

Background: Scap controls cholesterol by transporting SREBPs from ER to Golgi in a sterol-sensitive fashion. Two luminal loops of Scap (Loops 1 and 7) may play a regulatory role.
Results: A point mutation in Scap Loop 7 prevents interaction with Loop 1 and abolishes ER exit.
Conclusion: Scap movement and activation of SREBPs require interaction between Loops 1 and 7.
Significance: This work provides insight into how cells control cholesterol.
Keywords: Cholesterol-binding Protein, Cholesterol Regulation, Membrane Function, Membrane Lipids, Membrane Proteins, Scap, Sterol Regulatory Element-binding Protein (SREBP)
Abstract
Scap is a polytopic protein of the endoplasmic reticulum (ER) that controls cholesterol homeostasis by transporting sterol regulatory element-binding proteins (SREBPs) from the ER to the Golgi complex. Scap has eight transmembrane helices (TM) joined by four small hydrophilic loops and three large loops. Two of the large loops (Loops 1 and 7) are in the ER lumen, and the other large loop (Loop 6) faces the cytosol where it binds COPII proteins that initiate transport to Golgi. Cholesterol binding to Loop 1 alters the configuration of Loop 6, precluding COPII binding and preventing the exit of Scap from the ER. Here, we create a point mutation (Y640S) in luminal Loop 7 that prevents Scap movement to Golgi. Trypsin cleavage assays show that Loop 6 of Scap(Y640S) is always in the configuration that precludes COPII binding, even in the absence of cholesterol. When expressed separately by co-transfection, the NH2-terminal portion of Scap (containing TM helices 1–6, including Loop 1) binds to the COOH-terminal portion (containing TM helices 7–8 and Loop 7) as determined by co-immunoprecipitation. This binding does not occur when Loop 7 contains the Y640S mutation. Co-immunoprecipitation is also abolished by a point mutation in Loop 1 (Y234A) that also prevents Scap movement. These data suggest that Scap Loop 1 must interact with Loop 7 to maintain Loop 6 in the configuration that permits COPII binding. These results help explain the operation of Scap as a sterol sensor.
Introduction
Scap is a polytopic membrane protein that binds and transports sterol regulatory element-binding proteins (SREBPs)4 from their sites of origin in the endoplasmic reticulum (ER) to their sites of proteolytic processing in the Golgi complex (1). After their release in the Golgi complex, the active segments of SREBPs translocate to the nucleus, where they activate all of the genes necessary to produce cholesterol and unsaturated fatty acids (2). The transport activity of Scap is mediated by the binding of COPII to a cytosolic loop (Loop 6) of Scap (3). When cholesterol levels in the ER rise above 4–5% of ER lipids, Loop 6 undergoes a conformational change that precludes COPII protein binding (4, 5). This change is facilitated by the binding of Scap to another ER protein called Insig (6). The net result is prevention of the proteolytic processing of SREBPs. As a result, cholesterol synthesis declines. This feedback mechanism ensures a constant level of cholesterol in cell membranes.
Recent studies have begun to dissect the functional domains of Scap (see Fig. 1). The protein has eight transmembrane helices separated by luminal and cytosolic loops (7). Three of these loops are large enough to have structure: Loops 1 and 7, which are luminal, and Loop 6, which is cytosolic. The cholesterol binding site was recently localized to luminal Loop 1 (8). Somehow, this binding reaction in luminal Loop 1 must be communicated to cytosolic Loop 6 to change its conformation. One possibility is that luminal Loop 1 interacts with luminal Loop 7, which might alter Loop 6 through a direct action mediated by transmembrane helix 7.
FIGURE 1.

Topology model of the membrane domain of hamster Scap, showing its three functional domains and the sites of three point mutations (Y234A, D428A, and Y640S) that confer a constitutive cholesterol-bound conformation, even in the absence of sterols. Amino acids (aa) 40–284 correspond to the sequence of luminal Loop 1, the cholesterol-binding domain of Scap. The three hydrophobic patches in the Loop 1 sequence are shaded in purple, and the N-linked glycosylation site is denoted by the red box. The Insig-binding domain is localized to transmembrane helices 2–6, shown by the blue bracket. The COPII-binding site is localized to the MELADL sequence in Loop 6, shaded in orange. Amino acids 538–710 correspond to the sequence of luminal Loop 7; its two N-linked glycosylation sites are denoted by the red boxes. In membranes from sterol-deprived cells, trypsin cleaves Scap on its NH2-terminal side at Arg-496; in sterol-replete membranes, trypsin cleaves at Arg-503/Arg-505. The trypsin-cleavage site on the COOH-terminal side of Scap in both the absence and the presence of sterols occurs within a cluster of arginines (Arg-747–Arg-750).
We recently identified tyrosine 234 in Loop 1 as an essential residue for the transport activity of Scap. When this tyrosine is replaced by alanine (Y234A), Scap continues to bind SREBPs, but it can no longer carry the proteins to the Golgi complex (8). In the current studies, we have identified another crucial tyrosine at position 640 in luminal Loop 7. When this tyrosine is replaced by serine (Y640S), Scap also loses the ability to transport SREBPs. We provide evidence that these two tyrosines are necessary for the intramolecular binding between Scap segments that contain Loop 1 and those that contain Loop 7, an interaction that appears to be crucial for the transport activity of Scap.
EXPERIMENTAL PROCEDURES
Reagents
We obtained all sterols from Steraloids, Inc.; FuGENE 6 from Roche Applied Sciences; mouse anti-HSV monoclonal antibody and protease inhibitor mixture set III from Novagen; cycloheximide from Sigma; peptide-N-glycosidase F and recombinant endoglycosidase Hf from New England Biolabs; cyclodextrins from Trappsol; bovine serum albumin (catalog number 23209) from Thermo Scientific; and phRL-TK (encoding the Renilla luciferase gene) and Dual-Luciferase reporter assay system from Promega. Complexes of cholesterol with methyl-β-cyclodextrin were prepared at a stock concentration of 2.5 mm (9). Newborn calf lipoprotein-deficient serum (d >1.215 g/ml) was prepared by ultracentrifugation (10). Solutions of compactin and sodium mevalonate were prepared as described previously (11, 12). A stock solution of 10 mm sodium oleate-bovine serum albumin in 0.15 m NaC1 (final pH 7.6) was prepared as described previously (13). IgG-4H4, a mouse monoclonal antibody against hamster Scap (amino acids 1–767) (14), IgG-9E10, a mouse monoclonal antibody against c-Myc (15), and IgG-9D5, a mouse monoclonal antibody against hamster Scap (amino acids 540–707) (16) were previously described in the indicated references.
Buffers
Buffer A contained 10 mm Hepes-chloride (pH 7.6), 1.5 mm MgCl2, 10 mm KCl, 5 mm sodium EDTA, 5 mm sodium EGTA, and 250 mm sucrose. Buffer B contained 10 mm Tris-chloride (pH 6.8), 100 mm NaCl, and 0.5% (w/v) SDS. Buffer C contained 62.5 mm Tris-chloride (pH 6.8), 15% SDS, 8 m urea, 10% (v/v) glycerol, and 100 mm dithiothreitol. Buffer D contained 50 mm Tris-chloride (pH 7.4), 100 mm KCl, 1% (v/v) Nonidet P-40, and 1% (v/v) protease inhibitor mixture set III. Buffer E contained 50 mm Tris-chloride (pH 7.4), 135 mm NaCl, 10 mm KCl, 0.1% (v/v) Nonidet P-40, and 1% (v/v) protease inhibitor mixture set III.
Culture Medium
Medium A contained a 1:1 mixture of Ham's F-12 and Dulbecco's modified Eagle's medium (catalog number 10-090-CV, Mediatech, Inc.) supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin sulfate. Medium B contained medium A supplemented with 5% newborn calf lipoprotein-deficient serum, 50 μm sodium mevalonate, 50 μm compactin, and 1% (w/v) hydroxypropyl-β-cyclodextrin. Medium C contained medium A supplemented with 5% newborn calf lipoprotein-deficient serum, 50 μm sodium mevalonate, and 50 μm compactin. Medium D contained Dulbecco's modified Eagle's medium, low glucose (1000 mg/liter) supplemented with 10% fetal calf serum (FCS), 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate.
Plasmids
The following recombinant expression plasmids have been previously described: pCMV-Scap, encoding hamster Scap under control of the cytomegalovirus (CMV) promoter (16); pTK-Scap, encoding hamster Scap under control of the thymidine kinase (TK) promoter (17); pGFP-Scap, encoding GFP fused to hamster Scap under control of the CMV promoter (18); pTK-HSV-BP2, encoding HSV-tagged human SREBP-2 under control of the TK promoter (17); pTK-Insig1-Myc, encoding human Insig-1 followed by six tandem copies of c-Myc epitope tag under control of the TK promoter (19); pCMV-Insig1-Myc, encoding human Insig-1 followed by six tandem copies of a c-Myc epitope tag under control of the CMV promoter (20); pSRE-Luc, encoding three tandem copies of Repeat 2 + Repeat 3 of the human LDL receptor promoter, the adenovirus E1b TATA box, and the firefly luciferase gene (21); pCMV-Scap(TM1–6)-Myc, encoding hamster Scap (amino acids 1–464) followed by six tandem copies of a Myc tag under control of the CMV promoter (3); and pCMV-Scap(TM7-end), encoding hamster Scap (amino acids 504–1276) under control of the CMV promoter (3). Point mutations in the above Scap plasmids were produced by site-directed mutagenesis. The coding regions of all mutated plasmids were sequenced.
Cell Culture and Transfection
SRD-13A cells, a Scap-deficient cell line derived from CHO-7 cells (22), were grown in monolayer at 37 °C in 8–9% CO2 in medium A supplemented with 5% FCS, 1 mm sodium mevalonate, 20 mm sodium oleate-albumin, and 5 μg/ml cholesterol. SV589 cells, a line of SV-40 immortalized human fibroblasts (23), were grown in monolayer at 37 °C in 5% CO2 in medium D.
Trypsin Cleavage Assay of Scap
This assay was carried out as described previously (9, 24) with modifications in the culture and transfection conditions as described in the legend for Fig. 4B. On day 3, the cells were washed twice with PBS and switched to medium C. After incubation for 16 h at 37 °C, the cells were harvested for preparation of membranes for use in the trypsin cleavage assay.
FIGURE 4.
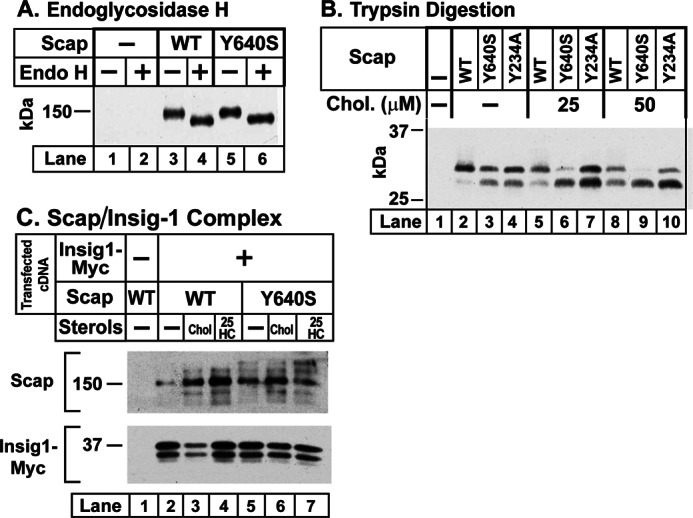
Biochemical characterization of Y640S Scap. A–C, on day 0, Scap-deficient SRD-13A cells were set up for experiments at a density of 2.5 × 105 cells/60-mm dish in 4 ml of medium A containing 5% FCS. On day 2, cells were transfected with the indicated plasmid using FuGENE 6 as the transfection agent. A, endoglycosidase H (Endo H) sensitivity. Transfected cDNAs were as follows: 2 μg of pcDNA mock vector (lanes 1 and 2), WT pTK-Scap (lanes 3 and 4), or mutant pTK-Scap(Y640S) (lanes 5 and 6) in 3 ml of medium A supplemented with 5% FCS. On day 3, five pooled dishes of cells were harvested per condition, washed with PBS, lysed in 1 ml buffer A containing protease inhibitors (30) by passing through a 22-gauge needle 30 times. The lysate was centrifuged at 1000 × g for 5 min at 4 °C, after which the supernatant was centrifuged at 20,000 × g for 30 min at 4 °C. The pellet was then resuspended in 100 μl buffer B and shaken for 30 min at room temperature. Reactions, in a final volume of 100 μl buffer B, contained 40 μg of solubilized membranes and 0.16 mm dithiothreitol in the absence or presence of 5000 units of endoglycosidase H. After incubation at 37 °C for 3 h, an equal volume of buffer C was added to each reaction and incubated at 37 °C for 30 min. Samples were then subjected to 8% SDS-PAGE and immunoblotted with 5 μg/ml IgG-4H4 (Scap). The film was exposed for 15 s. B, tryptic digestion. On day 2, SRD-13A cells were transfected with 1 μg of the indicated full-length WT or mutant version of pCMV-Scap plasmid. After incubation with lipoprotein-deficient serum for 16 h, three dishes of cells were harvested per condition and pooled for preparation of membranes. Aliquots of the 20,000 × g membrane fraction (100 μg) were incubated for 20 min at room temperature with the indicated concentration of cholesterol complexed with methyl-β-cyclodextrin (Chol.) followed by sequential treatments with 14 μg/ml trypsin (30 min at 30 °C) and 3000 units/ml peptide-N-glycosidase F (5 h at 37 °C). By removing N-linked sugars, peptide-N-glycosidase F increases the separation between the upper and lower trypsin-generated bands. The air-dried acetone-precipitated samples were then subjected to SDS-PAGE and immunoblot analysis with 5 μg/ml anti-Scap IgG-4H4. The film was exposed for 5 s. C, immune detection of Scap-Insig-1 complexes. On day 2, cells were co-transfected with 0.1 μg of pCMV-Insig1-Myc together with 0.3 μg of full-length WT pCMV-SCAP or its mutant Y640S version as indicated. On day 3, the cells were incubated for 1 h with 1% (w/v) hydroxypropyl-β-cyclodextrin, after which they received fresh medium containing one of the following sterols: none (−), 50 μm cholesterol complexed with methyl-β-cyclodextrin (Chol), or 1 μg/ml 25-hydroxycholesterol (25HC). After incubation at 37 °C for 4 h, the cells were harvested and lysed. Lysates from two pooled dishes were incubated with anti-Myc beads to trap Insig-1. After washing, the proteins were eluted by incubation for 5 min at 37 °C with 1× SDS sample buffer. The eluates were subjected to immunoblot analysis with either 10 μg/ml anti-IgG-4H4 (Scap) or 5 μg/ml anti-Myc IgG-9E10 (Insig-1 Myc). Films were exposed for 4–30 s.
Co-immunoprecipitation of Scap(TM1–6)-Myc and Scap(TM7-end)
This assay was carried out as described previously (8) with the use of an anti-C-Myc affinity gel (Sigma) except for the following modification. The solubilization and wash buffers consisted of buffer D (with Nonidet P-40) supplemented with one of the following sterols: none, 1 μg/ml 25-hydroxycholesterol (delivered in ethanol, final concentration of 0.1%), or 30 μm cholesterol complexed with methyl-β-cyclodextrin.
Assay for Scap-Insig Complex
This assay was carried out as described previously with the use of an anti-C-Myc affinity gel (8) except for the following modifications. The pellets from two 60-mm dishes of transfected SRD-13A cells were solubilized in 0.4 ml of Nonidet P-40-containing buffer E, and the washed beads were eluted in 1× SDS sample buffer.
SREBP-2 Processing in Cultured Cells
SRD-13A cells were set up for experiments as described in the figure legends. After transfection and incubation with sterols, nuclear and membrane fractions were prepared as described previously (17) and then subjected to immunoblot analysis.
Immunoblot Analysis
Unless otherwise indicated, samples for immunoblotting were subjected to 8 or 10% SDS-PAGE, after which the proteins were transferred to nitrocellulose filters that were incubated at 4 °C for 16 h with the following primary antibodies: 0.17 μg/ml mouse monoclonal anti-HSV antibody, 1–5 μg/ml mouse monoclonal anti-Myc IgG-9E10, 5–10 μg/ml mouse monoclonal anti-Scap IgG-4H4, or 2.5 μg/ml mouse monoclonal anti-Scap IgG-9D5. For all experiments except the assay for Scap-Insig complex, bound antibodies were visualized by SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) using a 1:5000 dilution of anti-mouse IgG conjugated to horseradish peroxidase (Jackson ImmunoResearch Laboratories). For the experiments detecting Scap-Insig complex, bound antibodies were visualized by SuperSignal West Dura extended duration substrate using a 1:3333 dilution of anti-mouse Ig HRP (mouse TrueBlot ULTRA, eBioscience). Immunoblots were exposed to Phenix Research Products Blue x-ray film (F-BX810) at room temperature.
RESULTS
Fig. 1 shows a diagram of the membrane domain of Scap. The protein has eight apparent transmembrane helices and three large loops (7). Loops 1 and 7 (245 amino acids and 173 amino acids, respectively) project into the lumen of the ER. Loop 6 projects into the cytosol. Loop 6 contains the MELADL sequence that is the recognition site for COPII proteins that cluster the Scap-SREBP complex into coated vesicles for transport to the Golgi (3). When cholesterol accumulates in ER membranes, Loop 6 undergoes a conformational change that precludes COPII protein binding and abrogates ER-to-Golgi transport (4). When expressed as a recombinant protein segment, Loop 1 binds cholesterol in vitro (8). The specificity of sterol binding to Loop 1 mimics the binding specificity of sterol binding to the entire membrane domain of Scap. A point mutation in Loop 1 (Y234A) renders Loop 6 of Scap in the closed COPII-inaccessible configuration even in sterol-depleted membranes and abrogates movement to the Golgi (8). The other large intraluminal loop (Loop 7) is the subject of the current study.
In an initial attempt to characterize the function of Loop 7, we conducted an alanine-scanning mutagenesis study of the entire loop by systematic replacement of every pair or trio of contiguous residues with alanines. Plasmids encoding the mutant Scaps were transfected into Scap-deficient SRD-13A CHO cells together with a plasmid encoding firefly luciferase under control of an SRE-dependent promoter. To control for transfection efficiency, we also transfected a plasmid encoding Renilla luciferase driven by a constitutive thymidine kinase promoter, and we included a plasmid encoding Insig-1 to assist in the sterol-mediated inhibition of Scap transport. As shown in Fig. 2A, most of the Loop 7 mutants behaved like wild-type Scap in restoring SREBP cleavage as measured by the sterol-regulated increase in luciferase activity. Four of the mutants showed a marked reduction in the ability to elicit activation of the SRE-dependent promoter (indicated by red asterisks). All of these defective mutants clustered in the region of Scap between residues 640 and 657.
FIGURE 2.

Alanine-scanning mutagenesis of Loop 7 region of hamster Scap. On day 0, SRD-13A cells were set up for experiments in 1 ml of medium A containing 5% FCS at a density of 2.5 × 104 cells/well in 24-well plates. On day 1, cells were co-transfected in 1 ml of medium A supplemented with 5% FCS containing 38 ng of pTK-Insig1-Myc, 125 ng of pSRE-firefly luciferase, 125 ng of pTK-Renilla luciferase, and 50 ng of WT or the indicated mutant pTK-Scap in which 2–3 contiguous residues were mutated to alanine. FuGENE 6 was used as the transfection agent. For each transfection, the total amount of DNA was adjusted to 338 ng/dish by the addition of pcDNA mock vector. On day 2 (after incubation with plasmids for 24 h), the cells were washed once with PBS and then treated with hydroxypropyl-β-cyclodextrin-containing medium B for 1 h. Cells were then washed twice with PBS and re-fed with 1 ml of medium C in the absence or presence of 15 μm cholesterol complexed with methyl-β-cyclodextrin. After incubation for 16 h, the cells were washed with PBS, after which luciferase activity was read on a Synergy 4 plate reader (BioTek) according to the Promega protocol. The amount of SRE-firefly luciferase activity in each dish was normalized to the amount of Renilla luciferase activity in the same dish. Relative SRE-luciferase activity of 1.0 represents the normalized luciferase value in dishes transfected with WT pTK-Scap in the absence of cholesterol. All values are the average of duplicate assays. A, red asterisks denote triple contiguous alanine mutations that produced a loss of SRE-luciferase activity in both the absence and the presence of cholesterol. The data in these graphs were obtained in four different experiments, each with its own WT control. B, deconvolution of triple mutants that showed loss of SRE-luciferase activity in A. Blue boxes denote single alanine mutations that show partial loss of SRE-luciferase activity. C, substituting serine, glutamine, aspartic acid, and lysine in place of Tyr-640, Tyr-648, and Ile-655. The red arrow denotes the Y640S mutation that was selected for further characterization. In B and C, single-letter codes are used to denote amino acids.
All four of the severely defective mutants in Fig. 2A consisted of three contiguous alanine substitutions. When any of the individual residues in these triplets were changed to alanine, there were only minor losses of function (Fig. 2B). Reasoning that the single alanine substitutions were not drastic enough to perturb function, we selected three hydrophobic residues (Tyr-640, Tyr-648, and Ile-655) and replaced them individually with hydrophilic or charged residues (Ser, Gln, Asp, or Lys). Several of these single substitutions severely reduced SREBP-dependent luciferase activity (Fig. 2C). For further study, we chose Y640S.
To study SREBP cleavage directly, we transfected plasmids encoding WT or Y640S Scap into Scap-deficient SRD-13A cells together with a plasmid containing epitope-tagged SREBP-2. Expression of Scap was driven by the weak TK promoter so as to produce low level expression of Scap, which does not overwhelm endogenous Insigs and thereby facilitates sterol-dependent inhibition of processing. Cells were incubated in sterol-depleted medium with or without added cholesterol. After cells were harvested, nuclear extracts and membrane fractions were prepared and subjected to SDS-PAGE and immunoblotting. As reported previously (25), in the absence of Scap, we failed to visualize a nuclear fragment of SREBP-2 (Fig. 3A, lane 1). SREBP-2 was also not observed in the membrane fraction because the membrane-bound precursor is rapidly degraded in the absence of Scap (22). When WT Scap was co-transfected, we visualized the membrane form of SREBP-2, indicating that the precursor was stabilized (lanes 2 and 3). We also observed the nuclear form of SREBP-2, and this was reduced by cholesterol. The Y640S Scap mutant stabilized the membrane precursor form of SREBP-2, indicating that it bound the precursor, but it did not generate any nuclear form (lanes 4 and 5). When Insig-1 was transfected together with WT Scap, we observed a more profound cholesterol-mediated reduction in nuclear SREBP-2 (lanes 6 and 7). Again, Scap Y640S stabilized the membrane form of SREBP-2, but it did not generate the nuclear form (lanes 8 and 9). We blotted the membrane fractions with antibodies to the epitope tags on Scap and Insig-1 to show that comparable levels were expressed in all of the transfected cells.
FIGURE 3.

Immunoblot analysis of SREBP-2 cleavage in Scap-deficient cells transfected with WT or Y640S mutant version of full-length Scap in the absence or presence of transfected Insig-1. A, Y640S mutant driven by TK promoter. On day 0, SRD-13A cells were set up for experiments at a density of 2.5 × 105 cells/60-mm dish in 4 ml of medium A containing 5% FCS. On day 2, cells were transfected with 2 μg of pTK-HSV-BP2 and 0.4 μg of full-length WT pTK-Scap or 0.6 μg of its Y640S version with or without 0.2 μg of pTK-Insig1-Myc in 3 ml of medium A supplemented with 5% FCS; FuGENE 6 was used as the transfection agent. For each transfection, the total amount of DNA was adjusted to 2.8 μg/dish by the addition of pcDNA mock vector. On day 3, cells were washed once with PBS and then switched to hydroxypropyl-β-cyclodextrin-containing medium B for 1 h. The cells were washed with PBS and then incubated with medium C containing 30 μm cholesterol complexed with methyl-β-cyclodextrin as indicated. After incubation for 4 h, two pooled dishes of cells per condition were harvested, and the isolated nuclear and membrane fractions were subjected to immunoblot analysis with 0.167 μg/ml anti-HSV (SREBP-2), 1 μg/ml anti-Myc IgG-9E10 (Insig-1), or 5 μg/ml IgG-4H4 (Scap). B, Y640S mutant driven by CMV promoter. Experimental design was as in A except for DNA transfections. Cells were transfected with 2 μg of pTK-HSV-BP2 together with one of the following additional plasmids: none (lane 1); 0.4 μg of full-length WT pTK-Scap (lanes 2, 3, 8, and 9); 0.6 μg of mutant pCMV-Scap(Y640S) (lanes 4, 5, 10, and 11); or 0.4 μg of WT pTK-Scap plus 0.6 μg of mutant pCMV-Scap(Y640S) (lanes 6, 7, 12, and 13) with or without 0.2 μg of pTK-Insig1-Myc as indicated. For each transfection, the total amount of DNA was adjusted to 3.2 μg/dish by the addition of pcDNA mock vector. Films were exposed for 1–20 s.
We previously showed that overexpression of WT Scap, driven by the strong CMV promoter, saturates Insig and prevents cholesterol from blocking the transport and cleavage of SREBP-2 (26). To test whether the Y640S mutant would also saturate Insig, we transfected a plasmid encoding Scap(Y640S) driven by the CMV promoter. As shown in Fig. 3B, transfection of TK-driven WT Scap stabilized the membrane SREBP-2 precursor and restored cleavage in the SRD-13A cells, and cleavage was reduced by cholesterol (lanes 1–3). Scap(Y640S) stabilized the SREBP-2 precursor, but it did not restore cleavage even when driven by the strong CMV promoter (lanes 4 and 5). Importantly, when co-transfected together with WT Scap, the CMV-driven Scap(Y640S) prevented cholesterol-mediated suppression of SREBP-2 cleavage (lanes 6 and 7). This prevention persisted even when we co-transfected a plasmid encoding Insig-1 (lanes 12 and 13). These data indicate that Scap(Y640S) is capable of binding and saturating Insig-1, a conclusion that was confirmed by direct co-immunoprecipitation (see below).
We previously showed that WT Scap becomes glycosylated in the ER, and the carbohydrates are sensitive to removal by endoglycosidase H (27). Fig. 4A repeats this result and shows that the Y640S mutant is similarly glycosylated. In Fig. 4B, we used our previously described trypsin digestion assay (9, 24) to detect the cholesterol-induced change in the conformation of Loop 6 of WT Scap and the Y640S and Y234A mutants. SRD-13A cells were transfected with plasmids encoding one of these proteins. Sealed membrane vesicles were prepared, incubated with varying amounts of cholesterol, and digested with trypsin followed by treatment with peptide-N-glycosidase F and SDS-PAGE. A trypsin-protected fragment of Scap was visualized by blotting with a monoclonal antibody directed against luminal Loop 7 (Fig. 1). In the absence of cholesterol, WT Scap gave a single band of 250 amino acids at ∼30 kDa (lane 2), which is produced by trypsin cleavage at arginine 496 and a cluster of arginines at residues 747–750 (9) (Fig. 1). In the presence of cholesterol, a new trypsin-protected band of ∼241 amino acids was detected (lanes 5 and 8). The lower band is generated by cleavage at arginines 503/505, which become exposed to trypsin as a result of a cholesterol-induced conformation change in Scap Loop 6 (9, 24). The Y640S and Y234A mutants both showed the lower band even in the absence of cholesterol (lanes 3 and 4). When cholesterol was added to the Y640S mutant, the upper band disappeared and the lower band increased (lanes 6 and 9). In contrast, the Y234A mutant showed little change in the presence of cholesterol (lanes 7 and 10).
To confirm the interaction of Scap(Y640S) with Insig-1, we performed a co-immunoprecipitation experiment (Fig. 4C). SRD-13A cells were transfected with plasmids encoding Myc-tagged Insig-1 and WT or Y640S Scap. The cells were incubated in the absence of sterols or in the presence of cholesterol or 25-hydroxycholesterol. Membranes were solubilized, and the Myc-tagged Insig-1 was precipitated on anti-Myc beads. The eluted proteins were blotted with anti-Scap or anti-Insig-1. In the absence of Insig-1 expression, no WT Scap was precipitated by the anti-Myc beads (lane 1). When Myc-Insig-1 was expressed, a small amount of Scap was co-immunoprecipitated (lane 2), and this increased when cholesterol or 25-hydroxycholesterol was added to the cells (lanes 3 and 4). In the absence of sterols, Scap(Y640S) was co-precipitated with Myc-Insig-1 (lane 5), and there was no further increase when cholesterol or 25-hydroxycholesterol was added (lanes 6 and 7). These data indicate that Scap(Y640S) retains the ability to bind to Insig-1, and it does so even in the absence of cholesterol.
The failure of Scap(Y640S) to reach the Golgi was confirmed by the immunofluorescence experiment of Fig. 5. SV589 cells were transfected with a plasmid encoding GFP-tagged WT Scap or the Y640S mutant. The cells were incubated with hydroxypropyl-β-cyclodextrin to remove cholesterol and stimulate Scap translocation to the Golgi. We included cycloheximide to minimize fluorescence due to newly synthesized Scap. The cells were permeabilized and immunostained for the Golgi marker GM130 using an antibody conjugated to Alexa Fluor 594, which fluoresces red. We also added Hoechst 33342, which stains nuclei blue. With WT Scap, the GFP signal was concentrated in discrete particles that stained for GM130, indicating that they represented Golgi stacks (upper panels). In sharp contrast, the GFP-tagged Scap(Y640S) showed a lacy distribution consistent with retention in the ER (lower panels). The images shown in Fig. 5 are representative of many transfected cells that were examined individually. Of the 1565 transfected WT cells examined, Scap was localized to the ER in 15% of cells and to the Golgi in 85% of cells, whereas in the 1000 transfected Y640S mutant cells examined, Scap was visualized in the ER in >98% of cells.
FIGURE 5.

Mutant GFP-Scap (Y640S), but not WT GFP-Scap, fails to reach the Golgi. On day 0, SV589 cells (23) were set up for experiments in medium D at a density of 1 × 105 cells/37-mm dish containing three 12-mm glass coverslips. On day 1, cells were transfected with 1 μg of full-length WT pGFP-Scap (top panels) or its mutant Y640S version (bottom panels) in 3 ml of medium A supplemented with 5% FCS. FuGENE 6 was used as the transfection reagent. On day 2, the cells were washed once with PBS and then incubated for 1 h at 37 °C with cyclodextrin-containing medium B supplemented with 50 μg/ml cycloheximide, after which each coverslip was fixed and permeabilized in methanol at −20 °C for 15 min. The cells were then incubated with rabbit polyclonal serum against the Golgi resident protein GM130 (31) followed by 6.7 μg/ml goat anti-rabbit antibody conjugated to Alexa Fluor 594 (Invitrogen). The nuclei were then stained with 1 μg/ml Hoechst 33342 (Invitrogen), and the coverslips were mounted in Mowiol 4-88 (Calbiochem) mounting solution (32). Fluorescence images were acquired using an LD Plan-Neofluar 40×/1.3 differential interference contrast objective, an Axiovert 200M microscope (Zeiss), an Orca 285 camera (Hamamatsu), and the software Openlab 4.0.2 (Improvision). Scale bar, 10 μm.
Previously, we transfected cells simultaneously with a plasmid encoding the TM1–6 portion of Scap and a second plasmid encoding the remainder of the protein (TM7 to the COOH-terminal end of the WD repeat domain) (4, 26). We refer to the latter portion as TM7-end (Fig. 6A). We showed that these two segments interact with each other as measured by co-immunoprecipitation assays and that they reconstitute SREBP transport activity. Fig. 6B shows a co-immunoprecipitation experiment in which we transfected SRD-13A cells with a plasmid encoding either WT or the Y234A mutant version of Myc-tagged TM1–6 together with a second plasmid encoding WT TM7-end. We also transfected the cells with WT TM1–6 together with Y640S(TM7-end). Cells were incubated in the absence of sterols or in the presence of cholesterol or 25-hydroxycholesterol. We isolated the Myc-tagged TM1–6 segment on anti-Myc beads, eluted the bound proteins, and probed immunoblots with an antibody against TM7-end. When both segments were WT, the TM7-end was co-immunoprecipitated with TM1–6 (see pellet in bottom panel, lane 3). There was little effect when the cells were incubated with 25-hydroxycholesterol (lane 5). In contrast, cholesterol reduced the amount of co-immunoprecipitated Scap(TM7-end) by about 50%. We observed a similar 50% reduction in more than 10 experiments in which we varied the ratio between the TM1–6 fragment and the TM7-end fragment and in which we performed the co-immunoprecipitation with or without co-transfection with a plasmid encoding Insig-1. All of these experiments were performed by exposing the cells to maximal concentrations of cholesterol/methyl-β-cyclodextrin (30 μm). In none of the experiments did 25-hydroxycholesterol reduce the co-immunoprecipitation.
FIGURE 6.

Co-immunoprecipitation of NH2- and COOH-terminal segments of WT Scap, but not mutant Scap(Y640S). A, membrane topology of hamster Scap denoting NH2- and COOH-terminal segments encoded by two different cDNAs. Scap(TM1–6)-Myc encodes hamster Scap (amino acids (aa) 1–464) followed by six tandem copies of a Myc tag under control of the CMV promoter (3). Scap(TM7-end) encodes hamster Scap (amino acids 504–1276) under control of the CMV promoter (3). B, co-immunoprecipitation (IP). On day 0, Scap-deficient SRD-13A cells were set up for experiments in 3 ml of medium A containing 5% FCS at a density of 2.5 × 105 cells/60-mm dish. On day 2, cells were transfected in 3 ml of medium A containing 5% FCS with the following plasmids: 2.5 μg of pTK-HSV-BP2 (lanes 1–11); 2 μg of pCMV-SCAP(TM1–6)-Myc (lanes 3–8); 2 μg of the mutant Y234A version of pCMV-SCAP(TM1–6)-Myc (lanes 9–11); 0.5 μg of pCMV-SCAP(TM7-end) (lanes 2–5 and 9–11); and 0.5 μg of the mutant Y640S version of pCMV-SCAP(TM7-end) (lanes 6–8). FuGENE 6 was used as the transfection agent. The total amount of DNA was adjusted to 5 μg/dish by the addition of pcDNA mock vector. 12 h after transfection, the cells were incubated in medium C in the absence (lanes 1–3, 6, and 9) or presence of either 1 μg/ml 25-hydroxycholesterol (25-HC) (delivered in ethanol, final concentration of 0.1%) (lanes 4, 7, and 10) or 30 μm cholesterol complexed with methyl-β-cyclodextrin (Chol.) (lanes 5, 8, and 11). After incubation for 12 h, each detergent-solubilized whole cell lysate from two pooled dishes was incubated with anti-Myc beads followed by washing and elution with Myc peptide as described under “Experimental Procedures.” The eluates were subjected to immunoblot analysis with either 1 μg/ml anti-Myc IgG-9E10 (Scap(TM1–6)-Myc) or 2.5 μg/ml anti-IgG-9D5 (Scap(TM7-end)). The film was exposed for 1–10 s. S, supernatant; P, pellet.
As shown in Fig. 6B, lanes 6–8, the Y640S mutant TM7-end segment failed to co-immunoprecipitate with the WT TM1–6 segment. Similarly, the Y234A mutant TM1–6 failed to co-immunoprecipitate with WT TM7-end (lanes 9–11). Thus, point mutations either in Loop 1 or in Loop 7 can abolish the interaction between the TM1–6 segment and the TM7-end segment of Scap.
DISCUSSION
In a previous study, we showed that cholesterol binds to luminal Loop 1 of Scap. We also created a point mutation in Loop 1 (Y234A) that rendered Scap unable to transport SREBP-2 to the Golgi complex even in the absence of sterols (8). We used trypsin digestion assays to assess the conformation of Loop 6, which contains the MELADL sequence that binds COPII proteins. These digestions revealed that Loop 6 was in the closed conformation (i.e. the conformation that does not bind COPII proteins) even in the absence of sterols. In the current studies, we have created a point mutation in the other large luminal domain of Scap (Loop 7) that has the same effects as the Loop 1 mutation. In cells expressing Scap with this Y640S mutation, Loop 6 also assumed the closed conformation in the absence of sterols (Fig. 4B). Scap(Y640S) also failed to transport SREBPs to the Golgi as revealed by SREBP processing assays (Fig. 3A) and by immunofluorescence localization (Fig. 5).
We considered the possibility that Scap(Y640S) is misfolded and therefore precluded from exiting the ER through the action of the ER quality control system. Several observations argue against this possibility. First, Scap(Y640S) stabilizes the membrane precursor of SREBP-2, which is rapidly degraded in the absence of Scap (Fig. 3A) (22). Second, Scap(Y640S) binds Insig-1 as determined by direct co-immunoprecipitation (Fig. 4C) and by the observation that overexpression of Scap(Y640S) saturated Insig-1 and allowed WT Scap to carry SREBPs to the Golgi even in the presence of cholesterol (Fig. 3B). We conclude that Scap(Y640S) is properly folded.
The fact that point mutations in Loop 1 and Loop 7 block Scap movement raises the possibility that these two loops must interact in order for this movement to occur. This hypothesis is supported by the co-immunoprecipitation experiment of Fig. 6. When we expressed WT Scap(TM1–6) and WT Scap(TM7-end) in the same cells, the two proteins bound to each other, and they could be co-immunoprecipitated. When either the mutant TM1–6 segment or the mutant TM7-end segment was substituted for the corresponding WT segment, co-immunoprecipitation was abolished. Cholesterol reduced the amount of co-immunoprecipitation by about 50% (Fig. 6B, lane 5). This result was reproduced in more than 10 other experiments. In all of these experiments, 25-hydroxycholesterol failed to reduce the co-immunoprecipitation. This failure persisted even when we co-expressed Insig-1 by transfection.
Interaction of luminal Loop 1 of Scap with luminal Loop 7 provides a potential mechanism by which binding of cholesterol to Loop 1 would alter the conformation of cytosolic Loop 6. If cholesterol binding changes the conformation of Loop 1, and if this is transmitted to Loop 7, then Loop 7 might alter the conformation of Loop 6 through some effect on transmembrane helix 7, which joins the two loops (Fig. 1). The interaction between Loops 1 and 7 might occur within a single Scap monomer or between adjacent molecules in the Scap tetramer (28). The failure of 25-hydroxycholesterol to reduce the co-immunoprecipitation between Scap(TM1–6) and Scap(TM7-end) might be explained by our previous observation that 25-hydroxycholesterol blocks Scap movement not by binding to Scap, but rather by binding to Insig (4, 29). Our findings therefore raise the possibility that the 25-hydroxycholesterol-Insig complex directly blocks COPII binding to the MELADL sequence in Scap Loop 6 without a requirement for dissociation between Loop 1 and Loop 7. Experiments are under way to test these hypotheses.
Acknowledgments
We thank our colleagues Nick Grishin, Lisa Kinch, Dan Rosenbaum, Arun Radhakrishnan, and Jin Ye for helpful comments; Dorothy Williams and William Warshauer for excellent technical assistance; and Lisa Beatty, Muleya Kapaale, and Ijeoma Dukes for invaluable help with tissue culture.
This work was supported, in whole or in part, by National Institutes of Health Grants HL20948 (to J. L. G. and M. S. B.) and GM096070 (to J. S.).
- SREBP
- sterol regulatory element-binding protein
- SRE
- sterol regulatory element
- ER
- endoplasmic reticulum
- HSV
- Herpes simplex virus
- TK
- thymidine kinase
- TM
- transmembrane.
REFERENCES
- 1. Brown M. S., Goldstein J. L. (1997) The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 [DOI] [PubMed] [Google Scholar]
- 2. Horton J. D., Goldstein J. L., Brown M. S. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sun L.-P., Li L., Goldstein J. L., Brown M. S. (2005) Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J. Biol. Chem. 280, 26483–26490 [DOI] [PubMed] [Google Scholar]
- 4. Sun L.-P., Seemann J., Brown M. S., Goldstein J. L. (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 6519–6526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Radhakrishnan A., Goldstein J. L., McDonald J. G., Brown M. S. (2008) Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 8, 512–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goldstein J. L., DeBose-Boyd R. A., Brown M. S. (2006) Protein sensors for membrane sterols. Cell 124, 35–46 [DOI] [PubMed] [Google Scholar]
- 7. Nohturfft A., Brown M. S., Goldstein J. L. (1998) Topology of SREBP cleavage-activating protein, a polytopic membrane protein with a sterol-sensing domain. J. Biol. Chem. 273, 17243–17250 [DOI] [PubMed] [Google Scholar]
- 8. Motamed M., Zhang Y., Wang M. L., Seemann J., Kwon H. J., Goldstein J. L., Brown M. S. (2011) Identification of luminal Loop 1 of Scap protein as the sterol sensor that maintains cholesterol homeostasis. J. Biol. Chem. 286, 18002–18012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown A. J., Sun L., Feramisco J. D., Brown M. S., Goldstein J. L. (2002) Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell 10, 237–245 [DOI] [PubMed] [Google Scholar]
- 10. Goldstein J. L., Basu S. K., Brown M. S. (1983) Receptor-mediated endocytosis of low density lipoprotein in cultured cells. Methods Enzymol. 98, 241–260 [DOI] [PubMed] [Google Scholar]
- 11. Brown M. S., Faust J. R., Goldstein J. L., Kaneko I., Endo A. (1978) Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J. Biol. Chem. 253, 1121–1128 [PubMed] [Google Scholar]
- 12. Kita T., Brown M. S., Goldstein J. L. (1980) Feedback regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in livers of mice treated with mevinolin, a competitive inhibitor of the reductase. J. Clin. Invest. 66, 1094–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hannah V. C., Ou J., Luong A., Goldstein J. L., Brown M. S. (2001) Unsaturated fatty acids down-regulate SREBP isoforms 1a and 1c by two mechanisms in HEK-293 cells. J. Biol. Chem. 276, 4365–4372 [DOI] [PubMed] [Google Scholar]
- 14. Ikeda Y., Demartino G. N., Brown M. S., Lee J. N., Goldstein J. L., Ye J. (2009) Regulated endoplasmic reticulum-associated degradation of a polytopic protein: p97 recruits proteasomes to Insig-1 before extraction from membranes. J. Biol. Chem. 284, 34889–34900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yabe D., Brown M. S., Goldstein J. L. (2002) Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 99, 12753–12758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sakai J., Nohturfft A., Cheng D., Ho Y. K., Brown M. S., Goldstein J. L. (1997) Identification of complexes between the COOH-terminal domains of sterol regulatory element binding proteins (SREBPs) and SREBP cleavage-activating protein (SCAP). J. Biol. Chem. 272, 20213–20221 [DOI] [PubMed] [Google Scholar]
- 17. Feramisco J. D., Radhakrishnan A., Ikeda Y., Reitz J., Brown M. S., Goldstein J. L. (2005) Intramembrane aspartic acid in SCAP protein governs cholesterol-induced conformational change. Proc. Natl. Acad. Sci. U.S.A. 102, 3242–3247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nohturfft A., Yabe D., Goldstein J. L., Brown M. S., Espenshade P. J. (2000) Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell 102, 315–323 [DOI] [PubMed] [Google Scholar]
- 19. Gong Y., Lee J. N., Lee P. C. W., Goldstein J. L., Brown M. S., Ye J. (2006) Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 3, 15–24 [DOI] [PubMed] [Google Scholar]
- 20. Yabe D., Xia Z.-P., Adams C. M., Rawson R. B. (2002) Three mutations in sterol-sensing domain of SCAP block interaction with Insig and render SREBP cleavage insensitive to sterols. Proc. Natl. Acad. Sci. U.S.A. 99, 16672–16677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hua X., Nohturfft A., Goldstein J. L., Brown M. S. (1996) Sterol resistance in CHO cells traced to point mutation in SREBP cleavage activating protein (SCAP). Cell 87, 415–426 [DOI] [PubMed] [Google Scholar]
- 22. Rawson R. B., DeBose-Boyd R., Goldstein J. L., Brown M. S. (1999) Failure to cleave sterol regulatory element-binding proteins (SREBPs) causes cholesterol auxotrophy in Chinese hamster ovary cells with genetic absence of SREBP cleavage-activating protein. J. Biol. Chem. 274, 28549–28556 [DOI] [PubMed] [Google Scholar]
- 23. Yamamoto T., Davis C. G., Brown M. S., Schneider W. J., Casey M. L., Goldstein J. L., Russell D. W. (1984) The human LDL receptor: A cysteine-rich protein with multiple Alu sequences in its mRNA. Cell 39, 27–38 [DOI] [PubMed] [Google Scholar]
- 24. Adams C. M., Reitz J., De Brabander J. K., Feramisco J. D., Li L., Brown M. S., Goldstein J. L. (2004) Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J. Biol. Chem. 279, 52772–52780 [DOI] [PubMed] [Google Scholar]
- 25. Yang T., Espenshade P. J., Wright M. E., Yabe D., Gong Y., Aebersold R., Goldstein J. L., Brown M. S. (2002) Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110, 489–500 [DOI] [PubMed] [Google Scholar]
- 26. Yang T., Goldstein J. L., Brown M. S. (2000) Overexpression of membrane domain of SCAP prevents sterols from inhibiting SCAP/SREBP exit from endoplasmic reticulum. J. Biol. Chem. 275, 29881–29886 [DOI] [PubMed] [Google Scholar]
- 27. Nohturfft A., Brown M. S., Goldstein J. L. (1998) Sterols regulate processing of carbohydrate chains of wild-type SREBP cleavage-activating protein (SCAP), but not sterol-resistant mutants Y298C or D443N. Proc. Natl. Acad. Sci. U.S.A. 95, 12848–12853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Radhakrishnan A., Sun L.-P., Kwon H. J., Brown M. S., Goldstein J. L. (2004) Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol. Cell 15, 259–268 [DOI] [PubMed] [Google Scholar]
- 29. Radhakrishnan A., Ikeda Y., Kwon H. J., Brown M. S., Goldstein J. L. (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. U.S.A. 104, 6511–6518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang M. L., Motamed M., Infante R. E., Abi-Mosleh L., Kwon H. J., Brown M. S., Goldstein J. L. (2010) Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 12, 166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wei J.-H., Seemann J. (2009) The mitotic spindle mediates inheritance of the Golgi ribbon structure. J. Cell Biol. 184, 391–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wei J.-H., Seemann J. (2009) Induction of asymmetrical cell division to analyze spindle-dependent organelle partitioning using correlative microscopy techniques. Nat. Protoc. 4, 1653–1662 [DOI] [PubMed] [Google Scholar]