

Abstract
MicroRNAs play an important role in plant development and plant responses to various biotic and abiotic stimuli. As one of the most important ornamental crops, rose (Rosa hybrida) possesses several specific morphological and physiological features, including recurrent flowering, highly divergent flower shapes, colors and volatiles. Ethylene plays an important role in regulating petal cell expansion during rose flower opening. Here, we report the population and expression profiles of miRNAs in rose petals during flower opening and in response to ethylene based on high throughput sequencing. We identified a total of 33 conserved miRNAs, as well as 47 putative novel miRNAs were identified from rose petals. The conserved and novel targets to those miRNAs were predicted using the rose floral transcriptome database. Expression profiling revealed that expression of 28 known (84.8% of known miRNAs) and 39 novel (83.0% of novel miRNAs) miRNAs was substantially changed in rose petals during the earlier opening period. We also found that 28 known and 22 novel miRNAs showed expression changes in response to ethylene treatment. Furthermore, we performed integrative analysis of expression profiles of miRNAs and their targets. We found that ethylene-caused expression changes of five miRNAs (miR156, miR164, miR166, miR5139 and rhy-miRC1) were inversely correlated to those of their seven target genes. These results indicate that these miRNA/target modules might be regulated by ethylene and were involved in ethylene-regulated petal growth.
Introduction
MicroRNAs (miRNAs) are 20–24 nucleotide (nt)-long non-coding RNA species that play profound roles in plant development and in plant responses to abiotic and biotic stimuli by regulating expression of their target genes, mainly at the post-transcriptional level. In plants, most miRNA genes are transcribed by Pol II to primary miRNAs (pri-miRNAs) which are partially self-complementary and possess the fold-back hairpin structure [1]. The pri-miRNAs are then processed to generate precursor miRNAs (pre-miRNAs) by a protein complex consisting of the Dicer-like 1 (DCL1), the C2H2-zinc finger protein SERRATE 11 (SE), and the double-stranded RNA-binding protein HYPONASTIC LEAVES1 (HYL1) [1]. Mature miRNA duplex (miRNA/miRNA*) is excised from pre-miRNAs by DCL1 and each strand is methylated by HEN1 protein. The miRNA strand is ultimately loaded into the Argonaute (AGO) protein of RNA-induced silencing complex (RISC) to carry out its function [1], [2]. Mature miRNAs are able to regulate their target genes through at least four mechanisms: 1) direct cleavage of the target mRNAs, 2) translational inhibition of the targets, 3) regulation of the targets through secondary siRNAs, and 4) sequestration of the miRNA and targets through target mimicry [3]. Although translational repression of targets is the most important way for miRNA-mediated regulation in animals, in plants cleavage of the targets is predominant [1], [3].
Bioinformatic analysis shows that 21 miRNA families are likely well conserved in angiosperms, including miR156, miR159, miR160, miR162, miR164, miR166, miR167, miR168, miR169, miR171, miR172, miR319, miR390, miR393, miR394, miR395, miR396, miR397, miR398, miR399 and miR408. Plants contain much more non-conserved miRNAs than conserved ones, for example, at least 48 non-conserved miRNAs have been found in Arabidopsis thaliana [4]. Recently, advances in high throughput DNA sequencing technology have enabled rapid and deeper discovery of non-conserved miRNAs from divergent plant species, including grape [5], barley [6], cucumber [7], olive [8], tomato [9], apple [10], and peach [11]. Currently, at least 4,677 mature miRNAs were identified from plants [12]. Moreover, many small RNA libraries were constructed from different plants subjected to hormonal and environmental treatments to identify novel and specific miRNAs in response to these stimuli. Resultantly, hundreds of miRNAs were found to be modulated by various hormones and stresses, including ABA, GA, auxin, pathogen, high-salinity, drought, cold, heat, mechanical stress, hypoxia and oxidative stresses [13]–[15].
Rosaceae is an economically important plant family that includes several important fruits and ornamental plants, such as apple, peach, strawberry and rose. Genome sequences of apple, peach and strawberry have been generated; however, research in small RNAs of the Rosaceae plant is still limited. Recently, miRNAs from apple and peach were reported [10], [11], [16], [17]. Unlike apple and peach which are important for their fresh fruits, rose is important for its beautiful and fragrant flowers. In the past century, rose has been the most important crop in the floriculture industry worldwide and cut roses account for approximately one third of cut flower trade in Europe [18]. In addition, rose possesses some unique morphological and physiological features, including recurrent flowering and highly divergent flower shapes, colors and volatiles, which are unable to be studied in other model plant systems, like Arabidopsis thaliana and tobacco [19].
Gaseous phytohormone ethylene is a crucial modulator in multiple biological processes, including seed germination, organ elongation, flowering, fruit ripening, organ senescence and abscission, as well as abiotic and biotic stress responses [20], [21]. It has been well known that ethylene can cause severe deterioration of flower quality in cut roses, mainly through the inhibition of petal expansion and acceleration of opening and senescence processes [22]. Although it has been extensively documented concerning the regulatory pattern of ethylene biosynthesis and signaling during flower opening and senescence in roses [23]–[27], the gene network downstream to ethylene signaling remains largely unknown. A recent study reported the identification of miRNAs in three modern rose cultivars and Rosa rugosa, and suggested that miRNAs could be involved in regulating genes related to coloring, like flavonoid biosynthetic genes [28]. However, the expression pattern of miRNAs in rose petals during flower opening and in response to ethylene still remains unexplored.
Here, we report small RNA profiling in rose petals during the rapid opening period and in response to ethylene treatment through high-throughput sequencing. In addition, we performed integral analysis of expression profiles of miRNAs and their predicted targets to further screen the bona fide miRNA-mRNA modules and discuss the possible biological roles of miRNAs differentially expressed in petals during flower opening and in response to ethylene.
Results
Construction and Sequencing of Small RNA Libraries from Rose Petals
The flower opening process is divided into seven stages in rose. The duration from unopened buds (stage 0) to partially opened flowers (Stage 3) is the rapid growth (RG) period [25], [29] and is important for the establishment of flower opening quality, especially the flower shape. Treatment with ethylene in the RG period can accelerate flower opening, but inhibit petal enlargement and even result in abnormal flower shapes [22], [27], [30].
To obtain a comprehensive survey of miRNAs in rose petals in the RG period and in response to ethylene treatment, we constructed and sequenced small RNA libraries from petals of unopened buds (stage 0, S0), opened buds (stage 2, S2), partially opened flowers (stage 2 flowers exposed to air for 24 h, C24), and ethylene-treated flowers (stage 2 flowers treated with 10 ppm ethylene for 24 h, E24).
We obtained 16,648,213, 6,069,761, 11,579,864, and 15,937,871 redundant reads of 10–40 nt from petals of S0, S2, C24, and E24 samples, respectively, after removing adaptors, low quality reads and contaminants (Table S1). The length distribution of the small RNAs ranged from 18 to 30 nt was examined and shown in Figure 1. In all four samples, 21-nt, 23-nt and 24-nt small RNAs were the major population, consistent with the size of Dicer-like protein cleavage products, and 24-nt was the most dominant, similar to the results obtained from most tested plants, such as Arabidopsis thaliana, rice, tomato, cucumber, apple and peach [7], [9], [10], [11], [16], [17], [31], [32]. The redundancy level of sRNAs was low for the 23-nt and 24-nt sRNAs, while the 21-nt sRNAs have the highest redundancy level, especially in the E24 library. This is different from that of cucumber, as the redundancy level of 22-nt sRNAs was reported to be the highest [7]. All the small RNA sequences have been deposited into NCBI SRA database under accession SRA066431.
Figure 1. Size distribution of small RNA (sRNA) sequences from rose petals.
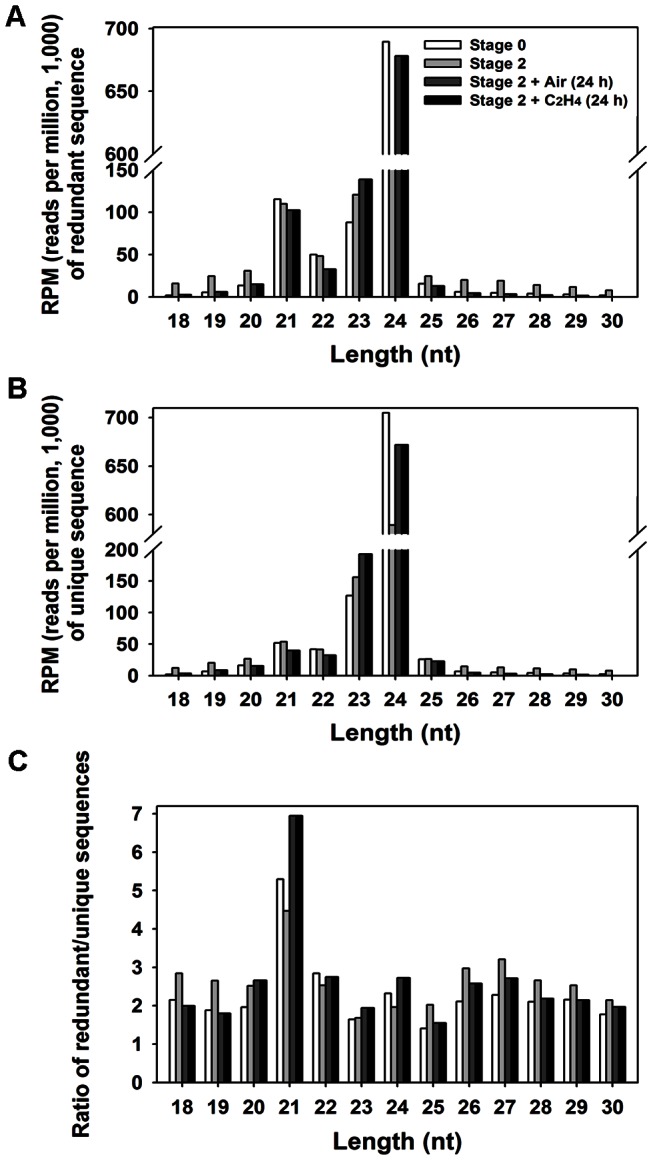
The number of redundant (A) and unique (B) sequences from rose petals. (C) Redundancy ratio for sRNAs from rose petals.
Identification of Conserved miRNAs in Rose Petals
It has been well known that miRNAs play critical roles in plant development and in plant responses to various abitotic and biotic stimuli [1]–[3]. To date, hundreds of plant miRNAs have been identified (miRBase release 18.0) [12]. Here, we explore the miRNAs in our sequencing data to systematically identify both conserved and species-specific miRNAs in rose.
To identify conserved miRNAs in rose, we aligned the small RNA sequences against the known plant mature miRNAs registered in the miRBase (Release 17, April 2011), and their corresponding precursor sequences were checked to insure the miRNAs have their expected secondary structures. The aligned sRNAs of 18–24 nt long and having abundance of no less than 5 RPM (reads per million) in at least one library were regarded as the miRNA candidates and used for further analysis. A total of 33 known miRNA families were identified and listed in Table 1. Among them, 27 families were known and well-conserved, including miR156/miR157, miR159, miR160, miR162, miR164, miR166, miR167, miR168, miR169, miR171, miR172, miR319, miR390, miR393, miR394, miR395, miR396, miR397, miR398, miR399, miR403, miR408, miR477, miR482, miR535, miR827, and miR858. Six families were known but not well-conserved, including miR2109, miR2478, miR4414, miR5072, miR5077, and miR5139. In addition, the corresponding miRNA* sequences were detected in 26 families, further supporting the existence of these families in rose (Table 1). Unexpectedly, rose has several less-conserved miRNAs which were found previously only in monocots, such as miR5072 and miR5077. In addition, miR2109 and miR4414, which were specific in legume plants in previous reports, were also discovered in rose. The 5′ end of 48.5% (16 of 33) and 18.2% known miRNA families appeared predominantly as uridine (U) and adenosine (A), which were specifically generated by different Dicer proteins and recognized by Argonaute 1 and 2 proteins, respectively [33], [34].
Table 1. Known microRNAs identified from rose petals. 5′ end indicated the base frequency at the miRNA 5′ end.
| Family | Size range | 5′ end | Star(*) | |||||||||
| A | U | C | G | ath | ctr | osa | ptc | vvi | zma | |||
| Well-conserved | ||||||||||||
| miR156/miR157 | 18–24 | 0.00 | 0.96 | 0.00 | 0.03 | Yes | Yes | No | Yes | Yes | Yes | Yes |
| miR159 | 18–26 | 0.01 | 0.86 | 0.13 | 0.01 | Yes | Yes | No | Yes | Yes | Yes | Yes |
| miR160 | 19–24 | 0.00 | 0.22 | 0.01 | 0.76 | Yes | Yes | No | No | Yes | Yes | Yes |
| miR162 | 18–23 | 0.01 | 0.90 | 0.00 | 0.09 | Yes | Yes | No | No | No | No | No |
| miR164 | 18–23 | 0.00 | 0.99 | 0.01 | 0.00 | Yes | Yes | No | Yes | Yes | No | Yes |
| miR166 | 18–24 | 0.00 | 0.97 | 0.00 | 0.03 | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| miR167 | 18–24 | 0.01 | 0.99 | 0.00 | 0.00 | Yes | Yes | No | Yes | Yes | Yes | Yes |
| miR168 | 18–24 | 0.00 | 0.94 | 0.06 | 0.00 | Yes | Yes | No | Yes | No | No | No |
| miR169 | 18–25 | 0.08 | 0.88 | 0.00 | 0.04 | Yes | Yes | No | Yes | Yes | Yes | Yes |
| miR171 | 19–24 | 0.03 | 0.96 | 0.00 | 0.00 | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| miR172 | 19–23 | 0.95 | 0.03 | 0.00 | 0.02 | Yes | Yes | No | Yes | Yes | Yes | Yes |
| miR319 | 20–22 | 0.08 | 0.92 | 0.00 | 0.00 | No | No | No | No | Yes | Yes | No |
| miR390 | 19–23 | 0.97 | 0.00 | 0.02 | 0.00 | Yes | Yes | No | No | No | No | No |
| miR393 | 19–22 | 0.93 | 0.07 | 0.00 | 0.00 | Yes | Yes | No | Yes | No | No | No |
| miR394 | 20–21 | 0.32 | 0.66 | 0.02 | 0.01 | Yes | Yes | No | No | Yes | Yes | Yes |
| miR395 | 18–24 | 0.01 | 0.10 | 0.78 | 0.11 | Yes | Yes | No | Yes | Yes | Yes | Yes |
| miR396 | 18–25 | 0.00 | 0.35 | 0.06 | 0.59 | Yes | Yes | No | Yes | Yes | Yes | Yes |
| miR397 | 19–24 | 0.30 | 0.59 | 0.11 | 0.00 | Yes | Yes | No | Yes | Yes | No | Yes |
| miR398 | 18–23 | 0.01 | 0.24 | 0.74 | 0.01 | Yes | Yes | No | Yes | No | No | No |
| miR399 | 18–24 | 0.03 | 0.86 | 0.01 | 0.10 | Yes | Yes | No | Yes | Yes | Yes | Yes |
| miR403 | 19–22 | 0.05 | 0.95 | 0.00 | 0.00 | Yes | Yes | No | No | No | No | No |
| miR408 | 18–24 | 0.91 | 0.01 | 0.09 | 0.00 | Yes | Yes | No | No | No | No | Yes |
| miR477 | 19–24 | 0.82 | 0.09 | 0.08 | 0.01 | Yes | No | No | No | Yes | Yes | No |
| miR482 | 18–24 | 0.00 | 0.97 | 0.03 | 0.00 | Yes | No | No | No | Yes | Yes | No |
| miR535 | 19–24 | 0.00 | 1.00 | 0.00 | 0.00 | No | No | No | No | No | No | No |
| miR827 | 21–24 | 0.00 | 1.00 | 0.00 | 0.00 | Yes | Yes | No | No | No | No | No |
| miR858 | 18–22 | 0.00 | 0.04 | 0.95 | 0.00 | Yes | Yes | No | No | No | No | No |
| Less-conserved | ||||||||||||
| miR2109 | 21 | 0.00 | 1.00 | 0.00 | 0.00 | No | No | No | No | No | No | No |
| miR2478 | 18–20 | 0.60 | 0.20 | 0.20 | 0.06 | No | No | No | No | No | No | No |
| miR4414 | 19–22 | 0.99 | 0.00 | 0.01 | 0.00 | No | No | No | No | No | No | No |
| miR5072 | 18–24 | 0.07 | 0.38 | 0.27 | 0.28 | No | No | No | Yes | No | No | No |
| miR5077 | 18–22 | 0.13 | 0.61 | 0.00 | 0.26 | No | No | No | Yes | No | No | No |
| miR5139 | 18–23 | 0.33 | 0.23 | 0.21 | 0.24 | No | No | No | No | No | No | No |
Ath, Arabidopsis thaliana; ctr, Citrus trifoliate; osa, Oryza sativa; ptc, Populus trichocarpa; vvi, Vitis vinifera; zma, Zea mays.
MiRNA precursor prediction is an important step to identify the authentic miRNAs. Since currently the genomic information of rose is not available, genome sequence of a closely related plant, strawberry (Fragaria vesca) [35], which belongs to the subfamily Rosoideae, was used as the reference to predict rose miRNA precursors. As shown in Table 2, 92 precursors of 21 known miRNA families were predicted. We also predicted precursors of known miRNAs using the rose floral transcriptome sequences we generated as the reference (http://bioinfo.bti.cornell.edu/rose). However, precursors of only two miRNAs, miR167 and miR482, were identified (Table 2 and Table S2). The failure of precursor prediction using the rose transcriptome database was mainly due to the fact that the database was constructed from poly (A) mRNAs, while miRNA precursors lack poly (A). The stem-loop structures of miRNA precursors of miR167 and miR482 predicted from rose and strawberry were almost identical (Figure 2), suggesting that they were highly conserved.
Table 2. Prediction of known miRNA precusors.
| ID | miRNA | family | Precusor | ||
| ID | chr | folding energy | |||
| S066 | UGACAGAAGAGAGUGAGCAC | miR156 | H000126 | LG3 | −46.4 |
| S066 | UGACAGAAGAGAGUGAGCAC | miR156 | H000127 | LG3 | −50.3 |
| S066 | UGACAGAAGAGAGUGAGCAC | miR156 | H000128 | LG2 | −44.2 |
| S066 | UGACAGAAGAGAGUGAGCAC | miR156 | H000129 | LG3 | −49.2 |
| S066 | UGACAGAAGAGAGUGAGCAC | miR156 | H000130 | LG2 | −46 |
| S067 | UGACAGAAGAGAGUGAGCACA | miR156 | H000131 | LG3 | −50.3 |
| S067 | UGACAGAAGAGAGUGAGCACA | miR156 | H000132 | LG2 | −44.2 |
| S067 | UGACAGAAGAGAGUGAGCACA | miR156 | H000133 | LG3 | −46.4 |
| S067 | UGACAGAAGAGAGUGAGCACA | miR156 | H000134 | LG3 | −49.2 |
| S067 | UGACAGAAGAGAGUGAGCACA | miR156 | H000135 | LG2 | −46 |
| S068 | UGACAGAAGAGAGUGAGCU | miR156 | H000136 | LG1 | −45.6 |
| S069 | UGACAGAAGAGAGUGAGCUC | miR156 | H000137 | LG1 | −45.6 |
| S070 | UGACAGAAGAGAGUGAGCUCA | miR156 | H000138 | LG1 | −45.6 |
| S071 | UGACAGAAGAUAGAGAGCAC | miR156 | H000139 | LG3 | −46.6 |
| S071 | UGACAGAAGAUAGAGAGCAC | miR156 | H000140 | LG3 | −42.8 |
| S071 | UGACAGAAGAUAGAGAGCAC | miR156 | H000141 | LG5 | −46.1 |
| S090 | UUGACAGAAGAGAGUGAGCAC | miR156 | H000165 | LG3 | −47.3 |
| S090 | UUGACAGAAGAGAGUGAGCAC | miR156 | H000166 | LG3 | −51.2 |
| S090 | UUGACAGAAGAGAGUGAGCAC | miR156 | H000167 | LG3 | −50.1 |
| S090 | UUGACAGAAGAGAGUGAGCAC | miR156 | H000168 | LG2 | −46.6 |
| S093 | UUGACAGAAGAUAGAGAGCACA | miR156 | H000173 | LG3 | −47.4 |
| S093 | UUGACAGAAGAUAGAGAGCACA | miR156 | H000174 | LG5 | −46.8 |
| S091 | UUGACAGAAGAUAGAGAGCA | miR156/miR157 | H000169 | LG3 | −47.4 |
| S091 | UUGACAGAAGAUAGAGAGCA | miR156/miR157 | H000170 | LG5 | −46.8 |
| S092 | UUGACAGAAGAUAGAGAGCAC | miR156/miR157 | H000171 | LG3 | −47.4 |
| S092 | UUGACAGAAGAUAGAGAGCAC | miR156/miR157 | H000172 | LG5 | −46.8 |
| S099 | UUUGGAUUGAAGGGAGCUCU | miR159 | H000181 | LG5 | −74.5 |
| S100 | UUUGGAUUGAAGGGAGCUCUA | miR159 | H000182 | LG5 | −75.5 |
| S079 | UGCCUGGCUCCCUGUAUGCCA | miR160 | H000153 | LG3 | −46.2 |
| S053 | UCGAUAAACCUCUGCAUCCAG | miR162 | H000101 | LG5 | −35.9 |
| S080 | UGGAGAAGCAGGGCACGUGCA | miR164 | H000154 | LG2 | −40.8 |
| S035 | CGGACCAGGCUUCAUUCCCC | miR166 | H000064 | LG7 | −40.36 |
| S035 | CGGACCAGGCUUCAUUCCCC | miR166 | H000065 | LG4 | −42.6 |
| S035 | CGGACCAGGCUUCAUUCCCC | miR166 | H000066 | LG4 | −43.2 |
| S035 | CGGACCAGGCUUCAUUCCCC | miR166 | H000067 | LG2 | −50.7 |
| S046 | GGACCAGGCUUCAUUCCCC | miR166 | H000084 | LG2 | −50.7 |
| S046 | GGACCAGGCUUCAUUCCCC | miR166 | H000085 | LG7 | −40.36 |
| S046 | GGACCAGGCUUCAUUCCCC | miR166 | H000086 | LG4 | −42.6 |
| S046 | GGACCAGGCUUCAUUCCCC | miR166 | H000087 | LG4 | −43.2 |
| S057 | UCGGACCAGGCUUCAUUCC | miR166 | H000105 | LG7 | −33.76 |
| S057 | UCGGACCAGGCUUCAUUCC | miR166 | H000106 | LG4 | −36 |
| S057 | UCGGACCAGGCUUCAUUCC | miR166 | H000107 | LG4 | −36.6 |
| S057 | UCGGACCAGGCUUCAUUCC | miR166 | H000108 | LG2 | −44.1 |
| S058 | UCGGACCAGGCUUCAUUCCC | miR166 | H000109 | LG7 | −37.06 |
| S058 | UCGGACCAGGCUUCAUUCCC | miR166 | H000110 | LG4 | −39.3 |
| S058 | UCGGACCAGGCUUCAUUCCC | miR166 | H000111 | LG4 | −39.9 |
| S058 | UCGGACCAGGCUUCAUUCCC | miR166 | H000112 | LG2 | −47.4 |
| S059 | UCGGACCAGGCUUCAUUCCCC | miR166 | H000113 | LG2 | −50.7 |
| S059 | UCGGACCAGGCUUCAUUCCCC | miR166 | H000114 | LG7 | −40.36 |
| S059 | UCGGACCAGGCUUCAUUCCCC | miR166 | H000115 | LG4 | −42.6 |
| S059 | UCGGACCAGGCUUCAUUCCCC | miR166 | H000116 | LG4 | −43.2 |
| S060 | UCGGACCAGGCUUCAUUCCCCU | miR166 | H000117 | LG7 | −41.96 |
| S060 | UCGGACCAGGCUUCAUUCCCCU | miR166 | H000118 | LG2 | −52.3 |
| S088 | UUCGGACCAGGCUUCAUUCCC | miR166 | H000163 | LG2 | −47.4 |
| S062 | UGAAGCUGCCAGCAUGAUCUA | miR167 | H000120 | LG1 | −32.2 |
| S062 | UGAAGCUGCCAGCAUGAUCUA | miR167 | H000121 | LG2 | −33.6 |
| S062 | UGAAGCUGCCAGCAUGAUCUA | miR167 | H000122 | LG2 | −43.4 |
| S063 | UGAAGCUGCCAGCAUGAUCUAA | miR167 | H000123 | LG1 | −32.2 |
| S064 | UGAAGCUGCCAGCAUGAUCUC | miR167 | H000124 | LG4 | −38.2 |
| S065 | UGAAGCUGCCAGCAUGAUCUCA | miR167 | H000125 | LG4 | −38.2 |
| R13 | UGAAGCUGCCAGCAUGAUCUC | miR167 | H000031 | RU29562 | −37.2 |
| R14 | UGAAGCUGCCAGCAUGAUCUCA | miR167 | H000032 | RU29562 | −37.2 |
| S034 | CGCUUGGUGCAGGUCGGGAA | miR168 | H000063 | LG5 | −43.2 |
| S054 | UCGCUUGGUGCAGGUCGGGA | miR168 | H000102 | LG5 | −44.7 |
| S055 | UCGCUUGGUGCAGGUCGGGAA | miR168 | H000103 | LG5 | −44.7 |
| S074 | UGAGCCAAGGAUGACUUGCCU | miR169 | H000144 | LG4 | −36.8 |
| S076 | UGAUUGAGCCGUGCCAAUAUC | miR171 | H000146 | LG5 | −37.3 |
| S076 | UGAUUGAGCCGUGCCAAUAUC | miR171 | H000147 | LG3 | −39.5 |
| S076 | UGAUUGAGCCGUGCCAAUAUC | miR171 | H000148 | LG5 | −43.1 |
| S076 | UGAUUGAGCCGUGCCAAUAUC | miR171 | H000149 | LG2 | −39.4 |
| S076 | UGAUUGAGCCGUGCCAAUAUC | miR171 | H000150 | LG6 | −38.2 |
| S095 | UUGAGCCGUGCCAAUAUCACA | miR171 | H000176 | LG5 | −45.5 |
| S095 | UUGAGCCGUGCCAAUAUCACA | miR171 | H000177 | LG3 | −41.4 |
| S039 | GAAUCUUGAUGAUGCUGCAU | miR172 | H000074 | LG7 | −45.5 |
| S039 | GAAUCUUGAUGAUGCUGCAU | miR172 | H000075 | LG3 | −42.5 |
| S039 | GAAUCUUGAUGAUGCUGCAU | miR172 | H000076 | LG2 | −35 |
| S003 | AAGCUCAGGAGGGAUAGCGCC | miR390 | H000005 | LG6 | −38.6 |
| S096 | UUGGCAUUCUGUCCACCUCC | miR394 | H000178 | LG2 | −38.5 |
| S086 | UUCCACAGCUUUCUUGAACUG | miR396 | H000160 | LG1 | −50.3 |
| S086 | UUCCACAGCUUUCUUGAACUG | miR396 | H000161 | LG3 | −36.7 |
| S087 | UUCCACAGCUUUCUUGAACUU | miR396 | H000162 | LG1 | −41 |
| S025 | AUUGAGUGCAGCGUUGAUGAA | miR397 | H000053 | LG3 | −49.5 |
| S030 | CAUUGAGUGCAGCGUUGAUGA | miR397 | H000059 | LG3 | −49.9 |
| S052 | UCAUUGAGUGCAGCGUUGAUG | miR397 | H000100 | LG3 | −49.9 |
| S036 | CGUGUUCUCAGGUCGCCCCUG | miR398 | H000068 | LG3 | −73.7 |
| S078 | UGCCAAAGGAGAGUUGCCCUG | miR399 | H000152 | LG5 | −43 |
| S024 | AUGCACUGCCUCUUCCCUGGC | miR408 | H000052 | LG1 | −50.6 |
| S077 | UGCACUGCCUCUUCCCUGGCU | miR408 | H000151 | LG1 | −50.6 |
| S011 | ACUCUCCCUCAAGGGCUUCUC | miR473 | H000019 | LG5 | −54 |
| S061 | UCUUUCCUAUUCCUCCCAUCCC | miR482 | H000119 | LG5 | −40.9 |
| R12 | UCUUGCCUAUGCCUCCCAUUCC | miR482 | H000030 | RU41075 | −28.5 |
| S084 | UUAGAUGACCAUCAACAAACA | miR827 | H000158 | LG1 | −31.8 |
The rose floral transcriptome database, and the genome sequence of strawberry (F.vesca) and transcriptome data of rose were used as reference, respectively. R, rose; S, strawberry. Detailed information is listed in Table S2.
Figure 2. Predicted precursor structures of miR167 and miR482 in rose.

The stem-loop structures were predicted by Vienna RNA software. miRNA sequences were highlighted in red.
Identification of Novel miRNAs in Rose Petals
The genome sequences of strawberry and transcriptome sequences of rose (http://bioinfo.bti.cornell.edu/rose) were also used to predict the potential novel miRNAs in rose. According to the criteria for miRNA annotation [36], we used 5 RPM as cutoff to get rid of miRNAs with low expression level. In addition, for the cases of novel miRNAs without star sequences, we required the candidate miRNAs to be present in all four independent libraries. A total of 47 novel miRNA families were obtained and named as rhy-miRC1 to rhy-miRC47 (Table 3). Of the 47 novel miRNAs, rhy-miRC1 was predicted from both rose and strawberry, while 8 and 38 miRNAs were predicted from rose and strawberry, respectively. The predicted hairpin structures of these novel miRNAs arranged from 61 to 242 nt in length and the folding energies were−18.4 to 106.6 ΔG (Table 3; Table S3). In addition, 16 out of the 47 novel miRNAs were predicted from more than one locus, suggesting that these miRNAs might be composed of multiple members (Table S3). Furthermore, the corresponding miRNA* sequences were identified for 27 novel miRNAs families, further supporting their existence as miRNAs (Table 3).
Table 3. Prediction of novel miRNA and their precusors.
| Family | Length (nt) | ID | Sequence | Star (*) |
| rhy-miRC1 | 24 | R1 | AAGGGACUAGCAAAAGCUAAGUGU | Yes |
| 24 | R5 | AGGGACUAGCAAAAGCUAAGUGUG | ||
| 24 | S004 | AAGGGACUAGCAAAAGCUAAGUGU | ||
| 24 | S019 | AGGGACUAGCAAAAGCUAAGUGUG | ||
| rhy-miRC2 | 21 | R3 | AGGGAAAAGCAUAGGAAUGAG | Yes |
| 22 | R4 | AGGGAAAAGCAUAGGAAUGAGU | Yes | |
| rhy-miRC3 | 21 | R16 | UGGGAUGGGAAGAAUGGCACG | |
| 22 | R17 | UGGGAUGGGAAGAAUGGCACGA | ||
| 23 | R18 | UGGGAUGGGAAGAAUGGCACGAA | ||
| 22 | R8 | AUGGGAUGGGAAGAAUGGCACG | ||
| rhy-miRC4 | 21 | S007 | AAUUUGGUGAUCGUUAAGGCA | |
| 23 | S015 | AGCCAAUUUGAUGAUCGUUAAGGC | Yes | |
| 24 | S016 | AGCCAAUUUGGUGAUCGUUAAGGCA | Yes | |
| 22 | S027 | CAAUUUGGUGAUCGUUAAGGCA | ||
| 23 | S042 | GCCAAUUUGGUGAUCGUUAAGGCA | Yes | |
| rhy-miRC5 | 24 | S013 | AGAUGAUCUAUACACUAGUACCUA | |
| 24 | S014 | AGAUGAUCUAUACAUUAGUACCUA | Yes | |
| rhy-miRC6 | 20 | S017 | AGGCAGUCACCUUGGCUAAC | Yes |
| 21 | S018 | AGGCAGUCACCUUGGCUAACU | Yes | |
| 19 | S048 | GGCAGUCACCUUGGCUAAC | Yes | |
| rhy-miRC7 | 20 | S038 | CUCAAGAAAGCUGUGGGACA | Yes |
| 21 | S044 | GCUCAAGAAAGCUGUGGGACA | Yes | |
| rhy-miRC8 | 21 | S040 | GAAUGUCGUCUGGUUCGAAAU | Yes |
| 22 | S041 | GAAUGUCGUCUGGUUCGAAAUC | Yes | |
| rhy-miRC9 | 20 | S072 | UGACGAUGAGAGAGAGCACG | |
| 21 | S073 | UGACGAUGAGAGAGAGCACGC | ||
| 21 | S094 | UUGACGAUGAGAGAGAGCACG | ||
| rhy-miRC10 | 19 | S082 | UGUAUGUUCGUCUCCAACU | |
| 21 | S083 | UGUAUGUUCGUCUCCAACUCU | ||
| rhy-miRC11 | 24 | S026 | AUUUUCAGCCAAAUUGAUGAUCGU | |
| 21 | S085 | UUCAGCCAAAUUGAUGAUCGU | ||
| 21 | S097 | UUUCAGCCAAAUUGAUGAUCG | Yes | |
| rhy-miRC12 | 21 | S102 | UUUUCUGAUUGAGCCGUGCCA | Yes |
| 21 | S103 | UUUUUCUGAUUGAGCCGUGCC | Yes | |
| rhy-miRC13 | 21 | R2 | ACAUGGAACACUACGACAUGG | Yes |
| rhy-miRC14 | 21 | R6 | AGUGGGAGGGUCGGCAAAAAA | Yes |
| rhy-miRC15 | 24 | R7 | AUGAUUGUGGAUAGAUUAAGCAUG | |
| rhy-miRC16 | 21 | R10 | GAGAUGGAGAUGGAGAGCUAG | |
| rhy-miRC17 | 21 | R11 | GCAUUCCUAUGCUUUUUCUCCA | Yes |
| rhy-miRC18 | 21 | R15 | UGGAUGCUUUGGAUGGAACGG | Yes |
| rhy-miRC19 | 21 | S001 | AAAUUGAUGAUCGUUAAGGUA | |
| rhy-miRC20 | 24 | S002 | AAGCCAAAUUGGUGAUCGUUAAGG | |
| rhy-miRC21 | 21 | S005 | AAUAAAGCUGUGGGAAGAUAC | Yes |
| rhy-miRC22 | 24 | S006 | AAUAUUACUAUUUUGAGGACUCAU | |
| rhy-miRC23 | 24 | S008 | ACAGGCGGUGGAACAAAUAUGAAU | |
| rhy-miRC24 | 21 | S009 | ACCUAGCUCUGAUACCAUGUG | Yes |
| rhy-miRC25 | 22 | S010 | ACUCUCCCUCAAGGGCUUCUAG | |
| rhy-miRC26 | 21 | S012 | AGAAUCUUGAUGAUGCUGCAU | Yes |
| rhy-miRC27 | 21 | S020 | AGUGGAGUUCUGGGAAAGAAG | |
| rhy-miRC28 | 24 | S021 | AGUUGGGACAAUAUCGGUACAAUG | |
| rhy-miRC29 | 24 | S022 | AGUUUUAAGGGACUGUGAGGGACA | |
| rhy-miRC30 | 21 | S023 | AUCAUGCUAUCCCUUUGGAUU | Yes |
| rhy-miRC31 | 21 | S028 | CAGGUCGGGAACUGCUUCGGU | |
| rhy-miRC32 | 21 | S029 | CAUCAACGCUGCACCCAAUUA | Yes |
| rhy-miRC33 | 21 | S031 | CCCGCCUUGCAUCAACUGAAU | Yes |
| rhy-miRC34 | 21 | S032 | CGAGCCGAACCAAUAUCACUC | |
| rhy-miRC35 | 21 | S033 | CGCUAUCCAUCCUGGGUUUCC | Yes |
| rhy-miRC36 | 21 | S037 | CUAGUCAUUGGUCAUAGCAUC | |
| rhy-miRC37 | 21 | S043 | GCGUACGAGGAGCCAAGCAUA | Yes |
| rhy-miRC38 | 21 | S045 | GCUCUCUAUGCUUCUGUCAUC | Yes |
| rhy-miRC39 | 24 | S047 | GGAGUGUGGAUUGUAAAAUGGGGA | |
| rhy-miRC40 | 21 | S049 | GUUCAAUAAAGCUGUGGGAAG | Yes |
| rhy-miRC41 | 22 | S050 | UAUGUCGCAGGAGAGAUGGUAC | |
| rhy-miRC42 | 22 | S051 | UCAAUAAAGCUGUGGGAAGAUA | Yes |
| rhy-miRC43 | 22 | S056 | UCGCUUGGUGCAGGUCGGGAAC | Yes |
| rhy-miRC44 | 21 | S081 | UGGGAUUUGGCGAAUUGUGGU | Yes |
| rhy-miRC45 | 22 | S089 | UUCGGACCAGGCUUCAUUCCCC | Yes |
| rhy-miRC46 | 21 | S098 | UUUGAAGUGGGAUUUGGCGAA | |
| rhy-miRC47 | 24 | S101 | UUUGGCUGAAAUUUUGCAGAGAUG |
The rose floral transcriptome database, and the genome sequence of strawberry (F.vesca) and transcriptome data of rose were used as reference, respectively. R, roses; S, strawberry.
We compared the stem-loop structures of rhy-miRC1 predicted from rose and strawberry. As shown in Figure 3A, the precursor structures were much more similar between these two species, indicating that rose and strawberry possess the same miRNAs which have not been reported in other plant species until now. In addition, the stem-loop structures of rhy-miRC2/11/42/43/44/45 were presented in Figure 3B.
Figure 3. Predicted precursor structures of novel miRNAs in rose.

Precursor stem-loop structures of novel miRNAs predicted based on rose transcriptome (A) or genome sequence of strawberry (F. vesca) (B) are displayed. The mature miRNA sequences are highlighted in red and miRNA* sequences in green.
Abundance of Conserved and Novel MiRNAs in Rose Petals
Since high-throughput sequencing provides the opportunity for quantitative profiling of sRNA populations, the sequencing frequency has been used to estimate the miRNA abundance in different samples. Nearly half of the conserved miRNA families were represented with more than 50 RPM in at least one library, including miR156, miR157, miR159, miR164, miR166, miR167, miR168, miR171, miR172, miR390, miR396, miR397, miR408, miR535, and miR827. On the other hand, all six known but less-conserved families (miR2109, miR2478, miR4414, miR5072, miR5077, and miR5139) exhibited high abundance, namely more than 50 RPM in at least one library (Table S4). MiR156, miR157, miR166 and miR168 were the most highly expressed conserved miRNAs. The highest expression level of miR159, miR164, miR166, miR167, miR397, miR408 and miR827 were observed in petals of unopened buds (S0), indicating that they may play roles in earlier period of flower opening; whereas, miR168, miR171 and miR390 were enriched in petals of opened flowers (C24). In addition, miR156, miR157, miR535 and miR2109 were highly accumulated in petals of ethylene-treated flowers (E24). Of the 47 novel miRNAs, 12 appeared to be highly expressed in petals (more than 50 RPM in at least one library) (Table S4). Rhy-miRC3, rhy-miRC9 and rhy-miRC26 were the most highly expressed novel miRNAs. Moreover, rhy-miRC3 and rhy-miRC26 were highly expressed in petals of unopened buds (S0), while rhy-miRC2, rhy-miRC9, rhy-miRC17, rhy-miRC34, and rhy-miRC41 were enriched in petals of ethylene-treated flowers (E24).
Generally, the miRNA* sequences are considered to be quickly degraded after their complementary miRNA sequences are selected from the miRNA/miRNA* duplex and loaded into the AGO protein [1], [2]. Therefore, the abundance of miRNA* is usually much lower than that of their corresponding miRNAs. However, we noticed that the abundance of miRNA* of two conserved miRNA families, miR171 and miR396, was much higher (Figure S1), which was consistent with the finding in Brassica [37].
In summary, our analysis showed that both known and novel miRNAs exhibited highly diverse expression patterns during flower opening and in response to ethylene, indicating that they may play different roles in these biological processes.
Prediction and Validation of MiRNA Targets in Rose Petals
To understand possible biological functions of the identified miRNAs in rose, we identified their putative targets using the rose floral transcriptome database as the reference transcript set (http://bioinfo.bti.cornell.edu/rose). Putative targets of 28 known miRNA families were predicted (Table 4 and 5; Table S5) and all the well-conserved miRNAs, such as miR156, miR159, miR160, miR164, miR167, miR172, miR396, miR397, and miR482 shared conserved target genes with their homologous miRNAs in other plants (Table 4), indicating that these miRNAs might play a fundamental role in plant development. Interestingly, we also identified some novel targets of both conserved and less-conserved known miRNA families (Table 5; Table S5). These putative novel targets included several regulatory proteins, such as protein kinase (miR156), E3 ubiquitin protein (miR159), enhancer of mRNA-decapping protein (miR169), zinc finger protein (miR172, miR394 and miR5139), and RING-H2 finger protein (miR397). Moreover, we also found a lot of structure and metabolism proteins, including DXPS3 (1-deoxy-D-xylulose-5-phosphate synthase 3) (miR156), cytokinin oxidase (miR159), pectin methylesterase (miR166), beta-galactosidase (miR166), hydrolase (miR167), pentatricopeptide (PPR) repeat-containing protein (miR2478), and expansin (miR5139) (Table S5).
Table 4. Predicted targets for conserved miRNAs in rose.
| miRNA family | Targets ID | Targets Annotation |
| miR156 | RU15050 | SQUAMOSA PROMOTER BINDING PROTEIN-LIKE |
| RU39321 | ||
| RU35697 | ||
| miR159 | RU13577 | R2R3-myb transcription factor |
| miR160 | RU26455 | Auxin response factor |
| miR164 | RU24899 | NAC domain protein |
| RU60879 | ||
| RU02822 | ||
| miR168 | RU01155 | Argonaute protein |
| miR172 | RU08179 | Transcription factor AHAP2 |
| RU01226 | AINTEGUMENTA protein | |
| miR319 | RU00790 | TCP transcription factor |
| miR397 | RU16913 | Laccase-like protein |
| miR482 | RU12701 | NBS-LRR type resistance protein |
Table 5. Predicted new targets for known miRNAs in rose.
| miRNA family | Targets ID | Targets Annotation |
| miR156 | RU23956 | E3 ubiquitin protein ligase UPL1, putative |
| miR156 | RU41939 | Nucleic acid binding protein, putative |
| miR156 | RU54762 | Protein kinase |
| miR156 | RU20461 | Timing of CAB expression 1 protein |
| miR156/miR157 | RU20216 | Guanine nucleotide-exchange-like protein |
| miR156/miR157 | RU06764 | RING/U-box domain-containing protein |
| miR159 | RU24195 | Cytokinin dehydrogenase |
| miR159 | RU48935 | ATP-binding region, ATPase-like domain-containing protein |
| miR159 | RU17672 | Glycosyl transferase family 17 protein |
| miR166 | RU06269 | beta-galactosidase |
| miR166 | RU10736 | Kinesin-like protein |
| miR166 | RU03738 | Pectin methylesterase 1 |
| miR166 | RU14941 | Rosa rugosa Rdr1 homologous region genomic sequence |
| miR168 | RU60766 | U-box domain-containing protein 43-like |
| miR169 | RU34168 | CCAAT-binding transcription factor subunit B |
| miR169 | RU00770 | Rosa multiflora breeding line 88/124–46 black spot resistance muRdr1 gene locus |
| miR169 | RU09780 | Nucleotide binding |
| miR171 | RU44711 | Ubiquitin-protein ligase |
| miR172 | RU24707 | Peptidyl-prolyl cis-trans isomerase |
| miR172 | RU60452 | Zinc finger, C2H2-type |
| miR394 | RU05839 | Dehydration-responsive protein-related |
| miR394 | RU21813 | E3 ubiquitin-protein ligase |
| miR394 | RU54433 | Rosa rugosa Rdr1 homologous region genomic sequence |
| miR394 | RU17225 | Zinc finger (C3HC4-type RING finger) |
| miR397 | RU01636 | Putative RING-H2 finger protein RHF2a [Arabidopsis thaliana] |
| miR398 | RU47741 | Receptor protein kinase-like protein |
| miR399 | RU52206 | Rosa multiflora breeding line 88/124–46 black spot resistance muRdr1 gene locus |
| miR482 | RU12701 | NBS-LRR type resistance protein |
| miR2109 | RU60115 | Rosa rugosa Rdr1 homologous region genomic sequence |
| miR2109 | RU27536 | TIR-NBS type disease resistance protein |
| miR4414 | RU22051 | Putative cyclic nucleotide-gated cation channel |
| miR4414 | RU44477 | SGS3 (SUPPRESSOR OF GENE SILENCING 3) |
| miR473 | RU33494 | Putative methyltransferase |
| miR5072 | RU23976 | Putative auxin influx carrier protein |
| miR5077 | RU25746 | ARF GTPase activator |
| miR5139 | RU22365 | Expansin |
| miR5139 | RU05060 | Zinc finger, C3HC4 type |
Putative targets of 26 out of the 47 novel miRNAs in rose were also predicted (Table S6). The predicted targets included several types of regulatory protein, such as transcription factor (rhy-miRC16/rhy-miRC26/rhy-miRC27), cell cycle factor (rhy-miRC16/rhy-miRC44) and component of ubiquitin-dependent protein degradation system (rhy-miRC46). Meanwhile, the novel targets also included a lot of functional proteins, such as glycosyl hydrolase (rhy-miRC2), ARPC1b (actin-related 18 protein C1b) (rhy-miRC7), plastid developmental protein (rhy-miRC9), UDP-glucuronate decarboxylase (rhy-miRC10) proton pump interactor (rhy-miRC14), auxin conjugate hydrolase (rhy-miRC14), phosphoenolpyruvate synthase (rhy-miRC16), acyl-CoA-binding protein (rhy-miRC18), dormancy/auxin associated protein (rhy-miRC22), ATPase (rhy-miRC41), argonaute protein (rhy-miRC43), and actin binding protein (rhy-miRC44). Candidate targets of eight novel miRNA families, including rhy-miRC1, rhy-miRC4, rhy-miRC5, rhy-miRC11, rhy-miRC19, rhy-miRC20, rhy-miRC24 and rhy-miRC47, failed to be annotated (Table S6). The lack of functional annotation of these putative targets indicated that they might be novel target genes which were specific in roses.
We then performed 5′-RNA ligase-mediated (RLM)-RACE analysis to validate the miRNA-guided cleavage of predicted target transcripts. As reported previously, squamosa-promoter-binding protein-like (SPL) family genes and R2R3 MYB transcription factors were predicted as targets of miR156 and miR159, respectively (Table 4) [3]. The putative miRNA-target sites of RU15050 (SPL 7 gene) and RU13577 (MYB) were fused with the ORF sequence of EGFP in frame and driven by a pSuper promoter to construct the SPL-sensor and MYB-sensor, respectively (Figure 4A). The sensor constructs were transformed into Agrobacterium and then co-infiltrated with 35S:miR156a (for SPL sensor) and 35S:miR159a (for MYB sensor), respectively. As expected, miR156- and miR159-mediated cleavage sites were detected in SPL and MYB sensors, respectively (Figure 4B). Meanwhile, we performed the 5′-RACE using total RNA extracted from rose petals and confirmed the miR156- and miR159-mediated cleavage of SPL (RU15050) and MYB (RU13577) gene in vivo, respectively (Figure 4C).
Figure 4. Validation of miRNA predicted targets.

(A) Constructs of miRNA target sensors. (B, C) Cleavage sites identified by 5′RLM-RACE assay in tobacco (B) and in rose petals (C). Positions of the cleavage sites are indicated by arrows with the proportion of sequenced clones.
MiRNA Profiling in Rose Petals during Early Flower Opening and in Response to Ethylene
Since the high-throughout sequencing data of small RNAs can be used to evaluate the miRNA expression profiles, we investigated miRNA profiles in petals during the early period of flower opening and in response to ethylene in rose. Except for miR160, miR390, miR482, miR858 and miR827, all known miRNAs exhibited substantial expression changes during early flower opening (stage 2/stage 0≤0.67 or≥1.50): abundance of 10 miRNAs decreased, while 18 increased (Table 6). Of the 28 changed miRNAs, the most pronounced expression decrease (≤10-fold) was found in miR4414 and miR159, while the increase≥10-fold) was found in miR5139, miR394, miR396 and miR473. Among the 47 novel miRNAs, expression of 17 and 22 miNRAs substantially decreased (stage 2/stage 0≤0.67) and increased (stage 2/stage 0≥1.50), respectively, during early flower opening. The novel miRNAs whose abundance decreased more than 10-fold included rhy-miRC45, rhy-miRC32 and rhy-miRC37, and the increased ones (≥10-fold) included rhy-miRC21, rhy-miRC42, rhy-miRC40, rhy-miRC13 and rhy-miRC30 (Table 6).
Table 6. Digital expression profiles of known and novel miRNAs in rose petals during earlier opening period and in response to ethylene.
| miRNA ID | Stage2/Stage 0 | +C2H4/−C2H4 | miRNA ID | Stage 2/Stage 0 | +C2H4/−C2H4 |
| miR156 | 1.80 | 1.81 | rhy-miRC1 | 0.48 | 0.53 |
| miR157 | 2.53 | 1.58 | rhy-miRC2 | 3.23 | 1.74 |
| miR159 | 0.07 | 1.03 | rhy-miRC3 | 0.35 | 1.34 |
| miR160 | 0.90 | 0.35 | Rhy-miRC4 | 2.94 | 0.67 |
| miR162 | 0.45 | 0.38 | rhy-miRC5 | 1.53 | 1.75 |
| miR164 | 0.25 | 0.47 | rhy-miRC6 | 3.98 | 0.48 |
| miR165 | 0.44 | 0.34 | rhy-miRC7 | 1.02 | 0.29 |
| miR166 | 0.28 | 0.39 | rhy-miRC8 | 0.81 | 0.52 |
| miR167 | 0.25 | 0.51 | Rhy-miRC9 | 1.81 | 1.11 |
| miR168 | 1.64 | 0.66 | rhy-miRC10 | 1.88 | 0.88 |
| miR169 | 1.59 | 0.53 | rhy-miRC11 | 0.50 | 0.25 |
| miR171 | 1.63 | 0.40 | rhy-miRC12 | 0.13 | 2.72 |
| miR172 | 6.30 | 0.61 | rhy-miRC13 | 5.84 | 0.98 |
| miR319 | 0.00 | / | rhy-miRC14 | 0.75 | 0.90 |
| miR390 | 1.08 | 0.83 | rhy-miRC15 | 0.23 | 2.52 |
| miR394 | 10.77 | 2.52 | rhy-miRC16 | 1.37 | 0.77 |
| miR395 | 5.65 | 1.05 | rhy-miRC17 | 2.20 | 1.15 |
| miR396 | 10.36 | 0.37 | rhy-miRC18 | 0.43 | 1.37 |
| miR397 | 0.16 | 0.33 | rhy-miRC19 | 0.30 | 0.73 |
| miR398 | 2.35 | 0.56 | rhy-miRC20 | 0.35 | 1.39 |
| miR399 | 3.25 | 2.19 | rhy-miRC21 | 43.92 | 1.08 |
| miR408 | 0.19 | 0.37 | rhy-miRC22 | 1.14 | 1.06 |
| miR473 | 10.23 | 2.31 | rhy-miRC23 | 3.57 | 2.22 |
| miR482 | 1.15 | 0.30 | rhy-miRC24 | 3.83 | 0.51 |
| miR535 | 3.41 | 3.38 | rhy-miRC25 | 7.17 | 0.42 |
| miR827 | 0.79 | 0.78 | rhy-miRC26 | 4.42 | 0.81 |
| miR858 | 0.78 | 0.38 | rhy-miRC27 | 0.40 | 2.18 |
| miR2109 | 4.47 | 5.71 | rhy-miRC28 | 0.30 | 0.88 |
| miR2478 | 5.91 | 0.27 | rhy-miRC29 | 0.54 | 0.43 |
| miR4414 | 0.00 | 0.12 | rhy-miRC30 | 14.90 | 1.17 |
| miR5072 | 7.75 | 0.33 | rhy-miRC31 | 2.26 | 0.49 |
| miR5077 | 3.30 | 0.33 | rhy-miRC32 | 0.06 | 1.12 |
| miR5139 | 22.93 | 0.49 | rhy-miRC33 | 4.47 | 0.75 |
| rhy-miRC34 | 1.12 | 1.55 | |||
| rhy-miRC35 | 4.01 | 0.73 | |||
| rhy-miRC36 | 1.17 | 0.35 | |||
| rhy-miRC37 | 0.08 | 4.33 | |||
| rhy-miRC38 | 4.03 | 1.19 | |||
| rhy-miRC39 | 5.91 | 0.94 | |||
| rhy-miRC40 | 22.17 | 0.56 | |||
| rhy-miRC41 | 1.17 | 45.72 | |||
| rhy-miRC42 | 30.67 | 0.86 | |||
| rhy-miRC43 | 1.50 | 0.93 | |||
| rhy-miRC44 | 0.42 | 0.92 | |||
| rhy-miRC45 | 0.01 | 0.34 | |||
| rhy-miRC46 | 0.50 | 1.40 | |||
| rhy-miRC47 | 0.64 | 0.97 |
For ethylene treatment, flowers (stage 2) were treated with 10 ppm ethylene in a sealed airtight chamber for 24 h, and flowers exposed to air were used as the control. The miRNAs in bold indicate miRNAs showing substantial expression changes in response to ethylene treatment (+C2H4/−C2H4≤0.67 or≥1.50).
To screen possible ethylene-sensitive miRNAs, we compared the expression level of miRNAs between flowers (stage 2) treated with or without 10 ppm ethylene for 24 h. We found that 28 out of 33 known miRNAs (84.8% of known miRNAs) showed substantial expression changes in response to ethylene treatment (+C2H4/−C2H4≤0.67 or≥1.50), while expression of 22 novel miRNAs (46.8% of novel miRNAs) was substantially changed after ethylene treatment. Interestingly, a novel miRNA, rhy-miRC41, exhibited a∼46-fold increase in its expression in ethylene-treated flower petals when compared to the control (Table 6).
We also performed quantitative RT-PCR to examine the expression of several known and novel miRNAs. The expression patterns obtained from qRT-PCR supported the sequencing data. More importantly, qRT-PCR results confirmed that expression of miR156, rhy-miRC2, rhy-miRC13, rhy-miRC24, and rhy-miRC35 was substantially increased and expression of miR159, miR164 and rhy-miRC32 was sharply reduced in petals during the early flower opening. After ethylene treatment, abundance of miR156 and rhy-miRC2 was substantially increased, while miR164 and rhy-miRC24 decreased (Figure 5). In addition, we also detected the expression of rhy-miRC2*, further confirming the authenticity of the predicted novel miRNAs.
Figure 5. qRT-PCR of selected known and novel miRNAs differentially expressed in petals during earlier flower opening or in response to ethylene.

5S rRNA was used as the internal control. Error bars indicate the SD of three biological replicates.
Putative mRNA/miRNA Modules Involved in Ethylene-Regulated Flower Opening of Rose
Recent reports showed that translation repression is an important and popular way for miRNAs to regulate their targets in plants, and it is likely that miRNAs repress their targets through both transcript cleavage and translation repression [38], [39]. However, integrative analysis of expression of miRNAs and their targets can still be helpful to identify miRNA/mRNA modules which might be involved in regulating specific biological processes. Here, we analyzed expression profiles of miRNAs and their predicted targets in rose petals treated with or without ethylene and identified 75 putative miRNA/mRNA modules, which included 21 known and 16 novel miRNAs (Table S7). Of these 75 miRNA/mRNA modules, expression of 5 miRNAs, including miR156, miR164, miR166, miR5139 and rhy-miRC1, were inversely correlated to that of their 7 corresponding targets in response to ethylene treatment (Table 7). We also tested the expression changes of predicted targets in response to ethylene in rose petals by using quantitative RT-PCR. As shown in Figure 6, qRT-PCR further confirmed that the expression of miRNAs and their predicted targets was inversely correlated.
Table 7. Integrated analysis of expression profiles of miRNAs and responsed targets in rose petals.
| miRNA ID | Target ID | Accession | Best Hit(nr_hit) | E-value | miRNA expression +C2H4/−C2H4 | Target expression +C2H4/−C2H4 | Score | |
| Fold change | FDR | |||||||
| miR156 | RU15050 | NP_175723 | SPL4 (SQUAMOSA PROMOTER BINDING PROTEIN-LIKE 4); DNA binding/transcription factor [Arabidopsis thaliana] | 8.00E-10 | 1.81 | 0.17 | 2.06E-03 | 3.0 |
| miR164 | RU02822 | CAO15010 | ANAC100 | 2.00E-58 | 0.47 | 15.83 | 3.41E-03 | 2.5 |
| RU24899 | ACI13682 | NAC domain protein [Malus x domestica] | 3.00E-60 | 0.47 | 2.51 | 2.86E-01 | 2.5 | |
| RU60879 | ACI13682 | NAC domain protein [Malus x domestica] | 1.00E-43 | 0.47 | 13.32 | 1.24E-08 | 2.5 | |
| miR166 | RU06269 | CAC44500 | beta-galactosidase [Fragaria x ananassa] | 2.00E-245 | 0.39 | 1.83 | 6.37E-02 | 3.0 |
| miR5139 | RU22365 | AAD44345 | expansin [Fragaria x ananassa] | 9.00E-17 | 0.49 | 3.07 | 7.08E-05 | 3.0 |
| rhy-miRC1 | RU25062 | No hit | 0.53 | 50.88 | 1.22E-03 | 0.5 | ||
For ethylene treatment, flowers (stage 2) were treated with 10 ppm ethylene in a sealed airtight chamber for 24 h, and flowers exposed to air were used as the control.
Figure 6. qRT-PCR of predicted miRNA targets (A) and expression ratio of miRNA/target modules in rose petals in response to ethylene (B).
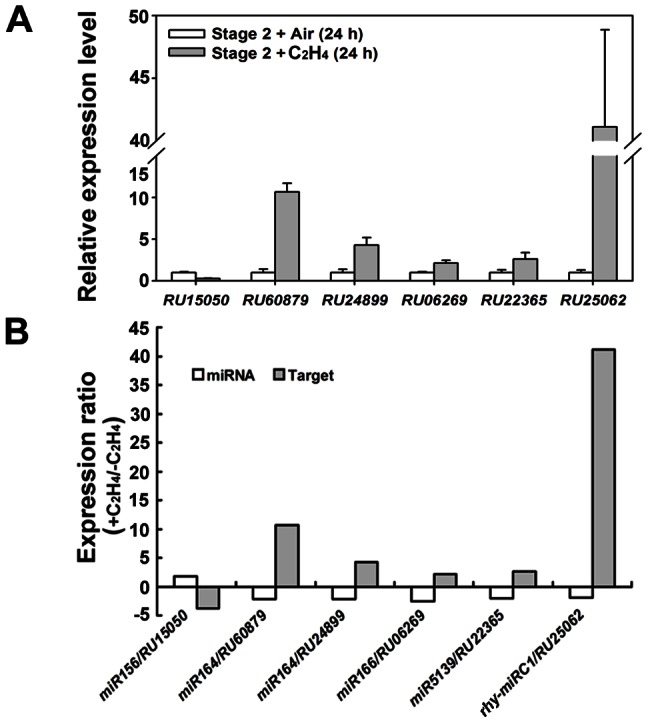
RhACT5 was used as the internal control. Error bars indicate the SD of three biological replicates.
Discussion
Known and Novel miRNAs in Rose Petals
Rosaceae plant is one of the six most economically important crop plant families, and includes many important fruits and ornamental plants, such as apple, pear, almond, peach, apricot, plum, cherry, strawberry, and rose [40]. As one of the most important ornamental crops, rose accounts for more than 30% of cut flower trade in Europe and China (Data from MOA, 2011) [18]. Aside from the economic significance, rose has also served as a model system to investigate some important biological processes, such as fragrance formation and flower opening. Therefore, understanding the molecular mechanism in shaping flower ornamental quality in rose will provide great value for rose production and breeding.
miRNAs have fundamental functions in regulating almost all aspects of plant development. In the present study, we performed small RNA sequencing and determined the expression profiles of miRNAs in rose petals in response to ethylene. We identified 47 novel and 33 known miRNA families, as well as their corresponding star sequences. Due to the lack of rose genome sequence, the precursors were predicted using both strawberry genome and our rose transcriptome sequences. Although we obtained precursors of miR166 and miR482 from the rose transcriptome sequences, their precursors were much more similar to those predicted using the strawberry genome.
A recent study reported 25 novel and 242 known miRNA sequences identified from flowers of three modern rose cultivars and Rosa rugosa [28]. Based on our analysis, we found that the reported 242 known miRNAs could be categorized into 37 miRNA families (Table S9), and 29 families were also identified by us. Five families (miR828, miR845, miR2111, miR2275 and miR2911) were not listed by us due to their low expression levels (<5 RPM), but found in our sequencing data. Four known miRNA families (miR2478, miR5072, miR5077 and miR5139) were identified from our sRNA dataset, but were not listed in Kim et al [28].
For novel miRNAs, 17 predicted novel miRNAs were reported by Kim et al [28], while 47 novel miRNAs were identified in our work. Although both Kim et al and us used strawberry (Fragaria vesca) genome to predict the novel miRNAs, only five (rhy-miRC4/ng9, rhy-miRC10/ng4, rhy-miRC24/ng8, rhy-miRC27/ng11 and rhy-miRC36/ng6) were presented in both works. In addition, seven other novel miRNAs (ng1, ng5, ng12, ng13, ng14, ng15 and ng16) in Kim et al [28] were also found in our sequencing data, but were not regarded as authentic miRNAs due to their low abundance (<5 RPM) in our samples (Table S9). Therefore, a total of 12 and 42 miRNAs were specific in Kim's and our dataset, respectively. Considering that the sampling strategies were different, the difference in novel miRNAs could also be attributed to the cultivar-, organ-, development- and/or condition-specific expression pattern of miRNAs. The five identical rose-specific miRNAs are likely to play conserved roles in different rose cultivars (Table S9).
Using a rose floral transcriptome database, we identified putative targets of rose miRNAs. The well known target genes of most conserved miRNAs, such as miR156, miR164, and miR172, have also been identified in rose. However, 32.5% (39/120) predicted targets of known miRNAs, and 48.4% (45/93) of novel miRNAs, were not homologous to any proteins in the GenBank nr database, indicating that they might be novel genes which were specific in rose. Interestingly, a novel miRNA, rhy-miRC27, was predicted to target an acyltransferase-like protein (RU23954) (Table S7), which was also identified as the target of ng11, a miRNA identical to rhy-miRC27, in a previous report [28]. These results suggested that rhy-miRC27 (ng11)/acyltransferase module might be conserved in Rosa sp.
MiRNA Profiling during Early Flower Opening and in Response to Ethylene
In the present study we investigated expression profiles of miRNAs during early flower opening and in response to ethylene in rose petals. We found that much larger portion of known miRNAs than novel ones appeared to be differentially expressed during early opening stages or regulated by ethylene treatment. This is consistent with recent findings that ethylene biosynthesis and signaling pathway emerged in earlier period of land plant evolution [41], [42], thus the non-conserved novel miRNAs are supposed to occur later evolutionarily [2], [4].
Recently, a report showed that ACC (1-aminocyclopropane-1-carboxylic acid), a precursor of ethylene, down-regulated miR159, miR164, miR319, miR390 and miR396 in roots of Medicago truncatula [43]. Except for miR159 and miR319, the expression of miR164, miR390 and miR396 were also repressed by ethylene in rose petals. Further studies are needed to investigate whether miR159 and miR319 were regulated by ethylene in an organ-specific manner.
Ethylene-Responsive miRNA/mRNA Modules in Rose Petals
Integrated analysis of expression of miRNAs and their targets can help to identify miRNA/mRNA modules involved in regulating specific biological processes, such as cold stress in wheat [44].
During the past decade, genomics approaches have been applied in rose and several EST libraries from rose petals were reported [45], [46], [47]. In 2008,∼10,000 ESTs were deposited in public databases, including GenBank and GDR (Genome database for the Rosaceae, http://www.rosaceae.org/). Based on these data, expression profiles of 4,765 transcripts were detected in roses from floral transition to flower senescence using a newly developed Affymetrix microarray [48]. Interestingly, we found that several genes, whose expression was changed during the early flower opening period, were potential targets of miRNAs, such as expansin (miR5139), MYB (miR159), and NAC (miR164). Functional analysis of miRNAs in the early flower opening period will be helpful for understanding this process.
Moreover, application of next-generation sequencing technologies greatly promoted the study on rose genomics. Kim et al reported a rose transcriptome database, which contained more than 30,000 transcripts. According to target prediction and gene expression analysis, several conserved miRNAs, such as miR156, miR159, and miR396, were proposed to be involved in regulating genes related to coloring, including those in the flavonoid biosynthetic pathway [28]. Lately, a transcriptome dataset containing 80,714 transcript clusters was generated by using the RNA from various tissues of R. chinensis cv. ‘Old Blush’ and in response to biotic and abiotic stresses [49].
We also constructed a floral transcriptome database containing 60,944 transcripts assembled from transcriptome sequences generated using the 454 sequencing technology (http://bioinfo.bti.cornell.edu/rose). Furthermore, based on microarray analysis, we identified 2,189 unique ethylene-responsive transcripts. In the present study, these transcripts were used to identify the ethylene-responsive miRNA/target modules. We were able to identify a total of seven ethylene-responsive miRNA/mRNA modules (Table 7). Quantitative RT-PCR confirmed that all identified miRNA/target modules exhibited negatively correlated expression profiles between miRNAs and their corresponding targets. Except miR164, all miRNAs have not been reported to be ethylene-responsive. Interestingly, the identified modules included well-conserved miRNAs (miR156, miR164 and miR166), a less-conserved miRNA (miR5139), and a novel miRNA (rhy-miRC1), suggesting profound and broad impacts of ethylene on plant development.
In the identified modules, targets of miR164 (NAC) and miR156 (SPL) are transcription factors. In Arabidopsis thaliana, miR164 regulates NAC1 and several NAC genes of the NAM subfamily. Consequently, it regulates many aspects of plant development. For instance, miR164 was found to be regulated by developmental cues and control organ boundary maintenance and leaf development [50]–[52], while it was also found to be auxin-responsive and regulate NAC1 to control lateral root initiation [53]. Recently, miR164 was found to be ethylene-responsive and regulated leaf senescence in Arabidopsis thaliana [54] and cell expansion in rose petals [Pei et al., unpublished data].
Like miR164, miR156 is also a well conserved miRNA in plants. The miR156/SPL module was previously reported to directly regulate FLOWERING LOCUS T (FT) expression to control ambient temperature-responsive flowering. A recent report showed that a miR156-targeted SPL protein could destabilize a MYB-bHLH-WD40 transcriptional activation complex to influence expression of anthocyanin biosynthetic genes in Arabidopsis thaliana [55]. High level of miR156 decreased the accumulation of anthocyanins, while low miR156 activity caused high levels of flavonols. Interestingly, ethylene increased expression levels of chalcone synthase (CHS), flavanone 3-hydroxylase (F3H), and UDP glucose-flavonoid 3-O-glucosyl transferase (UFGT), and consequently promoted the accumulation of anthocyanins in the skin of grape berries [56]. Since ethylene also significantly increased miR156 abundance in rose petals, whether ethylene regulates anthocyanin accumulation in rose petals through modulating the miR156/SPL module is worthy of future investigation.
In addition, miR166 and miR5139 appeared to target beta-galactosidase and expansin genes, respectively, in an ethylene-regulated manner. Beta-galactosidase and expansin genes are involved in cell-wall modification [57], [58], which has been proven to be very sensitive to ethylene treatment in Arabidopsis thaliana and tomato [59]–[61]. Whether miRNAs, such as miR166 and miR5139, are involved in ethylene-regulated cell wall modification in rose petals needs further investigation. Intriguingly, we identified an ethylene responsive module that included a novel miRNA, rhy-miRC1, and its target RU25062 that showed no homology to any known genes, indicating the function of rhy-miRC1/RU25062 might be specific to rose.
Conclusions
Here, we reported a set of miRNAs identified from rose (Rosa sp.) petals during early flower opening and in response to ethylene treatment. We found that expression of 28 known and 39 novel miRNAs was changed in rose petals during early opening process, and 28 known miRNAs and 22 novel miRNAs were responsive to ethylene treatment. Furthermore, integrated analysis of expression profiles of miRNAs and their targeted mRNAs in response to ethylene, an important factor influenced flower opening and senescence in rose, identified seven miRNA/mRNA modules. These modules might be important downstream regulatory components which facilitate the function of ethylene in flower opening, senescence, or both.
Materials and Methods
Flower Treatment, RNA Isolation, and Small RNA Sequencing
Flowers of cut roses (Rosa hybrida) cv Samantha were harvested at stage 0 and 2 of flower opening [25], respectively (We state clearly that no specific permissions were required for these locations/activities and confirm that the field studies did not involve endangered or protected species). The flowers were immediately put into tap water after harvest and then transported to the laboratory within 1 h. For ethylene treatment, stems of stage 2 flowers were cut to 25 cm under water, and then placed in deionized water (DW). The flowers were treated by 10 ppm ethylene in a sealed airtight chamber for 24 h, and flowers exposed to air were used as the control. Treatments were conducted at 23–25°C and 1 mol L−1 NaOH was placed in the chamber to prevent the accumulation of CO2 [62]. The samples were immediately collected after treatment. The 2nd and 3rd whorl petals of more than 15 flowers were collected and pooled together. Total RNAs were isolated using the pBiozol reagent (BioFlux, Hangzhou, China). Small RNA libraries were prepared according to the manufacturer's instructions and sequenced on an Illumina HiSeq2000 system.
Bioinformatics Analysis of sRNA Sequences
The raw sequencing data were processed to trim the adapter sequences and remove low quality sequences, and rRNA and tRNA sequences were also removed. The cleaned small RNA sequences with expression>5 RPM (reads per million) in at least one of the four libraries were aligned to the strawberry genome [33] and the rose transcriptome database (http://bioinfo.bti.cornell.edu/rose) using Bowtie [63] with perfect matches. Only sRNAs with no more than 20 hits were kept and their flanking sequences on the genome or transcriptome (200 bp on each side) were extracted and then folded in silico using the RNAfold program [64]. Resulting folded structures were checked with miRcheck [65] with default parameters. Candidate miRNAs whose precursors passed miRcheck were then aligned to the miRNA database, miRBase 17.0, using Bowtie [63] allowing up to 2 mismatches. The miRNAs shared homology to known miRNAs were identified as conserved miRNA candidates. Then, they were further confirmed by checking their corresponding precursor structures. Only the candidates with expected structures were identified as conserved miRNAs.
After identifying all candidate miRNAs, those which did not share homology to all known sequences in miRBase were regarded as novel miRNA candidates. And the novel miRNAs' precursor structures were further analyzed by miRcheck [66]. Potential miRNA star sequences were identified from the sRNA dataset to provide additional evidence supporting miRNA predictions. For novel miRNA candidates without corresponding miRNA star sequences identified, only those expressed in all four samples were kept. MiRNA targets were identified according to the scoring matrix described previously [66]. Briefly, all of the conserved and novel rose miRNAs were aligned against rose transcriptome dataset (http://bioinfo.bti.cornell.edu/rose) using a BLASTn search strategy. For evaluation of the complementary sites between predicted rose miRNAs and potential mRNA targets, no more than four mismatches between miRNAs and their potential mRNA targets (G:U was regarded as 0.5 mismatch), and no mismatch between positions 10 and 11.
Quantitative RT-PCR
The stem-loop RT-PCR was performed as described previously [67]. For each miRNA, 1 µg DNase I-treated total RNA was used in the reverse transcription reaction with SuperScript III (Invitrogen). 5S rRNA was used as the internal control. For quantitative RT-PCR of mRNAs, 1 µg DNase I-treated total RNA was used to synthesize cDNA by M-MLV (Promega) using poly(dT)18 oligonucleotides. RhACT5 was used as the internal control. SYBR Green PCR Master Mix (Applied Biosystems) was used in all quantitative RT-PCR experiments. The relative expression changes of miRNAs and genes were calculated using the 2 d-d Ct method [68]. Primers used in all quantitative RT-PCR experiments are listed in Table S8.
Plasmid Construction and Transformation
To construct sensor plasmid, the putative miRNA target site sequence was fused to the 5′-end of EGFP in frame. The resulted fusion sequences were inserted into a modified binary vector pCAMBIA 1300 harboring a Super promoter. The resulting sensor constructs were transformed into Agrobacterium strain GV3101 and then used to co-infiltrate the tobacco leaves with plasmid harboring corresponding miRNA foldbacks. After 3 days of co-infiltration, tobacco leaves were harvested and used to extract total RNA for RLM-RACE analysis.
RLM-RACE
The 5′ RLM-RACE was carried out using the FirstChoice RLM-RACE Kit (Ambion). Two microgram total RNA was directly ligated to 5′ RACE oligo adaptor without calf intestine alkaline phosphatase and tobacco acid pyrophosphatase treatments. The ligated RNA was used to synthesize the cDNA. The PCR products were gel-purified and cloned into the pGEM-T Easy vector (Premega, Madison, WI, USA), and randomly selected clones were sequenced. For the RU15050 and RU13577 sensor sequences, a set of general primers designed based on the EGFP sequence were used. For RLM-RACE using total RNA from rose petals, gene-specific primers were used. All primers were listed in Table S8.
Supporting Information
Digital expression profiles of miR171 , miR171 *, miR396 and miR396 * in rose petals during earlier opening period and in response to ethylene.
(DOC)
The sequencing results of small RNAs from 4 rose flower samples. The flowers of stage 2 were treated by 10 ppm ethylene in a sealed airtight chamber for 24 h, and flowers exposed to air were used as the control.
(XLS)
Prediction of known miRNA precusors. The rose floral transcriptome database, and the genome sequence of strawberry (F. vesca) and transcriptome data of rose were used as reference, respectively. R, rose; S, strawberry.
(XLS)
Prediction of novel miRNA and their precusors. The rose floral transcriptome database, and the genome sequence of strawberry (F.vesca) were used as reference, respectively. R, roses; S, strawberry.
(XLS)
Reads of known and novel miRNAs in rose petals. For each library, petals from 15 flowers were mixed and used to avoid the individual difference. The miRNAs in bold indicate highly expressed miRNA in petals (more than 50 RPM in at least one library).
(XLS)
Predicted targets of known miRNAs in roses.
(XLS)
Predicted targets of novel miRNAs in roses.
(XLS)
Integraded analysis of expression profiles of miRNAs and responsed targets in rose petals.
(XLS)
Oligonucleotide primer sequences.
(XLS)
Comparison of known and novel miRNAs identified by Kim et al and in our study.
(XLSX)
Acknowledgments
We thank Dr. Jia-Wei Wang (Shanghai Institutes for Biological Sciences, China) for providing miR156-ox plasmid and Dr. Patrick Laufs (Institut Jean-Pierre Bourgin, France) for providing miR164-ox plasmid.
Funding Statement
This work was supported by the National Natural Science Foundation of China Grants (Grant no. 31071827 and 31130359). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Chen X (2009) Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol 25: 21–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Voinnet O (2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136: 669–687. [DOI] [PubMed] [Google Scholar]
- 3. Jones-Rhoades MW (2011) Conservation and divergence in plant microRNAs. Plant Mol Biol 80: 3–16. [DOI] [PubMed] [Google Scholar]
- 4. Axtell MJ, Bowman JL (2008) Evolution of plant microRNAs and their targets. Trends Plant Sci 13: 343–349. [DOI] [PubMed] [Google Scholar]
- 5. Mica E, Piccolo V, Delledonne M, Ferrarini A, Pezzotti M, et al. (2009) High throughput approaches reveal splicing of primary microRNA transcripts and tissue specific expression of mature microRNAs in Vitis vinifera . BMC Genomics 10: 558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schreiber AW, Shi BJ, Huang CY, Langridge P, Baumann U (2011) Discovery of barley miRNAs through deep sequencing of short reads. BMC Genomics 12: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martinez G, Forment J, Llave C, Pallas V, Gomez G (2011) High-throughput sequencing, characterization and detection of new and conserved cucumber miRNAs. PLoS One 6: e19523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Donaire L, Pedrola L, Rosa R, Llave C (2011) High-throughput sequencing of RNA silencing-associated small RNAs in olive (Olea europaea L.). PLoS One 6: e27916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zuo J, Zhu B, Fu D, Zhu Y, Ma Y, et al. (2012) Sculpting the maturation, softening and ethylene pathway: The influences of microRNAs on tomato fruits. BMC Genomics 13: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xia R, Zhu H, An Y, Beers E, Liu Z (2012) Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol 13: R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu H, Xia R, Zhao B, An Y, Dardick C, et al. (2012) Unique expression, processing regulation, and regulatory network of peach (Prunus persica) miRNAs. BMC Plant Biol 12: 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res 36: D154–D158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ruiz-Ferrer V, Voinnet O (2009) Roles of plant small RNAs in biotic stress responses. Annu Rev Plant Biol 60: 485–510. [DOI] [PubMed] [Google Scholar]
- 14. Jay F, Renou J-P, Voinnet O, Navarro L (2010) Biotic stress-associated microRNAs: identification, detection, regulation, and functional analysis. Methods Mol Biol 592: 183–202. [DOI] [PubMed] [Google Scholar]
- 15. Khraiwesh B, Zhu JK, Zhu J (2012) Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim Biophys Acta 1819: 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang Y, Yu M, Yu H, Han J, Song C, et al. (2011) Computational identification of microRNAs in peach expressed sequence tags and validation of their precise sequences by miR-RACE. Mol Biol Rep 39: 1975–1987. [DOI] [PubMed] [Google Scholar]
- 17. Gao Z, Luo X, Shi T, Cai B, Zhang Z, et al. (2012) Identification and validation of potential conserved microRNAs and their targets in peach (Prunus persica). Mol Cells 34: 239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heinrichs F (2008) International statistics flowers and plants. AIPH/Union Fleurs 56: 16–90. [Google Scholar]
- 19. Debener Th, Linde M (2009) Exploring complex ornamental genomes: The rose as a model plant. Critical Rev Plant Sci 28: 267–280. [Google Scholar]
- 20.Abeles FB, Morgan PW, Saltveit ME (1992) Ethylene in Plant Biology, Ed 2. Academic Press, San Diego.
- 21. Wang K, Li H, Ecker J (2002) Ethylene biosynthesis and signaling networks. Plant Cell 14: S131–S151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reid MS, Dodge LL, Mor Y, Evans RY (1989) Effects of ethylene on rose opening. Acta Horti 261: 215–220. [Google Scholar]
- 23. Muller R, Lind-Iversen S, Stummann BM, Serek M (2000) Expression of genes for ethylene biosynthetic enzymes and an ethylene receptor in senescing flowers of miniature potted roses. J Hort Sci Biotech 75: 12–18. [Google Scholar]
- 24. Muller R, Stummann BM (2003) Genetic regulation of ethylene perception and signal transduction related to flower senescence. J. Food Agr Environ 1: 87–94. [Google Scholar]
- 25. Ma N, Cai W, Lu W, Tan H, Gao J (2005) Exogenous ethylene influences flower opening of cut roses (Rosa hybrida) by regulating the gene encoding ethylene biosynthesis enzymes. Sci China C Life Sci 48: 434–444. [DOI] [PubMed] [Google Scholar]
- 26. Tan H, Liu X, Ma N, Xue J, Lu W, et al. (2006) Ethylene influenced flower opening and expression of genes encoding Etrs, Ctrs, and Ein3s in two cut rose cultivars. Postharvest Biol Tech 40: 97–105. [Google Scholar]
- 27. Xue JQ, Li YH, Tan H, Yang F, Ma N, et al. (2008) Expression of ethylene biosynthetic and receptor genes in rose floral tissues during ethylene-enhanced flower opening. J Exp Bot 59: 2161–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim J, Park JH, Lim CJ, Lim JY, Ryu JY, et al. (2012) Small RNA and transcriptome deep sequencing proffers insight into floral gene regulation in Rosa cultivars. BMC Genomics 13: 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang D, Fan J, Ranu RS (2004) Cloning and expression of 1-aminocyclopropane-1 -carboxylate synthase cDNA from rosa (Rosa x hybrida). Plant Cell Rep 22: 422–429. [DOI] [PubMed] [Google Scholar]
- 30. Ma N, Xue J, Li Y, Liu X, Dai F, et al. (2008) Rh-PIP2;1, a rose aquaporin gene, is involved in ethylene-regulated petal expansion. Plant Physiol 148: 894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kasschau KD, Fahlgren N, Chapman EJ, Sullivan CM, Cumbie JS, et al. (2007) Genome-wide profiling and analysis of Arabidopsis siRNAs . PLoS Biol 5: e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jeong DH, Park S, Zhai J, Gurazada SG, De Paoli E, et al. (2012) Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 23: 4185–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mi S, Cai T, Hu Y, Chen Y, Hodges E, et al. (2008) Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133: 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Czech B, Hannon GJ (2011) Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet 12: 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, et al. (2011) The genome of woodland strawberry (Fragaria vesca). Nat Genet 43: 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, et al. (2008) Criteria for Annotation of Plant MicroRNAs. Plant Cell 20: 3186–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao YT, Wang M, Fu SX, Yang WC, Qi CK, et al. (2012) Small RNA profiling in two Brassica napus cultivars identifies microRNAs with oil production and developmental correlated expressions and new small RNA classes. Plant Physiol 158: 813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, et al. (2002) Prediction of plant microRNA targets. Cell 110: 513–520. [DOI] [PubMed] [Google Scholar]
- 39. Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, et al. (2008) Widespread translational inhibition by plant miRNAs and siRNAs. Science 320: 1185–1190. [DOI] [PubMed] [Google Scholar]
- 40.Bennett BC Economic Botany: Twenty-five economically important plant families. Encyclopedia of Life Support Systems (EOLSS) e-book.
- 41. Yasumura Y, Pierik R, Fricker MD, Voesenek LACJ, Harberd NP (2012) Studies of Physcomitrella patens reveal that ethylene mediated submergence responses arose relatively early in land-plant evolution. Plant J 72: 947–959. [DOI] [PubMed] [Google Scholar]
- 42. Zhang T-C, Qiao Q, Zhong Y (2012) Detecting adaptive evolution and functional divergence in aminocyclopropane-1-carboxylate synthase (ACS) gene family. Comput Biol Chem 38: 10–16. [DOI] [PubMed] [Google Scholar]
- 43. Chen L, Wang T, Zhao M, Zhang W (2011) Ethylene-responsive miRNAs in roots of Medicago truncatula identified by high-throughput sequencing at whole genome level. Plant Sci 184: 14–19. [DOI] [PubMed] [Google Scholar]
- 44. Tang Z, Zhang L, Xu C, Yuan S, Zhang F, et al. (2012) Uncovering small RNA-mediated responses to cold stress in a wheat thermosensitive genic male-sterile line by deep sequencing. Plant Physiol 159: 721–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Channeliere S, Riviere S, Scalliet G, Jullien F, Szecsi J, et al. (2002) Analysis of gene expression in rose petals using expressed sequence tags. FEBS Lett 515: 35–38. [DOI] [PubMed] [Google Scholar]
- 46. Guterman I, Shalit M, Menda M, Piestun D, Dafny-Yelin M, et al. (2002) Rose scent: genomics approach to discovering novel floral fragrance-related genes. Plant Cell 14: 2325–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bendahmane M, Dubois A, Raymond O, Le Bris M (2013) Genetics and genomics of flower initiation and development in roses. J Exp Bot 64: 847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dubois A, Remay A, Raymond O, Balzergue S, Chauvet A, et al. (2011) Genomic approach to study floral development genes in Rosa sp . PLoS One 6: e28455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dubois A, Carrere S, Raymond O, Pouvreau B, Cottret L, et al. (2012) Transcriptome database resource and gene expression atlas for the rose. BMC Genomics 13: 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Laufs P, Peaucelle A, Morin H, Traas J (2004) MicroRNA regulation of the CUC genes is required for boundary size control in Arabidopsis meristems. Development 131: 4311–4322. [DOI] [PubMed] [Google Scholar]
- 51. Peaucelle A, Morin H, Traas J, Laufs P (2007) Plants expressing a miR164-resistant CUC2 gene reveal the importance of post-meristematic maintenance of phyllotaxy in Arabidopsis . Development 134: 1045–1050. [DOI] [PubMed] [Google Scholar]
- 52. Raman S, Greb T, Peaucelle A, Blein T, Laufs P, et al. (2008) Interplay of miR164, CUP-SHAPED COTYLEDON genes and LATERAL SUPPRESSOR controls axillary meristem formation in Arabidopsis thaliana . Plant J 55: 65–76. [DOI] [PubMed] [Google Scholar]
- 53. He XJ, Mu RL, Cao WH, Zhang ZG, Zhang JS, et al. (2005) AtNAC2, a transcription factor downstream of ethylene and auxin signaling pathways, is involved in salt stress response and lateral root development. Plant J 44: 903–916. [DOI] [PubMed] [Google Scholar]
- 54. Kim JH, Woo HR, Kim J, Lim PO, Lee IC, et al. (2009) Trifurcate feed-forward regulation of age-dependent cell death involving miR164 in Arabidopsis . Science 323: 1053–1057. [DOI] [PubMed] [Google Scholar]
- 55. Gou JY, Felippes FF, Liu CJ, Weigel D, Wang JW (2011) Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 23: 1512–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. El-Kereamy A, Chervin C, Roustan JP, Cheynier V, Souquet J-M, et al. (2003) Exogenous ethylene stimulates the long-term expression of genes related to anthocyanin biosynthesis in grape berries. Physiologia Plantarum 119: 175–182. [Google Scholar]
- 57. Gantulga D, Ahn YO, Zhou C, Battogtokh D, Bevan DR, et al. (2009) Comparative characterization of the Arabidopsis subfamily a1 β-galactosidases. Phytochemistry 70: 1999–2009. [DOI] [PubMed] [Google Scholar]
- 58. Cosgrove DJ (2005) Growth of the plant cell wall. Nat Rev Mol Cell Biol 6: 850–861. [DOI] [PubMed] [Google Scholar]
- 59. Zhong GV, Burns JK (2003) Profiling ethylene-regulated gene expression in Arabidopsis thaliana by microarray analysis. Plant Mol Biol 53: 117–131. [DOI] [PubMed] [Google Scholar]
- 60. De Paepe A, Vuylsteke M, Van Hummelen P, Zabeau M, Van Der Straeten D (2004) Transcriptional profiling by cDNA-AFLP and microarray analysis reveals novel insights into the early response to ethylene in Arabidopsis. Plant J 39: 537–559. [DOI] [PubMed] [Google Scholar]
- 61. Alba R, Payton P, Fei ZJ, McQuinn R, Debbie P, et al. (2005) Transcriptome and selected metabolite analyses reveal multiple points of ethylene control during tomato fruit development. Plant Cell 17: 2954–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ma N, Tan H, Liu XH, Xue JQ, Li YH, et al. (2006) Transcriptional regulation of ethylene receptor and CTR genes involved in ethylene-induced flower opening in cut rose (Rosa hybrida) cv. Samantha. J Exp Bot 57: 2763–2773. [DOI] [PubMed] [Google Scholar]
- 63. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hofacker IL (2003) Vienna RNA secondary structure server. Nucleic Acids Res 31: 3429–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rajagopalan R, Vaucheret H, Trejo J, Bartel DP (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana . Genes Dev 20: 3407–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jones-Rhoades MW, Bartel DP (2004) Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell 14: 787–799. [DOI] [PubMed] [Google Scholar]
- 67. Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP (2007) Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Digital expression profiles of miR171 , miR171 *, miR396 and miR396 * in rose petals during earlier opening period and in response to ethylene.
(DOC)
The sequencing results of small RNAs from 4 rose flower samples. The flowers of stage 2 were treated by 10 ppm ethylene in a sealed airtight chamber for 24 h, and flowers exposed to air were used as the control.
(XLS)
Prediction of known miRNA precusors. The rose floral transcriptome database, and the genome sequence of strawberry (F. vesca) and transcriptome data of rose were used as reference, respectively. R, rose; S, strawberry.
(XLS)
Prediction of novel miRNA and their precusors. The rose floral transcriptome database, and the genome sequence of strawberry (F.vesca) were used as reference, respectively. R, roses; S, strawberry.
(XLS)
Reads of known and novel miRNAs in rose petals. For each library, petals from 15 flowers were mixed and used to avoid the individual difference. The miRNAs in bold indicate highly expressed miRNA in petals (more than 50 RPM in at least one library).
(XLS)
Predicted targets of known miRNAs in roses.
(XLS)
Predicted targets of novel miRNAs in roses.
(XLS)
Integraded analysis of expression profiles of miRNAs and responsed targets in rose petals.
(XLS)
Oligonucleotide primer sequences.
(XLS)
Comparison of known and novel miRNAs identified by Kim et al and in our study.
(XLSX)