

Background: H2O2 oxidizes peroxiredoxins (Prxs) to sulfenic acid intermediates which form disulfides or become hyperoxidized.
Results: Rate constants for hyperoxidation and disulfide formation were obtained for Prx2 and Prx3.
Conclusions: Prx2 is more susceptible than Prx3 to hyperoxidation due to slower disulfide formation.
Significance: H2O2 reacts with Prx sulfenic acid faster than with most reduced thiols.
Keywords: Hydrogen Peroxide, Oxidation-Reduction, Peroxiredoxin, Redox Signaling, Signal Transduction, Thiol, Hyperoxidation, Sulfenic Acid
Abstract
Typical 2-Cys peroxiredoxins (Prxs) react rapidly with H2O2 to form a sulfenic acid, which then condenses with the resolving cysteine of the adjacent Prx in the homodimer or reacts with another H2O2 to become hyperoxidized. Hyperoxidation inactivates the Prx and is implicated in cell signaling. Prxs vary in susceptibility to hyperoxidation. We determined rate constants for disulfide formation and hyperoxidation for human recombinant Prx2 and Prx3 by analyzing the relative proportions of hyperoxidized and dimeric products using mass spectrometry as a function of H2O2 concentration (in the absence of reductive cycling) and in competition with catalase at a fixed concentration of H2O2. This gave a second order rate constant for hyperoxidation of 12,000 m−1 s−1 and a rate constant for disulfide formation of 2 s−1 for Prx2. A similar hyperoxidation rate constant for Prx3 was measured, but its rate of disulfide formation was ∼10-fold higher, making it is more resistant than Prx2 to hyperoxidation. There are two active sites within the homodimer, and at low H2O2 concentrations one site was hyperoxidized and the other present as a disulfide. Prx with two hyperoxidized sites formed progressively at higher H2O2 concentrations. Although the sulfenic acid forms of Prx2 and Prx3 are ∼1000-fold less reactive with H2O2 than their active site thiols, they react several orders of magnitude faster than most reduced thiol proteins. This observation has important implications for understanding the mechanism of peroxide sensing in cells.
Introduction
Human peroxiredoxin 2 (Prx2)2 and Prx3 are typical 2-Cys peroxiredoxins belonging to a family of ubiquitous cysteine-dependent peroxidases (1). Prx2 is a cytosolic protein whereas Prx3 resides in mitochondria. They react with H2O2 as efficiently as catalase or glutathione peroxidase (2–4) and are able to reduce peroxynitrite, alkylhydroperoxides, and peroxides formed on amino acids and proteins (5). The enzymatic cycle of typical 2-Cys Prxs involves oxidation of their peroxidatic cysteine (SP) to a sulfenic acid, followed by condensation with the resolving cysteine (SR) of another subunit to form a head to tail disulfide-linked homodimer, which can be reduced back by the thioredoxin-thioredoxin reductase system. The oxidation sequence for one of the active sites is shown in Fig. 1. Although the dimer is the primary catalytic unit, the dimers can associate to form noncovalent decamers (6). Another feature of the Prxs is that the sulfenic acid on the peroxidatic Cys can undergo further oxidation (hyperoxidation) to form the sulfinic acid. The sulfinic acid cannot be reduced by thioredoxin but is a substrate for sulfiredoxin. However, the reduction by sulfiredoxin is slow and highly energy consuming. The mammalian enzyme requires one ATP and two GSH or thioredoxin molecules to reduce a single hyperoxidized cysteine (7). In yeast, thioredoxin rather than GSH is the preferred cellular reductant (8). Although hyperoxidation could be an unavoidable consequence of the high reactivity of the peroxidatic cysteine, there is good reason to believe that it has a physiological function (1). In support of this notion, eukaryotic Prxs are more sensitive to hyperoxidation than their prokaryotic counterparts, and this is due to acquisition of a C-terminal extension that facilitates the reaction (9). Hyperoxidation has been proposed as a mechanism for controlling local levels of H2O2 in cell signaling (9, 10). An example of this is regulation of steroid synthesis in adrenal cells by hyperoxidation of Prx3, with a proposed mechanism of allowing mitochondrial H2O2 to escape and act as a signal in the cytoplasm (11). Hyperoxidation increases the affinity constant of the decamer and may also enhance the ability of Prxs to act as chaperones (12). It is readily observed in cells treated with H2O2 and can be induced by stresses such as high ethanol exposure (13). The physiological relevance of hyperoxidation can also be inferred from findings of sulfiredoxin acting synergistically with Prxs in protecting cells from oxidative stress (14).
FIGURE 1.
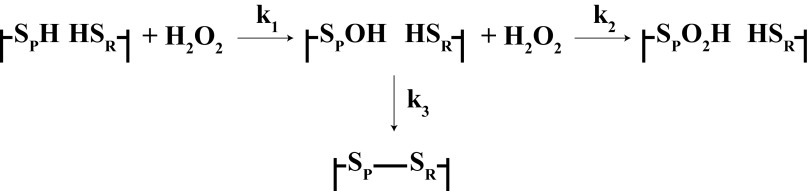
Scheme for catalytic cycle of a 2-Cys Prx showing reactions of one of the two active sites, as used in the kinetic analysis.
To appreciate the pathophysiological relevance of hyperoxidation, an understanding of the kinetics and regulation of the process is needed. Based on observations by Yang et al. (15) that Prx1 undergoes progressive inactivation in an enzymatic assay, a commonly held view is that catalytic turnover is needed for Prx hyperoxidation to occur. However, based on the most straightforward kinetic analysis of the reactions in Fig. 1, it would be expected that, after an initial fast reaction to form the sulfenic acid (reaction 1), hyperoxidation (reaction 2) would occur in competition with disulfide formation (reaction 3) and increase proportionately with increasing H2O2 concentration, without the need for recycling. No detailed kinetic analysis of this mechanism for a 2-Cys Prx has been performed. Therefore, we have quantified hyperoxidation as a function of H2O2 concentration and used these data plus results obtained from competition experiments with varying concentrations of catalase to determine rate constants for hyperoxidation and disulfide bond formation. We have studied Prx2 and Prx3, which we have shown previously to differ in their sensitivity to hyperoxidation (16). Our results are consistent with the sulfenic acid forms of both proteins reacting with H2O2 at similar rates and with the greater resistance of Prx3 to hyperoxidation being due to a faster rate of disulfide formation.
EXPERIMENTAL PROCEDURES
Preparation of Prx2 and Prx3
Recombinant untagged Prx2 was prepared as described (17). Human Prx 3 cDNA (Origene) was amplified using primers to encode a FXa protease cleavage site immediately before amino acid 62 (forward primer, 5′-GCGGAATTCATCGAAGGTCGTGCACCTGCTGTCACCCAGCATGC-3′; reverse primer, 5′-GCGCTCGAGTCACTGATTTACCTTCTGAAAGTAC-3′) and subcloned with EcoI-XhoI into a pET28a vector (Novagen) in-frame to express an N-terminal His6-tagged Prx 3 protein. His-tagged Prx 3 was expressed and purified as in Ref. 17, and the histidine tag was cleaved off using FXa (Roche Applied Science) and removed with His Select Cobalt Affinity Gel (Sigma). Despite the FXa cleavage site being engineered adjacent to Ala62, an additional 8 amino acids were present at the N terminus.
Each preparation gave a single band on reducing PAGE and only one peak by mass spectrometry. Immediately before experiments, the Prxs were reduced by 50 mm β-mercaptoethanol. Excess reductant was removed using Micro Bio-Spin 6 columns (Bio-Rad), which were prewashed with deionized water and then with 100 μl of 10 mg/ml catalase followed by 5 ml of 50 mm phosphate buffer, pH 7.4, containing 0.1 mm diethylenetriamine-pentaacetic acid. The phosphate buffer was pretreated with 10 μg/ml catalase, which was removed by passage through an Amicon Ultra-15 10K filter. This procedure was adopted to retain the Prxs in their fully reduced form. Final protein Prx concentrations were measured using the Bio-Rad DC Protein Assay Reagent with bovine serum albumin as standard and were converted into molar concentrations using molecular mass 21,892 Da for Prx2 and 22,418 Da for Prx3.
Reactions of Prxs with H2O2
Reactions were carried out in pH 7.4 phosphate buffer pretreated with catalase as described above and started by the addition of 1 μl of H2O2 to 20 μl of protein during vigorous vortex mixing at 20 °C. After 5 min, 1 μl of catalase (0.5 mg/ml) was added to scavenge excess H2O2 and stop any further oxidation. For catalase competition experiments, the catalase was added to the Prx before mixing with H2O2. For gel analysis 30 mm N-ethylmaleimide (NEM) was added to the reacted samples to block the remaining thiols, followed by loading buffer (62.5 mm Tris-HCl, pH 6.8, 4% (w/v) SDS, and 10% (v/v) glycerol). Liquid chromatography/mass spectrometry (LC/MS) analysis of the Prxs was performed both with and without reduction, after thiol derivatization. Reduced proteins were prepared by incubating with 1 m guanidine hydrochloride for 30 min and 10 mm dithiothreitol (DTT) followed by incubation with 30 mm NEM for 30 min. DTT (50 mm final concentration) was then added to quench unreacted NEM.
Bovine catalase was from Sigma-Aldrich. Its protein concentration was estimated spectrophotometrically (ϵ405 = 120 mm−1 cm−1), and activity was measured by following the loss of added H2O2 at 240 nm as advised by Sigma. From these data we calculated a specific activity of 4700 units/mg, which translates to a second order rate constant of k = 8 × 106 m−1 s−1. This compares well with the reported values of 5 × 106 m−1 s−1 and 1 × 107 m−1 s−1 for the ferric and compound I forms reported by Chance (18).
PAGE and Western Blotting
Samples were analyzed by 12% SDS-PAGE without reducing agent. Gels were either stained with Coomassie Blue or Western blotted and probed with an antibody to Prx2 (Sigma), Prx3 (Abfrontier, Seoul, Korea), or hyperoxidized Prxs (PrxSO2/3) (Abfrontier). A horseradish peroxidase-conjugated secondary antibody (Dako) was used to visualize the immunoblots through enhanced chemiluminescence using the ECLPlus Western Blotting Detection System (GE Healthcare). Stained gels and immunoblots were scanned using a ChemiDoc® XRS (Bio-Rad).
LC/MS Analysis
LC/MS was carried out on samples after reduction and alkylation with NEM to analyze the reduced and hyperoxidized, monomeric forms of Prx. Nonreduced samples were analyzed for disulfide-linked dimer, dimer with one site hyperoxidized, and hyperoxidized monomer. Fifty μl of sample containing 5 μg of protein was loaded onto a Jupiter C18 HPLC column (150 × 2 mm, 5 μm, 100 Å; Phenomenex). An acetonitrile gradient from 90% solvent A (0.1% formic acid in water)/10% solvent B (0.1% formic acid in acetonitrile) to 50% solvent B was run over 8 min at a flow rate of 200 μl/min. Solvent B was held at 50% for 8 min followed by column equilibration over 8 min with 90% solvent A. The HPLC was coupled inline to an electrospray ionization source of a Velos Pro mass spectrometer (Thermo Scientific). Voltage was 5 kV, and nitrogen gas flow was 20 arbitrary units. The temperature of the heated capillary was 275 °C, and the vaporizer temperature was 400 °C. Mass spectral data were acquired from 8 to 16 min of each chromatographic separation scanning between m/z 410 and 2000 in positive mode at a normal scan rate. These chromatographic conditions did not separate the different Prx species, but relative intensities of each could be obtained from the spectral data. Spectra were averaged over the full-length of each protein peak and deconvoluted to yield the molecular masses and relative intensities using ProMass for Xcalibur (version 2.8; Novatia LLC, Monmouth Junction, NJ). The accuracy of the deconvoluted masses was observed to be ±5 Da compared with the theoretical. To establish differences in sensitivity for the different Prx species, standard curves were generated by injecting 0.25, 0.5, 1, and 2 μg of reduced monomer and hyperoxidized monomer of Prx. The detector response was linear over this concentration range with an R2 of >0.98, and there was no difference in sensitivity between the two species. Disulfide-linked dimer also gave a linear response, but because of the more complex mixture of spectra in the nonreduced samples, accurate quantification of the different species was not possible.
Kinetic Analysis
A kinetic model of the scheme in Fig. 1 was constructed, containing known rate constants for H2O2 reacting with the reduced form of the Prx and catalase, and measured concentrations of the proteins and H2O2. The k2/k3 ratio was obtained from the H2O2 dependence data in the absence of catalase. The catalase dependence of the product distribution was analyzed using Berkeley Madonna, with simulations performed at different catalase concentrations and values of k2 and k3 varied, whereas the ratio remained constant, to obtain the best fit.
RESULTS
Prx Hyperoxidation Detected by SDS-PAGE
Oxidation of Prx2 (Fig. 2, left panels) and Prx3 (right panels) was initially monitored by nonreducing SDS-PAGE. Both Coomassie Blue staining and Western blotting showed that reduced Prx2 and Prx3 preparations (first lane, 0 mm H2O2) ran as a major band corresponding to the 22-kDa monomer and a minor band at ∼40 kDa corresponding to the disulfide-linked homodimer. As observed previously (16), H2O2 at lower concentrations caused transition of the monomer to the dimer, then the monomer reappeared as the H2O2 concentration increased. Corresponding blots with an antibody against hyperoxidized Prxs showed staining initially in the dimer, with hyperoxidized monomer appearing only at the higher H2O2 concentrations. Hyperoxidized Prx2 was detectable with even a slight excess of H2O2; but consistent with previous observations (16), Prx3 hyperoxidation required 10-fold higher H2O2 concentrations.
FIGURE 2.

SDS-PAGE analysis of hyperoxidation of purified Prx2 (left panels) and Prx3 (right panels). Reduced Prx (5 μm) was treated with the indicated concentrations of H2O2 and separated under nonreducing conditions. Gels were Coomassie Blue-stained or Western blotted with antibodies to Prx2, Prx3, or PrxSO2. Lines indicate positions of 20-, 25-, 37-, and 50-kDa marker proteins.
Dimeric Prx2 ran as two bands, the faster of which accumulated then disappeared with increasing H2O2 exposure. These could represent the dimeric species containing one and two disulfide bonds, with the latter being more compact and running faster. The presence of a small amount of dimer with one disulfide and one pair of reduced Cys residues would explain the presence of the slower band in the untreated Prx2. However, this should not accumulate on H2O2 treatment, and the upper dimer band seen under these conditions is more likely to represent one disulfide and one hyperoxidized peroxidatic cysteine. Although the same transformation would be expected for Prx3, dimer bands were not separated under our conditions.
Detection of Prx Species by LC/MS
To confirm the identity of the Prx oxidation products and obtain kinetic information, reaction mixtures were analyzed by LC/MS. The Prxs each contain 3 Cys residues. The theoretical masses of their reduced forms and potential disulfide and hyperoxidized (sulfinic acid) oxidation products (after blocking free thiols with NEM) are presented in Table 1. Initially, analyses were performed without reducing the proteins after H2O2 treatment, so as to detect monomeric and disulfide-linked dimeric species. As shown in Fig. 3, prior to reaction with H2O2, Prx2 was predominantly monomeric with a mass close to theoretical for the reduced protein. It also showed a small peak equivalent to the dimer with one disulfide and the other active site cysteines reduced, which is consistent with the interpretation of the gel pattern. As predicted from the gels, treatment with lower concentrations of H2O2 gave products with masses corresponding to dimer with two disulfides or with one disulfide containing hyperoxidized SP. At higher concentrations, the double disulfide decreased, and the hyperoxidized monomer became more prominent. Any other products, including the sulfonic acid, corresponded to <5% of the total peak area. Similar data were obtained with Prx3 (data not shown).
TABLE 1.
Theoretical masses (Da) of Prx2 and Prx3 adducts with NEM
In NEM-treated samples, there were also minor amounts of under- and overalkylated species present. For example the peak at 44,324 Da in the bottom panel of Fig. 3A is consistent with the hyperoxidized dimer with 1 extra NEM. All adducts were included for quantitative analysis. The mass of Prx3 corresponds to the processed protein starting at Ala62 with an additional 8 amino acids at the N terminus.
| Sample | Prx2 (21,892) + NEM (125) | Prx3 (22,418) + NEM (125) |
|---|---|---|
| Reduced monomer | 22,267 | 22,793 |
| (+3 NEM) | ||
| Hyperoxidized monomer | 22,174 | 22,700 |
| (+2 NEM) | ||
| Dimer with one disulfide bond | 44,282 | 45,334 |
| (+4 NEM) | ||
| Dimer with 2 disulfide bond | 44,030 | 45082 |
| (+2 NEM) | ||
| Hyperoxidized dimer | 44,189 | 45,243 |
| (+2 NEM) |
FIGURE 3.

LC/MS analysis of Prx2 treated with H2O2. A, Prx2 (5 μm) was reduced, treated with none, 10 μm, or 120 μm H2O2, derivatized with NEM, and analyzed by LC/MS. Peak masses are identified in Table 1. Note the presence of a peak at 44,324 Da in the 120 μm H2O2 sample, corresponding to hyperoxidized dimer with one extra NEM (overalkylated). B, the profile of Prx species present as a function of H2O2 concentration is shown. ○, dimer; ▵, hyperoxidized dimer; ■, hyperoxidized monomer.
Determination of Rate Constants for the Reactions of Hyperoxidation and Disulfide Bond Formation
The results in Fig. 3 indicate that H2O2-dependent hyperoxidation is competitive with dimerization. We therefore analyzed the data kinetically to obtain estimates for the rate constants for the two reactions. We used the scheme in Fig. 1 for a single active site and assumed that the kinetic properties of the two active sites of the dimer were the same. The products were analyzed either as a disulfide or sulfinic acid, which were obtained by reducing the dimeric species in the reaction mixtures and NEM blocking before LC/MS analysis, without considering the complexity of mixed dimers. Thus, the double disulfide would give two reduced monomers, and the hyperoxidized dimer would give one reduced and one hyperoxidized monomer. Kinetically, reaction 1 would proceed rapidly (k ∼2 × 107 m−1 s−1 (2, 4)). Based on data with other small molecule or protein thiols (19), oxidation of the sulfenic acid to sulfinic acid would be slow compared with its rate of formation. Therefore, we assumed that equimolar H2O2 would rapidly generate the sulfenic acid, which would then react with the surplus H2O2 to form the sulfinic acid or condense with the resolving cysteine to form the disulfide. Hyperoxidation to the sulfinic acid is a second order reaction represented by Equation 1.
Because disulfide bond formation is internal within the functional dimeric unit of the Prx it should be a first order reaction as represented by Equation 2.
From these equations, provided the H2O2 is in sufficient excess for its concentration to change little during the reaction, the following relationship can be derived.
Therefore, if this mechanism holds, a plot of the product ratio versus surplus H2O2 concentration should be linear with slope k2/k3.
The data obtained from reacting 5 μm Prx2 with H2O2 for 5 min (after which the reaction is complete) gave a good fit to this relationship and a k2/k3 value of 6700 m−1 (Fig. 4A). Data obtained with 50 μm Prx2 also gave a linear plot with similar slope (data not shown). Thus, 50% hyperoxidation of 5 μm Prx2 was observed with ∼150 μm H2O2 whereas 50 μm Prx2 required ∼200 μm H2O2. A linear plot was also observed for Prx3 (Fig. 4B); but in accordance with the higher H2O2 concentration required for hyperoxidation, the k2/k3 ratio of 540 m−1 is ∼12-fold less.
FIGURE 4.

Kinetic analysis of the H2O2 dependence of hyperoxidation of Prx2 (A) and Prx3 (B). Each reduced Prx (5 μm) was treated with the indicated concentration of H2O2, excess of H2O2 was removed by adding catalase, then the proteins were reduced by DTT and alkylated with NEM before analysis by LC/MS. Results are means ± S.D. (error bars) for triplicate experiments.
To determine the individual rate constants, competition experiments with bovine catalase were performed. Catalase was used at concentrations sufficient to compete with reaction 2 but too low to inhibit reaction 1. SDS-PAGE and blotting with anti-PrxSO2 showed concentration-dependent inhibition of Prx hyperoxidation by catalase (Fig. 5).
FIGURE 5.

Inhibition of Prx2 and Prx3 hyperoxidation by catalase. Reduced Prx2 or Prx3 (5 μm) was treated with 240 μm H2O2 in the presence of increasing concentrations of catalase, separated by SDS-PAGE under reducing conditions, and probed with anti-PrxSO2/3.
Disulfide and hyperoxidized products were quantified by LC/MS following reduction and alkylation. Kinetic modeling of the data was performed, incorporating the reactions in Fig. 1 plus the reactions of H2O2 with compounds I and II of catalase and their respective rate constants (5 × 106 m−1 s−1 and 1 × 107 m−1 s−1, respectively (18)).
For Prx2, treatment with 120 μm H2O2 (Fig. 6A) or 240 μm H2O2 (Fig. 6B) showed progressive inhibition of hyperoxidation with increasing catalase concentration. Best fit curves (solid lines) give k2 and k3 values that are comparable for the two H2O2 concentrations, with average values (±range) of 1.2 (±0.2) × 104 m−1 s−1 and 1.7 (±0.3) s−1, respectively. Plots calculated for a 2-fold increase or decrease in k2 and k3 (dashed lines in Fig. 6) suggest that the values fall within these limits.
FIGURE 6.

Kinetic analysis of the inhibition by catalase of hyperoxidation of Prx2 (A and B) or Prx3 (C). Each Prx (5 μm) was treated with 120 μm (A), 240 μm (B), or 800 μm H2O2 (C) in the presence of various concentrations of catalase. NEM-derivatized samples were treated, and products were analyzed by LC/MS as in Fig. 4. Data points represent means ± S.E. (error bars) for three analyses (A), means and range of duplicates (B), or means ± S.E. for four analyses (C). In A and B, solid lines represent the best fit for experimental data, which correspond to k2 = 14,000 m−1 s−1, k3 = 2 s−1 (A), and k2 = 10,000 m−1 s−1, k3 = 1.4 s−1 (B); dashed lines represent calculated plots with k2 and k3 two times higher or lower. In C, solid line represents the fit corresponding to k2 = 12,000 m−1 s−1, k3 = 22 s−1. The dotted line corresponds to both values being two times lower, and the dashed line corresponds to k2 = 1100 m−1 s−1, k3 = 2 s−1.
For Prx3, a higher H2O2 concentration was required to cause hyperoxidation and correspondingly higher catalase concentrations for inhibition (Fig. 6C). Because of the relatively low proportion of hyperoxidized protein formed (we were reluctant to use more H2O2 because of the possibility of nonspecific effects), quantifying changes in the presence of catalase were less accurate than for Prx2. Therefore, rather than finding the best fit for k2 and k3, we modeled the data using the k2/k3 ratio of 540 m−1 obtained from Fig. 4B and either the k2 or k3 value determined for Prx2. As shown by the solid line in Fig. 6C, there is good fit with the same k2 value for the two proteins (1.2 × 104 m−1 s−1), giving a value for k3 of 22 s−1 for Prx3. The plot obtained by setting k3 equal to that for Prx2 (and k2 = 1100 m−1 s−1) is well outside the range of the experimental data (Fig. 6C, dashed line). The dotted line for 2-fold lower values for both constants lies outside the data points, suggesting that the true values vary from our estimates by less than this amount. Our data are therefore consistent with the rate constant for hyperoxidation of the sulfenic acid being similar for the two proteins, with the lower susceptibility of Prx3 to hyperoxidation being due to the >10-fold higher rate of dimerization.
DISCUSSION
Susceptibility to Hyperoxidation
We have shown that Prx2 and Prx3 undergo hyperoxidation on exposure to relatively low concentrations of H2O2 without requiring recycling of the disulfide. With Prx2, hyperoxidation was evident with a slight molar excess of H2O2. In agreement with previous observations (16), Prx3 was more resistant, and higher H2O2 concentrations were required. MS analysis showed that the initial product was a covalent dimer with one disulfide and one hyperoxidized SP residue, with a hyperoxidized monomer appearing with higher H2O2 concentrations. We also distinguished the two dimeric forms of Prx2 by SDS-PAGE; and by comparing gels with MS results, we established that the faster band contains two disulfides whereas the slower band has one disulfide and the other SP either reduced or hyperoxidized. The identification of the hyperoxidized dimer as a major product has implications for physiological situations of oxidative stress where there is partial Prx hyperoxidation. Under these conditions the predominant catalytic unit would be a dimer with one active site hyperoxidized, and redox cycling would occur at the other.
Kinetics of Hyperoxidation
The simplest mechanistic representation of hyperoxidation can be described by the scheme in Fig. 1, in which equimolar H2O2 reacts rapidly with the peroxidatic cysteine (k1 = ∼2 × 107 m−1 s−1) to form the sulfenic acid, which then undergoes competitive reactions with either the remaining H2O2 or the resolving cysteine. Our analysis of the MS data gave a good fit with this competitive model and enabled us to estimate rate constants for both reactions. The value obtained for the ratio k2/k3 was 12-fold higher for Prx2 than for Prx3. This reflects the greater resistance of Prx3 to hyperoxidation and translates into 50% hyperoxidation of Prx2 occurring at a H2O2 concentration of ∼150 μm greater than the Prx concentration and Prx3 requiring an excess of ∼2 mm. Individual values for k2 and k3 were obtained using catalase competition, a method that has not previously been used in this context. Kinetic analysis gave good fit to the Prx2 data at two H2O2 concentrations, with curves generated using 2-fold higher or lower rate constants falling outside the data range. Allowing for any uncertainties in absolute catalase concentrations and rate constants under our conditions, we have rounded the calculated values to 12,000 m−1 s−1 for k2 and 2 s−1 for k3. The catalase inhibition data for Prx3 were not as precise but fitted well with the same k2 value as Prx2 and a rounded value of 20 s−1 (an order of magnitude higher) for the dimerization rate constant. These k3 values correspond to half-times for dimerization of 0.4 s for Prx2 and 0.03 s for Prx3.
Although we have used a single site kinetic model, Prx2 and Prx3 function as dimers with two active sites, and it is possible that oxidation of one might affect the reactivity of the other. To make a preliminary assessment of this we used product data for Prx2 that included the hyperoxidized dimer and a kinetic model that incorporated rate equations for the individual steps. Analysis using the same rate constants for each site suggested that there is not a major difference in reactivity between them. However, more accurate quantification of relative concentrations from the MS data is required before concluding whether or not the sites are independent.
Our data indicate that H2O2 reacts with the sulfenic acid of SP in Prx2 and Prx3 approximately 1000 times more slowly than with its thiol. Nevertheless, this is still a very fast reaction. For comparison, rate constants for the sulfenic acids of serum albumin and free cysteine are >104-fold lower (20, 21). There are limited data available on rates of hyperoxidation of other Prxs. AhpE, a 1-Cys Prx from mycobacteria, has been studied most because oxidation can be monitored by following changes in fluorescence of a tryptophan residue near the active site. Although k2 for this Prx is only 40 m−1 s−1, the reduced form is also much less reactive than Prxs 2 and 3 with H2O2, and the difference in rate constants is again approximately 1000 (22). AhpE is approximately 1000 times more reactive with fatty acid hydroperoxides than H2O2, and it is interesting that rate constants for hyperoxidation by these peroxides are also high (∼105 m−1 s−1) and about 3 orders of magnitude less than for the thiolate (23).
Determinants of k2 and k3
The reduced forms of Prxs 2 and 3 react with H2O2 in their fully folded (FF) state. The high reactivity is determined both by the low pKa of the peroxidatic Cys and by structural features that enable the peroxide to align in the transition state in such a way as to lower the electron density of its oxygen atoms via H-bonding and make it a better electrophile (17, 24). If the second H2O2 reacts with the sulfenic acid while it is still in the FF state, the same positioning could explain why this reaction is also fast. However, the presence of the extra oxygen could affect the active site geometry, and further structural studies are required to test this proposal.
Formation of the disulfide bond in the typical 2-Cys Prxs requires transition from the FF to the locally unfolded (LU) state and movement of the sulfenic acid form of SP toward SR (the two sulfur atoms are 14 Å apart in the reduced form) (25). The reaction can be considered as two steps, the first being unfolding from the FF to the LU state and displacement of SP from the active site and the second the reaction of the sulfenic acid with the SR thiol. The rate constant k3 would apply to the slower of the two. If the reaction between the sulfenic acid and H2O2 occurred in the active site, this would imply that k3 applied to the FF to LU transition. However, further structural and kinetic studies are required to establish if this is the case. The rate of unfolding has been shown to be controlled by two sequence motifs, GGLG and YF, which allow the C terminus to wrap over the active site in the FF conformation and facilitate hyperoxidation (9). Even though Prx2 is more susceptible to hyperoxidation than Prx3, the GGLG and YF motifs are conserved between the two proteins. There are other variations in the C-terminal region that might be responsible for this effect (16).
Significance of the Fast Reaction of H2O2 with SPOH
Table 2 compares k1, k2, and k3 values for Prx2 and Prx3 with a range of other thiols. Reaction 3 (k3) is fast for low molecular mass thiol/sulfenic acid reactions, but much slower for proteins such as albumin where the sulfenic acid is protected. The rate of internal condensation of OxyR is within the range of the Prxs. The sulfenic acids of Prx2 and Prx3 are much more reactive than other sulfenic acids with H2O2. Although H2O2 reacts much more slowly with the sulfenic acids of Prx2 and Prx3 than with the reduced forms, the second order rate constants of ∼104 m−1 s−1 are still 100–1000-fold higher than rate constants measured for low pKa cysteine thiols in other proteins, including phosphatases (26). Reduced Prxs, glutathione peroxidases, catalase, and heme peroxidases are the only mammalian proteins known to react faster than these sulfenic acids with H2O2. This high reactivity has biological ramifications. Hyperoxidation of Prxs has been observed in cells and tissues subjected to a variety of stresses. This would require the Prx to be exposed to high or sustained H2O2 exposure, most likely at a localized site where the majority of the Prx becomes oxidized. In these situations the Prx sulfenic acid intermediate should be a favored over other thiol proteins as a target for H2O2, and reformation of the sulfenic acid during turnover would enable the hyperoxidized form to accumulate. Hyperoxidation of eukaryotic Prxs appears to impart a gain of function, in some cases conferring chaperone activity. It may also play a regulatory role by allowing other reactions to occur, as proposed in the “floodgate” response (9) and observed during steroidogenesis (11). The unusually high reactivity of the sulfenic acid would facilitate this process.
TABLE 2.
Rate constants for the reactions of thiol (k1) and corresponding sulfenic acid (k2) with H2O2 and condensation of sulfenic acid (k3)
Sources of the rate constants not measured in this paper are given in parentheses.
| k1 | k2 | k3a | |
|---|---|---|---|
| m−1 s−1 | m−1 s−1 | ||
| Prx2b | 20,000,000 (4) | 12,000 | 2 s−1 |
| Prx3c | 20,000,000 (2) | 12,000 | 20 s−1 |
| Cysteine | 1(27) | <1(21) | >10,000 m−1 s−1(21) |
| Cdk A and B | 120–160 (28) | 60–110 (28) | 0.012–0.16 s−1 (28) |
| Human serum albumin | 3 (29) | 0.4 (20) | 3 m−1 s−1 (with GSH) (20) |
| 22 m−1 s−1 (with Cys) (20) | |||
| OxyR | >100,000 (30) | 9.7 s−1 (30) |
a Reaction 3 is first order (s−1) when the disulfide is formed within a protein or second order (m−1 s−1) when two reactants are involved.
b k3 for Prx2 has been rounded to a single figure to reflect the accuracy of the kinetic analysis.
c For Prx3, k2 was taken as the same as the value for Prx2 on the basis that it gave a good fit of the data in Fig. 6C; k3 has been rounded to a single figure to reflect the accuracy of the analysis.
Acknowledgment
We thank Andrea Betz for advice in the preparation of Prx3.
This work was supported by the Marsden Fund and the Health Research Council of New Zealand.
- Prx
- peroxiredoxin
- FF
- fully folded
- LU
- locally unfolded
- NEM
- N-ethylmaleimide
- SP
- peroxidatic cysteine
- SR
- resolving cysteine.
REFERENCES
- 1. Rhee S. G., Woo H. A. (2011) Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger HO, and protein chaperones. Antioxid. Redox Signal. 15, 781–794 [DOI] [PubMed] [Google Scholar]
- 2. Peskin A. V., Low F. M., Paton L. N., Maghzal G. J., Hampton M. B., Winterbourn C. C. (2007) The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 282, 11885–11892 [DOI] [PubMed] [Google Scholar]
- 3. Ogusucu R., Rettori D., Munhoz D. C., Netto L. E., Augusto O. (2007) Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: rate constants by competitive kinetics. Free Radic. Biol. Med. 42, 326–334 [DOI] [PubMed] [Google Scholar]
- 4. Cox A. G., Peskin A. V., Paton L. N., Winterbourn C. C., Hampton M. B. (2009) Redox potential and peroxide reactivity of human peroxiredoxin 3. Biochemistry 48, 6495–6501 [DOI] [PubMed] [Google Scholar]
- 5. Peskin A. V., Cox A. G., Nagy P., Morgan P. E., Hampton M. B., Davies M. J., Winterbourn C. C. (2010) Removal of amino acid, peptide and protein hydroperoxides by reaction with peroxiredoxins 2 and 3. Biochem. J. 432, 313–321 [DOI] [PubMed] [Google Scholar]
- 6. Wood Z. A., Schröder E., Robin Harris J., Poole L. B. (2003) Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 28, 32–40 [DOI] [PubMed] [Google Scholar]
- 7. Jeong W., Park S. J., Chang T. S., Lee D. Y., Rhee S. G. (2006) Molecular mechanism of the reduction of cysteine sulfinic acid of peroxiredoxin to cysteine by mammalian sulfiredoxin. J. Biol. Chem. 281, 14400–14407 [DOI] [PubMed] [Google Scholar]
- 8. Roussel X., Kriznik A., Richard C., Rahuel-Clermont S., Branlant G. (2009) Catalytic mechanism of sulfiredoxin from Saccharomyces cerevisiae passes through an oxidized disulfide sulfiredoxin intermediate that is reduced by thioredoxin. J. Biol. Chem. 284, 33048–33055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wood Z. A., Poole L. B., Karplus P. A. (2003) Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300, 650–653 [DOI] [PubMed] [Google Scholar]
- 10. Woo H. A., Chae H. Z., Hwang S. C., Yang K. S., Kang S. W., Kim K., Rhee S. G. (2003) Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science 300, 653–656 [DOI] [PubMed] [Google Scholar]
- 11. Kil I. S., Lee S. K., Ryu K. W., Woo H. A., Hu M. C., Bae S. H., Rhee S. G. (2012) Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol. Cell 46, 584–594 [DOI] [PubMed] [Google Scholar]
- 12. Neumann C. A., Cao J., Manevich Y. (2009) Peroxiredoxin 1 and its role in cell signaling. Cell Cycle 8, 4072–4078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jeong W., Bae S. H., Toledano M. B., Rhee S. G. (2012) Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic. Biol. Med. 53, 447–456 [DOI] [PubMed] [Google Scholar]
- 14. Bae S. H., Sung S. H., Cho E. J., Lee S. K., Lee H. E., Woo H. A., Yu D. Y., Kil I. S., Rhee S. G. (2011) Concerted action of sulfiredoxin and peroxiredoxin I protects against alcohol-induced oxidative injury in mouse liver. Hepatology 53, 945–953 [DOI] [PubMed] [Google Scholar]
- 15. Yang K. S., Kang S. W., Woo H. A., Hwang S. C., Chae H. Z., Kim K., Rhee S. G. (2002) Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J. Biol. Chem. 277, 38029–38036 [DOI] [PubMed] [Google Scholar]
- 16. Cox A. G., Pearson A. G., Pullar J. M., Jönsson T. J., Lowther W. T., Winterbourn C. C., Hampton M. B. (2009) Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem. J. 421, 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nagy P., Karton A., Betz A., Peskin A. V., Pace P., O'Reilly R. J., Hampton M. B., Radom L., Winterbourn C. C. (2011) Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: a kinetic and computational study. J. Biol. Chem. 286, 18048–18055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chance B. (1949) The primary and secondary compounds of catalase and methyl or ethyl hydrogen peroxide: reactions with hydrogen peroxide. J. Biol. Chem. 180, 947–959 [PubMed] [Google Scholar]
- 19. Nagy P. (2013) Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid. Redox Signal. 18, 1623–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Turell L., Botti H., Carballal S., Ferrer-Sueta G., Souza J. M., Durán R., Freeman B. A., Radi R., Alvarez B. (2008) Reactivity of sulfenic acid in human serum albumin. Biochemistry 47, 358–367 [DOI] [PubMed] [Google Scholar]
- 21. Ashby M. T., Nagy P. (2006) On the kinetics and mechanism of the reaction of cysteine and hydrogen peroxide in aqueous solution. J. Pharm. Sci. 95, 15–18 [DOI] [PubMed] [Google Scholar]
- 22. Hall A., Parsonage D., Poole L. B., Karplus P. A. (2010) Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J. Mol. Biol. 402, 194–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hugo M., Turell L., Manta B., Botti H., Monteiro G., Netto L. E., Alvarez B., Radi R., Trujillo M. (2009) Thiol and sulfenic acid oxidation of AhpE, the one-cysteine peroxiredoxin from Mycobacterium tuberculosis: kinetics, acidity constants, and conformational dynamics. Biochemistry 48, 9416–9426 [DOI] [PubMed] [Google Scholar]
- 24. Selles B., Hugo M., Trujillo M., Srivastava V., Wingsle G., Jacquot J. P., Radi R., Rouhier N. (2012) Hydroperoxide and peroxynitrite reductase activity of poplar thioredoxin-dependent glutathione peroxidase 5: kinetics, catalytic mechanism and oxidative inactivation. Biochem. J. 442, 369–380 [DOI] [PubMed] [Google Scholar]
- 25. Hall A., Karplus P. A., Poole L. B. (2009) Typical 2-Cys peroxiredoxins: structures, mechanisms and functions. FEBS J. 276, 2469–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Winterbourn C. C., Hampton M. B. (2008) Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 45, 549–561 [DOI] [PubMed] [Google Scholar]
- 27. Winterbourn C. C., Metodiewa D. (1999) Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 27, 322–328 [DOI] [PubMed] [Google Scholar]
- 28. Sohn J., Rudolph J. (2003) Catalytic and chemical competence of regulation of cdc25 phosphatase by oxidation/reduction. Biochemistry 42, 10060–10070 [DOI] [PubMed] [Google Scholar]
- 29. Carballal S., Radi R., Kirk M. C., Barnes S., Freeman B. A., Alvarez B. (2003) Sulfenic acid formation in human serum albumin by hydrogen peroxide and peroxynitrite. Biochemistry 42, 9906–9914 [DOI] [PubMed] [Google Scholar]
- 30. Lee C., Lee S. M., Mukhopadhyay P., Kim S. J., Lee S. C., Ahn W. S., Yu M. H., Storz G., Ryu S. E. (2004) Redox regulation of OxyR requires specific disulfide bond formation involving a rapid kinetic reaction path. Nat. Struct. Mol. Biol. 11, 1179–1185 [DOI] [PubMed] [Google Scholar]