

Abstract
Munro’s microabscesses contain polymorphonuclear leukocytes and form specifically in the epidermis of psoriasis patients. The mechanism whereby the neutrophils are recruited into the epidermis is poorly understood. Using a combination of human and mouse primary keratinocyte cell cultures and the imiquimod-induced psoriasis-like mouse model of skin inflammation we explored the role of interleukin-1 (IL-1) signaling in microabscess formation. In vitro imiquimod stimulated production of IL-1α and neutrophil recruiting chemokines. Imiquimod activated chemokine expression was dependent upon adenosine signaling and independent of IL-1α and IL-1 receptor type 1 (IL-1R1); nevertheless, IL-1α could enhance chemokine expression initiated by imiquimod. Topical application of imiquimod in vivo led to epidermal microabscess formation, acanthosis and increased IL-1α and chemokine expression in the skin of wild type mice. However, in IL-1R1 deficient mice these responses were either absent or dramatically reduced. These results demonstrate that IL-1α and IL-1R1 signaling is essential for microabscess formation, neutrophil recruiting chemokine expression and acanthosis in psoriasis-like skin inflammation induced by imiquimod.
INTRODUCTION
Munro’s microabscesses are a characteristic hallmark of psoriasis pathology (Munro, 1898; Steffen, 2002). These sites of inflammation contain polymorphonuclear leukocytes and form specifically in the epidermal layer of the skin (Munro, 1898; Steffen, 2002). Given the localization of these microabscesses within the epidermis keratinocytes are likely to play a role in their development; however, the mechanism whereby this interesting phenomenon develops is incompletely characterized. Psoriasis is a chronic inflammatory condition of the skin. In addition to the presence of Munro’s microabscesses the disease is characterized by acanthosis, hyperkeratosis, and parakeratosis of the epidermis as well as a mixed inflammatory infiltrate and increased vascularization in the dermis. Genome-wide expression profiling has identified several chemokines, e.g. CXCL1 and interleukin-8 (IL-8), which are expressed at higher levels in psoriatic skin compared to normal skin (Bowcock et al., 2001; Lew et al., 2004; Zhou et al., 2003).
IL-1 represents the two pleiotropic cytokines IL-1α and IL-1β of which IL-1α is the predominant form expressed in keratinocytes ((Lian et al., 2012; Olaru and Jensen, 2010b) and refs. therein). Extracellular IL-1α and IL-1β activate gene expression via the IL-1 receptor type I (IL-1R1) and a complex signaling cascade leading to activation of transcription factors such as NF-κB and AP-1 (Jensen, 2010). Interestingly, the gene expression profiles of skin biopsies from psoriasis patients strongly resemble that of the keratinocyte transcriptome induced in response to IL-1α (Mee et al., 2007; Yano et al., 2008). Previously IL-1α and IL-1β have been linked to several of the phenotypes observed in psoriasis, e.g. proliferation and cell differentiation (see (Sanmiguel et al., 2009) for refs.), but their potential role in formation of Munro’s microabscesses have not been explored.
Aldara (imiquimod cream) is FDA approved for treatment of human papillomavirus genital warts, actinic keratoses and basal cell carcinomas. The drug has been known for some time to cause psoriasis-like disease in some patients and trigger outbreaks in patients with psoriasis (Fanti et al., 2006; Gilliet et al., 2004; Patel et al., 2011; Rajan and Langtry, 2006; van der Fits et al., 2009; Wu et al., 2004). In 2009 van der Fitz et al. reported that application of imiquimod cream to the back skin of mice caused phenotypes with remarkable resemblance to human psoriasis pathology including the presence of Munro’s microabscesses (van der Fits et al., 2009). It was further established that the disease in mice was dependent upon the IL-23•IL-17 axis (van der Fits et al., 2009), which is now widely recognized for its involvement in human psoriasis (Tonel et al., 2010; Zaba et al., 2009). Subsequent studies of the model using microarray expression analyses have revealed a striking overlap with the expression profiles of human psoriatic skin (Swindell et al., 2011). Imiquimod is best known as a ligand for TLR7 and TLR8; however, the drug can also modulate an adenosine receptor signaling pathway (Schön et al., 2006) and has cytotoxic effects upon cells at high concentrations (reviewed in (Schön and Schön, 2007)). We recently characterized the role of IL-1α as an intermediate signaling molecule between TLR2 and neutrophil targeting chemokine expression in keratinocytes (Olaru and Jensen, 2010b) and therefore hypothesized that one or more forms of IL-1 may play a role in Munro’s microabscess formation. Here we demonstrate that in the imiquimod-induced psoriasis-like skin disease mouse model Munro’s microabscess formation is dependent upon IL-1R1 signaling. Furthermore, we show that keratinocytes express neutrophil targeting chemokines in response to imiquimod. Activated keratinocytes also release IL-1α, and IL-1α can enhance chemokine expression in response to imiquimod.
RESULTS
Human and mouse keratinocytes produce neutrophil targeting chemokines in response to imiquimod in vitro
Since Munro’s microabscesses specifically occur in the epidermis comprised of keratinocytes we wished to identify signaling pathways governing the mechanism whereby keratinocytes recruit neutrophils to these sites of inflammation. The two neutrophil targeting chemokines CXCL1 and IL-8 have been reported to be overexpressed in psoriatic skin (Bowcock et al., 2001; Lew et al., 2004; Zhou et al., 2003). Our previous studies of the role of keratinocytes as the first responders to stress and injury have established that human keratinocytes produce the neutrophil chemotactic CXCL1, CXCL2 and IL-8 when sensing TLR ligands (Olaru and Jensen, 2010b) and therefore we focused our analyses on this subset of chemokines. Initial studies established that at concentrations up to 50 μg/ml, imiquimod induced expression of the CXCL1, CXCL2 and IL-8 mRNAs in concentration dependent manners (Figure 1, upper panels). The gene expression appeared to be biphasic as decreases in mRNA levels were observed between the 3- and 24-hour time-points (Figure 1, upper panels). Expression of the chemokines was also concentration dependent and levels in the medium increased throughout the duration of the experiment (Figure 1, lower panels). Similar observations were made for CXCL1 and CXCL2 using mouse primary keratinocytes (data not shown, mice do not have IL-8).
Figure 1. Expression of neutrophil targeting chemokines is elevated in imiquimod treated human primary keratinocytes in vitro.
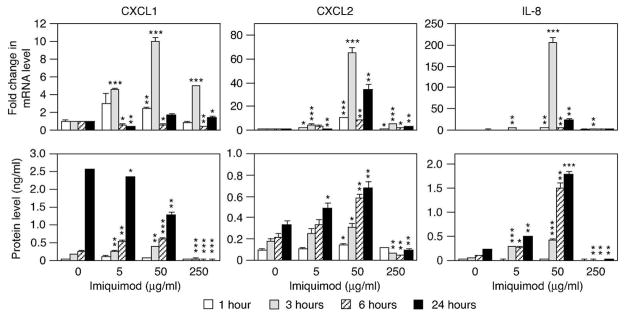
Cells were treated with medium only or imiquimod and RNA and conditioned medium collected as indicated. Levels of CXCL1, CXCL2 and IL-8 mRNA (top row) and protein (bottom row) were determined using real-time RT-PCR and ELISA, respectively. Levels of mRNA are represented as fold change (mean ± SD) compared to medium only treated samples at the same time-point. Chemokine levels are shown as protein concentration in the conditioned medium (mean ± SD). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to medium only at the same time-point).
While chemokine expression was observed with lower levels of imiquimod, at higher concentrations imiquimod was clearly toxic to cells as protein levels actually significantly decreased when cells were treated with imiquimod (Figure 1, 250 μg/ml). This is in agreement with previously reported cytotoxicity of imiquimod (Schön and Schön, 2007).
IL-1α is released by imiquimod treated human and mouse keratinocytes in vitro
Previously we reported that keratinocytes express and release IL-1α and IL-1β in response to TLR ligands (Olaru and Jensen, 2010b). We therefore examined IL-1α and IL-1β expression by keratinocytes treated with imiquimod. As described above, the highest levels of imiquimod led to reduced mRNA and protein expression (Figure 2a). This is likely due to the cytotoxic effect of imiquimod and relatively short half-lives of the IL-1α and IL-1β mRNAs (Kang et al., 1996). At lower concentrations of imiquimod both cytokine mRNAs were induced by imiquimod in concentration and time-dependent manners (Figure 2a (human keratinocytes) and data not shown (mouse keratinocytes)). Increased levels of the IL-1α cytokine could be detected in the culture medium as early as 3 hours after addition of imiquimod (Figure 2a). This appears to be earlier than the loss of membrane potential as determined by measuring lactate dehydrogenase LDH (Figure 2b). Furthermore, reduced cell density was observed after 24 hours (data not shown). IL-1β could not be detected in conditioned medium from the above keratinocyte cultures (data not shown).
Figure 2. Human primary keratinocytes produce and release elevated levels of IL-1α in response to imiquimod in vitro.

Cells were treated with medium or imiquimod as indicated. (a) Expression of IL-1α and IL-1β mRNA and protein was examined using real-time RT-PCR and ELISA, respectively. Levels of mRNA are represented as fold change (mean ± SD) compared to medium only treated samples at the same time-points. IL-1 cytokine levels are shown as protein concentration in the culture conditioned medium (mean ± SD). (b) Levels of LDH in the culture medium were determined and represented as fold change (means ± SD) compared to medium only treated control conditioned medium. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to medium only at the same time-point).
Imiquimod induced chemokine expression is independent of IL-1α signaling in human and mouse keratinocytes in vitro
Our previous studies have established that IL-1α is an intermediate signaling molecule between TLR2 and the neutrophil targeting chemokines CXCL1, CXCL2 and IL-8 (Olaru and Jensen, 2010b). Therefore we hypothesized that a similar mechanism would be responsible for the imiquimod induced chemokine expression, i.e. that chemokine expression would be dependent upon IL-1 release from cells and subsequent engagement of the IL-1 signaling receptor IL-1R1. IL-1 and poly(I:C) induced chemokine expression was significantly reduced when human keratinocytes were co-treated with an IL-1α neutralizing antibody compared to cells receiving an isotype matched control antibody (Figure 3a). Surprisingly, cells treated with imiquimod and the IL-1α neutralizing antibody responded equally well as control cells treated with isotype matched immunoglobulin and imiquimod (Figure 3a). Interestingly, the constitutive (medium only) production of CXCL1, CXCL2 and IL-8 was reduced in the presence of the IL-1α neutralizing antibody when cultures were continued for 24 hours (Figure 3b). The role of IL-1R1 signaling was further explored in wild type and IL-1R1 knockout (KO) mouse primary keratinocytes. Constitutive chemokine production by IL-1R1 KO cells was lower than that observed from wild type cells (Figure 3c, medium and Supplementary Figure S1). Interestingly, the fold increase in chemokine expression in response to imiquimod was not significantly different between the wild type (2- and 5-fold changes in CXCL1 and CXCL2, respectively) and IL-1R1 KO cells (3- and 8-fold changes in CXCL1 and CXCL2, respectively, Figure 3c). These observations are in agreement with the experiments involving human cells and IL-1α neutralizing antibodies (Figure 3a–b). Overall, the data suggest that cultured keratinocytes can express neutrophil targeting chemokines in response to imiquimod independent of their capacity to signal through IL-1R1 expression. However, IL-1 signaling appears to be important for the constitutive baseline chemokine production.
Figure 3. Imiquimod and IL-1 have independent and additive effects upon chemokine expression in vitro.

(a) Primary human keratinocytes were pre-treated for 30 min with neutralizing anti-human IL-1α IgG (black bars) or isotype matched IgG (white bars) before the addition of medium only, 50 μg/ml imiquimod, 10 ng/ml human IL-1α or 25 μg/ml poly(I:C). Chemokine expression was examined using ELISA (mean ± SD) after 6 hours. (b) Human keratinocytes were incubated with neutralizing anti-human IL-1α IgG or isotype matched IgG for 24 hours after which chemokine expression was examined using ELISA. (c) Mouse primary keratinocytes isolated from wild type (WT, white bars) or IL-1R1 KO mice (KO, black bars) were treated with medium only, 50 μg/ml imiquimod or 10 ng/ml mouse IL-1α for 6 hours. Levels of CXCL1 and CXCLL2 were determined using ELISA (mean ± SD). # Low levels of CXCL2 were detected. Primary human (d) and mouse (e) keratinocytes were treated with medium only, imiquimod, IL-1α (species matched) or a combination of imiquimod and IL-1α for 6 hours and chemokine expression evaluated using ELISA (mean ± SD). (f–g) Human primary keratinocytes were pre-treated with 10 μM forskolin, 80 μM CV 1808 or vehicle control for 30 min before addition of medium only (white bars) or 50 μg/ml imiquimod (black bars). Protein secretion was determined using ELISA (mean ± SD). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to medium only (a–e), equivalent vehicle control (f–g) or as indicated).
IL-1α and imiquimod have additive effects upon chemokine expression in human and mouse keratinocytes in vitro
Our earlier studies have demonstrated that IL-1 can cooperate with interferon-γ and tumor necrosis factor-α to drive chemokine expression in keratinocytes (Sanmiguel et al., 2009). We therefore conducted experiments in which cells were treated with medium, imiquimod alone, IL-1α alone or imiquimod with IL-1α. Curiously, these studies revealed that the single agents (imiquimod versus IL-1α) affected the different chemokines in distinct manners (Figure 3d–e). While CXCL1 was most potently induced by IL-1α, IL-8 was more dramatically regulated by imiquimod (Figure 3d) and CXCL2 appeared to be equally regulated by the two agents (Figure 3e). This may suggest that expression of the chemokines is activated through different mechanisms and imiquimod and IL-1α modulate these in distinct ways.
Further analyses of co-stimulation of cells with both imiquimod and IL-1α (Figure 3d–e) revealed that CXCL1, CXCL2 and IL-8 expression was enhanced compared to cells treated with a single agent. The enhancement appeared to be additive as, for example, 125 and 225 pg/ml of CXCL1 was expressed in response to imiquimod and IL-1α, respectively, and 345 pg/ml was expressed when imiquimod and IL-1α were added together (Figure 3d). The additive effects of imiquimod and IL-1α upon chemokine expression may further suggest that these activators of gene expression act through independent signaling pathways.
Chemokine, but not IL-1α, secretion is induced by imiquimod via modulation of an adenosine receptor/adenylate cyclase pathway in human keratinocytes in vitro
It has previously been reported that imiquimod can activate gene expression in epithelial cells, including keratinocytes, via its association with adenosine receptors and modulation of downstream adenylate cyclase activity (Kan et al., 2012; Schön et al., 2006). We found that ligands for the adenosine A2 receptor (CV 1808) and adenylate cyclase (forskolin) significantly reduced imiquimod induced chemokine expression compared to vehicle control treated cells (Figure 3f). One exception from this was CV 1808 which enhanced expression of CXCL1 (Figure 3f). This may suggest involvement of multiple adenosine receptors. The ligands affected constitutive expression of chemokines in some experiments (CXCL1 and CXCL2, Figure 3f); however, this effect was not consistently observed in all experiments (data not shown). Interestingly, IL-1α expression was not affected by either CV 1808 or forskolin (Figure 3g). This suggests that imiquimod activates several different pathways with differential effects upon cytokine and chemokine expression.
Topical application of imiquimod in vivo stimulates release of IL-1α and IL-1β from mouse skin
To examine the in vivo role of IL-1 signaling and neutrophil targeting chemokines such as CXCL1 and CXCL2 in formation of Munro’s microabscesses we employed the imiquimod-induced psoriasis-like mouse model of skin inflammation (van der Fits et al., 2009). It has previously been shown that aged (> 18 months) mice produce elevated levels of IL-1α in the skin following a 7-day regiment of imiquimod application (Barland et al., 2004). We first examined secretion of IL-1α and IL-1β from explanted skin collected from young wild type mice treated with imiquimod for 2 days (Figure 4a–b). Levels of IL-1α released from imiquimod treated skin were significantly higher than that from untreated mice (Figure 4b). A modest, but statistically significant, increase in IL-1β was also observed after 4 days; however, levels of IL-1β were dramaticall y lower than the IL-1α levels (Figure 4b). This opens the possibility that extracellular IL-1 is involved in initiating pathology in imiquimod-induced skin inflammation.
Figure 4. IL-1α• IL-1R1 signaling is involved in formation of Munro’s microabscesses in vivo.

(a) Timeline for imiquimod-induced psoriasis-like skin inflammation model. Denuded mice were treated with imiquimod on days 1 and 2 (arrowheads) and skin collected (arrows) either 6 hours post-treatment on day 2 or 2 days post-treatment (day 4). Untreated skin from control mice was collected on day 1. (b) Levels of IL-1α (black symbols) and IL-1β (white symbols) released from wild type explanted skin were measured using ELISA. Circles, triangles and diamonds indicate untreated, day 2 or day 4 time points, respectively. (c–d) Wild type (WT) and IL-1R1 KO (KO) mice were treated with imiquimod on days 1 and 2 and skin collected on day 4. Control untreated skin was collected on day 1 (n = 3–4 per group). (c) Skin sections were examined using hematoxylin and eosin (H&E) staining or immunohistochemistry for the granulocyte specific antigen Ly-6G/Ly-6C. Arrows indicate positions of Munro’s microabscesses at the top of the epidermis. Arrowhead indicates presence of intradermal granulocytes. Bars indicate 100 μm. (d) Wild type (WT, black symbols) and IL-1R1 KO (KO, white symbols) mice were treated with imiquimod (squares) or left untreated (circles). Levels of IL-1α and IL-1β released from the skin were examined using ELISA. *, P < 0.05; ***, P < 0.001.
Epidermal microabscess formation in mice is dependent upon IL-1R1 in vivo
Since imiquimod treated skin released elevated levels of IL-1 (Figure 4b), we next examined the role of IL-1 signaling in formation of Munro’s microabscesses in wild type and IL-1R1 KO mice. Following imiquimod treatment the presence of cells in the stratum corneum of wild type mice could be identified by H&E staining (Figure 4c, WT, day 4, H&E, arrow). These cells were not present in untreated mice (Figure 4c, WT, day 1) or IL-1R1 KO mice (Figure 4c, KO, day 4, H&E). Further immunohistochemical analyses using the granulocyte specific cell marker Ly-6G/Ly-6C revealed that these epidermis/stratum corneum infiltrating cells were neutrophils (Figure 4c, day 4, Ly-6G/Ly-6C, arrows). Granulocytes could also be identified in the dermis of imiquimod treated wild type mice (Figure 4c, arrowhead), but not in IL-1R1 KO mice. These observations establish that IL-1R1 signaling is required for the formation of Munro’s microabscesses.
IL-1R1 signaling regulates additional psoriasis related phenotypes in mice in vivo
Further examination of imiquimod treated skin revealed that IL-1R1 KO mice had reduced acanthosis and parakeratosis compared to wild type mice (Figure 4c). Reduced proliferation of the IL-1R1 KO basal keratinocytes was confirmed through detection of Proliferating Cell Nuclear Antigen (Supplementary Figure S2). Antibodies for keratin 1 did not stain the bottom two layers of proliferating cells in wild type mice; however, in IL-1R1 only a single layer of cells remained unstained (Supplementary Figure S2). IL-1R1 deficiency did not affect vascularization of the dermis (Supplementary Figure S2). The numbers of dermal macrophages and epidermal T cells also appeared similar in wild type and IL-1R1 KO mice (Supplementary Figure S2).
It has previously been shown that IL-1 plays an essential role in co-stimulating, in conjunction with IL-23, dermal γδ T cells to produce IL-17 (Cai et al., 2011). We observed a few dermal T cells in wild type mice and possibly fewer cells were found in IL-1R1 KO mice (Supplementary Figure S2). However, the level of dermal infiltration by T cells was too low to make a definitive conclusion regarding differences between mouse strains. It should be noted that we here examined an early stage of skin inflammation in the employed mouse model. Hence, the observed inflammation was less than that observed when mice are treated for 5–6 days as reported previously (van der Fits et al., 2009) and our unpublished observations. Overall, IL-1R1 appears to differentially regulate several, but not all, psoriasiform phenotypes in the imiquimod-induced skin inflammation model.
IL-1α and IL-1β expression is regulated via IL-1R1 in vivo in mice and in vitro in human keratinocytes
Previous array studies performed by others and in our laboratory have suggested that IL-1 can stimulate expression of the IL-1α and IL-1β mRNAs in keratinocytes; however, the array data were not validated (Sanmiguel et al., 2009; Yano et al., 2008). Here we found that following imiquimod treatment secreted levels of IL-1α and IL-1β were lower in IL-1R1 KO mice than in wild type mice (Figure 4d). There were no significant differences between constitutive Interestingly, levels production of the IL-1 cytokines in untreated mice (Figure 4d). of IL-1β were not induced by imiquimod in IL-1R1 KO mice (Figure 4d). This could suggest that the source of IL-1β protein found in vivo, but not in vitro, is the neutrophils that are poorly recruited to the skin in IL-1R1 KO mice. In vitro experiments confirmed that IL-1 treated keratinocytes express elevated levels of both IL-1α and IL-1β mRNAs (Supplementary Figure S3). To summarize, IL-1R1 signaling plays an important role in regulating expression of IL-1 cytokines.
IL-1R1 KO mice produce reduced levels of CXCL1 and CXCL2 in vivo
To further explore the in vivo mechanism whereby IL-1 signaling may facilitate recruitment of neutrophils to the epidermis, we measured levels of CXCL1 and CXCL2 secreted from untreated and imiquimod treated wild type and IL-1R1 KO skin. Levels of both CXCL1 and CXCL2 were up-regulated when wild type mice were treated with imiquimod (Figure 5a, black symbols). This increase in chemokine expression was not observed in IL-1R1 KO mice (Figure 5a, white symbols). Interestingly, CXCL1 and CXCL2 levels in untreated IL-1R1 KO mice were significantly lower than those observed in wild type mice (Figure 5a, circles). This is in agreement with our in vitro observations above demonstrating that neutralization of IL-1α or elimination of IL-1R1 reduces the constitutive production of chemokines (Figure 3b–c). Overall, these experiments establish that IL-1•IL-1R1 signaling plays an essential role in regulation of chemokine expression in vivo.
Figure 5. IL-1R1 signaling is essential for constitutive and imiquimod induced chemokine expression required for granulocyte recruitment in vivo.

(a) Wild type (WT, black symbols) and IL-1R1 KO (KO, white symbols) mice were treated with imiquimod (squares) or left untreated (circles) as described in Figure 4c (n = 4). Levels of CXCL1 and CXCL2 released from the skin were examined using ELISA. *, P < 0.05; ***, P < 0.001. (b) IL-1R1 KO mice were treated with imiquimod containing, PBS, CXCL1 or CXCL2 (25 ng protein/mouse) following the schedule described in Figure 4c. Neutrophils were identified using immunohistochemistry for Ly-6G/Ly-6C. Arrows indicate positions of Munro’s microabscesses in the epidermis. Bars indicate 100 μm.
Topically applied CXCL1 and CXCL2 can rescue Munro’s microabscess formation in vivo in IL-1R1 KO mice
Since we observed that CXCL1 and CXXCL2 levels were significantly lower in IL-1R1 KO mice than in wild type mice we tested whether addition of recombinant chemokines to the imiquimod cream could rescue the recruitment of neutrophils to the epidermis. Based on the data presented in Figure 5a we estimated that the constitutive chemokine production in wild type mice was 8 ng/cm2 and we therefore treated the mice with physiologically relevant 4 ng of chemokine per cm2. Ly-6G/Ly-6C positive cells were observed in the upper layers of the epidermis of IL-1R1 KO mice treated with either CXCL1 or CXCL2 in combination with imiquimod, but not in mice treated with imiquimod and PBS (Figure 5b). This suggests that the levels of CXCL1 and CXCL2 chemokines produced by IL-1R1 KO are insufficient to recruit neutrophils to the epidermis and further establishes IL-1 signaling as an essential component of this process.
DISCUSSION
Psoriasis lesions typically contain Munro’s microabscesses in the epidermis (Munro, 1898; Steffen, 2002). We have here established that in the imiquimod-induced psoriasis-like skin inflammation mouse model formation of neutrophil containing microabscesses is dependent upon IL-1R1 (Figure 4c). Our data also demonstrate that IL-1 signaling through IL-1R1 regulates both constitutive and imiquimod-induced chemokine expression involved in neutrophil recruitment in vivo (Figures 4–5). Neutrophils express IL-1R1 and it could be speculated that ablation of IL-1R1 would impair functionality of the neutrophils. However, this does not appear to be the case as topical application of CXCL1 or CXCL2 can rescue the formation of microabscesses (Figure 5b). Interestingly, while this manuscript was under revision an advance online publication reported that mice lacking the IL-17 receptor A (IL-17RA) still develop psoriasiform disease in the imiquimod-induced skin inflammation model (El Malki et al., 2012). The involvement of the IL-1R1 signaling pathway in disease pathology as shown here in Figure 4 and Supplementary Figure S2 explain, at least in part, why IL-17RA KO mice still exhibit phenotypes.
Our in vitro studies revealed that imiquimod directly induces chemokine expression independent of IL-1 and/or IL-1R1 in keratinocytes through an adenosine receptor-signaling pathway (Figure 3). Curiously, in vivo this IL-1R1-independent pathway appears to be absent as IL-1R1 KO mice fail to increase their chemokine expression in response to imiquimod (Figure 5a). It has been demonstrated that the skin commensal bacteria Staphylococcus epidermidis can suppress signaling from TLR3 via up-regulation of TRAF1 (Lai et al., 2009). The pharmaceutical formulation of imiquimod used here is designed to carry compounds into the skin. During disease progression the barrier function of the skin is significantly compromised, e.g. the skin becomes dry and flaky. Hence, it is plausible that the cream carries S. epidermidis, or byproducts from the bacteria, into the skin with anti-inflammatory outcome, e.g. up-regulation of TRAF1. Further studies are required to determine whether the IL-1R1-independent imiquimod-induced signaling pathway is absent or present, but potentially suppressed, in vivo.
Given the apparent absence of the IL-1R1-independent pathway in vivo an interesting remaining question is how imiquimod stimulates release of IL-1α from the keratinocytes. Keratinocytes do not express TLR7 and TLR8 ((Olaru and Jensen, 2010a) and refs. therein); the best known receptors for imiquimod. Our in vitro data illustrates that high concentrations (250 μg/ml) of imiquimod are toxic to the keratinocytes (Figures 1–2); however, this toxicity does not give rise to elevated extracellular IL-1α (Figure 2a, levels are actually decreased). At lower concentrations loss of cell membrane potential is observed during extended incubation times (Figure 2b, 24 hours). However, extracellular IL-1α can be observed as early as 3–6 hours post-treatment (Figure 2a), suggesting that this cell death is not the cause of IL-1α release. In vivo there are no apparent signs of damage to the epidermis; in fact acanthosis of the epidermis is observed in the employed model (Figure 4c and Supplementary Figure S2). It is unlikely that the elease of IL-1α is mediated through the adenosine receptor pathway(s) as ligands for both adenosine receptors and adenylate cyclase failed to affect secretion (Figure 3f–g). It appears that imiquimod has effects upon cellular mechanisms which are yet to be discovered.
The imiquimod-induced mouse model of psoriasis-like disease represents exciting new opportunities for exploring mechanisms of skin inflammation. The model is clearly highly complex and involves multiple independent pathways and mechanisms. Further characterization of the model is needed to fully reveal its potential for improving our understanding of human disease and testing new therapeutic modalities.
MATERIALS AND METHODS
Cell culture and viability assays
Pooled human primary keratinocytes were obtained from Life Technologies (Grand Island, NY) and maintained in EpiLife or Defined Keratinocyte Serum Free medium (Life Technologies) with gentamicin (25 μg/ml, Life Technologies). Cells were adapted to Defined Keratinocyte Serum Free medium for at least 24 hours prior to treatments.
Mouse primary keratinocytes were isolated from newborn (24–72 hours) mice. The epidermis and dermis were separated by enzymatic digestion with 0.25% trypsin (Life Technologies) at 37°C for 45 min. The epidermal sheets were nutated to separate the keratinocytes at 4°C for 45 min. Cells were grown at 34°C with 5% CO2 in Keratinocyte Serum Free Medium (Life Technologies) supplemented with epidermal growth factor (10 ng/ml, Sigma-Aldrich), bovine pituitary extract (140 μg/ml, Life Technologies), hydrocortisone (50 μM, Sigma-Aldrich, St. Louis, MO), calcium chloride (45 μM) and gentamicin on rat tail type I collagen (BD Bioscience, San Jose, CA) coated plates.
Cell permeabilization was determined by measuring release of lactate dehydrogenase (LDH, Cytotoxicity Detection kit, Roche Applied Science, Indianapolis, IN).
Culture treatments and neutralization/inhibition experiments
Confluent cells were treated with medium only, imiquimod (Invivogen, San Diego, CA), polyinosinic-polycytidylic acid (poly(I:C), Sigma-Aldrich) and/or IL-1α (species matched, PeproTech, Rocky Hill, NJ) at the indicated concentrations. Activity of IL-1α was blocked using the neutralizing antibody MAB200 (R&D Systems, Minneapolis, MN) as described elsewhere (Olaru and Jensen, 2010b). Control cells were treated with isotype matched IgG2a (R&D Systems). Adenosine signaling was modulated using the adenylate cyclase ligand forskolin and the adenosine A2 receptor agonist CV 1808 (both from Santa Cruz Biotechnology, Santa Cruz, CA).
Real-time RT-PCR
RNA isolation, reverse transcription and real-time PCR primers for relative expression analyses of human CXCL1, CXCL2, IL-1α, IL-1β IL-8 and GAPDH are described elsewhere Olaru and Jensen, 2010b). The following primers were used for analyses of gene expression in mouse cells: mCXCL1-F, 5′ACCCAAACCGAAGTCATAGC; mC×CL1-R, 5′TTTCTCCGTTACTTGGGGAC; mCXCL2-F, 5′CAGACAGAAGTCATAGCCAC; mCXCL2-R; 5′TTCCAGGTCAGTTAGCCTTG; mGAPDH-R2A, 5′GCCCAATACGGCCAAATCC; mGAPDH-F2B, 5′CTTGTGCAGTGCCAGCC. Real-time PCR assays were validated as previously described (Sanmiguel et al., 2009). GAPDH levels were used for normalization of mRNA levels.
ELISA
Human CXCL1, CXCL2, IL-1α and IL-1β were detected as described elsewhere (Olaru and Jensen, 2010b). Mouse CXCL1 (also known as KC), CXCL2 (alternative name: MIP-2), IL-1α and IL-1β levels were determined using ELISA Development kits from PeproTech according to the manufacturer’s instructions.
Mice
C57BL/6J and IL-1R1 KO (C57BL/6 background) mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and bred in house in a specific pathogen free animal facility. All housing, breeding and experimental procedures involving mice were approved by the Temple University Institutional Animal Care and Use Committee and in compliance with the U.S. Department of Health and Human Services Guide for the Care and Use of Laboratory Animals.
In vivo imiquimod treatment and cytokine reconstitution
Mice were used at 8–12 weeks of age. KO background matched C57BL/6J control mice were matched for age and gender in each experiment. Fur on the back of the mice was removed by shaving followed by depilatory cream treatment. Cream was thoroughly removed with water. Each of the following two days (day 1 and 2) 62.5 mg Aldara cream (5% imiquimod, Medicis, Scottsdale, AZ) was applied to an approximately 2 × 3 cm area of the back. On day 4 back skin was collected for histology, immunohistochemistry and explant cultures. Control untreated mice were euthanized and skin collected on day 1. For explant cultures 4 pieces of skin were excised per mouse using circular punch biopsy tools (4 mm diameter each) generating full thickness skin samples of identical surface area. Each explant was incubated in 200 μl tissue culture medium for 24 hours at 34°C and the 4 cultures (per mouse) pooled. For cytokine reconstitution, CXCL1 or CCL2 (25 ng/mouse approximately equivalent to 4 ng/cm2, PeproTech) was mixed into the imiquimod cream and applied as described above on day 1 and day 2.
Histology and immunohistochemistry
Mouse skin was fixed in PBS buffered 4% formaldehyde and processed for standard hematoxylin and eosin staining at the Histotechnology Facility at The Wistar Institute (Philadelphia, PA). For immunohistochemistry, mouse skin was embedded in OCT compound and stored at −80°C until use. Cryosections (7 μm) were fixed in acetone/methanol (1:1) and incubated with normal mouse serum to reduce non-specific binding. Tissue sections were incubated with rat anti-mouse Ly-6G/Ly-6C antibody (BD Biosciences) overnight in a humidified chamber at 4°C. Tissues were subsequently labeled with biotinylated mouse anti-rat IgG2b antibody (BD Biosciences) for 1 hour at room temperature, streptavidin-HRP (Lab Vision, Thermo Fisher Scientific, Fremont, CA) for 30 min, and DAB Chromogen (Lab Vision). Tissue sections were counterstained with Mayer’s Hematoxylin (Sigma-Aldrich).
Statistical analyses
Arithmetic mean and standard deviations (SD) from one representative experiment of at least three independent experiments (n=2 per independent time-point and treatment within each individual experiment unless stated/indicated otherwise) are shown in figures. Significance was determined using the Student’s t test.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institute of Arthritis, Musculoskeletal and Skin Diseases (AR053672 to L.E.J.).
Abbreviations used in this article
- IL
interleukin
- IL-1R1
IL-1 receptor type I
- KO
knockout
- LDH
lactate dehydrogenase
- poly(I:C)
polyinosinic-polycytidylic acid
- TLR
Toll-like receptor
Footnotes
CONFLICT OF INTEREST
The authors state no conflicts of interest.
References
- Barland CO, Zettersten E, Brown BS, et al. Imiquimod-induced interleukin-1alpha stimulation improves barrier homeostasis in aged murine epidermis. J Invest Dermatol. 2004;122:330–336. doi: 10.1046/j.0022-202X.2004.22203.x. [DOI] [PubMed] [Google Scholar]
- Bowcock AM, Shannon W, Du F, et al. Insights into psoriasis and other inflammatory diseases from large-scale gene expression studies. Hum Mol Genet. 2001;10:1793–1805. doi: 10.1093/hmg/10.17.1793. [DOI] [PubMed] [Google Scholar]
- Cai Y, Shen X, Ding C, et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity. 2011;35:596–610. doi: 10.1016/j.immuni.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Malki K, Karbach SH, Huppert J, et al. An alternative pathway of imiquimod-induced psoriasis-like skin inflammation in the absence of interleukin-17 receptor A signaling. J Invest Dermatol. 2012 doi: 10.1038/jid.2012.318. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Fanti PA, Dika E, Vaccari S, et al. Generalized psoriasis induced by topical treatment of actinic keratosis with imiquimod. Int J Dermatol. 2006;45:1464–1465. doi: 10.1111/j.1365-4632.2006.02980.x. [DOI] [PubMed] [Google Scholar]
- Gilliet M, Conrad C, Geiges M, et al. Psoriasis triggered by Toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol. 2004;140:1490–1495. doi: 10.1001/archderm.140.12.1490. [DOI] [PubMed] [Google Scholar]
- Jensen LE. Targeting the IL-1 family members in skin inflammation. Curr Opin Investig Drugs. 2010;11:1211–1220. [PMC free article] [PubMed] [Google Scholar]
- Kan Y, Okabayashi T, Yokota S-i, et al. Imiquimod suppresses propagation of herpes simplex virus 1 by upregulation of cystatin A via the adenosine receptor A1 pathway. J Virology. 2012;86:10338–10346. doi: 10.1128/JVI.01196-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang K, Hammerberg C, Cooper KD. Differential regulation of IL-1 and IL-1 receptor antagonist in HaCaT keratinocytes by tumor necrosis factor-alpha and transforming growth factor-beta 1. Exp Dermatol. 1996;5:218–226. doi: 10.1111/j.1600-0625.1996.tb00120.x. [DOI] [PubMed] [Google Scholar]
- Lai Y, Di Nardo A, Nakatsuji T, et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med. 2009;15:1377–1382. doi: 10.1038/nm.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew W, Lee E, Krueger JG. Psoriasis genomics: analysis of proinflammatory (type 1) gene expression in large plaque (Western) and small plaque (Asian) psoriasis vulgaris. Br J Dermatol. 2004;150:668–676. doi: 10.1111/j.0007-0963.2004.05891.x. [DOI] [PubMed] [Google Scholar]
- Lian L-H, Milora K, Manupipatpong KK, et al. The double-stranded RNA analogue polyinosinic-polycytidylic acid induces keratinocyte pyroptosis and release of interleukin-36γ. J Invest Dermatol. 2012;132:1346–1353. doi: 10.1038/jid.2011.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mee JB, Johnson CM, Morar N, et al. The psoriatic transcriptome closely resembles that induced by interleukin-1 in cultured keratinocytes: dominance of innate immune responses in psoriasis. Am J Pathol. 2007;171:32–42. doi: 10.2353/ajpath.2007.061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro WJ. Note sur l’histopathologie du psoriasis. Ann Dermatol Syph. 1898;9:961–967. [Google Scholar]
- Olaru F, Jensen LE. Chemokine expression by human keratinocyte cell lines after activation of Toll-like receptors (TLRs) Exp Dermatol. 2010a;19:e314–316. doi: 10.1111/j.1600-0625.2009.01026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaru F, Jensen LE. Staphylococcus aureus stimulates neutrophil targeting chemokine expression in keratinocytes through an autocrine IL-1alpha signaling loop. J Invest Dermatol. 2010b;130:1866–1876. doi: 10.1038/jid.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel U, Mark NM, Machler BC, et al. Imiquimod 5% cream induced psoriasis: a case report, summary of the literature and mechanism. Br J Dermatol. 2011;164:670–672. doi: 10.1111/j.1365-2133.2010.10124.x. [DOI] [PubMed] [Google Scholar]
- Rajan N, Langtry JAA. Generalized exacerbation of psoriasis associated with imiquimod cream treatment of superficial basal cell carcinomas. Clin Exp Dermatol. 2006;31:140–141. doi: 10.1111/j.1365-2230.2005.01938.x. [DOI] [PubMed] [Google Scholar]
- Sanmiguel JC, Olaru F, Li J, et al. Interleukin-1 regulates keratinocyte expression of T cell targeting chemokines through interleukin-1 receptor associated kinase-1 (IRAK1) dependent and independent pathways. Cell Signal. 2009;21:685–694. doi: 10.1016/j.cellsig.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schön MP, Schön M. Imiquimod: mode of action. Br J Dermatol. 2007;157:8–13. doi: 10.1111/j.1365-2133.2007.08265.x. [DOI] [PubMed] [Google Scholar]
- Schön MP, Schön M, Klotz K-N. The small antitumoral immune response modifier imiquimod interacts with adenosine receptor signaling in a TLR7- and TLR8-independent fashion. J Invest Dermatol. 2006;126:1338–1347. doi: 10.1038/sj.jid.5700286. [DOI] [PubMed] [Google Scholar]
- Steffen C. William John Munro and Munro’s abscess, and Franz Kogoj and Kogoj’s spongiform pustule. Am J Dermatopathol. 2002;24:364–368. doi: 10.1097/00000372-200208000-00016. [DOI] [PubMed] [Google Scholar]
- Swindell WR, Johnston A, Carbajal S, et al. Genome-wide expression profiling of five mouse models identifies similarities and differences with human psoriasis. PLoS ONE. 2011;6:e18266. doi: 10.1371/journal.pone.0018266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonel G, Conrad C, Laggner U, et al. Cutting edge: A critical functional role for IL-23 in psoriasis. J Immunol. 2010;185:5688–5691. doi: 10.4049/jimmunol.1001538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Fits L, Mourits S, Voerman JSA, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 2009;182:5836–5845. doi: 10.4049/jimmunol.0802999. [DOI] [PubMed] [Google Scholar]
- Wu JK, Siller G, Strutton G. Psoriasis induced by topical imiquimod. Australas J Dermatol. 2004;45:47–50. doi: 10.1111/j.1440-0960.2004.00030.x. [DOI] [PubMed] [Google Scholar]
- Yano S, Bann T, Walsh R, et al. Transcriptional responses of human epidermal keratinocytes to cytokine interleukin-1. J Cell Physiol. 2008;214:1–13. doi: 10.1002/jcp.21300. [DOI] [PubMed] [Google Scholar]
- Zaba LC, Suárez-Farinas M, Fuentes-Duculan J, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124:1022–1030.e1395. doi: 10.1016/j.jaci.2009.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Krueger JG, Kao M-CJ, et al. Novel mechanisms of T-cell and dendritic cell activation revealed by profiling of psoriasis on the 63,100-element oligonucleotide array. Physiol Genomics. 2003;13:69–78. doi: 10.1152/physiolgenomics.00157.2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.