aid for the Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP) Exploratory Research from Japan Science and Technology Agency (JST) to S.H.; Research Grants for Nervous and Mental Disorder (21B-5), Health and Labor Science Research Grant (21210301 and KB220001), and a Grant-in-aid for the Research on Measures for Intractable Diseases (No. H22-Nanji-Ippan-49) from the Ministry of Health, Labor and Welfare (MHLW) to S.H.; the Project for the Realization of Regenerative Medicine from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) to H.O.; The Program for Intractable Disease Research utilizing Disease-specific iPS Cells from JST to H.O. Research grants from The Japan Foundation for Pediatric Research (10–003), The Clinical Research Promotion Foundation, and Kaibara Morikazu Medical Science Promotion Foundation to N.H.; Research grants from The Japan Epilepsy Research Foundation to N.H. and T.U.; and Research grants from the 2013–2017 “Central Research Institute for the Molecular Pathomechanisms of Epilepsy of Fukuoka University” and Recommended Projects of Fukuoka University (117016) to S.H.
References
- Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. In: Epileptic syndromes in infancy, childhood and adolescence. 4. Roger J, Bureau M, Dravet C, Genton P, Tassinari CA, Wolf P, editor. Montrouge: John Libbey Eurotext; 2005. Severe myoclonic epilepsy in infancy (Dravet syndrome) pp. 89–113. [Google Scholar]
- Genton P, Velizarova R, Dravet C. Dravet syndrome: the long-term outcome. Epilepsia. 2011;52(Suppl 2):44–49. doi: 10.1111/j.1528-1167.2011.03001.x. [DOI] [PubMed] [Google Scholar]
- Sakauchi M, Oguni H, Kato I, Osawa M, Hirose S, Kaneko S, Takahashi Y, Takayama R, Fujiwara T. Retrospective multiinstitutional study of the prevalence of early death in Dravet syndrome. Epilepsia. 2011;52:1144–1149. doi: 10.1111/j.1528-1167.2011.03053.x. [DOI] [PubMed] [Google Scholar]
- Depienne C, Trouillard O, Saint-Martin C, Gourfinkel-An I, Bouteiller D, Carpentier W, Keren B, Abert B, Gautier A, Baulac S. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet. 2009;46:183–274. doi: 10.1136/jmg.2008.062323. [DOI] [PubMed] [Google Scholar]
- Marini C, Scheffer I, Nabbout R, Suls A, De Jonghe P, Zara F, Guerrini R. The genetics of Dravet syndrome. Epilepsia. 2011;52(Suppl 2):24–33. doi: 10.1111/j.1528-1167.2011.02997.x. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Escayg A, Meisler M, Goldin A. Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. J Neurosci. 2001;21:7481–7571. doi: 10.1523/JNEUROSCI.21-19-07481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossin C, Rhodes T, Desai R, Vanoye C, Wang D, Carniciu S, Devinsky O, George A. Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel SCN1A. J Neurosci. 2003;23:11289–11384. doi: 10.1523/JNEUROSCI.23-36-11289.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Mantegazza M, Westenbroek R, Robbins C, Kalume F, Burton K, Spain W, McKnight G, Scheuer T, Catterall W. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1151. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5917. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, Rubenstein JL, Catterall WA. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA. 2012;109:14646–14651. doi: 10.1073/pnas.1211591109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, Catterall WA. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489:385–390. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler M, Kearney J. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115:2010–2017. doi: 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suls A, Claeys K, Goossens D, Harding B, Van Luijk R, Scheers S, Deprez L, Audenaert D, Van Dyck T, Beeckmans S. Microdeletions involving the SCN1A gene may be common in SCN1A-mutation-negative SMEI patients. Hum Mutat. 2006;27:914–934. doi: 10.1002/humu.20350. [DOI] [PubMed] [Google Scholar]
- Bechi G, Scalmani P, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Pure haploinsufficiency for Dravet syndrome Na(V)1.1 (SCN1A) sodium channel truncating mutations. Epilepsia. 2012;53:87–187. doi: 10.1111/j.1528-1167.2011.03346.x. [DOI] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Sci (New York, NY) 2010;328:906–915. doi: 10.1126/science.1187958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Martin M, Dutt K, Papale L, Dubé C, Dutton S, de Haan G, Shankar A, Tufik S, Meisler M, Baram T. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J Biol Chem. 2010;285:9823–9857. doi: 10.1074/jbc.M109.078568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Hockemeyer D, Beard C, Gao Q, Bell G, Cook E, Hargus G, Blak A, Cooper O, Mitalipova M. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–1041. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- Marchetto M, Carromeu C, Acab A, Yu D, Yeo G, Mu Y, Chen G, Gage F, Muotri A. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–566. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand K, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–226. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi Y, Okada Y, Akamatsu W, Koike M, Kuzumaki N, Hayakawa H, Nihira T, Kobayashi T, Ohyama M, Sato S. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain. 2012;5:35. doi: 10.1186/1756-6606-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nihei Y, Ito D, Okada Y, Akamatsu W, Yagi T, Yoshizaki T, Okano H, Suzuki N. Enhanced aggregation of androgen receptor in induced pluripotent stem cell-derived neurons from spinal and bulbar muscular atrophy. J Biol Chem. 2013;288:8043–8052. doi: 10.1074/jbc.M112.408211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakai S, Ikehata M, Nihira H, Ito N, Sueoka H, Kawamoto Y, Hayasaka H, Chiba S. “Obtundation status (Dravet)” caused by complex partial status epilepticus in a patient with severe myoclonic epilepsy in infancy. Epilepsia. 1996;37:1020–1022. doi: 10.1111/j.1528-1157.1996.tb00542.x. [DOI] [PubMed] [Google Scholar]
- Fukuma G, Oguni H, Shirasaka Y, Watanabe K, Miyajima T, Yasumoto S, Ohfu M, Inoue T, Watanachai A, Kira R. Mutations of neuronal voltage-gated Na+ channel alpha 1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB) Epilepsia. 2004;45:140–148. doi: 10.1111/j.0013-9580.2004.15103.x. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Maquat LE. Nonsense-mediated mRNA decay in mammals. J Cell Sci. 2005;118:1773–1776. doi: 10.1242/jcs.01701. [DOI] [PubMed] [Google Scholar]
- Kole M, Ilschner S, Kampa B, Williams S, Ruben P, Stuart G. Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci. 2008;11:178–264. doi: 10.1038/nn2040. [DOI] [PubMed] [Google Scholar]
- Yamakawa K. Molecular and cellular basis: insights from experimental models of Dravet syndrome. Epilepsia. 2011;52(Suppl 2):70–71. doi: 10.1111/j.1528-1167.2011.03006.x. [DOI] [PubMed] [Google Scholar]
- Xu Q, Guo L, Moore H, Waclaw R, Campbell K, Anderson S. Sonic hedgehog signaling confers ventral telencephalic progenitors with distinct cortical interneuron fates. Neuron. 2010;65:328–368. doi: 10.1016/j.neuron.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Chen J. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat Chem Biol. 2006;2:29–59. doi: 10.1038/nchembio753. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay A, McGuire T, Peng C-Y, Kessler J. Differential effects of BMP signaling on parvalbumin and somatostatin interneuron differentiation. Development. 2009;136:2633–2675. doi: 10.1242/dev.034439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Tam M, Anderson S. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J Comp Neurol. 2008;506:16–45. doi: 10.1002/cne.21529. [DOI] [PubMed] [Google Scholar]
- Xu Q, Cobos I, De La Cruz E, Rubenstein J, Anderson S. Origins of cortical interneuron subtypes. J Neurosci. 2004;24:2612–2634. doi: 10.1523/JNEUROSCI.5667-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Wonders C, Anderson S. Sonic hedgehog maintains the identity of cortical interneuron progenitors in the ventral telencephalon. Development. 2005;132:4987–5085. doi: 10.1242/dev.02090. [DOI] [PubMed] [Google Scholar]
- Gulacsi A, Anderson SA. Shh maintains Nkx2.1 in the MGE by a Gli3-independent mechanism. Cereb Cortex. 2006;16(Suppl 1):i89–i95. doi: 10.1093/cercor/bhk018. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Ogiwara I, Ito K, Kaneda M, Mazaki E, Osaka H, Ohtani H, Inoue Y, Fujiwara T, Uematsu M. Deletions of SCN1A 5′ genomic region with promoter activity in Dravet syndrome. Hum Mutat. 2010;31:820–829. doi: 10.1002/humu.21275. [DOI] [PubMed] [Google Scholar]
- Kalume F, Yu F, Westenbroek R, Scheuer T, Catterall W. Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci. 2007;27:11065–11139. doi: 10.1523/JNEUROSCI.2162-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duflocq A, Le Bras B, Bullier E, Couraud F, Davenne M. Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments. Mol Cell Neurosci. 2008;39:180–272. doi: 10.1016/j.mcn.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Vacher H, Mohapatra D, Trimmer J. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev. 2008;88:1407–1447. doi: 10.1152/physrev.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T, Ohmori I, Ouchida M, Ohno Y, Tsurumi T, Miki T, Wakamori M, Ishihara S, Yoshida T, Takizawa A. A missense mutation of the gene encoding voltage-dependent sodium channel (Nav1.1) confers susceptibility to febrile seizures in rats. J Neurosci. 2010;30:5744–5797. doi: 10.1523/JNEUROSCI.3360-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek R, Merrick D, Catterall W. Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron. 1989;3:695–1399. doi: 10.1016/0896-6273(89)90238-9. [DOI] [PubMed] [Google Scholar]
- Wenze W, Sachio T, Yoshie S, Masayuki I, Xiuyu S, Su-Kyeong H, Kazuki N, Morishige T, Shinichi H. The developmental changes of Nav1.1 and Nav1.2 expression in the human hippocampus and temporal lobe. Brain Res. 2011;1389:61–70. doi: 10.1016/j.brainres.2011.02.083. [DOI] [PubMed] [Google Scholar]
- Trimmer JS, Rhodes KJ. Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol. 2004;66:477–996. doi: 10.1146/annurev.physiol.66.032102.113328. [DOI] [PubMed] [Google Scholar]
- Amoh Y, Li L, Katsuoka K, Penman S, Hoffman RM. Multipotent nestin-positive, keratin-negative hair-follicle bulge stem cells can form neurons. Proc Natl Acad Sci USA. 2005;102:5530–5534. doi: 10.1073/pnas.0501263102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoh Y, Kanoh M, Niiyama S, Hamada Y, Kawahara K, Sato Y, Hoffman RM, Katsuoka K. Human hair follicle pluripotent stem (hfPS) cells promote regeneration of peripheral-nerve injury: an advantageous alternative to ES and iPS cells. J Cell Biochem. 2009;107:1016–1020. doi: 10.1002/jcb.22204. [DOI] [PubMed] [Google Scholar]
- Ohta S, Imaizumi Y, Okada Y, Akamatsu W, Kuwahara R, Ohyama M, Amagai M, Matsuzaki Y, Yamanaka S, Okano H, Kawakami Y. Generation of human melanocytes from induced pluripotent stem cells. PLoS One. 2011;6:e16182. doi: 10.1371/journal.pone.0016182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Matsumoto A, Shimazaki T, Enoki R, Koizumi A, Ishii S, Itoyama Y, Sobue G, Okano H. Spatiotemporal recapitulation of central nervous system development by murine embryonic stem cell-derived neural stem/progenitor cells. Stem Cells. 2008;26:3086–3098. doi: 10.1634/stemcells.2008-0293. [DOI] [PubMed] [Google Scholar]
- Nori S, Okada Y, Yasuda A, Tsuji O, Takahashi Y, Kobayashi Y, Fujiyoshi K, Koike M, Uchiyama Y, Ikeda E. Grafted human-induced pluripotent stem-cell-derived neurospheres promote motor functional recovery after spinal cord injury in mice. Proc Natl Acad Sci USA. 2011;108:16825–16855. doi: 10.1073/pnas.1108077108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Tang B, Ta N, Escayg A. Characterization of 5′ untranslated regions of the voltage-gated sodium channels SCN1A, SCN2A, and SCN3A and identification of cis-conserved noncoding sequences. Genomics. 2007;90:225–260. doi: 10.1016/j.ygeno.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic representation of Nav1.1 topology. The typical Nav channel complex consists of one main, pore-forming α subunit (Nav1.1–Nav1.9) and one or more auxiliary β subunits. The α subunit is made up of four homologous domains (D1–D4) with six transmembrane regions each (S1–S6). Voltage sensitivity is mediated by positively charged residues in the S4 regions that move in the electrical field upon depolarization to cause a conformational change that favors opening of the channel. The antigenic regions for the Nav antibodies are shown as grey boxes; the site of the truncating mutation in D4/S4 is highlighted in red. Terminated at the R1645 residue, the Nav1.1 protein looses the faded protein portion (i.e., part of the voltage sensor, pore-lining residues and the entire C- terminus) and thereby its ability to function.
Real-time PCR efficiency, Nav genes.
Nav1.1 antibody selectivity. Representative images of Nav1.1 immunostaining (red). The image on the right was acquired after the Nav1.1 antibody had been pre-treated with epitope peptide. Scale bar, 50 μm. Nuclei are stained blue with Hoechst33342 to facilitate cell identification.
Characterization of patient-derived neurons. (A) Intense expressions of PAN-Nav in the axon initial segment (solid arrowheads) of SCN1A Venus-positive neurons. (B) Co-localization of Nav1.1 and GAD67 staining. (C) VGlut1-positive neuron co-localized with SCN1A Venus (solid arrowheads). Scale bars: 30 μm (A), 200 μm (B), and 50 μm (C).
Characterization of Nav1.1-positive neurons. (a) Calretinin-positive neurons with (arrowhead) and without Nav1.1 staining (open arrowhead). Scale bar, 50 μm. (b) Somatostatin-positive neurons are negative for Nav1.1. Scale bar, 100 μm.
RT-PCR of parvalbumin mRNA from iPSCs-derived neurons. 180-bp bands are indicated beta-actin mRNA expression. 85-bp bands demark parvalbumin (PV). When total RNA was used as template (RT-), no product was generated.
Increase in Nkx2.1 mRNA expression following treatment with sonic hedgehog (SHH) or purmorphamine. (a) During embryoid body formation (approx. 20–30 days) of cell line D1-1, the growth medium was supplemented with SHH to the indicated concentrations. This resulted in a dose-related increase in Nkx2.1 mRNA expression. Data from two different setups were averaged and normalized to the control (0 nM SHH); error bars are S.E.M. (b) Similar setup as in Panel (a), but SHH was added during neurosphere (NS) formation; cell line D1-6. This produced an increase in Nkx2.1 mRNA expression, although apparently not in dose-dependent fashion, which may relate to SHH only maintaining Nkx2.1 expression rather than inducing new ventral neuronal precursors. (c) Setup similar to Panel (a), albeit with purmorphamine treatment.
Nav1.1 and GABA expression in SCN1A Venus-positive patient neurons. Venus was detected using a GFP antibody. (A) Venus-positive neurons lacking Nav1.1 staining, open arrowheads. (B) Venus-positive neurons with (solid arrowheads) and without GABA staining (open arrowhead). Scale bars: 200 μm.
Electrophysiological characteristics of all recruited iPSC-derived neurons. (A) Capacitance & resting membrane potential (RMP) and (B) action potential (AP) firing threshold & voltage peak. No statistical differences were found in all items (P >0.05, Kruskal-Wallis test). Error bars indicate S.E.M.
Input–output relationship of large (≥100 pF) control neurons. Current clamping as in Figure 4. This produced a set number of action potentials per 500-ms stimulation period, which was plotted against the injected current amplitude. Note the size-dependent increase in the current required to trigger the same number of action potentials compared to smaller neurons (average for Figure 4C depicted in bold).
Illustration of “action potential attenuation” and “depolarization block” in current-clamped neurons. (A) The number of action potentials increased with stronger current injections but a simultaneous tapering of action potential amplitude was apparent. (B) Action potential tapering reached a state where further firing was prevented despite continued stimulation. Rectangular pulses represent current injection periods (500 ms) at the indicated intensities.
Example current-clamp traces of 201B7 control neurons with immature voltage responses. 500-ms depolarizing currents were injected at the indicated intensities. Transverse dotted lines demark 0 mV membrane potential. Scale bars, 20 mV vs. 100 ms.
Individual input–output relationship plots for control and Dravet-derived neurons. Experimental setup and plotting as in Additional file 6. Each line plot represents one cell. Current injections of <100 pA frequently maxed out the number of action potentials triggered in patient neurons (D1-1 and D1-6), but only rarely in the control neurons (201B7).