

Abstract
The endothelin-A receptor (Ednra) is involved in several physiological, pathological, and developmental pathways. Known for its function in vasoconstriction after being activated by endothelin-1, Ednra also controls cephalic neural crest cell development and appears to play a role in several pathologies, including cancer and periodontitis. However, the mechanisms regulating Ednra expression have not been identified despite its important functions. In this study, we investigated the role progesterone plays in Ednra gene expression in vivo and in vitro. In mice, pregnancy promotes Ednra expression in the heart, kidney, lung, uterus, and placenta, and the up-regulation is mediated by progesterone. We determined that the conserved region between −5.7 and −4.2 kb upstream of the mouse Ednra gene is necessary for the progesterone response. We also found that progesterone mediates Ednra activation through progesterone receptor B activation by its recruitment to PRE6, one of the 6 progesterone response elements found in that locus. However, gene activation by means of a GATA2 site was also necessary for the progesterone response. The Gata2 transcription factor enhances the progesterone response mediated by the progesterone receptor B. Together these results indicate that progesterone regulates Ednra expression by synergizing with Gata2 activity, a previously unknown mechanism. This mechanism may have an impact on pathologies involving the endothelin signaling.
Endothelin signaling involving endothelin-1 (Edn1) and the endothelin-A receptor (Ednra) is implicated in several physiological, pathological, and developmental processes (1, 2). The secreted bioactive 21-amino-acid peptide Edn1 is produced by the successive cleavage of the pre-pro-protein by furin-like endopeptidases to produce pro-Edn1 (big Edn1), which is then cleaved by the endothelin-converting enzymes 1 or 2 into Edn1. The peptide preferably binds the 7-pass transmembrane G protein-coupled receptor Ednra to activate numerous signaling cascades (1) through the recruitment of different Gα proteins (3). The activation of Ednra signaling via Gαq/11 is associated with cardiac hypertrophy and causes the contraction of smooth muscles via a calcium-dependent mechanism, whereas Gα12/13 induce contraction via a calcium-independent mechanism (2, 3). Secondary messenger signaling by Gαs/i promotes smooth muscle relaxation (2, 3). During embryonic development, the specific conditional inactivation of Gαq and Gα11 genes or Gα12 and Gα13 genes in the neural crest cells causes craniofacial and cardiovascular defects, respectively (4).
Inactivation of the Ednra gene in mice causes lethal craniofacial and cardiovascular defects (5, 6). Specific inactivation of the gene in neural crest cells causes similar defects, including the homeotic transformation of the lower jaw into upper jaw-like structures (7). Craniofacial and cardiovascular defects are similar to those observed in Gαq/11 and α12/13 protein mutant embryos, respectively (4), confirming the role of these receptor mediators activated by Ednra signaling in these cells. The defects indicate that endothelin signaling is crucial for neural crest cell patterning and development during embryogenesis.
Under normal physiological conditions, the activation of Ednra causes localized vasoconstriction, in conjunction with the renin/angiotensin system upregulating Edn1 production (1, 8, 9). However, being the favored receptor for Edn1, Ednra is also involved in pathological conditions such as congestive heart failure, cardiac hypertrophy, chronic kidney disease, systemic hypertension, pulmonary hypertension, and tissue fibrosis (1, 2, 10). In the kidney, Ednra is found in the medullar and cortical vessels, mesangial cells, pericytes of descending vasa recta, distal tubular epithelial cells, epithelial cells of cortical collecting ducts, glomeruli, and interstitial cells of the inner and outer medulla (11, 12). The Edn1/Ednra axis is also involved in cancer, mainly in metastases that localize in bone (13–15). In addition, Ednra expression is upregulated in the periodontal tissues of patients with periodontitis (16). The signaling system is involved in hypertensive pregnancy, including pre-eclampsia and eclampsia (17). In some of these problematic pregnancies, an activating autoantibody against the angiotensin II receptor stimulates Edn1 production (9). In the uterus, Ednra is expressed in the endometrial stroma and myometrium (18, 19). Its expression is influenced by steroid hormones, including during pregnancy (20, 21).
Despite the important functions of the Ednra gene, the molecular mechanisms regulating its expression are unknown. A previous study indicated that fibroblast growth factors 1 and 2 and platelet-derived growth factors stimulate Ednra expression by a MAP kinase MEK-dependent mechanism without providing molecular evidence of the gene promoter involvement (22). ERK activation via protein kinase C appears to play a role also (23). Glucocorticoids, including the receptor agonist dexamethasone, decrease Ednra expression in the aorta and kidneys but increase its expression in the placenta (24–26). In this study, we report that the pregnancy-associated hormone progesterone, through the activation and binding of the progesterone receptor B (PRB) to specific cis-regulatory elements in the gene promoter, regulates Ednra expression. Progesterone elicits the activation of Ednra expression by means of a classical mechanism involving the recruitment of progesterone response elements (PREs) and a nonclassical activation mechanism via a GATA2 element enhancing the promoter activity. The synergy between progesterone receptors and the transcription factor Gata2 appears indispensable for the in vitro upregulation of Ednra expression by progesterone.
Materials and Methods
Animals
The protocol for the use of animals was approved by the Institutional Animal Care and Use Committee at Texas A&M University Baylor College of Dentistry, and the animals were euthanized following National Institutes of Health guidelines. Virgin and ovariectomized C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, Maine). Tissues were collected from mice aged from 58 to 78 days old. After acclimatizing, mice were bred, and 12:00 noon of the day that a vaginal plug was observed was designated as day 0.5 of pregnancy. Maternal tissues were collected at 10.5, 15.5, and 18.5 days of pregnancy. Collection of tissues from virgin control mice was spread throughout the same time period for age matching. Five females were used in each group. Ovariectomized mice were given 2 mg progesterone by ip injection (27, 28). Progesterone was dissolved in ethanol and then resuspended in canola oil. Mock-injected control females were given the same amount of alcohol and oil. Females were killed and tissues collected 12 and 24 hours after administration. Four females were used in each group, including for the 2 control groups. Our gene expression analysis did not reveal any differences between these 2 control groups.
Real-time PCR
Ednra expression was analyzed in mouse tissues by quantitative real-time PCR (qRT-PCR), performed using the Bio-Rad CFX96 apparatus. Total RNA was obtained from the tissues using the Trizol method (Invitrogen, Carlsbad, California) and treated with deoxyribonuclease before reverse transcription using the Moloney murine leukemia virus reverse transcriptase and Primer 9 following the manufacturer's instructions (New England Biolabs, Beverly, Massachusetts). After dilution of the cDNAs, the amplification was measured for 40 cycles using the DyNAmo SYBR Green qPCR reagent and compared with 18S rRNA (Invitrogen AM1716M). The oligonucleotide primers for Ednra were 5′-TGCTGGTTCCCTCTTCACTT-3′ and 5′-ACAGCAACAGAGGCAGGACT-3′ and for Gata2 were 5′-TGCATGCAAGAGAAGTCACC-3′ and 5′-ACCACCCTTGATGTCCATGT-3′. These primers were from 2 different exons. These primers (along with 18S rRNA) produced a single amplification product as confirmed by their respective single peak dissociation curve and gel electrophoresis. Each sample was analyzed in triplicate.
Primary cell cultures
Cells for the primary myometrium and kidney cell cultures were obtained from 2-month-old female mice. For the myometrium cells, uteruses were minced and digested in trypsin (29). After washing to remove the epithelial, endometrial, and stromal cells, uterus fragments were incubated with 0.1% collagenase in complete medium overnight at 37°C and then cultured after washing as described by others (30). The uterine smooth muscle cells were cultured in DMEM supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 50 μg/ml ascorbic acid at 37°C in a humidified incubator with 5% carbon dioxide. Kidney cells were obtained by trypsin digestion as previously described by other investigators (31). Renal cells were cultured as detailed above but without ascorbic acid. Cells were treated as described below for the progesterone response analysis.
Plasmids construction
The pGL4.26-luciferase vector was purchased from Promega (Madison, Wisconsin). The large Ednra-luciferase expression clone pGL4.26 Ednra−8.0;−0.5-luc was generated using the Gateway Vector Cloning System (Invitrogen) according to the manufacturer's instructions. Briefly, an attBEdnra−8;−0.5 fragment was generated by PCR amplification from the BAC RPCI·23-99o4 using the high-fidelity PfuTurbo DNA polymerase (Stratagene, La Jolla, California) and cloned into the donor vector pDONR221 to produce the entry clone pDONR211 Ednra−8;−0.5. The primers used were Ednra-8up attB1, 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTGTCCTGTCTGCCCTTCTTTA-3′, and Ednra-0.5dn RV attB2, 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTGATATCCATCTCCAGCACCACAGAGA-3′.
The pGL4.26 attB-RfB-luc destination vector was converted from the pGL4.26-luc vector following the manufacturer's instructions. Then the Ednra−8.0;−0.5 fragment was delivered from the entry clone pDONR211Ednra−8;−0.5 into the destination vector pGL4.26 attB-RfB-luc by recombination with Clonase (Invitrogen) to generate the expression clone pGL4.26 Ednra−8.0;−0.5-luc. The clones were tested by restriction digestion and PCR analysis for integrity and partially sequenced. Other Ednra-luciferase vectors were obtained by restriction enzyme digestions and religations from the original pGL4.26 Ednra−8.0;−0.5-luc construct.
Site-directed mutagenesis
PRE sites were mutated using the QuikChange II site-directed mutagenesis kit (Stratagene) with high-fidelity PfuTurbo DNA polymerase. The primers used are listed in Supplemental Table 1 (published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org), and the Ednra−5.7;−4.2-luc vector was the template. Clones were confirmed by restriction enzyme digestions and/or sequencing. The GATA2 and AP2 sites were mutated using the same method. We converted the PRE sites into an EcoRI restriction site and the GATA2 and AP2 sites into an XhoI restriction site.
Cell transfection and luciferase activity quantification
For the luciferase assays and chromatin immunoprecipitation (ChIP) assays, HEK-293E cells were transfected using the calcium phosphate coprecipitation method (32). The cells were seeded into plates the day before and grown to 70% confluency before transfection. They were then transfected with the luciferase vector along with the pSG5-hPRB vector encoding the PRB or pFLAG-Gata2 (Addgene clone 1418) (33). A cotransfected pCMV-lacZ vector encoding the β-galactosidase (β-gal) enzyme was used as a transfection control and a standard for the luciferase assays. After 6 to 8 hours incubation with the DNA/calcium phosphate solution, the cells were cultured with Cellgro Complete serum-free medium (Mediatech, Manassas, Virginia). One day later, serum-free medium with 2 ng/ml (6.26μM) progesterone (P8783; Sigma Chemical Co, St Louis, Missouri) or the same amount of carrier (ethanol) was added to the cells. After 24 h, the cells were collected in the reporter lysis buffer, and luciferase assays were performed following the manufacturer's instructions (Promega). The enzymatic activity was quantified using a BMG FLUOstar OPTIMA microplate reader. Standardization was done using the β-gal assay by measuring the cleavage of ortho-nitrophenyl-β-d-galactopyranoside) at 420 nm after stopping the enzymatic reaction (34). Each sample was analyzed in triplicate, and each construct was tested a minimum of 3 times. Small interfering RNA (siRNA) transfection was performed using Lipofectamine 2000 following the manufacturer's instructions (Invitrogen).
ChIP and coimmunoprecipitation assays
The anti-PRB monoclonal antibody (35) from Fisher Scientific (Pittsburgh, Pennsylvania) or anti-FLAG rabbit antibody (Cell Signaling, Danvers, Massachusetts) were tested by Western blotting using 293E cells transfected with and without pSG5-hPRB or pFLAG-Gata2 as previously described (34). The ChIP assays were performed with little modification of Basic Protocol 2 described by Aparicio and colleagues (36); the buffer composition is described in the protocol, beginning with transfected cells. All the buffers used after Tris-buffered saline washing were supplemented with 2mM phenylmethylsulfonyl fluoride (Sigma) and 1× Complete protease inhibitors (Roche, Indianapolis, Indiana). Sonication was performed using the Branson Sonifier 450. Most the DNA fragments were between 200 and 300 bp, as verified by gel electrophoresis. The same amount of cell lysate was incubated with 1) FA lysis buffer prewashed protein A/G plus-agarose beads (Santa Cruz Biotechnology, Santa Cruz, California) and 2) the anti-PRB, FLAG, or transcription factor IID (TFIID; Santa Cruz sc-204) antibodies, an unrelated antibody, or IgG overnight at 4°C. The antibody-linked chromatin was precipitated with the protein A/G plus beads by centrifugation. After washing the beads, the cross-linking was reversed in the ChIP elution buffer by incubation at 67°C overnight. The chromatin was extracted using the phenol/chloroform method after treatment with ribonuclease A and proteinase K. The DNA obtained was resuspended in 10mM Tris (pH 8.5). PRB or Gata2 binding to Ednra gene sequences was analyzed by PCR. The primers used are listed in Supplemental Table 2. Each experiment was repeated a minimum of 3 times. Coimmunoprecipitation assays were performed using unfixed transfected cells in NTM buffer (200 mM NaCl, 50 mM Tris [pH 8.0], 0.5% Nonidet P-40, and protease inhibitors as above) overnight at 4°C. Assays were performed with 2 ng/ml progesterone with either the PRB or FLAG antibody with similar results.
Electrophoretic mobility shift assays
293E cells were transfected and treated with progesterone as described above. Nuclear proteins were extracted (37) and EMSA performed as described by Ye and colleagues (38), using salmon sperm DNA instead of poly(dI-dC) and with the addition of 2 ng/ml progesterone during the formation of protein-DNA complexes. Extracted proteins were incubated with biotin-11-dUTP-labeled (Fisher Scientific) PRE6 or GATA2 probes before the addition of the antibody when supershifting the DNA-protein complexes. Probes were generated by high-fidelity PCR with the oligos ATTCTTTCCCAGGGGCTAGA and AAGATGGGGAGTGGGAGG (from −4713 to −4525) for PRE6 or synthesized (20 nucleotides on each side of the binding site; from −4545 to −4502) for GATA2. Probes were labeled using the tdt terminal transferase enzyme (New England Biolabs). After separation in a nondenaturing acrylamide gel, protein-DNA complexes were transferred onto a membrane for detection with the horseradish peroxidase-conjugated streptavidin, following manufacturer's recommendations (Fisher, Scientific).
Western blotting
SDS-PAGE separation and immunodetection of proteins on polyvinylidene difluoride membranes was carried as previously described (34), with 40 to 50 μg of total protein extracts for PRB, FLAG-Gata2, and actin and 100 μg of proteins for Ednra and native Gata2. The actin mouse monoclonal antibody was purchased from Santa Cruz Biotechnology, and the rabbit polyclonal antibodies for Ednra and Gata2 were purchased from Epitomics (Burlingame, California) and Cell Signaling, respectively. Protein-antibody complexes were revealed with horseradish peroxidase-conjugated secondary antibodies on x-ray films using ECL peroxidase reagents (GE Healthcare, Piscataway, New Jersey) or Li-Cor secondary antibodies for Ednra and Gata2 proteins from cultured primary myometrium cell extracts (Li-Cor, Lincoln, Nebraska). For the latter, a Li-Cor Odyssey apparatus was used to detect the signals.
Statistical analysis
Two-tailed Student's t test analysis or 2-way ANOVA was used to evaluate the statistical significance of the results. The multiway analyses included the Kruskal-Wallis test followed by the Mann-Whitney U test. P < .05 was considered significant, but trends (P < .1) were also noted.
Results
Progesterone elevates Ednra expression during pregnancy
Despite the important roles played by Ednra during embryonic development, in physiological processes and pathological conditions, the mechanisms regulating its expression are unknown. The rVista program (39) was used to analyze upstream (in 5′ of the ATG start codon) sequences of the Ednra gene (mouse chromosome 8 ENSEMBL EMSMUSG00000031606 and NCBI GI: 94383902) to identify possible mechanisms regulating its expression. The analyses revealed the significant presence of conserved putative PREs (data not shown). We transfected bacterial artificial chromosomes (BACs) containing the Ednra gene locus (RPI 23-99O3 and RPI 24-289K16) to test whether progesterone alters Ednra expression. The transfected BACs contain a lacZ reporter gene encoding the β-gal enzyme introduced by recombineering (40) to replace exon 2. The activities from CHO, Cos7, NIH 3T3, and 293E cells transfected with the BAC constructs were used as controls. Cotransfection of the PRB along with the BACs increased the enzymatic activity in the cells (Supplemental Figure 1, only Cos7 shown) in response to the progesterone contained in the fetal bovine serum. No enzymatic activity was measured in the cells transfected with PRB alone. Upon obtaining a positive response, we investigated the function of progesterone in the regulation of Ednra expression.
We analyzed the expression of the Ednra gene in mouse maternal tissues at 10.5, 15.5, and 18.5 days of pregnancy because the hormone progesterone is associated with pregnancy and is found at higher levels in late pregnancy and declining before parturition (41). The gene expression in tissues from pregnant mice (n = 5 for each group) was analyzed by qRT-PCR and compared with the same tissues from virgin females of the same age (58–78 days old). The analysis revealed a significant increase (P < .05) in the gene expression in some pregnant female tissues (Figure 1A). In the kidneys, the expression initially decreased significantly (P < .05) by day 10.5 and then significantly increased (P < .05; day 18.5 vs 10.5) in late pregnancy. No significant changes in the expression of the Ednra gene were observed in the liver of pregnant mice, whereas the expression rapidly increased about 2-fold in the lungs by day 10.5 (P < .1) and remained elevated during pregnancy (P < .05 at day 18.5). In the heart, a significant 2-fold increase (P < .05) in the expression of Ednra occurred in late pregnancy. In the placenta, the expression increased about 2.5-fold at days 15.5 and 18.5 (P < .05). Comparison was made to the day 10.5 placenta. We also analyzed the expression of the gene in the uterus because progesterone mainly targets this tissue (41, 42). The findings revealed an ∼17-fold increase at day 15.5 and an ∼25-fold increase at day 18.5 in the gene expression in the uterus from pregnant mice (P < .05) (Figure 1B), which correlates with the known expression of the PRB to prevent parturition (43, 44).
Figure 1.

Relative Ednra expression in tissues from pregnant or progesterone-treated mice. The qRT-PCR results were normalized with 18S rRNA. Data are displayed as fold inductions of mRNA after normalization compared with the virgin or carrier controls. For the placentas, results were compared with day 10.5 of pregnancy. A, qRT-PCR quantification of Ednra expression in C57BL/6J mice pregnant for 10.5, 15.5, and 18.5 days. B, Relative Ednra expression in the uterus of pregnant mice; same legend as in A. C, Relative expression of the gene in tissues from ovariectomized mice (ovx) treated with the carrier (c) or progesterone (p) for 12 and 24 hours. D and E, Relative Ednra expression in cultured primary myometrium (D) and kidney (E) cells treated with 2 and 4 ng/ml progesterone for up to 24 hours. For statistical analysis (ANOVA), comparisons were made with virgin controls unless otherwise indicated. #, P < .01; *, P < .05; and ^^, P < .1. Error bars represent SEs.
Because these results indicated that pregnancy influences Ednra expression, we treated ovariectomized mice with progesterone to confirm that the hormone was the factor responsible for the effects observed in pregnant mice. Ovariectomized females were used because of their steady hormonal status and absence of progesterone (28). They (n = 4) were injected ip with 2 mg progesterone dissolved in ethanol and then canola oil to achieve a level of circulating progesterone similar to that observed in late pregnancy (27, 28). The control females (n = 4) were injected ip with canola oil containing the same amount of alcohol. The heart, lung, liver, and kidney tissues were collected after 12 and 24 hours of treatment, and Ednra expression was analyzed as described above. The results showed that Ednra expression had increased in all the analyzed tissues (Figure 1C). However, the temporal response differed among the tissues. In the kidneys, the gene expression increased significantly at both time points (P < .05), whereas the response in the liver was slower and the expression significantly increased only after 24 hours of treatment (P < .05). Although the response in the lung and heart significantly increased in tissues from pregnant mice, it was moderate but not statistically significant (P < .1) and not sustained in the ovariectomized mice treated with progesterone. Elevation of Ednra expression was observed only after 12 hours of treatment. Unfortunately, we could not analyze the gene expression in the uterus because the uterine horns were removed by the vendor during the ovarian resection procedure (due to the young age of the animals). To overcome this problem, we treated cultured primary myometrium cells and used primary kidney cells as a control. In both cell types, progesterone caused a rapid and significant (P < .05 for 2 vs 0 hours) increase of Ednra expression eliciting a hormonal negative feedback response to decrease Ednra expression afterward (P < .05 for 4 and 8 vs 2 hours; Figure 1, D and E). The negative feedback was eventually released, and Ednra expression increased again by 24 hours after treatment in the myometrial cells (both treatments) and in the kidney cells (4 ng/ml only). Based on these findings and the pregnancy results, we concluded that progesterone can regulate Ednra expression. We then investigated whether progesterone regulates directly the expression of the gene or via the activation of another signaling mechanism.
The −5.7- to −4.2-kb region regulates the progesterone-response activity
The Ednra gene is structurally complex. This unusual complexity appears to be conserved among different species, including chicken, mouse, rat, and human (Vista and ENSMBL analyses, data not shown). Structurally, the basic gene promoter appears to be located between 4.2 and 4 kb upstream of the start (ATG) codon (Figure 2A). The 5′-untranslated region (UTR) is encoded by 2 different segments separated by an intron about 3.6 kb long (from −3648 to −56). Exon 1 and exon 2 are separated by a conserved intron spanning more than 30 kb. To investigate the molecular mechanisms employed by progesterone to regulate Ednra expression, the sequence located between −7950 to −564 bp was cloned into the pGL4.26 luciferase vector (hereinafter referred to as Ednra−8;−0.5). The vector was transfected into HEK293E cells either alone or with a vector encoding the human PRB (pCMV-hPRB, henceforth hPRB), essential for the progesterone transcriptional response (45). We confirmed the efficacy of the hPRB transfection by Western blotting (Figure 2B), which also revealed that the receptor was not endogenously expressed in the 293E cells albeit that the receptor is normally expressed in kidney cells (46, 47). This absence of PRB protein is advantageous to test the specificity of the hormonal response. These kidney cells were chosen because of the positive response observed in the kidneys of ovariectomized mice and cultured primary cells, and they are easily transfected. Furthermore, the Ednra gene is also expressed in the kidney in different epithelial cell populations (11, 12). Cells transfected with the Ednra−8;−0.5 construct alone and treated with progesterone or cotransfected with hPRB did not respond and displayed similar luciferase activity levels. Cells cotransfected with Ednra−8;−0.5 and PRB and treated with progesterone responded with significant increases in their levels of luciferase activity (Figure 2C). A similar response was observed in other cell lines such as CHO-1 and Cos7 (Supplemental Figure 2).
Figure 2.

Identification of the Ednra gene 5′-flanking region responding to progesterone stimulation. A, Schematic representation of the murine Ednra gene structural organization. Ex, exon; P, basal/core promoter; u1 and 2, 5′-UTR fragments 1 and 2; R1 and X1, EcoR1 and Xho1 restriction sites, respectively. B, Western blot analysis confirming the transfection of hPRB in 295E cells. C, Identification of the upstream region of the gene responding to progesterone stimulation. Cells were transfected with the described vectors and treated with the carrier (vector) or with progesterone (+P), cotransfected with hPRB (+hPRB) or cotransfected with hPRB, and treated with progesterone (+P+hPRB). The relative luciferase activity was measured to indicate the promoter activity, and comparisons were made to each untreated vector for statistical analysis (ANOVA). #, P < .01; *, P < .05. Error bars represent SEs.
The Ednra−8;−0.5 fragment cloned into the pGL4.26 luciferase vector was digested with different restriction enzymes to identify the region responding to progesterone. A summary of different clones generated through this promoter-bashing process are presented in Figure 2C. Each one was tested for its response to progesterone stimulation through measurements of the resulting luciferase enzymatic activities representing the promoter activity. The analyses revealed that the −4.2;−0.5-kb promoter region did not have a significant response, whereas the −8;−4.2-kb region responded significantly (P < .05) to progesterone treatment (Figure 2C). Further analyses revealed that the −8- to −5.7-kb sequence did not respond to progesterone stimulation. The −6.3;−3.0 fragment responded positively to progesterone, but when this region was deleted from the −8;−0.5 construct, the response did not take place. After further refinements, the analyses indicated that the −5.7- to −4.2-kb region (EcoRI/XhoI fragment) controlled the progesterone response (Figure 2C). This region is just upstream of the basal promoter region located between −4.2 and −4.0 kb.
PREs partly mediate the activation of Ednra expression by progesterone
We analyzed the Ednra −5.7;−4.2-kb region with the MatInspector (www.Genomatix.de) (48) and TESS (http://www.cbil.upenn.edu/tess/) (49) programs to identify the location of a PRE (PRE) in the conserved sequence. MatInspector identified 4 potential sites that were also recognized by TESS, which identified 2 additional putative sites. We named these 6 sites PRE1 to -6 in the 5′ to 3′ direction. PRE1 is located from −5572 to −5567 bp and PRE6 from −4658 and −4653 bp, with the remaining sites located between them (Figure 3A). Except for PRE2 with its alternative PRE sequence (TESS analysis), all the other PRE sites have the core consensus TGTTCT sequence (Figure 3B), although PRE1 was not identified with the MatInspector analysis.
Figure 3.

Presence of PREs in the −5.7;−4.2-kb region of the mouse Ednra gene. A, Sequence indicating the location of the 6 PREs and also the GATA2 and AP2 sites in the −5.7;−4.2 fragment from the 5′-flanking region of the gene. B, Alignment of the PRE sequences with the consensus sequence from MatInspector.
Because 6 putative PRE sites were identified, we performed ChIP assays to determine to which ones liganded PRB was binding. For these assays, 293E cells were transfected with the vector encoding PRB and then treated with progesterone or the carrier for 24 hours. Precipitation with an unrelated antibody or IgG was used as a negative control. Our results indicated that PRB can bind PRE1 and PRE3 to site 6 (n = 4), which means that PRE2 is not used to activate Ednra expression (Figure 4 and Supplemental Figure 3). At sites 1, 3, 5, and 6, PRB can be seen binding these PRE sites at a low level in the absence of progesterone. Treatment with progesterone significantly increased this attachment (from 2.65- to 9.36-fold increase). However, such low-level binding in the absence of progesterone does not stimulate a response, as indicated by the luciferase activities.
Figure 4.
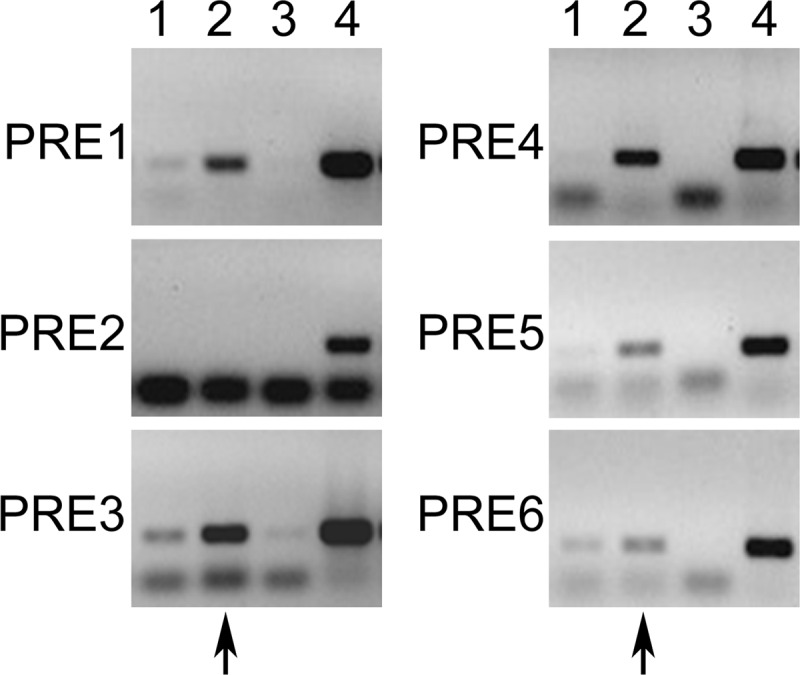
ChIP PCR results from the different PRE sites present in the −5.7;−4.2-kb region. PRE1 to -6 were tested individually, and immunoprecipitation was carried out with the anti-PRB antibody. Cells were transfected with hPRB and treated with the carrier (lane 1) or progesterone (lanes 2, 3, and 4). Lane 3 is a negative control with an unrelated antibody or IgG, and lane 4 is an input control. The observed lower bands are the oligonucleotide dimers. The arrows indicate the lane where a positive PCR amplification is expected.
Because all the PRE sites, with the exception of PRE2, were bound by PRB, we mutated each core consensus TGT-T/c-CT element to determine which one is essential for regulating the Ednra promoter activity. Mutations were introduced by site-directed mutagenesis, in which each core sequence was replaced by an EcoRI site (GAATTC). A map showing the location of these sites is presented at the bottom of Figure 5E for reference. Mutation of PRE2 to PRE5 had no significant effects on the promoter activity (Figure 5A). These results confirmed that PRE2 did not control the Ednra expression and also revealed that PRE3, -4, and -5 were likely not needed to regulate the expression of Ednra by progesterone. Mutating PRE1 and PRE6 significantly decreased the luciferase activity by about 40%, but never below 50% of the control levels (intact construct) (Figure 5A). Double PRE1 and PRE6 mutations did not further reduce the promoter activity, indicating that their functions were not additive. No other double, triple or quadruple mutation combinations changed the response (data not shown) but did suggest that PRE6 was central to the progesterone response.
Figure 5.

Identification of the elements essential for the progesterone response. Mutations were introduced by site-directed mutagenesis, and sequential deletions were produced by using the restriction sites introduced by site-directed mutagenesis. A, Relative promoter activity, measured by luciferase assays, in mutant (m) and double-mutant (dm) constructs for the different PRE sites. B, Relative promoter activity after the sequential deletion of the −5.7;−4.2 region. C, Relative activity after the mutation of the GATA2 and AP2 sites. In A, B, and C, results were compared with the intact Ednra−5.7;−4.2-luc construct. D, ChIP PCR results from the GATA2 site using the PRB antibody. The lower bands observed in some PCR are the dimers of primers. Lane 1, hPRB; lane 2, hPRB and progesterone; lane 3, as in lane 2 but with an unrelated antibody; lane 4, input control. E, Schematic representation of the sites mutated or deleted in A, B, and C. PRE sites are indicated by their respective number. G, GATA2; A, AP2. #, P < .01; *, P < .05 with ANOVA, comparisons made with control vector.
To test this hypothesis, we sequentially deleted the PRE sites from a 5′ to 3′ direction, judiciously using the EcoRI site introduced by site-directed mutagenesis. Deletion of −5.7 kb to PRE1 increased the promoter activity, further supporting the assumption that PRE6 is essential for the progesterone response (Figure 5B) and suggesting that the upstream region contains binding sites for repression. Removal of the other PRE sites 2, 3, and 4 gradually decreased the luciferase activity to the same level of the intact Ednra−5.7;−4.2 construct. When the −5.7 kb to PRE5 region was removed, the activity of the promoter was significantly reduced by ∼40% (Figure 5B). These results suggest that the activation of the Ednra expression by liganded PRB requires PRE6 and another PRE site. Synergy between 2 or more PRE sites has been observed previously (50–52). Removal of all PRE sites did not significantly reduce any further the activity of the luciferase reporter gene. The gradual decrease observed with the serial deletions could be explained by the likely reduction of a physical hindrance, but as the sites are removed, the availability of other sites is likewise reduced, eventually preventing any interactions by PRB between PRE6 and another site. These results suggested that PRE6 was central to the positive progesterone response and, along with combinatorial mutation analysis, they indicate that PRE6 activation elicits the recruitment of the PRE1, -3, -4, or -5 sites to sustain the response (see further analysis below). However, the results indicated the possibility of other progesterone-dependent mechanisms regulating Ednra expression because the luciferase activity never dropped below 50% of its original level. Removal of the downstream segment of the −5.7;−4.2 fragment (ΔGata2;−4.2) significantly reduced the luciferase activity (Figure 5B).
The progesterone response is also mediated by a GATA2 site synergizing with PRE6 recruitment
We tested the hypothesis that another mechanism was used by examining whether a nonclassical progesterone response also controlled the Ednra expression. We defined the nonclassical progesterone response (53) as the capacity of the hormone to stimulate a response, which could occur even in absence of PRE binding (co-response elements) and involves the cooperation of other transcription factors (54–56). Because a positive response was still observed when the entire region −5.7 kb to PRE6 (−4653) was deleted, we suspected that the remaining ∼450 bp contained elements allowing the nonclassical response. A previous report indicated that Gata2 could participate in this type of response (57). In the study by Magklara and Smith (57), a GATA2 site was found within a PRE element. A similar site was found within PRE6 (reverse orientation) and 2 AGGTC sequences are located a few nucleotides on each side of the PRE6 element, nearly identical to the AGATC sequence of the FKBP5 GATA2 element. These sites were not recognized as Gata-binding elements by TESS and MatInspector, and our analyses revealed that these 3 sites were not involved. However, the analysis of the promoter region 3′ of the PRE6 element revealed the presence of another GATA2-binding site (Figure 3A). An AP2 site, which can potentially be used for progesterone signaling (58), was also found. Site-directed mutation of the AP2 site did not decrease the reporter activity (Figure 5C), indicating that the site is not involved in the activation of Ednra expression by progesterone. The elevation of luciferase activity suggests that AP2 may repress Ednra expression, but when the transcription factor was tested, the luciferase activity was similar to the negative control (data not shown), indicating that steric hindrance might be involved instead.
Inactivation of the GATA2 site significantly reduced the luciferase activity by about 40%, showing that this site had responded to progesterone stimulation (Figure 5C). ChIP analysis revealed that liganded PRB interacts directly or indirectly with the GATA2 site (Figure 5D). When both the GATA2 and PRE6 sites were mutated, the response to progesterone stimulation was reduced to ∼20% of the control level (no significant differences with the unstimulated control). These results demonstrate the additive effects of these 2 response elements and the fact that progesterone controls Ednra expression using both classical and nonclassical mechanisms. Furthermore, these results confirm that PRE6 is central to the regulation of Ednra expression in response to progesterone stimulation. The remaining low promoter activity may represent the noise level generated by the other PRE sites.
We investigated whether PRB activation and binding to the PRE6 and GATA2 sites are required before PRB binding to other PRE sites to identify the mechanistic activation of Ednra expression by the liganded PRB. For these experiments, the mutated PRE6 and PRE6/GATA2 constructs were transfected, along with the hPRB construct for ChIP assays. The assays indicated that activated PRB failed to attach the mutated PRE6 and GATA2 sites, confirming the efficacy of the mutations (Figure 6). Mutation of PRE6 alone reduced PRB from binding, presumably indirectly, to the GATA2 site. Activated PRB binding to PRE1 was reduced (2.9-fold reduction by qRT-PCR) and abrogated for PRE5 (12.3-fold reduction) (Figure 6, compared with Figure 4). The double mutation of the PRE6 and GATA2 sites eliminated PRB binding to PRE5 (14.4-fold reduction) and reduced binding to PRE1 (3.1-fold reduction) (Figure 6). However, the PRE6 or PRE6/GATA2 mutations did not prevent the attachment of the unliganded and liganded PRB to PRE3 and -4, as revealed by their positive PCR amplification. These results thus show that the binding to these 2 sites is independent of PRB binding to PRE6 and GATA2 sites. However, it is unclear why PRB binding to these sites does not elicit a positive response to progesterone stimulation, as revealed by the above mutation and luciferase assays.
Figure 6.
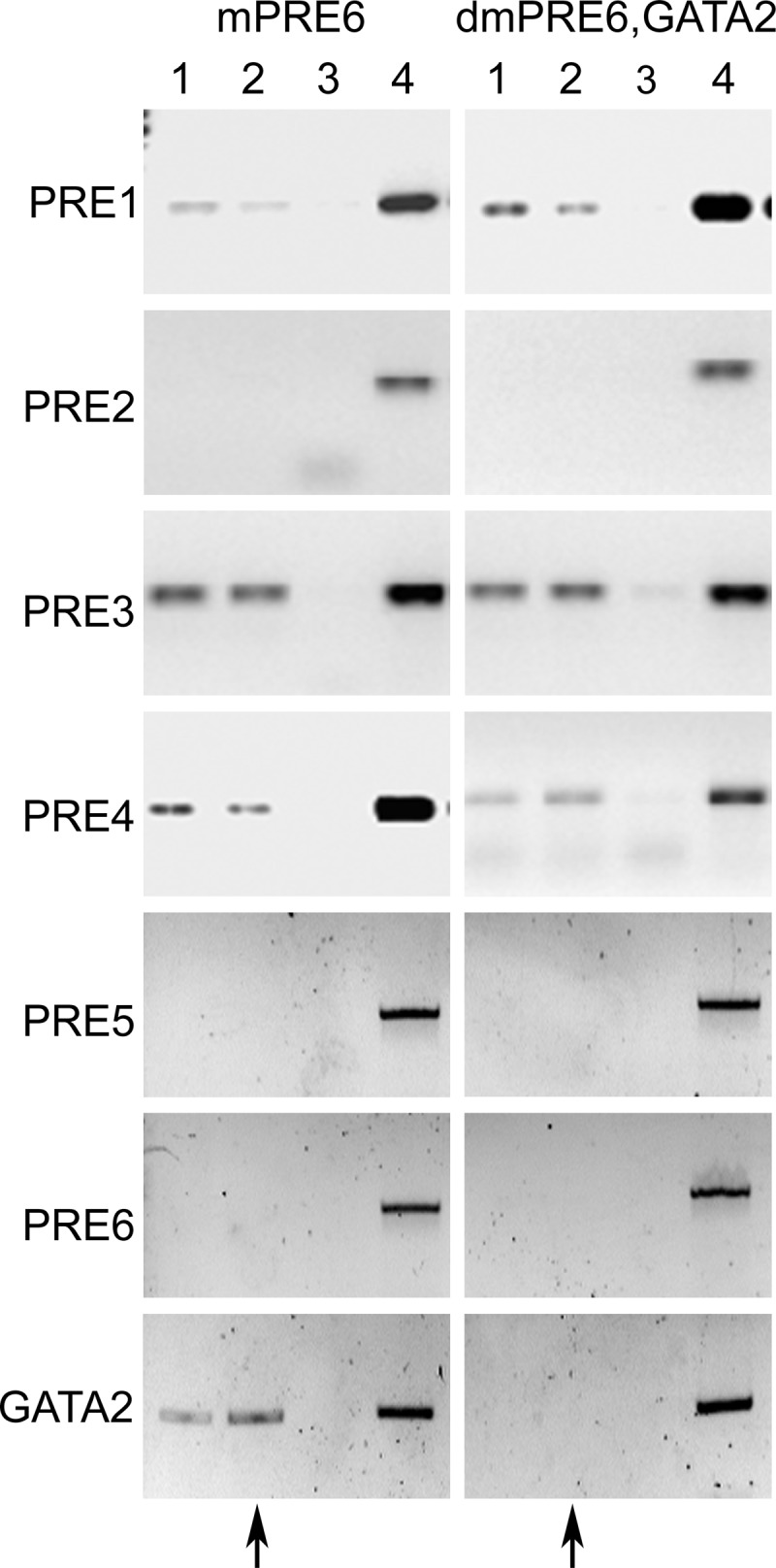
Effect of PRE6 and GATA2 mutations on PRB binding to other sites. ChIP PCR results for the different sites when PRE6 or PRE6 and GATA2 sites were mutated. Immunoprecipitations were carried with the anti-PRB antibody. Lane 1, hPRB; lane 2, hPRB and treated with progesterone; lane 3, as in lane 2 but with an unrelated antibody or IgG; lane 4, input control. The lower bands observed in some lanes are the dimers of oligonucleotides. The arrows indicate the lane where a positive PCR amplification is expected.
Gata2 enhances the progesterone response
The GATA2 site appeared to play an important role in the response to progesterone stimulation. We tested whether the Gata2 transcription factor was also important for that response by transfecting a FLAG-Gata2 expression vector along with the Ednra−5.7;−4.2-luc construct. Gata2 alone was sufficient to stimulate the promoter activity (Figure 7A). However, when progesterone was added to the cultured cells, the promoter activity did not increase further. When the PRB-encoding vector was transfected along with the Gata2 vector, the promoter activity remained similar. However, the promoter activity increased dramatically more than 600-fold when progesterone was added to the cell culture medium (Figure 7A). We then confirmed the specificity of the response using the vectors containing mutated PRE6, GATA2, or both sites. These experiments were performed in the presence of PRB and progesterone for all the samples. In the samples from cells transfected with the mutated PRE6 vector, the luciferase activity decreased (Figure 7B) to the same level as that of Gata2 stimulation in the absence of progesterone stimulation (Figure 7A). Mutation of PRE6 and GATA2 sites abrogated the luciferase activity to less than 10% of the original levels (no significant difference with control without Gata2), confirming the specificity of the response (Figure 7B). We examined whether progesterone induced native Gata2 expression in the transfected cells. A study on the effect of progesterone on the uterus indicated that Gata2 expression was regulated by the signaling mechanism (59). Transfection of PRB and treatment with progesterone increased Gata2 protein levels about 2-fold (2.1 ± 0.3) (Figure 7C). The rise appears sufficient to increase the luciferase activity as indicated above. We concluded that PRB is needed to stimulate the promoter activity in response to progesterone stimulation, and the synergy with Gata2 is needed to enhance the activity.
Figure 7.

Gata2 enhances the progesterone response without interacting directly with PRB. A, Luciferase assays revealing that Gata2 enhances the response to progesterone stimulation in the presence of PRB; ns, not significant. B, Mutating the PRE6 and GATA2 sites in the Ednra−5.7;−4.2 region abrogated the promoter activity. All samples were transfected with PRB and treated with progesterone. C, Progesterone (Progest.) treatment increases Gata2 protein levels about 2-fold after 24 hours of treatment in 293E cells transfected with PRB. Control cells treated with progesterone in the absence of PRB were transfected with the empty pcDNA3 expression vector. An extract from native 293E cells is loaded in the first lane. D, Immunoprecipitation (IP) assays revealed that PRB and Gata2 do not physically interact, even in presence of progesterone. The FLAG antibody (for Gata2) was used for the IP assays. The top image reveals the detection of PRB, actin, and indirectly Ig by Western blotting. Actin was used to reveal the input controls. The Gata2 input control is shown in the lower image.
Mechanistic analysis of PRB and Gata2 activity
We first tested whether PRB and Gata2 were physically interacting. Both proteins failed to coimmunoprecipitate, even in presence of progesterone (Figure 7D). Our ChIP results indicated that PRB was somehow interacting with the GATA2 site. We suspected that the PRB attached to PRE6 was interacting with the transcriptional complex associated with Gata2 after DNA folding. We tested this hypothesis using the EMSA. Assays were performed in presence of progesterone and both PRB and Gata2 proteins. PRB bound the PRE6 probe, and addition of the PRB antibody supershifted the complex (gray arrow in Figure 8A). Competitive assays by addition of cold probe in excess (10×) confirmed the specificity of PRB interaction with the probe. Addition of the FLAG antibody (for Gata2) did not supershift the complexes, indicating that Gata2 was not part of the protein complexes attached to the probe. When the GATA2 probe was assayed, a band appeared in the lane with the native 293E cell extract (Figure 8B, black arrow). The size or intensity of the complex increased in the lanes with cellular extracts from 293E cells transfected with Gata2-FLAG. Cellular extracts from cells transfected with PRB did not change the pattern. Addition of the FLAG or Gata2 antibody supershifted the protein-DNA complexes (gray arrow) and confirmed the identity of the Gata2-DNA complexes. The addition of PRB antibody failed to supershift the complexes. Competitive assays by addition of cold probe in excess (10×) confirmed the specificity of Gata2 interaction with the probe. The EMSA results suggested that the PRB interaction between the PRE6 and GATA2 sites observed in the earlier ChIP experiments was achieved by DNA folding and not by the direct attachment of PRB to the transcriptional complex sitting on the GATA2 site or vice versa. To test this hypothesis, we performed ChIP assays using the FLAG antibody for Gata2. Cells were treated with progesterone for these assays. The assay confirmed Gata2 binding to the GATA2 site, and mutation of this site (mGATA2 construct) prevented the attachment of the transcription factor, confirming the specificity of the assay (Figure 8C). We then assayed whether the PRE6 site was also immunoprecipitated along with the Gata2-DNA complexes. The PCR results revealed that PRE6 was pulled along only when PRB was present and the site was intact (Figure 8C). The interaction ceased when the PRE6 or GATA2 elements were mutated. Inactivation of the PRE6 site reduced (∼60%) Gata2 interaction with the GATA2 site, which suggests that DNA folding could stabilize the complex sitting on the GATA2 site and explain the ∼2-fold (2.2 ± 0.2) increase of Gata2 binding to the GATA2 site in presence of PRB (Figure 8C). Because the PRE6 element was not precipitated when the GATA2 site was mutated, we concluded that the identical and 2 almost identical sites to the FKBP5 GATA2 element (located on each side of the PRE6 element) were not involved in the progesterone-stimulated Gata2 response. Because PRB and Gata2 are not physically interacting, we examined whether the apparent interaction between PRB and Gata2 was mediated indirectly by the transcription complex assembled on the promoter. For these experiments, we performed ChIP assays using the TFIID antibody; TFIID is part of the RNA polymerase II preinitiation complex needed for gene transcription (60). We transfected the full-length Ednra −5.7;−4.2 luciferase vector or mutant PRE6, GATA2, and double-mutant constructs in 293E cells to avoid false-positive interference from genomic DNA during PCR amplification. In the cells transfected with the −5.7;−4.2 vector, amplification was seen only for the GATA2 region (Figure 8D). PRE6 amplification occurred in the samples from cells cotransfected with PRB and treated with progesterone. The GATA2 region was also amplified in these samples, and the small amplification increase may indicate that PRB stabilizes Gata2 interaction with the preinitiation complex as proposed earlier. Transfection of Gata2-FLAG along with PRB slightly increased GATA2 amplification but not PRE6. Amplification of PRE6 ceased when the site was mutated, but the GATA2 site was still pulled along with TFIID. The amplification pattern was reversed when the GATA2 site was mutated. When both the PRE6 and GATA2 sites were mutated, immunoprecipitation of TFIID failed to pull along these 2 sites. From these and other EMSA and ChIP results presented above, we concluded that the interaction between PRB and Gata2 is mediated by the preinitiation complex.
Figure 8.

The preinitiation complex mediates the interaction between PRB and Gata2. A and B, EMSA confirming PRB interaction with the PRE6 site (black arrows in A) and Gata2 interaction with the GATA2 site (black arrow in B). The DNA-protein complexes supershifted with the addition of PRB antibody (Ab) (gray arrow in A) or the addition of the FLAG or Gata2 antibodies (gray arrow in B). The competitive addition of excess cold probe (10×) displaced the labeled probe from the DNA-protein complexes (*, probe artifact). Cell extracts were obtained from transfected 293E cells. C, ChIP assays with the FLAG antibody for Gata2 confirmed Gata2 interaction with the GATA2 site in 293E cells cotransfected with the Ednra−5.7;−4.2 construct. Cells were also treated with progesterone. D, ChIP assays with TFIID antibody confirmed PRB and Gata2 interaction with the preinitiation complex. mGATA2, mPRE6, and dmPRE6;GATA2 are vectors containing mutated GATA2, PRE6, and PRE6 and GATA2 sites, respectively.
Respective contribution of PRE6 and GATA2 to the progesterone response
We analyzed what was the respective contribution of the PRE6 and GATA2 sites to the progesterone response. For these experiments, we used a specific siRNA to reduce Gata2 protein levels. The siRNA efficiently reduced Gata2 levels, particularly in 293E cells stimulated by progesterone (Figure 9A). We cotransfected the cells with the siRNA or control scrambled sequence, PRB, and different luciferase constructs and treated them with progesterone. The addition of the Gata2 siRNA significantly reduced the luciferase activity but not as efficiently as mutating the GATA2 site (Figure 9B). Comparisons of the intact construct with the mutated constructs indicated that the direct action of PRB on the enhancer contributes to about half of the response and the indirect action of PRB via Gata2 expression increase contributes to the other half of the progesterone response. The results also revealed that no other GATA sites were involved in the progesterone response. The experiments also confirmed the pivotal role of PRE6 in the progesterone response. Together these results revealed that DNA folding is the likely mechanism allowing the interaction between PRE6 and GATA2 sites, via PRB and Gata2 binding their respective sites.
Figure 9.

Evaluation of PRB and Gata2 contribution to the progesterone regulation of Ednra expression. A, The Gata2 siRNA efficiently reduced Gata2 protein levels in 293E cells at 200μM concentration, including in cells transfected with PRB and treated with progesterone for 24 hours. The scrambled sequence (Scr.) control siRNA had no effect on Gata2 protein levels and did not prevent the increase of Gata2 protein in PRB-transfected cells treated with progesterone. B, The Gata2 siRNA decreased the luciferase activity similarly to GATA2 mutation (mGATA2) in cells cotransfected with the Ednra−5.7;−4.2 construct or with specific mutated sites (mPRE6, mGATA2, and dmPRE6;GATA2) or missing all the PRE elements (ΔPRE1;PRE6). However, the siRNA failed to decrease further the activity in cells transfected with the mGATA2 vector or in cells transfected with the ΔPRE1;PRE6 construct compared with the dmPRE6;GATA2 control. Control cells were cotransfected with the scrambled control siRNA with the exception of the cells transfected with the dmPRE6;GATA2 vector. Two-way ANOVA: #, P < .001; *, P < .01; ^^, P < .05; ns, not significant. Error bars represent SEs.
Gata2 participates in the regulation of Ednra expression in vivo
We used cultured primary myometrium and kidney cells to test whether Gata2 was regulating Ednra expression. Cells were cultured in the presence of progesterone and collected for qRT-PCR analysis of Gata2 and Ednra expression. An opposite response to the early increase of Ednra expression was observed for Gata2, suggesting that the down-regulation of Gata2 may be a component of the negative feedback response mechanism to control Ednra expression (Figure 10, A and B). Afterward, the increase of Gata2 expression was preceding or matching the increase of Ednra expression at 24 hours. The expression analyses were then correlated with Ednra and Gata2 protein levels in these cells. Proteins were analyzed by immunoblotting. After 6 hours of treatment with progesterone, Ednra abundance was increased in the primary kidney cells, whereas Gata2 protein levels were greatly decreased (Figure 10D). Treatment with the carrier slightly but not significantly altered the abundance of these 2 proteins. Similar results were observed with the primary myometrium cells (data not shown). A time-lapse analysis of the Ednra and Gata2 protein levels was also performed in the myometrial cells (Figure 10C). Ednra levels increased at first upon treatment with progesterone. Eventually, the levels gradually decreased and then increased, following the same pattern observed with the gene expression. The levels of Gata2 protein abundance were matching the expression of the Ednra gene. Initially, upon treatment with progesterone, Gata2 levels remained elevated, matching the increased Ednra expression. Then the protein levels decreased, correlating with the reduced Ednra expression. As Ednra protein levels were decreased, Gata2 expression and protein abundance increased, which were then matched by an elevation of Ednra expression.
Figure 10.

Regulation of Gata2 expression by progesterone in primary cultured cells. A and B, Relative qRT-PCR quantification of Gata2 and Ednra expression in cultured primary myometrium (A) and kidney (B) cells treated with 4 ng/ml progesterone for up to 24 hours. Control cells were treated with the carrier, and results were standardized with 18S rRNA. Error bars represent SEs. C, Time-lapse analysis of Ednra and Gata2 proteins in cultured primary myometrium cells treated with 4 ng/ml progesterone. Actin was used as a loading control. The numbers below the blots represent the relative abundance of the proteins (compared with time 0). D, Analysis of Ednra and Gata2 proteins in cultured primary kidney cells after 6 hours of treatment with progesterone (Progest.) or carrier. E, Relative luciferase activity in primary myometrium and kidney cells transfected with the Ednra−5.7;−4.2-luc vector and treated with the carrier (black bars) or progesterone (white bars; #, P < .01 vs carrier) or in progesterone-treated cells transfected with the mutated GATA2 site (mGATA2, gray bars; *, P < .05 vs native vector). F, ChIP assay using the TFIID antibody revealed that the PRE1, PRE5, PRE6, and GATA2 sites are recruited in the uterus of 18-day pregnant mice. IgG was used as a negative control (input with PRE3 for that sample).
We also transfected the primary cells with the −5.7;−4.2 luciferase vector to confirm that progesterone stimulates the Ednra promoter in these cells and that Gata2 was involved in that response. The addition of progesterone to the cultured primary cells stimulated the luciferase activity about 12- and 8-fold in the uterine and renal cells, respectively (Figure 10E). We transfected the primary cells with the construct containing the mutated GATA2-binding site to evaluate the contribution of Gata2 to the progesterone response. The mutation significantly reduced the luciferase activity by ∼40% (P = .021) in the myometrial cells and ∼35% (P = .032) in the renal cells. These results indicate that Gata2 is normally part of the regulatory mechanisms controlling Ednra expression. We then analyzed whether PRB and Gata2 roles were biologically significant for Ednra expression. We used the uterus of 18-day pregnant mice for these ChIP assays, and we used the TFIID antibody to immunoprecipitate the DNA-protein complexes. The pulled-down DNA was amplified by PCR. The analysis revealed that the PRE1, -5, and -6 and GATA2 sites were pulled along with the preinitiation complexes (Figure 10F). Quantitative PCR analysis and band intensity indicated that PRE1 was not as commonly used as PRE5 as the second PRE site working with PRE6. Surprisingly, PRE3 and 4 were not precipitated, which may explain why these 2 sites did not appear to have any function in the mutation analysis. These results confirm that PRB and Gata2 synergize to regulate Ednra expression.
Discussion
Ednra is a 7-pass transmembrane G protein-coupled receptor (1, 2). It plays a role in the physiological control of blood pressure/vasoconstriction and regulation of neural crest cell development during embryogenesis and may also be a key to different unrelated pathologies, including cancer, eclamptic pregnancy, and periodontitis. Despite its important functions, the molecular mechanisms regulating its expression are unknown. Elucidating these mechanisms may help understand the etiology of some pathology. In our attempt to distinguish some of these regulatory mechanisms, we identified progesterone signaling as a possibility. Elevated progesterone levels are associated with pregnancy (41). Our results indicate that pregnancy positively influences the expression of Ednra in mouse maternal tissues. Our experiments in ovariectomized mice revealed that progesterone is the factor altering Ednra expression during pregnancy.
We identified a crucial region upstream of the mouse Ednra gene responding directly to progesterone stimulation. This conserved region between −5.7 and −4.2 kb is just upstream of the region containing the putative basal/core promoter of the gene, which is, according to ENSEMBL and Vista analyses, located in the −4.2- to −4.0-kb region. The position of the basal promoter is consistent with the 5′-UTR transcription start site at −4005 bp; the location appears conserved in other species, including rat and human (Vista and ENSEMBL analyses). The −5.7- to −4.2-kb region contains 6 PREs. The presence of more than 1 PRE site has been observed previously in the regulatory sequences of other genes (50, 52, 57). None of these PRE elements are a palindrome, but PRE half-sites or intermediates between half-sites with a few conserved palindromic nucleotides are more commonly found than palindromic PRE elements themselves (58). Interestingly, PRE elements are similar to glucocorticoid response elements with their TGTTCT core sequence (61), which could explain the regulation of the receptor's expression by glucocorticoids (24–26). The sequential deletion of PRE sites suggests that the spatial organization of these sites may be important for the activation by progesterone. Our experiments identified PRE6 as the most important regulatory element for the progesterone response. However, when only this PRE6 remained, the progesterone response was greatly reduced, suggesting that at least 2 PRE sites were needed to sustain the response. From our results, it appears that PRE1 and PRE5 have supporting roles in the activation of Ednra expression by progesterone.
Other transcription factors may interact with PRB sitting on a PRE element (ie, c-Fos) (54) or with PRB not interacting with a PRE site (ie, Sp1) (55). Our results revealed that a GATA2 site is also essential for the activation of Ednra expression by progesterone. The use of a GATA2 site, in addition to the classical PRE element, is unusual for gene expression regulation by progesterone. A mechanism using Gata2 was previously observed in the activation of FKBP5 and appears complementary to the classical mechanism of other nuclear receptors (57). For the FKBP5 gene, Gata2 appears to bind the PRB dimer sitting on the intronic PRE site and not another site; the GATA2 binding element is located within the PRE element. Two almost identical sites are found 6 nucleotides away on each side of the PRE6 element, and 1 identical site was found within PRE6. However, these 3 sites are not involved in the progesterone response as revealed by the ChiP, EMSA, and luciferase results. It is possible that the reverse orientation of the identical GATA2 sequence within PRE6 prevented Gata2 from binding to that site with the PRB dimer present. Instead, the recruitment of a GATA2 element located 127 nucleotides away was essential for the maximal response of the enhancer to progesterone stimulation. We could not find another gene whose expression was regulated similarly by PRB and Gata2. The recruitment of a distant GATA2 element and recruitment of Gata2 to enhance a progesterone response represent is a novel mechanism previously unknown. From our results, it appears that the interaction between PRB and Gata2 with the preinitiation complex occurs independently, but PRB may stabilize Gata2 interaction with the complex (Figure 11). Our next step will be to determine whether the involvement of Gata2 leads to the formation of a different transcription complex assembly and whether distinct PRB dimers bind PRE6 and the other PRE site needed for the progesterone response.
Figure 11.

Mechanistic model of Ednra regulation by progesterone. PRB dimer binds the PRE6 site and possibly another PRE element and interacts, along with Gata2 attached to the GATA2 site, with the preinitiation complex (PIC) containing the RNA polymerase II (Pol II) and the TFIID. These interactions may involve the mediator complex or other factors serving as intermediates between PRB/Gata2 and the PIC.
Interestingly, progesterone is a known activator of Gata2 expression (59). We hypothesize this may help sustain Ednra expression, thus complementing the activation regulated by PRE6, as indicated by our results. The activation of Gata2 expression by progesterone can also explain why an indirect progesterone response was observed in presence of PRB when all PRE sites were removed. This regulation by PRB and Gata2 may be conserved because PRE and GATA sites are also present in the same region of the human EDNRA gene (Supplemental Figure 4), although these sites are not always perfectly aligning with the murine elements. Their presence suggests that the mechanism may have a function in different pathological and physiological conditions, including parturition.
The uterus is well known for its response to progesterone stimulation. Endothelin signaling may regulate uterine smooth muscle contractions, as hypothesized by others (62, 63). The observed elevation of Ednra expression in late pregnancy may help trigger the onset of parturition, but what mechanism prevents their premature onset is unknown. However, whereas progesterone levels are high in late pregnancy, they drop just before parturition. We hypothesize that progesterone regulates the expression of another factor inhibiting Ednra activity. The hormonal changes in late pregnancy could alleviate the repression, thus allowing the activation of Ednra. The control of Ednra expression by progesterone may have other functions in uterine cells because the expression of the gene is up-regulated in uterine leiomyoma, a common benign tumor (64–66). Activation of endothelin signaling in these transformed cells protects them from apoptotic death and increases their proliferation. In regard to other tumors, no direct link between uterine cancer and endothelin signaling has been reported, but a link with other cancers was established, particularly with female cancers (67). Our discovery of progesterone regulating Ednra expression may provide an explanation for that link. Furthermore, endothelin signaling, including the upregulation of the Ednra expression, is particularly linked to a poor prognosis for breast cancer (14, 68, 69). This poor prognosis may be related to the osteoblastic metastasis function associated with endothelin signaling (13, 15). Based on our results, we anticipate that progesterone may be involved in the process by modulating Ednra expression, thus favoring the establishment of metastases in their new environment.
With progesterone, adverse conditions associated with pregnancy may be more relevant. Overactivation of the endothelin system is observed with some pre-eclamptic and eclamptic pregnancies (17, 20, 70). Kidney functions are often affected in eclamptic pregnancies, which could explain the similar response in kidney cells. Although this type of pregnancy complication is mostly associated with the upregulation of Edn1 production, the link between progesterone and Ednra may also be involved in the pathological condition by increasing tissue response to the overproduction of Edn1. Interestingly, one of the associated risk factors for eclamptic pregnancies is periodontitis (71). Increased Ednra expression has been observed in the gingival tissues of afflicted patients (16). Intake of progesterone or progesterone-like substances in female contraceptives has been associated with periodontitis for a long period of time (72, 73) or with circulatory complications, including stroke, cardiac infraction, and thrombosis (74–76). These complications can involve endothelin signaling (1, 2). Because the incidence of periodontitis and its progression increases during pregnancy (77, 78), the link between progesterone, Ednra, and periodontitis is worth investigating in the future. The association between progesterone and Ednra may provide a molecular link between pre-eclampsia/eclampsia and periodontitis and clarify why periodontal treatments during pregnancy do not decrease the risks of developing the gestational complications too often associated with premature birth (79, 80). In conclusion, investigating the connection between progesterone, Ednra, and diseases is important and may help understanding the etiology of some pathological conditions.
Supplementary Material
Acknowledgments
We thank Dr Hany Abdel-Hafiz (University of Colorado Health Science Center Aurora, Aurora, Colorado) for the PRB clone and Dr Lisa Chen for the use of her FLUOStar microplate reader. We thank Ms Jeanne Santa Cruz for her help with the manuscript and the other members of the Ruest laboratory for their support.
This work was supported by funding from the National Institutes of Health (NIH), U24 DE16472 (startup funds to L.B.R.), and a research development grant from the Texas A&M Health Science Center Office of the Vice-President for Research and Graduate Studies (to L.B.R.). The work was partially supported by fellowships from the NIH, T32-DE018380 (to Y.Z.) and the Baylor Oral Health Foundation summer student research program (to M.D.B. and G.R.K.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BAC
- bacterial artificial chromosome
- ChIP
- chromatin immunoprecipitation
- Edn1
- endothelin-1
- Ednra
- endothelin-A receptor
- β-gal
- β-galactosidase
- PRB
- progesterone receptor B
- PRE
- progesterone response element
- qRT-PCR
- quantitative real-time PCR
- siRNA
- small interfering RNA
- TFIID
- transcription factor IID
- UTR
- untranslated region.
References
- 1. Kedzierski RM, Yanagisawa M. Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol. 2001;41:851–876 [DOI] [PubMed] [Google Scholar]
- 2. Barton M, Yanagisawa M. Endothelin: 20 years from discovery to therapy. Can J Physiol Pharmacol. 2008;86:485–498 [DOI] [PubMed] [Google Scholar]
- 3. Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005;85:1159–1204 [DOI] [PubMed] [Google Scholar]
- 4. Dettlaff-Swiercz DA, Wettschureck N, Moers A, Huber K, Offermanns S. Characteristic defects in neural crest cell-specific Gαq/Gα11- and Gα12/Gα13-deficient mice. Dev Biol. 2005;282:174–182 [DOI] [PubMed] [Google Scholar]
- 5. Clouthier DE, Hosoda K, Richardson JA, et al. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998;125:813–824 [DOI] [PubMed] [Google Scholar]
- 6. Ruest LB, Xiang X, Lim KC, Levi G, Clouthier DE. Endothelin-A receptor-dependent and -independent signaling pathways in establishing mandibular identity. Development. 2004;131:4413–4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ruest LB, Clouthier DE. Elucidating timing and function of endothelin-A receptor signaling during craniofacial development using neural crest cell-specific gene deletion and receptor antagonism. Dev Biol. 2009;328:94–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hirata Y. Endothelin-1 receptors in cultured vascular smooth muscle cells and cardiocytes of rats. J Cardiovasc Pharmacol. 1989;13(Suppl 5):S157–S158 [DOI] [PubMed] [Google Scholar]
- 9. LaMarca B, Parrish M, Ray LF, et al. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin-1. Hypertension. 2009;54:905–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kohan DE. Endothelin, hypertension and chronic kidney disease: new insights. Curr Opin Nephrol Hypertens. 2010;19:134–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wendel M, Knels L, Kummer W, Koch T. Distribution of endothelin receptor subtypes ETA and ETB in the rat kidney. J Histochem Cytochem. 2006;54:1193–1203 [DOI] [PubMed] [Google Scholar]
- 12. Neuhofer W, Pittrow D. Role of endothelin and endothelin receptor antagonists in renal disease. Eur J Clin Invest. 2006;36(Suppl 3):78–88 [DOI] [PubMed] [Google Scholar]
- 13. Guise TA, Yin JJ, Mohammad KS. Role of endothelin-1 in osteoblastic bone metastases. Cancer. 2003;97:779–784 [DOI] [PubMed] [Google Scholar]
- 14. Wülfing P, Diallo R, Kersting C, et al. Expression of endothelin-1, endothelin-A, and endothelin-B receptor in human breast cancer and correlation with long-term follow-up. Clin Cancer Res. 2003;9:4125–4131 [PubMed] [Google Scholar]
- 15. Yin JJ, Mohammad KS, Käkönen SM, et al. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc Natl Acad Sci U S A. 2003;100:10954–10959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fujioka D, Nakamura S, Yoshino H, et al. Expression of endothelins and their receptors in cells from human periodontal tissues. J Periodontal Res. 2003;38:269–275 [DOI] [PubMed] [Google Scholar]
- 17. George EM, Granger JP. Endothelin: key mediator of hypertension in preeclampsia. Am J Hypertens. 2011;24:964–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Collett GP, Kohnen G, Campbell S, Davenport AP, Jeffers MD, Cameron IT. Localization of endothelin receptors in human uterus throughout the menstrual cycle. Mol Hum Reprod. 1996;2:439–444 [DOI] [PubMed] [Google Scholar]
- 19. Bacon CR, Morrison JJ, O'Reilly G, Cameron IT, Davenport AP. ETA and ETB endothelin receptors in human myometrium characterized by the subtype selective ligands BQ123, BQ3020, FR139317 and PD151242. J Endocrinol. 1995;144:127–134 [DOI] [PubMed] [Google Scholar]
- 20. Wolff K, Faxén M, Lunell NO, Nisell H, Lindblom B. Endothelin receptor type A and B gene expression in human nonpregnant, term pregnant, and preeclamptic uterus. Am J Obstet Gynecol. 1996;175:1295–1300 [DOI] [PubMed] [Google Scholar]
- 21. O'Reilly G, Charnock-Jones DS, Davenport AP, Cameron IT, Smith SK. Presence of messenger ribonucleic acid for endothelin-1, endothelin-2, and endothelin-3 in human endometrium and a change in the ratio of ETA and ETB receptor subtype across the menstrual cycle. J Clin Endocrinol Metab. 1992;75:1545–1549 [DOI] [PubMed] [Google Scholar]
- 22. Li P, Oparil S, Sun JZ, Thompson JA, Chen YF. Fibroblast growth factor mediates hypoxia-induced endothelin: a receptor expression in lung artery smooth muscle cells. J Appl Physiol. 2003;95:643–651; discussion 863 [DOI] [PubMed] [Google Scholar]
- 23. Nilsson D, Wackenfors A, Gustafsson L, et al. PKC and MAPK signalling pathways regulate vascular endothelin receptor expression. Eur J Pharmacol. 2008;580:190–200 [DOI] [PubMed] [Google Scholar]
- 24. Villeneuve A, Bernier C, Provencher PH. Regulation of endothelin-1 and ET-A receptor expression by glucocorticoids: implications in blood pressure control. Curr Topics Steroid Res. 1999;2:69–80 [Google Scholar]
- 25. Villeneuve A, Gignac S, Provencher PH. Glucocorticoids decrease endothelin-A- and -B-receptor expression in the kidney. J Cardiovasc Pharmacol. 2000;36:S238–240 [PubMed] [Google Scholar]
- 26. Kutzler MA, Molnar J, Schlafer DH, Kuc RE, Davenport AP, Nathanielsz PW. Maternal dexamethasone increases endothelin-1 sensitivity and endothelin a receptor expression in ovine foetal placental arteries. Placenta. 2003;24:392–402 [DOI] [PubMed] [Google Scholar]
- 27. Piekorz RP, Gingras S, Hoffmeyer A, Ihle JN, Weinstein Y. Regulation of progesterone levels during pregnancy and parturition by signal transducer and activator of transcription 5 and 20α-hydroxysteroid dehydrogenase. Mol Endocrinol. 2005;19:431–440 [DOI] [PubMed] [Google Scholar]
- 28. Sharma R, Bulmer D. The effects of ovariectomy and subsequent progesterone replacement on the uterus of the pregnant mouse. J Anat. 1983;137 (Pt 4):695–703 [PMC free article] [PubMed] [Google Scholar]
- 29. Inada K, Hayashi S, Iguchi T, Sato T. Establishment of a primary culture model of mouse uterine and vaginal stroma for studying in vitro estrogen effects. Exp Biol Med (Maywood). 2006;231:303–310 [DOI] [PubMed] [Google Scholar]
- 30. Palmberg L, Thyberg J. Uterine smooth muscle cells in primary culture. Alterations in fine structure, cytoskeletal organization and growth characteristics. Cell Tissue Res. 1986;246:253–262 [DOI] [PubMed] [Google Scholar]
- 31. Rybak SL, Murphy RF. Primary cell cultures from murine kidney and heart differ in endosomal pH. J Cell Physiol. 1998;176:216–222 [DOI] [PubMed] [Google Scholar]
- 32. Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd ed Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001 [Google Scholar]
- 33. Tong Q, Tsai J, Tan G, Dalgin G, Hotamisligil GS. Interaction between GATA and the C/EBP family of transcription factors is critical in GATA-mediated suppression of adipocyte differentiation. Mol Cell Biol. 2005;25:706–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ruest LB, Marcotte R, Wang E. Peptide elongation factor eEF1A-2/S1 expression in cultured differentiated myotubes and its protective effect against caspase-3-mediated apoptosis. J Biol Chem. 2002;277:5418–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bergeron C, Ferenczy A, Toft DO, Schneider W, Shyamala G. Immunocytochemical study of progesterone receptors in the human endometrium during the menstrual cycle. Lab Invest. 1988;59:862–869 [PubMed] [Google Scholar]
- 36. Aparicio O, Geisberg JV, Sekinger E, Yang A, Moqtaderi Z, Struhl K. Chromatin immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo. Curr Protoc Mol Biol. 2005;Chapter 21:Unit 21.23 [DOI] [PubMed] [Google Scholar]
- 37. Willis D, Zhang Y, Molloy GR. Transcription of brain creatine kinase in U87-MG glioblastoma is modulated by factor AP2. Biochim Biophys Acta. 2005;1728:18–33 [DOI] [PubMed] [Google Scholar]
- 38. Ye J, Cippitelli M, Dorman L, Ortaldo JR, Young HA. The nuclear factor YY1 suppresses the human γ interferon promoter through two mechanisms: inhibition of AP1 binding and activation of a silencer element. Mol Cell Biol. 1996;16:4744–4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Loots GG, Ovcharenko I. rVISTA 2.0: evolutionary analysis of transcription factor binding sites. Nucleic Acids Res. 2004;32:W217–W221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee EC, Yu D, Martinez de Velasco J, et al. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73:56–65 [DOI] [PubMed] [Google Scholar]
- 41. Boron WF, Boulpaep EL. Medical Physiology: A Cellular and Molecular Approach. Philadelphia, PA: Elsevier Saunders; 2005 [Google Scholar]
- 42. Mesiano S, Wang Y, Norwitz ER. Progesterone receptors in the human pregnancy uterus: do they hold the key to birth timing? Reprod Sci. 2011;18:6–19 [DOI] [PubMed] [Google Scholar]
- 43. Davies IJ, Ryan KJ. The uptake of progesterone by the uterus of the pregnant rat in vivo and its relationship to cytoplasmic progesterone-binding protein. Endocrinology. 1972;90:507–515 [DOI] [PubMed] [Google Scholar]
- 44. Haukkamaa M. Binding of progesterone by rat myometrium during pregnancy and by human myometrium in late pregnancy. J Steroid Biochem. 1974;5:73–79 [DOI] [PubMed] [Google Scholar]
- 45. Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451–486 [DOI] [PubMed] [Google Scholar]
- 46. McDonald MW, Diokno AC, Seski JC, Menon KM. Measurement of progesterone receptor in human renal cell carcinoma and normal renal tissue. J Surg Oncol. 1983;22:164–166 [DOI] [PubMed] [Google Scholar]
- 47. Bumke-Vogt C, Bahr V, Diederich S, et al. Expression of the progesterone receptor and progesterone: metabolising enzymes in the female and male human kidney. J Endocrinol. 2002;175:349–364 [DOI] [PubMed] [Google Scholar]
- 48. Cartharius K, Frech K, Grote K, et al. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942 [DOI] [PubMed] [Google Scholar]
- 49. Schug J. Using TESS to Predict Transcription Factors Binding Sites in DNA Sequence. In: Baxevanis AD, ed. Current Protocols in Bioinformatics. New York, NY: J Wiley, Sons; 2003 [DOI] [PubMed] [Google Scholar]
- 50. Chalepakis G, Arnemann J, Slater E, Brüller HJ, Gross B, Beato M. Differential gene activation by glucocorticoids and progestins through the hormone regulatory element of mouse mammary tumor virus. Cell. 1988;53:371–382 [DOI] [PubMed] [Google Scholar]
- 51. Connaghan-Jones KD, Heneghan AF, Miura MT, Bain DL. Thermodynamic dissection of progesterone receptor interactions at the mouse mammary tumor virus promoter: monomer binding and strong cooperativity dominate the assembly reaction. J Mol Biol. 2008;377:1144–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jacobsen BM, Jambal P, Schittone SA, Horwitz KB. ALU repeats in promoters are position-dependent co-response elements (coRE) that enhance or repress transcription by dimeric and monomeric progesterone receptors. Mol Endocrinol. 2009;23:989–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Connor EE, Meyer MJ, Li RW, Van Amburgh ME, Boisclair YR, Capuco AV. Regulation of gene expression in the bovine mammary gland by ovarian steroids. J Dairy Sci. 2007;90(Suppl 1):E55–E65 [DOI] [PubMed] [Google Scholar]
- 54. Musgrove EA, Lee CS, Sutherland RL. Progestins both stimulate and inhibit breast cancer cell cycle progression while increasing expression of transforming growth factor α, epidermal growth factor receptor, c-fos, and c-myc genes. Mol Cell Biol. 1991;11:5032–5043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. Progesterone regulates transcription of the p21(WAF1) cyclin-dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem. 1998;273:10696–10701 [DOI] [PubMed] [Google Scholar]
- 56. Bamberger AM, Bamberger CM, Gellersen B, Schulte HM. Modulation of AP-1 activity by the human progesterone receptor in endometrial adenocarcinoma cells. Proc Natl Acad Sci U S A. 1996;93:6169–6174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Magklara A, Smith CL. A composite intronic element directs dynamic binding of the progesterone receptor and GATA-2. Mol Endocrinol. 2009;23:61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yin P, Roqueiro D, Huang L, et al. Genome-wide progesterone receptor binding: cell type-specific and shared mechanisms in T47D breast cancer cells and primary leiomyoma cells. PLoS One. 2012;7:e29021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jeong JW, Lee KY, Kwak I, et al. Identification of murine uterine genes regulated in a ligand-dependent manner by the progesterone receptor. Endocrinology. 2005;146:3490–3505 [DOI] [PubMed] [Google Scholar]
- 60. Kornberg RD. The molecular basis of eukaryotic transcription. Proc Natl Acad Sci U S A. 2007;104:12955–12961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nelson CC, Hendy SC, Shukin RJ, et al. Determinants of DNA sequence specificity of the androgen, progesterone, and glucocorticoid receptors: evidence for differential steroid receptor response elements. Mol Endocrinol. 1999;13:2090–2107 [DOI] [PubMed] [Google Scholar]
- 62. Dallot E, Pouchelet M, Gouhier N, Cabrol D, Ferré F, Breuiller-Fouché M. Contraction of cultured human uterine smooth muscle cells after stimulation with endothelin-1. Biol Reprod. 2003;68:937–942 [DOI] [PubMed] [Google Scholar]
- 63. Di Liberto G, Dallot E, Eude-Le Parco I, Cabrol D, Ferré F, Breuiller-Fouché M. A critical role for PKCζ in endothelin-1-induced uterine contractions at the end of pregnancy. Am J Physiol Cell Physiol. 2003;285:C599–C607 [DOI] [PubMed] [Google Scholar]
- 64. Raymond MN, Bole-Feysot C, Banno Y, Tanfin Z, Robin P. Endothelin-1 inhibits apoptosis through a sphingosine kinase 1-dependent mechanism in uterine leiomyoma ELT3 cells. Endocrinology. 2006;147:5873–5882 [DOI] [PubMed] [Google Scholar]
- 65. Raymond MN, Robin P, De Zen F, Vilain G, Tanfin Z. Differential endothelin receptor expression and function in rat myometrial cells and leiomyoma ELT3 cells. Endocrinology. 2009;150:4766–4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Weston G, Trajstman AC, Gargett CE, Manuelpillai U, Vollenhoven BJ, Rogers PA. Fibroids display an anti-angiogenic gene expression profile when compared with adjacent myometrium. Mol Hum Reprod. 2003;9:541–549 [DOI] [PubMed] [Google Scholar]
- 67. Smollich M, Wülfing P. Targeting the endothelin system: novel therapeutic options in gynecological, urological and breast cancers. Expert Rev Anticancer Ther. 2008;8:1481–1493 [DOI] [PubMed] [Google Scholar]
- 68. Ha NH, Nair VS, Reddy DN, et al. Lactoferrin-endothelin-1 axis contributes to the development and invasiveness of triple-negative breast cancer phenotypes. Cancer Res. 2011;71:7259–7269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Smollich M, Wülfing P. The endothelin axis: a novel target for pharmacotherapy of female malignancies. Curr Vasc Pharmacol. 2007;5:239–248 [DOI] [PubMed] [Google Scholar]
- 70. Margarit L, Griffiths A, Tsapanos V, Decavalas G, Gumenos D. Second trimester amniotic fluid endothelin concentration. A possible predictor for pre-eclampsia. J Obstet Gynaecol. 2005;25:18–20 [DOI] [PubMed] [Google Scholar]
- 71. Conde-Agudelo A, Villar J, Lindheimer M. Maternal infection and risk of preeclampsia: systematic review and metaanalysis. Am J Obstet Gynecol. 2008;198:7–22 [DOI] [PubMed] [Google Scholar]
- 72. Sooriyamoorthy M, Gower DB. Hormonal influences on gingival tissue: relationship to periodontal disease. J Clin Periodontol. 1989;16:201–208 [DOI] [PubMed] [Google Scholar]
- 73. Deasy MJ, Vogel RI. Female sex hormonal factors in periodontal disease. Ann Dent. 1976;35:42–46 [PubMed] [Google Scholar]
- 74. Shapiro S. Oral contraceptives, hormone therapy and cardiovascular risk. Climacteric. 2008;11:355–363 [DOI] [PubMed] [Google Scholar]
- 75. Lidegaard Ø, Nielsen LH, Skovlund CW, Skjeldestad FE, Løkkegaard E. Risk of venous thromboembolism from use of oral contraceptives containing different progestogens and oestrogen doses: Danish cohort study, 2001–9. BMJ. 2011;343:d6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dunn N. The risk of deep venous thrombosis with oral contraceptives containing drospirenone. BMJ. 2011;342:d2519. [DOI] [PubMed] [Google Scholar]
- 77. Cohen DW, Friedman L, Shapiro J, Kyle GC. A longitudinal investigation of the periodontal changes during pregnancy. J Periodontol. 1969;40:563–570 [DOI] [PubMed] [Google Scholar]
- 78. Cohen DW, Shapiro J, Friedman L, Kyle GC, Franklin S. A longitudinal investigation of the periodontal changes during pregnancy and fifteen months post-partum. II. J Periodontol. 1971;42:653–657 [DOI] [PubMed] [Google Scholar]
- 79. Michalowicz BS, Hodges JS, DiAngelis AJ, et al. Treatment of periodontal disease and the risk of preterm birth. N Engl J Med. 2006;355:1885–1894 [DOI] [PubMed] [Google Scholar]
- 80. Offenbacher S, Beck JD, Jared HL, et al. Effects of periodontal therapy on rate of preterm delivery: a randomized controlled trial. Obstet Gynecol. 2009;114:551–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.