

Abstract
Although the nature of solvent-protein interactions is generally weak and non-specific, addition of cosolvents such as denaturants and osmolytes strengthens protein-protein interactions for some proteins, whereas it weakens protein-protein interactions for others. This is exemplified by the puzzling observation that addition of glycerol oppositely affects the association constants of two antibodies, D1.3 and D44.1, with lysozyme. To resolve this conundrum, we develop a methodology based on the thermodynamic principles of preferential interaction theory and the quantitative characterization of local protein solvation from molecular dynamics simulations. We find that changes of preferential solvent interactions at the protein-protein interface quantitatively account for the opposite effects of glycerol on the antibody-antigen association constants. Detailed characterization of local protein solvation in the free and associated protein states reveals how opposite solvent effects on protein-protein interactions depend on the extent of dewetting of the protein-protein contact region and on structural changes that alter cooperative solvent-protein interactions at the periphery of the protein-protein interface. These results demonstrate the direct relationship between macroscopic solvent effects on protein-protein interactions and atom-scale solvent-protein interactions, and establish a general methodology for predicting and understanding solvent effects on protein-protein interactions in diverse biological environments.
Author Summary
Solvents play a fundamental role in living systems where they mediate the interactions between proteins and other biomolecules. Besides water, biological solvents often contain high concentrations of small molecular compounds known as cosolvents. Although many studies have reported specific and opposite effects of cosolvents on protein-protein interactions, the molecular origins of this phenomenon remain unknown. In this study, we develop a methodology to predict solvent effects on protein-protein interactions by computational characterization of local protein solvation. We use this methodology to explain the opposite effects of glycerol on the binding affinity of two antibodies. Quantitative characterization of local solvation near the protein-protein interface reveals that solvation changes not only depend on the extent of dewetting of the protein-protein contact region, but also on specific protein structural changes at the periphery of the protein-protein interface. Our results demonstrate the direct relationship between solvent effects on protein-protein interactions and local solvent-protein interactions, and establish a general methodology for predicting and understanding cosolvent effects on protein-protein interactions in diverse biological environments.
Introduction
Cosolvents such as denaturants, salts, amino acids and polyols play an important role in many protein processes as they modify the strength of intra- and intermolecular interactions of proteins in various cellular and biochemical environments [1]–[5]. Cosolvents that strengthen protein-protein interactions induce macromolecular assembly and increase the conformational stability of proteins [4], [6]; cosolvents that weaken protein-protein interactions generally increase protein solubility and may prevent the formation of protein aggregates with undesired immunological or pathological properties [7], [8]. Despite the growing evidence for the importance of cosolvents in regulating biological processes [9]–[11] and the widespread use of cosolvents in protein formulation and refolding [1], [2], [12]–[16], general understanding of cosolvent effects on protein interactions is lacking and optimizing solvent conditions for a particular protein process typically requires laborious empirical screening of various cosolvents.
Preferentially excluded cosolvents generally stabilize proteins, whereas cosolvents that preferentially interact with the protein surface often destabilize and denature proteins [6], [17]. Similarly, it is often implied that preferentially excluded cosolvents increase protein-protein interactions, whereas cosolvents that preferentially interact with the protein surface weaken protein-protein interactions. This dichotomy is, however, irreconcilable with many studies in literature that report specific – and even opposite – effects of cosolvents on protein-protein interactions [9], [18]–[23]. For instance, osmolytes such as glycerol and TMAO increase fibril formation of Aβ-peptide involved in Alzheimer's disease, but decrease aggregation of ataxin-3 involved in Machado-Joseph disease [18]. Another study reports that glycerol promotes the association of cytochrome c with cytochrome b5 but inhibits the association of cytochrome c and cytochrome c oxidase [19]. Yet another study reports a more than tenfold decrease of antibody-antigen binding affinity measured in vivo compared to the corresponding value measured in vitro [24]. This example not only illustrates how protein-protein interactions differ in distinct solution environments, but also calls for caution in correlating pharmacological properties to protein-protein interactions data measured in vitro [25]. Taken together, these studies highlight that a general approach for understanding cosolvent effects on protein interactions should account for specific solvent-protein interactions.
Current understanding of cosolvent effects on protein interactions is largely derived from the principles of linked functions [26] and the thermodynamic theory of preferential interactions in multicomponent solutions [27]–[36]. These principles dictate that the addition of cosolvent will shift the association constant KA of two proteins towards the protein state with the highest preferential interaction coefficient 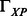 [6], [37], [38]:
[6], [37], [38]:
| (1) |
In Eq. 1, 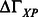 is the difference of the preferential interaction coefficients of the associated and free protein states, and ax is the activity of the cosolvent. This equation directly relates cosolvent effects on the association constant with solvation changes upon association. Unfortunately, application of Eq. 1 for understanding cosolvent effects on protein processes has been incapacitated because of the difficulty to obtain precise values of preferential interaction coefficients
is the difference of the preferential interaction coefficients of the associated and free protein states, and ax is the activity of the cosolvent. This equation directly relates cosolvent effects on the association constant with solvation changes upon association. Unfortunately, application of Eq. 1 for understanding cosolvent effects on protein processes has been incapacitated because of the difficulty to obtain precise values of preferential interaction coefficients 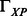 for distinct protein states [39], [40], and because
for distinct protein states [39], [40], and because 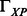 , which quantifies preferential interactions averaged over the entire protein surface, does not provide information on local solvation properties at distinct loci of the protein surface [6], [40].
, which quantifies preferential interactions averaged over the entire protein surface, does not provide information on local solvation properties at distinct loci of the protein surface [6], [40].
Here we develop and validate a methodology to quantify the molecular origins of opposite solvent effects on protein-protein interactions. By combining the thermodynamic principles of preferential interaction theory with surface plasmon resonance experiments and computational characterization of local protein solvation, we demonstrate the direct relationship between macroscopic solvent effects on protein-protein interactions and atom-scale solvent-protein interactions. We apply this methodology to understand the opposite effects of glycerol on the association constants of two antibodies - D1.3 and D44.1 - with lysozyme, and we find that cosolvent-effects on protein-protein interactions critically depend on the extent of dewetting of the protein-protein contact region and on local structural changes of the protein that alter cooperative solvent-protein interactions through multiple hydrogen-bonds.
Results
Opposite effects of glycerol on protein-protein interactions
To gain understanding in the molecular origins of opposite solvent effects on protein-protein interactions, we focus on a pertinent example of opposite effects of glycerol on the association constants of two different antibodies with lysozyme [21]. We use surface plasmon resonance to characterize the opposite effects of glycerol on the association constants of antibody fragments D1.3 and D44.1 over a wide concentration range (0–9 molal glycerol). Figure 1 shows that the association constant of the D1.3-lysozyme complex decreases exponentially with respect to glycerol molality, whereas the association constant KA of the D44.1-lysozyme complex increases exponentially with glycerol molality. Exponential responses of equilibrium constants with respect to cosolvent concentrations have also been observed for other protein binding and unfolding reactions, and it has been suggested that the underlying mechanisms are closely related [17]. Such a common mechanism could stem from the thermodynamic principles of preferential interaction theory, yet evidence for this hypothesis is lacking.
Figure 1. Opposite effects of glycerol on the association constant KA of Fab D44.1 and scFv D1.3 with lysozyme.
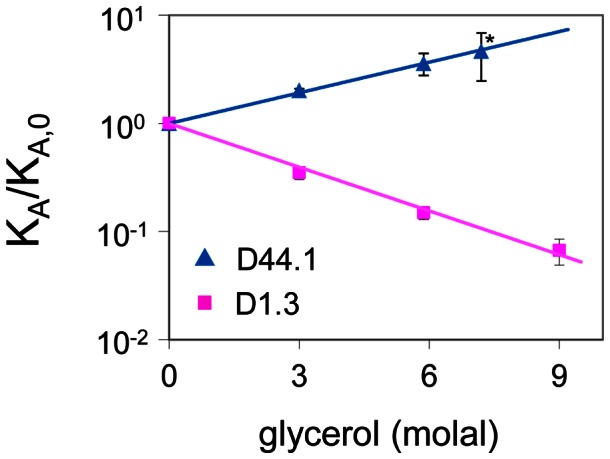
KA/KA,0 is the ratio of the association constants with and without glycerol. The data point marked with an asterisk is derived from Goldbaum et al. [21] and all other data points are determined by surface plasmon resonance.
To find out whether opposite solvent effects on protein-protein interactions can be understood from preferential interaction theory, we investigate whether Eq. 1 is able to explain the opposite effects of glycerol on the association constants of D1.3 and D44.1. Taking into account the exponential responses of the association constants with respect to glycerol molality (Figure 1), Eq. 1 can be simplified into the following equation (Text S1):
| (2) |
This equation dictates that the change of the logarithms of the association constant KA upon addition of glycerol equals the difference of preferential interaction coefficients 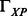 of the associated and free protein states. Application of Eq. 2 thus requires
of the associated and free protein states. Application of Eq. 2 thus requires 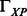 -values of the associated and free states of D1.3, D44.1 and lysozyme in aqueous glycerol.
-values of the associated and free states of D1.3, D44.1 and lysozyme in aqueous glycerol.
Preferential solvent interactions of free and associated proteins
To quantify 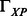 -values of the free and associated protein states of D1.3, D44.1 and lysozyme in aqueous glycerol, we performed six independent molecular dynamics simulations for the respective protein systems.
-values of the free and associated protein states of D1.3, D44.1 and lysozyme in aqueous glycerol, we performed six independent molecular dynamics simulations for the respective protein systems. 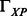 -values of all proteins and protein-complexes are negative (Table 1), indicating overall exclusion of glycerol for all proteins. Differences of
-values of all proteins and protein-complexes are negative (Table 1), indicating overall exclusion of glycerol for all proteins. Differences of 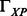 -values between the associated and free protein states are relatively small and subject to large standard errors (Table 1). To improve the precision of computed
-values between the associated and free protein states are relatively small and subject to large standard errors (Table 1). To improve the precision of computed 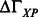 -values, we identified protein surface regions where local solvation differs in the associated and free protein state. Local concentration maps of the free and associated protein states differ markedly near the protein-protein interface region, but not for the rest of the protein surface (Figure 2 and Figure S1). This indicates that protein-protein association only affects solvation near the protein-protein interface.
-values, we identified protein surface regions where local solvation differs in the associated and free protein state. Local concentration maps of the free and associated protein states differ markedly near the protein-protein interface region, but not for the rest of the protein surface (Figure 2 and Figure S1). This indicates that protein-protein association only affects solvation near the protein-protein interface.
Table 1. Preferential interaction coefficients of free and associated proteins in 6 molal glycerol.
| D1.3 | lysozyme | D1.3-lysozyme | D44.1 | Lysozyme | D44.1-lysoyzme | |
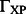
|
−11.1±0.4 | −5.2±0.8 | −20.6±1.5 | −28.2±1.5 | −6.9±0.8 | −33.5±1.9 |
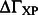 a
a
|
−4.3±1.7 | 1.7±2.5 | ||||
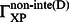 b
b
|
−9.2±0.4 | −3.5±0.5 | −13.9±1.3 | −26.0±1.6 | −4.7±0.7 | −31.2±1.7 |
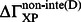 a
a
|
−1.2±1.5 | −0.5±2.4 | ||||
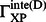 b
b
|
−1.9±0.2 | −1.8±0.2 | −6.7±0.5 | −2.1±0.4 | −2.2±0.4 | −2.3±0.6 |
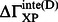 a
a
|
−3.0±0.6 | 2.0±0.9 | ||||
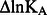 c
c
|
−2.3±0.1 | 1.2±0.3 |
Difference of preferential interaction coefficients between associated and free proteins.
Regional preferential interaction coefficients at the protein-protein interface region inte(D) and the complementary protein surface region non-inte(D). The distance D is 7 Å for the D1.3-lysozyme complex and 9 Å for the D44.1-lysozyme complex.
Calculated from experimental KA-values at 6 molal glycerol (Figure 1).
Figure 2. Local concentration maps of lysozyme and D1.3 in the associated and free states.
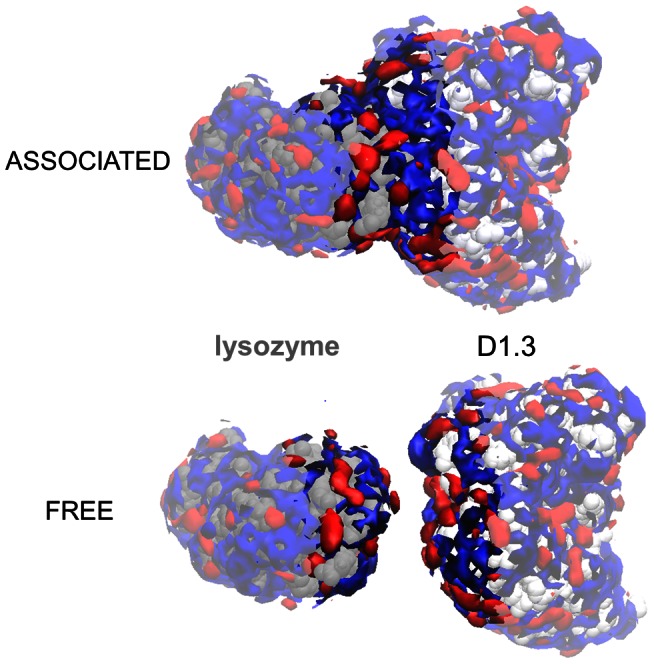
Solvent regions that are preferentially solvated by glycerol and water are colored in red and blue respectively, and solvent regions near the interface region are highlighted.
Since solvation changes upon protein-protein association are limited to protein surface regions near the protein-protein surface, the difference of 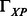 -values between the associated and free protein states could be calculated from local preferential interaction coefficients near the protein-protein interface. We define the protein-protein interface region inte(D) as the contiguous protein surface region comprising all residues of the protein-protein complex with at least one atom within a distance D from the associated protein (Figure 3). All protein residues outside inte(D) are grouped into the complementary region non-inte(D), and the following equation is automatically met [40]:
-values between the associated and free protein states could be calculated from local preferential interaction coefficients near the protein-protein interface. We define the protein-protein interface region inte(D) as the contiguous protein surface region comprising all residues of the protein-protein complex with at least one atom within a distance D from the associated protein (Figure 3). All protein residues outside inte(D) are grouped into the complementary region non-inte(D), and the following equation is automatically met [40]:
| (3) |
In the above equation, 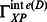 and
and 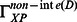 are the regional preferential interaction coefficients of the interface region inte(D) and the complementary surface region, respectively. The distance D is determined as the minimal distance at which values of
are the regional preferential interaction coefficients of the interface region inte(D) and the complementary surface region, respectively. The distance D is determined as the minimal distance at which values of 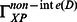 do not significantly differ between the free and associated proteins (Table 1), and we get
do not significantly differ between the free and associated proteins (Table 1), and we get 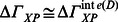 . Notably,
. Notably, 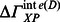 -values have a higher precision than the corresponding
-values have a higher precision than the corresponding 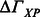 -values (Table 1).
-values (Table 1).
Figure 3. Definition of the interface region inte(D) of a protein-protein complex.
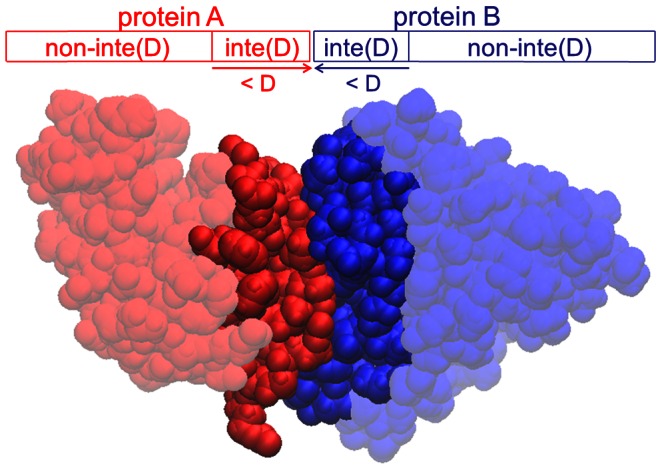
The interface region inte(D) of protein A is defined as the continuous protein surface region comprising all residues with at least one atom within a distance D from protein B. All other residues of protein A belong to the complementary region, non-inte(D).
The association of D1.3 with lysozyme results in an overall decrease in preferential interaction coefficients (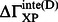 <0), whereas the association of D44.1 with lysozyme results in an overall increase in preferential interaction coefficients upon protein-protein association (
<0), whereas the association of D44.1 with lysozyme results in an overall increase in preferential interaction coefficients upon protein-protein association (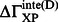 >0) (Table 1). Strikingly, the values of
>0) (Table 1). Strikingly, the values of 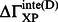 quantitatively agree with experimentally determined changes of the association constant,
quantitatively agree with experimentally determined changes of the association constant, 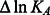 (Table 1). This agreement conforms with Eq. 2 and establishes the direct relationship between protein solvation and solvent effects on protein-protein interactions. Although the theoretical foundations of this relationship – i.e. the thermodynamic principles of linked function and preferential interactions theory - have been established over the past decades [26]–[36], empirical evidence supporting this relationship is lacking and the extent to which other solvent-related factors, such as the dielectric constant and viscosity of the solvent [41], (co-)determine cosolvent effects on protein-protein interactions remain unknown. Our finding that cosolvent effects on protein-protein association constants quantitatively agree with changes in preferential interaction coefficients between the associated and free protein states pinpoints the predominant role of preferential solvent interactions in determining the effects of cosolvents on protein-protein interactions.
(Table 1). This agreement conforms with Eq. 2 and establishes the direct relationship between protein solvation and solvent effects on protein-protein interactions. Although the theoretical foundations of this relationship – i.e. the thermodynamic principles of linked function and preferential interactions theory - have been established over the past decades [26]–[36], empirical evidence supporting this relationship is lacking and the extent to which other solvent-related factors, such as the dielectric constant and viscosity of the solvent [41], (co-)determine cosolvent effects on protein-protein interactions remain unknown. Our finding that cosolvent effects on protein-protein association constants quantitatively agree with changes in preferential interaction coefficients between the associated and free protein states pinpoints the predominant role of preferential solvent interactions in determining the effects of cosolvents on protein-protein interactions.
Having established the direct relationship between solvent effects on protein-protein interactions and preferential solvent interactions at the protein-protein interface , we can now address the first part of the conundrum of opposite glycerol effects on the association constants of D1.3 and D44.1: glycerol weakens binding of D1.3 with lysozyme because of the overall decrease of preferential interaction coefficients upon antibody-antigen association, but glycerol strengthens binding of D44.1 with lysozyme because of the overall increase of preferential interaction coefficients upon antibody-antigen association (Table 1). This raises, however, another pertinent question: why does the association of D1.3 with lysozyme result in an overall decrease of preferential interactions with glycerol, whereas the association of D44.1 with lysozyme results in an overall increase of preferential interactions with glycerol? To address this question, we further analyze protein-association related changes of local solvation near the protein-protein interface of D1.3, D44.1 and lysozyme.
Solvation at the protein-protein interface
The global preferential interaction coefficient of a protein, 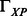 , is the sum of the local preferential interaction coefficients
, is the sum of the local preferential interaction coefficients 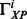 of all protein residues that comprise the protein surface [40], [42]. Changes of
of all protein residues that comprise the protein surface [40], [42]. Changes of 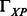 upon protein association can therefore be attributed to differences of
upon protein association can therefore be attributed to differences of 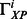 in the free and associated protein states. For the D1.3-lysozyme complex, protein-protein association leads to a decrease of
in the free and associated protein states. For the D1.3-lysozyme complex, protein-protein association leads to a decrease of 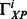 for all residues that are buried at the protein-protein contact region (Figure 4 and Figure S2). This is because, unlike water, glycerol is totally excluded from the protein-protein contact (Figure 5). Similarly, most residues at the periphery of the contact region of the D1.3-lysozyme complex see a decrease of
for all residues that are buried at the protein-protein contact region (Figure 4 and Figure S2). This is because, unlike water, glycerol is totally excluded from the protein-protein contact (Figure 5). Similarly, most residues at the periphery of the contact region of the D1.3-lysozyme complex see a decrease of 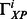 -values in the associated state (colored in blue Figure 4). The only exception is Asp54 of the VH-chain of D1.3, which is strongly preferentially hydrated in the free state but only moderately preferentially hydrated as its side chain becomes partially buried in the associated state (Figure 6 and Figure S2). The positive contribution of Asp54 to
-values in the associated state (colored in blue Figure 4). The only exception is Asp54 of the VH-chain of D1.3, which is strongly preferentially hydrated in the free state but only moderately preferentially hydrated as its side chain becomes partially buried in the associated state (Figure 6 and Figure S2). The positive contribution of Asp54 to 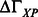 is, however, significantly smaller than the sum of the negative contributions of the other interface residues. As a result,
is, however, significantly smaller than the sum of the negative contributions of the other interface residues. As a result, 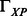 decreases upon association of D1.3 with lysozyme.
decreases upon association of D1.3 with lysozyme.
Figure 4. Local changes in preferential interactions upon protein-protein association of D1.3 and D44.1 with lysozyme.

Residues for which the local preferential interaction coefficient 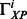 is greater (smaller) for the associated than for the free proteins are colored red (blue). For clarity, only the VH and VL regions of the antibody fragments are displayed.
is greater (smaller) for the associated than for the free proteins are colored red (blue). For clarity, only the VH and VL regions of the antibody fragments are displayed.
Figure 5. Solvation of the interface regions of D1.3, D44.1 and lysozyme in the associated and free states.

Solvent regions that are preferentially solvated by glycerol (water) are colored in red (blue). The yellow circle indicates the protein surface locus near the N-terminus of lysozyme.
Figure 6. Snapshot of solvent molecules near Asp54 of D1.3 VH in the free state (A) and associated to lysozyme (B).
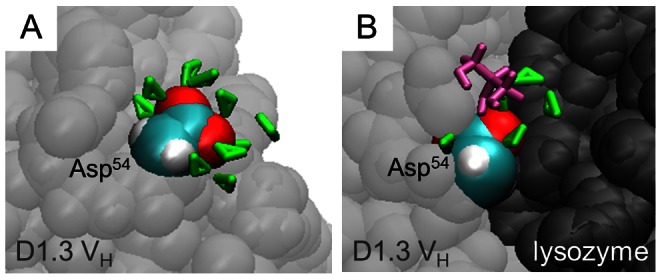
Water and glycerol molecules within 5 Å from Asp54 are represented in green and purple, respectively. C- and O-atoms of the side-chain of Asp54 are highlighted in cyan and red, respectively.
For the D44.1-lysozyme complex, changes of local preferential interactions upon protein-protein association are more balanced with values of 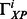 increasing for some residues and decreasing for others (Figure 4 and Figure S3). Similar to the D1.3-lysozyme complex, most residues with significant changes of
increasing for some residues and decreasing for others (Figure 4 and Figure S3). Similar to the D1.3-lysozyme complex, most residues with significant changes of 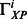 are found near the protein-protein contact region (Figure 4 and Figure S3). However, unlike the D1.3-lysozyme complex, the contact region of the D44.1-lysozyme complex is mostly dry (Figure 5). Changes of
are found near the protein-protein contact region (Figure 4 and Figure S3). However, unlike the D1.3-lysozyme complex, the contact region of the D44.1-lysozyme complex is mostly dry (Figure 5). Changes of 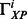 for residues at the contact region of the D44.1-lysozyme complex thus reflect the loss of preferential solvent interactions when protein residues become (partially) buried at the dry contact region. Values of
for residues at the contact region of the D44.1-lysozyme complex thus reflect the loss of preferential solvent interactions when protein residues become (partially) buried at the dry contact region. Values of 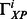 for residues at the contact region of D44.1 and lysozyme in the free states are balanced (Figure S3), such that the combined contribution of contact residues to the protein-associated change of
for residues at the contact region of D44.1 and lysozyme in the free states are balanced (Figure S3), such that the combined contribution of contact residues to the protein-associated change of 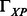 is negligible
is negligible
Another distinctive feature of the D44.1-lysozyme complex is that several residues with significant changes of 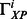 are located further from the protein-protein contact region (Figure 4 and Figure S3). Closer examination of local protein solvation near these residues reveals that changes of
are located further from the protein-protein contact region (Figure 4 and Figure S3). Closer examination of local protein solvation near these residues reveals that changes of 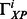 are caused by the specific rearrangement of protein side-chains upon protein-protein association. This is illustrated for the protein surface region near the N-terminus of lysozyme, which is preferentially hydrated in the free state, but becomes preferentially solvated by glycerol in the associated state (Figure 5 and Figure S3). In the free state of lysozyme, Gln41 forms intramolecular hydrogen-bonds with adjacent residues including the N-terminus (Figure 7A), but in the D44.1-lysozyme complex, Gln41 adopts extended orientations as it forms hydrogen-bonds with D44.1 (Figure 7B). Extended orientations of Gln41 favor the formation of multiple hydrogen-bonds between glycerol and several lysozyme-residues including Gln41, Ser86 and the N-terminus (Figure 7B and Movie S1). This leads to strong preferential solvation of the corresponding protein locus in the D44.1-lysozyme complex.
are caused by the specific rearrangement of protein side-chains upon protein-protein association. This is illustrated for the protein surface region near the N-terminus of lysozyme, which is preferentially hydrated in the free state, but becomes preferentially solvated by glycerol in the associated state (Figure 5 and Figure S3). In the free state of lysozyme, Gln41 forms intramolecular hydrogen-bonds with adjacent residues including the N-terminus (Figure 7A), but in the D44.1-lysozyme complex, Gln41 adopts extended orientations as it forms hydrogen-bonds with D44.1 (Figure 7B). Extended orientations of Gln41 favor the formation of multiple hydrogen-bonds between glycerol and several lysozyme-residues including Gln41, Ser86 and the N-terminus (Figure 7B and Movie S1). This leads to strong preferential solvation of the corresponding protein locus in the D44.1-lysozyme complex.
Figure 7. Snapshot of glycerol molecules near the N-terminus of lysozyme in the free state (A) and associated to D44.1 (B).
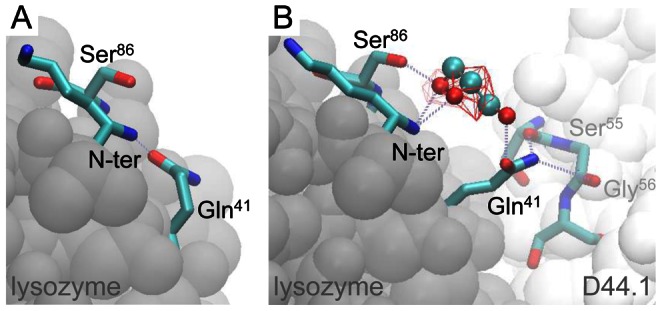
Hydrogen-bonds are indicated by dotted lines and red wireframes demark solvent regions with high local glycerol concentrations (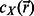 >
>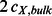 ).
).
Discussion
In this study, we have characterized the opposite effects of glycerol on the association constants of two antibodies against lysozyme using surface plasmon resonance, and we have used molecular dynamics simulations to quantify preferential interaction coefficients of the corresponding proteins in the free and associated states. Our results indicate that glycerol weakens the association of D1.3 with lysozyme because of the overall decrease in preferential interactions as a result of the total exclusion of glycerol, but not of water, from the protein-protein contact region (Table 1, Figure 4 and Figure 5). Conversely, glycerol strengthens the association of D44.1 with lysozyme because of the overall increase in preferential interactions due to (1) exclusion of water from the dry protein-protein contact region (Figure 5) and (2) rearrangement of specific protein side-chains at the periphery of the D44.1-lysozyme interface resulting in local preferential binding of glycerol through multiple hydrogen-bonding (Figure 7). These results demonstrate the direct relationship between macroscopic solvent effects on protein-protein interactions and atom-scale solvent-protein interactions, and show that cosolvent-effects on protein-protein interactions critically depend on the extent of dewetting of the protein-protein contact region and on local protein structural changes that alter cooperative solvent interactions with adjacent residues.
Our surface plasmon resonance data showed that the association constants of both antibodies change exponentially with glycerol molality over the entire concentration range investigated (0–9 molal glycerol) (Figure 1). Exponential responses of equilibrium constants KA with respect to cosolvent molality have been observed for many biomolecular reactions [21], [43]–[54], and it has been suggested that the underlying mechanisms are closely related [17]. Considering the direct relationship between solvent-protein interactions and solvent effects on protein reactions (Eq. 1 and Eq. 2), exponential responses of KA can be attributed to the linear behavior of 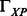 with respect to cosolvent molality. Linear behavior of
with respect to cosolvent molality. Linear behavior of 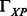 with respect to cosolvent molality has been observed for a wide range of proteins and cosolvents [55]–[58], and can be explained by considering solvent exchange equilibria at protein surface sites that weakly interact with solvent molecules [59]. Taken together, these points support the notion that exponential responses of biomolecular equilibria with respect to cosolvent molality reflect linear changes of
with respect to cosolvent molality has been observed for a wide range of proteins and cosolvents [55]–[58], and can be explained by considering solvent exchange equilibria at protein surface sites that weakly interact with solvent molecules [59]. Taken together, these points support the notion that exponential responses of biomolecular equilibria with respect to cosolvent molality reflect linear changes of 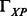 caused by differences in weak solvent-protein interactions between different biomolecular states.
caused by differences in weak solvent-protein interactions between different biomolecular states.
Our methodology for quantifying the molecular origins of solvent effects on protein-protein interactions comprises the following steps: (1) run extended molecular dynamics simulations of free and associated proteins with constrained backbone coordinates, (2) calculate global, regional and residue-based preferential interaction coefficients and local concentration maps of free and associated proteins, (3) determine the protein-protein interface region inte(D) where protein solvation changes occur, (4) quantify cosolvent effects on the protein-protein association constant KA from regional preferential interaction coefficients at the interface region inte(D), (5) identify and map protein residues for which residue-based preferential interaction coefficients significantly differ between associated and free proteins, (6) analyze local solvation changes near these residues by inspecting local concentration maps and solvent trajectories. We found that Step 3 of our methodology is critical as it enables the calculation of protein association-induced changes of preferential interaction coefficients with high precision (Table 1). Such high precision is needed for Step 4, and can generally not be obtained from experiment [56], [57]. Another important feature of our methodology is the identification of specific loci at the protein surface that contribute to macroscopic solvent effects on protein-protein interactions (Step 5). This enables the user to locate and quantify local solvation changes that determine macroscopic solvent effects on protein-protein interactions.
In a previous molecular dynamics study with unconstrained protein coordinates, we found that large conformational changes of the protein backbone result in large changes of the preferential interaction coefficient 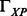 [58]. Trajectory-dependent sampling of the protein conformational ensemble caused large differences of
[58]. Trajectory-dependent sampling of the protein conformational ensemble caused large differences of 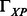 -values obtained from independent simulations, and
-values obtained from independent simulations, and 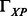 -values of specific protein conformations sampled within nanoseconds differed by several units [58]. Such large differences of
-values of specific protein conformations sampled within nanoseconds differed by several units [58]. Such large differences of 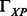 are of similar magnitude as the differences of
are of similar magnitude as the differences of 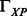 between free and associated proteins (Table 1), and differentiating protein-association induced changes of
between free and associated proteins (Table 1), and differentiating protein-association induced changes of 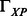 from trajectory-dependent conformational sampling effects would be extremely challenging. Moreover, quantitative characterization of local protein solvation is currently only possible for simulations with constrained backbone coordinates [40]. Constraining backbone coordinates is therefore an essential feature of our methodology. An arguable limitation of using constrained backbone coordinates is that protein-association induced conformational changes of the protein backbone that could significantly affect solvent preferential interactions are not accounted for. However, such conformational changes are expected to be rare since backbone conformations for most protein complexes differ little between the free and associated protein states [60], [61].
from trajectory-dependent conformational sampling effects would be extremely challenging. Moreover, quantitative characterization of local protein solvation is currently only possible for simulations with constrained backbone coordinates [40]. Constraining backbone coordinates is therefore an essential feature of our methodology. An arguable limitation of using constrained backbone coordinates is that protein-association induced conformational changes of the protein backbone that could significantly affect solvent preferential interactions are not accounted for. However, such conformational changes are expected to be rare since backbone conformations for most protein complexes differ little between the free and associated protein states [60], [61].
Owing to the important role of water in protein binding [62]–[70], much recent research effort has evolved in fairly accurate methods for predicting the location of crystallographically observed waters at the interface of protein cavities and small molecule ligands [65], [71]–[74]. Hydration sites at protein-protein interfaces may be more difficult to predict, and studies on hydration of protein-protein interfaces have been mainly limited to the analysis of crystal waters [75]–[78]. In this study, we obtained good agreement between the location of high-occupancy water sites and crystal waters at the protein-protein interface (Figure S4). Over the course of the simulation, all water molecules at the protein-protein interface undergo dynamic interchange between different solvation sites (Movie S2), and the protein-protein interface region contacts many more water molecules than the waters resolved in the crystal structure (Figure S4). All these waters contribute to the overall preferential interaction coefficient, and it is therefore not surprising that crystallographic studies of protein solvation fail to explain cosolvent effects on protein-protein interactions [79].
To this day, cosolvent effects on protein reactions are commonly interpreted based on the global preferential interaction coefficient of the native free protein state and the change of surface area involved in the reaction [6], [56]. Thereby, it is – often implicitly - assumed that local protein solvation is homogeneous over the entire protein surface. Based on this assumption, one would conclude that glycerol – which is, on average, preferentially excluded from the protein surface – would always strengthen protein-protein interactions. The flaw of the underlying assumption is evidenced by our results which reveal a remarkable heterogeneity of differences between local preferential solvent interactions in the free and associated protein states (Figure 4 and Figure S2). A more detailed approach for predicting solvent effects on protein reactions was pioneered by Tanford, who quantified thermodynamic solvent effects on smaller constituent groups of a protein molecule and hypothesized the additivity of individual contributions of the constituent groups [80]. Group transfer models, however, cannot account for hydration changes at the protein-protein contact regions and cooperative interactions of cosolvent molecules with adjacent protein residues. We find that these features play a key role in determining solvent effects on protein-protein interactions, and we conclude that quantitative characterization of local protein solvation is prerequisite for understanding cosolvent effects on protein-protein interactions.
Quantitative characterization of local protein solvation requires atomic protein structures, accurate force fields and computational resources for running long protein simulations (>100 ns) [40]. Atomic protein structures can be retrieved from the Protein Data Bank (PDB) which covers more than 25% of the human genome and includes more than 10,000 protein complexes [81], [82]. Force fields validated against experimental values of protein preferential interaction coefficients are available for several cosolvents [42], [58], [83], and future research is expected to increase this list. Computational resources for running long all-atom simulations of large protein complexes may appear daunting at first sight. However, since protein-protein association only affects solvation near the protein-protein interface (Figure 1 and Table 1), computational costs could be significantly reduced by truncating the simulation system around the protein-protein interface region. In this way, sufficiently long simulations may be achieved using standard high performance clusters.
Granted the availability of accurate force fields, our methodology may also be used to study crowding effects on protein association. Similar to small-molecule cosolvents, effects of macromolecular crowders on protein association are protein-dependent [84] and appear to be the balanced result of steric exclusion and specific crowder-protein interactions [23], [85], [86]. By including chemical details of the protein and the macromolecular crowder, our methodology could significantly improve current crowding models which generally fail to quantitatively reproduce crowding effects on protein association [87]. Finally, we would like to point out that the scope of our methodology is not restricted to protein-protein interactions, but extends to any molecular recognition process that involves the formation of supramolecular complexes with well-defined atomic structures. Our methodology may therefore prove an important tool to elucidate solvent effects on molecular recognition processes and protein function in diverse biological environments.
Materials and Methods
Protein expression and purification
The genes of scFv D1.3 and Fab D44.1 were cloned into pET-39b(+) vectors (Novagen) and expressed in E. Coli BL21(DE3). scFv D1.3 was recovered from the periplasmic fraction by osmotic shock, and Fab D44.1 was refolded from the insoluble cell fraction. The recombinant proteins were purified by affinity chromatography using CnBr-Sepharose FF resin (GE Healthcare) coupled to lysozyme. The purity of the proteins was estimated to be >95% as judged by SDS-PAGE. Protein concentrations were calculated using a UV280 nm absorption coefficient (mL.mg−1.cm−1) of 1.80 for scFv D1.3 and 1.60 for Fab D44.1. Further details are described in Text S1.
Surface plasmon resonance
The effects of glycerol on the binding affinity of scFv D1.3 and Fab D44.1 with lysozyme were measured by surface plasmon resonance using a BIACORE 3000 system (GE Healthcare). Lysozyme was coupled to a CM5 sensor chip (GE Healthcare) using amine coupling. Antibody fragments were diluted in buffer with 0–9 molal glycerol to concentrations ranging from 10–2000 nM, and injected into the sensor chip for 7.5 minutes. Associated antibody fragments were subsequently dissociated by flowing buffer with 0–9 molal over the chip for 8 minutes. The chip was then regenerated by injecting 10 mM HCl for 30 seconds. For each glycerol concentration, association constants (KA) were determined from Scatchard analysis by measuring steady-state-responses at 6 different protein concentrations. Further details are described in Text S1.
Molecular dynamics simulations
Six independent molecular dynamics simulations were run for Fv D1.3, Fab D44.1 and lysozyme in the free and associated states in a 6 molal aqueous solution of glycerol. Protein structures for the D1.3-lysozyme and D44.1-lysozyme complexes were retrieved from PDB-structures 1VFB [63] and 1MLC [88], respectively, and crystal waters at the protein-protein interface were included in the starting structures of the associated states. For all simulations, a minimum of 10 Å between the protein and the boundary of the solvent box was kept. The CHARMM22 parameter set [89] was used to model protein atoms, water was modeled by the TIP3-model [90] and force field parameters for glycerol were taken from the carbohydrate hydrate parameters developed by Liang and Brady (the parameters are available at http://mackerell.umaryland.edu/CHARMM_ff_params.html under the link toppar_c32b1.tar.gz in the file par_all22_sugar.inp) with partial charges published by Reiling et al. [91]. Simulations were run with NAMD v2.7 [92] with constrained protein backbone coordinates for at least 160 ns, which is longer than the minimum simulation time for characterizing local protein solvation in mixed solvents [40]. Further details are described in Text S1.
Characterization of local protein solvation
Local protein solvation of D1.3, D44.1 and lysozyme in the free and associated states was analyzed from the respective MD simulations following a newly developed method for quantitative characterization of local protein solvation [40]. Local concentrations were calculated based on the solvent occupancy of a three-dimensional grid and visualized with the software VMD 1.9 [93]. Global preferential interaction coefficients 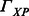 , residue-based preferential interactions coefficients
, residue-based preferential interactions coefficients 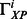 , and regional preferential interactions coefficients
, and regional preferential interactions coefficients 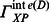 and
and 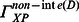 , were calculated from the average number of water and glycerol molecules within 5 Å from the corresponding protein van der Waals surfaces [40], [94]. Standard errors of preferential interaction coefficients were calculated by dividing the simulation trajectories in time blocks of increasing length followed by systematic analysis of the corresponding standard deviations [94]. Further details are described in Text S1.
, were calculated from the average number of water and glycerol molecules within 5 Å from the corresponding protein van der Waals surfaces [40], [94]. Standard errors of preferential interaction coefficients were calculated by dividing the simulation trajectories in time blocks of increasing length followed by systematic analysis of the corresponding standard deviations [94]. Further details are described in Text S1.
Supporting Information
Local concentration maps of the associated and free proteins of the D44.1-lysozyme complex. Solvent regions that are preferentially solvated by glycerol or water are colored red and blue respectively, and solvent regions near the interface region are highlighted.
(TIF)
Local preferential interaction coefficients 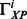 of interface residues of free (green squares) and associated (blue diamonds) proteins of the D1.3-lysozyme complex. Interface residues are indicated by grey bars on the X-axis, and
of interface residues of free (green squares) and associated (blue diamonds) proteins of the D1.3-lysozyme complex. Interface residues are indicated by grey bars on the X-axis, and 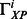 -values are only depicted for residues for which
-values are only depicted for residues for which 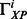 significantly differs between free and associated proteins.
significantly differs between free and associated proteins. 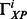 -values corresponding with Asp54 of D1.3 VH in the free and associated states are indicated by red arrows.
-values corresponding with Asp54 of D1.3 VH in the free and associated states are indicated by red arrows.
(TIF)
Local preferential interaction coefficients 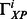 of interface residues of free and associated proteins for the lysozyme-D44.1 complex. Interface residues are indicated by grey bars on the X-axis, and
of interface residues of free and associated proteins for the lysozyme-D44.1 complex. Interface residues are indicated by grey bars on the X-axis, and 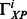 -values are only depicted for residues for which
-values are only depicted for residues for which 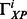 significantly differs between free and associated proteins.
significantly differs between free and associated proteins.
(TIF)
Hydration at the protein-protein interface in the D1.3-lysozyme complex. A) Waters resolved in the crystal structure. B) Snapshot of interface waters after 100 ns of simulation. C) Local concentration map of water calculated from the entire simulation.
(TIF)
Local protein solvation by glycerol molecules near the N-terminus of lysozyme in the D44.1-lysozyme complex. Image frames were rendered every 1 ns with VMD 1.9 [93] and include glycerol molecules with at least one atom within 4 Å of the N-terminus and Gln41 of lysozyme. The red wireframe demarks a solvent region with high local glycerol concentration (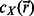 >
>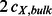 ). Note that this region is occupied by glycerol more than half the time.
). Note that this region is occupied by glycerol more than half the time.
(AVI)
Hydration of the protein-protein interface region of lysozyme in the D1.3-lysozyme complex. Out of 48 crystal waters, only three crystal waters remain at the protein-protein interface during the entire simulation (represented as colored spheres). All other water molecules within 4 Å of the protein-protein interface region are represented as blue spheres.
(AVI)
Detailed description of experimental and computational methods, and derivation of thermodynamic equations.
(DOC)
Funding Statement
This research was supported in part by the National Science Foundation through TeraGrid resources provided by Texas Advanced Computing Centre under grant number TG-MCB100058, the A*STAR Computational Resource Centre (A*CRC) and the Biomedical Research Council of A*STAR (Agency for Science, Technology and Research), Singapore. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kamerzell TJ, Esfandiary R, Joshi SB, Middaugh CR, Volkin DB (2011) Protein-excipient interactions: Mechanisms and biophysical characterization applied to protein formulation development. Advanced Drug Delivery Reviews 63: 1118–1159. [DOI] [PubMed] [Google Scholar]
- 2. Ohtake S, Kita Y, Arakawa T (2011) Interactions of formulation excipients with proteins in solution and in the dried state. Advanced Drug Delivery Reviews 63: 1053–1073. [DOI] [PubMed] [Google Scholar]
- 3. Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN (1982) Living with Water-Stress - Evolution of Osmolyte Systems. Science 217: 1214–1222. [DOI] [PubMed] [Google Scholar]
- 4. Kumar A, Venkatesu P (2012) Overview of the Stability of alpha-Chymotrypsin in Different Solvent Media. Chemical Reviews 112: 4283–4307. [DOI] [PubMed] [Google Scholar]
- 5. Roccatano D (2008) Computer simulations study of biomolecules in non-aqueous or cosolvent/water mixture solutions. Current Protein & Peptide Science 9: 407–426. [DOI] [PubMed] [Google Scholar]
- 6. Timasheff SN (1998) Control of protein stability and reactions by weakly interacting cosolvents: The simplicity of the complicated. Advances in Protein Chemistry 51: 355–432. [DOI] [PubMed] [Google Scholar]
- 7. Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS (2010) Stability of Protein Pharmaceuticals: An Update. Pharmaceutical Research 27: 544–575. [DOI] [PubMed] [Google Scholar]
- 8. Cohen FE, Kelly JW (2003) Therapeutic approaches to protein-misfolding diseases. Nature 426: 905–909. [DOI] [PubMed] [Google Scholar]
- 9. Singh LR, Poddar NK, Dar TA, Kumar R, Ahmad F (2011) Protein and DNA destabilization by osmolytes: The other side of the coin. Life Sciences 88: 117–125. [DOI] [PubMed] [Google Scholar]
- 10. Gierasch LM, Gershenson A (2009) Post-reductionist protein science, or putting Humpty Dumpty back together again. Nature Chemical Biology 5: 774–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bandyopadhyay A, Saxena K, Kasturia N, Dalal V, Bhatt N, et al. (2012) Chemical chaperones assist intracellular folding to buffer mutational variations. Nature Chemical Biology 8: 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frokjaer S, Otzen DE (2005) Protein drug stability: A formulation challenge. Nature Reviews Drug Discovery 4: 298–306. [DOI] [PubMed] [Google Scholar]
- 13. Shukla D, Schneider CP, Trout BL (2011) Molecular level insight into intra-solvent interaction effects on protein stability and aggregation. Advanced Drug Delivery Reviews 63: 1074–1085. [DOI] [PubMed] [Google Scholar]
- 14. Jungbauer A, Kaar W (2007) Current status of technical protein refolding. Journal of Biotechnology 128: 587–596. [DOI] [PubMed] [Google Scholar]
- 15. Tsumoto K, Ejima D, Kumagai I, Arakawa T (2003) Practical considerations in refolding proteins from inclusion bodies. Protein Expression and Purification 28: 1–8. [DOI] [PubMed] [Google Scholar]
- 16. Clark EDB (1998) Refolding of recombinant proteins. Current Opinion in Biotechnology 9: 157–163. [DOI] [PubMed] [Google Scholar]
- 17.Harries D, Rosgen J (2008) A practical guide on how osmolytes modulate macromolecular properties. Biophysical Tools for Biologists: Vol 1, in Vitro Techniques. pp. 679–735. [DOI] [PubMed]
- 18. Melo EP, Estrela N, Lopes C, Matias AC, Tavares E, et al. (2010) Compacting Proteins: Pros and Cons of Osmolyte-Induced Folding. Current Protein & Peptide Science 11: 744–751. [DOI] [PubMed] [Google Scholar]
- 19. Kornblatt JA, Kornblatt MJ, Hoa GHB, Mauk AG (1993) Responses of 2 Protein-Protein Complexes to Solvent Stress - Does Water Play a Role at the Interface. Biophysical Journal 65: 1059–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burgess RR, Thompson NE (2002) Advances in gentle immunoaffinity chromatography. Current Opinion in Biotechnology 13: 304–308. [DOI] [PubMed] [Google Scholar]
- 21. Goldbaum FA, Schwarz FP, Eisenstein E, Cauerhff A, Mariuzza RA, et al. (1996) The effect of water activity on the association constant and the enthalpy of reaction between lysozyme and the specific antibodies D1.3 and D44.1. Journal of Molecular Recognition 9: 6–12. [DOI] [PubMed] [Google Scholar]
- 22. Faber-Barata J, Sola-Penna M (2005) Opposing effects of two osmolytes - trehalose and glycerol - on thermal inactivation of rabbit muscle 6-phosphofructo-1-kinase. Molecular and Cellular Biochemistry 269: 203–207. [DOI] [PubMed] [Google Scholar]
- 23. Sukenik S, Politi R, Ziserman L, Danino D, Friedler A, et al. (2011) Crowding Alone Cannot Account for Cosolute Effect on Amyloid Aggregation. PLoS One 6: e15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pond SM, Pentel PR, Keyler DE, Winzor DJ (1991) Determination of the in vivo antigen-antibody affinity constant from the redistribution of desipramine in rats following administration of a desipramine-specific monoclonal antibody. Biochemical Pharmacology 41: 473–476. [DOI] [PubMed] [Google Scholar]
- 25. Laue T, Demeler B (2011) A postreductionist framework for protein biochemistry. Nature Chemical Biology 7: 331–334. [DOI] [PubMed] [Google Scholar]
- 26. Wyman J (1964) Linked functions and reciprocal effect in hemoglobin: a second look. Advances in Protein Chemistry 19: 223–286. [DOI] [PubMed] [Google Scholar]
- 27. Scatchard G (1946) Physical chemistry of protein solutions. I. Derivation of the equations for the osmotic pressure. Journal of the American Chemical Society 68: 2315–2319. [DOI] [PubMed] [Google Scholar]
- 28. Kirkwood JG, Buff FP (1951) The statistical mechanical theory of solutions. I. Journal of Chemical Physics 19: 774–777. [Google Scholar]
- 29. Casassa EF, Eisenberg H (1964) Thermodynamic analysis of multicomponent solutions. Advances in Protein Chemistry 19: 287–395. [DOI] [PubMed] [Google Scholar]
- 30. Tanford C (1969) Extension of the theory of linked functions to incorporate the effects of protein hydration. journal of molecular biology 39: 539–544. [DOI] [PubMed] [Google Scholar]
- 31. Shimizu S, Boon CL (2004) The Kirkwood-Buff theory and the effect of cosolvents on biochemical reactions. Journal of Chemical Physics 121: 9147–9155. [DOI] [PubMed] [Google Scholar]
- 32. Schurr JM, Rangel DP, Aragon SR (2005) A contribution to the theory of preferential interaction coefficients. Biophysical Journal 89: 2258–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shulgin IL, Ruckenstein E (2005) A protein molecule in an aqueous mixed solvent: Fluctuation theory outlook. Journal of Chemical Physics 123: 054909. [DOI] [PubMed] [Google Scholar]
- 34. Shimizu S, Matubayasi N (2006) Preferential hydration of proteins: A Kirkwood-Buff approach. Chemical Physics Letters 420: 518–522. [Google Scholar]
- 35. Smith PE (2006) Chemical potential derivatives and preferential interaction parameters in biological systems from Kirkwood-Buff theory. Biophysical Journal 91: 849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiao Y, Smith PE (2011) Fluctuation theory of molecular association and conformational equilibria. Journal of Chemical Physics 135: 014502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Record MT, Zhang WT, Anderson CF (1998) Analysis of effects of salts and uncharged solutes on protein and nucleic acid equilibria and processes: A practical guide to recognizing and interpreting polyelectrolyte effects, Hofmeister effects, and osmotic effects of salts. Advances in Protein Chemistry 51: 281–353. [DOI] [PubMed] [Google Scholar]
- 38. Parsegian VA, Rand RP, Rau DC (2000) Osmotic stress, crowding, preferential hydration, and binding: A comparison of perspectives. Proceedings of the National Academy of Sciences of the United States of America 97: 3987–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xie GF, Timasheff SN (1997) The thermodynamic mechanism of protein stabilization by trehalose. Biophysical Chemistry 64: 25–43. [DOI] [PubMed] [Google Scholar]
- 40. Vagenende V, Trout BL (2012) Quantitative characterization of local protein solvation to predict solvent effects on protein structure. Biophysical Journal 103: 1354–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schreiber G, Haran G, Zhou HX (2009) Fundamental Aspects of Protein-Protein Association Kinetics. Chemical Reviews 109: 839–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baynes BM, Trout BL (2003) Proteins in mixed solvents: A molecular-level perspective. Journal of Physical Chemistry B 107: 14058–14067. [Google Scholar]
- 43. Colombo MF, Rau DC, Parsegian VA (1992) PROTEIN SOLVATION IN ALLOSTERIC REGULATION - A WATER EFFECT ON HEMOGLOBIN. Science 256: 655–659. [DOI] [PubMed] [Google Scholar]
- 44. Garner MM, Rau DC (1995) WATER RELEASE ASSOCIATED WITH SPECIFIC BINDING OF GAL REPRESSOR. EMBO Journal 14: 1257–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xavier KA, Shick KA, SmithGill SJ, Willson RC (1997) Involvement of water molecules in the association of monoclonal antibody HyHEL-5 with bobwhite quail lysozyme. Biophysical Journal 73: 2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Poon J, Bailey M, Winzor DJ, Davidson BE, Sawyer WH (1997) Effects of molecular crowding on the interaction between DNA and the Escherichia coli regulatory protein TyrR. Biophysical Journal 73: 3257–3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vossen KM, Wolz R, Daugherty MA, Fried MG (1997) Role of macromolecular hydration in the binding of the Escherichia coli cyclic AMP receptor to DNA. Biochemistry 36: 11640–11647. [DOI] [PubMed] [Google Scholar]
- 48. Robinson CR, Sligar SG (1998) Changes in solvation during DNA binding and cleavage are critical to altered specificity of the EcoRI endonuclease. Proceedings of the National Academy of Sciences of the United States of America 95: 2186–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fried MG, Stickle DF, Smirnakis KV, Adams C, MacDonald D, et al. (2002) Role of hydration in the binding of lac repressor to DNA. Journal of Biological Chemistry 277: 50676–50682. [DOI] [PubMed] [Google Scholar]
- 50. Patel CN, Noble SM, Weatherly GT, Tripathy A, Winzor DJ, et al. (2002) Effects of molecular crowding by saccharides on alpha-chymotrypsin dimerization. Protein Science 11: 997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chopra S, Dooling RM, Horner CG, Howell EE (2008) A balancing act between net uptake of water during dihydrofolate binding and net release of water upon NADPH binding in R67 dihydrofolate reductase. Journal of Biological Chemistry 283: 4690–4698. [DOI] [PubMed] [Google Scholar]
- 52. Grubbs J, Rahmanian S, DeLuca A, Padmashali C, Jackson M, et al. (2011) Thermodynamics and Solvent Effects on Substrate and Cofactor Binding in Escherichia coli Chromosomal Dihydrofolate Reductase. Biochemistry 50: 3673–3685. [DOI] [PubMed] [Google Scholar]
- 53. Sidorova NY, Muradymov S, Rau DC (2011) Solution parameters modulating DNA binding specificity of the restriction endonuclease EcoRV. FEBS Journal 278: 2713–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Duff MR Jr, Grubbs J, Serpersu E, Howell EE (2012) Weak Interactions between Folate and Osmolytes in Solution. Biochemistry 51: 2309–2318. [DOI] [PubMed] [Google Scholar]
- 55. Gekko K, Timasheff SN (1981) Mechanism of Protein Stabilization by Glycerol - Preferential Hydration in Glycerol-Water Mixtures. Biochemistry 20: 4667–4676. [DOI] [PubMed] [Google Scholar]
- 56. Courtenay ES, Capp MW, Anderson CF, Record MT (2000) Vapor pressure osmometry studies of osmolyte-protein interactions: Implications for the action of osmoprotectants in vivo and for the interpretation of “osmotic stress” experiments in vitro. Biochemistry 39: 4455–4471. [DOI] [PubMed] [Google Scholar]
- 57. Schneider CP, Trout BL (2009) Investigation of Cosolute-Protein Preferential Interaction Coefficients: New Insight into the Mechanism by Which Arginine Inhibits Aggregation. Journal of Physical Chemistry B 113: 2050–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vagenende V, Yap MGS, Trout BL (2009) Mechanisms of Protein Stabilization and Prevention of Protein Aggregation by Glycerol. Biochemistry 48: 11084–11096. [DOI] [PubMed] [Google Scholar]
- 59. Schellman JA (2003) Protein stability in mixed solvents: A balance of contact interaction and excluded volume. Biophysical Journal 85: 108–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hwang H, Pierce B, Mintseris J, Janin J, Weng Z (2008) Protein-protein docking benchmark version 3.0. Proteins-Structure Function and Bioinformatics 73: 705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ruvinsky AM, Kirys T, Tuzikov AV, Vakser IA (2011) Side-Chain Conformational Changes upon Protein-Protein Association. Journal of Molecular Biology 408: 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Quiocho FA, Wilson DK, Vyas NK (1989) SUBSTRATE-SPECIFICITY AND AFFINITY OF A PROTEIN MODULATED BY BOUND WATER-MOLECULES. Nature 340: 404–407. [DOI] [PubMed] [Google Scholar]
- 63. Bhat TN, Bentley GA, Boulot G, Greene MI, Tello D, et al. (1994) Bound Water-Molecules and Conformational Stabilization Help Mediate an Antigen-Antibody Association. Proceedings of the National Academy of Sciences of the United States of America 91: 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li Z, Lazaridis T (2007) Water at biomolecular binding interfaces. Physical Chemistry Chemical Physics 9: 573–581. [DOI] [PubMed] [Google Scholar]
- 65. Abel R, Young T, Farid R, Berne BJ, Friesner RA (2008) Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. Journal of the American Chemical Society 130: 2817–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Michel J, Tirado-Rives J, Jorgensen WL (2009) Energetics of Displacing Water Molecules from Protein Binding Sites: Consequences for Ligand Optimization. Journal of the American Chemical Society 131: 15403–15411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Baron R, Setny P, McCammon JA (2010) Water in Cavity-Ligand Recognition. Journal of the American Chemical Society 132: 12091–12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hummer G (2010) MOLECULAR BINDING Under water's influence. Nature Chemistry 2: 906–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ahmad M, Gu W, Geyer T, Helms V (2011) Adhesive water networks facilitate binding of protein interfaces. Nature Communications 2: 261. [DOI] [PubMed] [Google Scholar]
- 70. Wereszczynski J, McCammon JA (2012) Statistical mechanics and molecular dynamics in evaluating thermodynamic properties of biomolecular recognition. Quarterly Reviews of Biophysics 45: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Michel J, Tirado-Rives J, Jorgensen WL (2009) Prediction of the Water Content in Protein Binding Sites. Journal of Physical Chemistry B 113: 13337–13346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cappel D, Wahlstroem R, Brenk R, Sotriffer CA (2011) Probing the Dynamic Nature of Water Molecules and Their Influences on Ligand Binding in a Model Binding Site. Journal of Chemical Information and Modeling 51: 2581–2594. [DOI] [PubMed] [Google Scholar]
- 73. Rossato G, Ernst B, Vedani A, Smiesko M (2011) AcquaAlta: A Directional Approach to the Solvation of Ligand-Protein Complexes. Journal of Chemical Information and Modeling 51: 1867–1881. [DOI] [PubMed] [Google Scholar]
- 74. Wang L, Berne BJ, Friesner RA (2011) Ligand binding to protein-binding pockets with wet and dry regions. Proceedings of the National Academy of Sciences of the United States of America 108: 1326–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rodier F, Bahadur RP, Chakrabarti P, Janin J (2005) Hydration of protein-protein interfaces. Proteins-Structure Function and Bioinformatics 60: 36–45. [DOI] [PubMed] [Google Scholar]
- 76. Ahmed MH, Spyrakis F, Cozzini P, Tripathi PK, Mozzarelli A, et al. (2011) Bound Water at Protein-Protein Interfaces: Partners, Roles and Hydrophobic Bubbles as a Conserved Motif. PLoS One 6: e24712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li Z, He Y, Wong L, Li J (2012) Progressive dry-core-wet-rim hydration trend in a nested-ring topology of protein binding interfaces. BMC Bioinformatics 13: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Meenan NAG, Sharma A, Fleishman SJ, MacDonald CJ, Morel B, et al. (2010) The structural and energetic basis for high selectivity in a high-affinity protein-protein interaction. Proceedings of the National Academy of Sciences of the United States of America 107: 10080–10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cohen GH, Silverton EW, Padlan EA, Dyda F, Wibbenmeyer JA, et al. (2005) Water molecules in the antibody-antigen interface of the structure of the Fab HyHEL-5-lysozyme complex at 1.7 angstrom resolution: comparison with results from isothermal titration calorimetry. Acta Crystallographica Section D-Biological Crystallography 61: 628–633. [DOI] [PubMed] [Google Scholar]
- 80. Tanford C (1964) Isothermal unfolding of globular proteins in aqueous urea solutions. Journal of the American Chemical Society 86: 2050–2059. [Google Scholar]
- 81. Levy ED, Pereira-Leal JB, Chothia C, Teichmann SA (2006) 3D complex: A structural classification of protein complexes. PLoS Computational Biology 2: 1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xie L, Bourne PE (2005) Functional coverage of the human genome by existing structures, structural genomics targets, and homology models. PLoS Computational Biology 1: 222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vagenende V, Han AX, Mueller M, Trout BL (2012) Protein-Associated Cation Clusters in Aqueous Arginine Solutions and Their Effects on Protein Stability and Size. ACS Chemical Biology 8: 416–422. [DOI] [PubMed] [Google Scholar]
- 84. Ma Q, Fan J-B, Zhou Z, Zhou B-R, Meng S-R, et al. (2012) The Contrasting Effect of Macromolecular Crowding on Amyloid Fibril Formation. PLoS One 7: e36288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jiao M, Li H-T, Chen J, Minton AP, Liang Y (2010) Attractive Protein-Polymer Interactions Markedly Alter the Effect of Macromolecular Crowding on Protein Association Equilibria. Biophysical Journal 99: 914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rosen J, Kim YC, Mittal J (2011) Modest Protein-Crowder Attractive Interactions Can Counteract Enhancement of Protein Association by Intermolecular Excluded Volume Interactions. Journal of Physical Chemistry B 115: 2683–2689. [DOI] [PubMed] [Google Scholar]
- 87. Elcock AH (2010) Models of macromolecular crowding effects and the need for quantitative comparisons with experiment. Current Opinion in Structural Biology 20: 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Braden BC, Souchon H, Eisele JL, Bentley GA, Bhat TN, et al. (1994) 3-dimensional structures of the free and the antigen-complex Fab from monoclonal antilysozyme antibody - D44.1. Journal of Molecular Biology 243: 767–781. [DOI] [PubMed] [Google Scholar]
- 89. MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. Journal of Physical Chemistry B 102: 3586–3616. [DOI] [PubMed] [Google Scholar]
- 90. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of Simple Potential Functions for Simulating Liquid Water. Journal of Chemical Physics 79: 926–935. [Google Scholar]
- 91. Reiling S, Schlenkrich M, Brickmann J (1996) Force field parameters for carbohydrates. Journal of Computational Chemistry 17: 450–468. [Google Scholar]
- 92. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, et al. (2005) Scalable molecular dynamics with NAMD. Journal of Computational Chemistry 26: 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Humphrey W, Dalke A, Schulten K (1996) VMD: Visual molecular dynamics. Journal of Molecular Graphics 14: 33–38. [DOI] [PubMed] [Google Scholar]
- 94. Vagenende V, Yap MGS, Trout BL (2009) Molecular Anatomy of Preferential Interaction Coefficients by Elucidating Protein Solvation in Mixed Solvents: Methodology and Application for Lysozyme in Aqueous Glycerol. Journal of Physical Chemistry B 113: 11743–11753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Local concentration maps of the associated and free proteins of the D44.1-lysozyme complex. Solvent regions that are preferentially solvated by glycerol or water are colored red and blue respectively, and solvent regions near the interface region are highlighted.
(TIF)
Local preferential interaction coefficients of interface residues of free (green squares) and associated (blue diamonds) proteins of the D1.3-lysozyme complex. Interface residues are indicated by grey bars on the X-axis, and -values are only depicted for residues for which significantly differs between free and associated proteins. -values corresponding with Asp54 of D1.3 VH in the free and associated states are indicated by red arrows.
(TIF)
Local preferential interaction coefficients of interface residues of free and associated proteins for the lysozyme-D44.1 complex. Interface residues are indicated by grey bars on the X-axis, and -values are only depicted for residues for which significantly differs between free and associated proteins.
(TIF)
Hydration at the protein-protein interface in the D1.3-lysozyme complex. A) Waters resolved in the crystal structure. B) Snapshot of interface waters after 100 ns of simulation. C) Local concentration map of water calculated from the entire simulation.
(TIF)
Local protein solvation by glycerol molecules near the N-terminus of lysozyme in the D44.1-lysozyme complex. Image frames were rendered every 1 ns with VMD 1.9 [93] and include glycerol molecules with at least one atom within 4 Å of the N-terminus and Gln41 of lysozyme. The red wireframe demarks a solvent region with high local glycerol concentration (>). Note that this region is occupied by glycerol more than half the time.
(AVI)
Hydration of the protein-protein interface region of lysozyme in the D1.3-lysozyme complex. Out of 48 crystal waters, only three crystal waters remain at the protein-protein interface during the entire simulation (represented as colored spheres). All other water molecules within 4 Å of the protein-protein interface region are represented as blue spheres.
(AVI)
Detailed description of experimental and computational methods, and derivation of thermodynamic equations.
(DOC)