

Background: The mechanism for association and dissociation of Psb27 and CP43 is poorly understood.
Results: Loop E of CP43 undergoes significant conformational change upon D1 processing.
Conclusion: D1 processing initiates the dissociation of Psb27 from CP43.
Significance: The structural dynamics of the lumenal domain of CP43 plays a critical role in the assembly of functional Photosystem II centers.
Keywords: Electron Transfer, Mass Spectrometry (MS), Metalloenzymes, Photosynthesis, Photosystem II, CP43 Loop E Conformational Rearrangement, PSII Assembly Intermediate, Protein Cross-linking, Protein Footprinting, Psb27
Abstract
The PSII repair cycle is required for sustainable photosynthesis in oxygenic photosynthetic organisms. In cyanobacteria and higher plants, proteolysis of the precursor D1 protein (pD1) to expose a C-terminal carboxylate group is an essential step leading to coordination of the Mn4CaO5 cluster, the site of water oxidation. Psb27 appears to associate with both pD1- and D1-containing PSII assembly intermediates by closely interacting with CP43. Here, we report that reduced binding affinity between CP43 and Psb27 is triggered by the removal of the C-terminal extension of the pD1 protein. A mass spectrometry-based footprinting strategy was adopted to probe solvent-exposed aspartic and glutamic acid residues on the CP43 protein. By comparing the extent of footprinting between HT3ΔctpAΔ27PSII and HT3ΔctpAPSII, two genetically modified PSII assembly complexes, we found that Psb27 binds to CP43 on the side of Loop E distal to the pseudo-symmetrical D1-D2 axis. By comparing a second pair of PSII assembly complexes, we discovered that Loop E of CP43 undergoes a significant conformational rearrangement due to the removal of the pD1 C-terminal extension, altering the Psb27-CP43 binding interface. The significance of this conformational rearrangement is discussed in the context of recruitment of the PSII lumenal extrinsic proteins and Mn4CaO5 cluster assembly. In addition to CP43's previously known function as one of the core PSII antenna proteins, this work demonstrates that Loop E of CP43 plays an important role in the functional assembly of the Water Oxidizing Center (WOC) during PSII biogenesis.
Introduction
Photosynthesis harvests solar energy to power life on Earth. The corresponding energy transduction and storage processes are accomplished by a diverse family of pigment-protein complexes, of which Photosystem II (PSII)2 is one of the most ubiquitous. The reactions that PSII mediate include light-induced oxidation of water and reduction of plastoquinone. This integral membrane protein complex contains more than 20 subunit proteins and numerous cofactors. An x-ray structural model of the active PSII dimer from a cyanobacterium was recently described at 1.9 Å resolution (1), providing molecular-level detail of the interactions between the subunits present in this solid-state form of PSII.
An unusual property of PSII is that, under various light conditions, photoinactivation of PSII takes place, because of its demanding photochemistry. Sustainable photosynthesis, consequently, must rely on an efficient PSII repair cycle. At any given instant, active PSII complexes and many inactive PSII assembly intermediates coexist. These intermediates are especially difficult to capture and characterize by using traditional biochemical techniques, because of their low abundance, structural heterogeneity, and thermodynamic instability.
Psb27, present in multiple PSII assembly intermediates but not in the active PSII dimer, appears to be involved in the repair cycle of PSII (2, 3) and in the assembly of the Mn4CaO5 cluster (4). Psb27-containing PSII assembly intermediates are not capable of oxygen evolution (2). We have determined that PsbO, but not other extrinsic proteins, is able to bind to the lumenal side of PSII in a Psb27-containing PSII complex (5). The absence of a functional Mn4CaO5 cluster in this complex might be necessary to decouple light harvesting from the reaction centers (6) and thus minimize photodamage to non-functional PSII assembly intermediates. Given the apparent role of Psb27 in PSII assembly, significant efforts have been made to determine its location within PSII assembly intermediates (7–9). Our recent study using chemical cross-linking analysis and mass spectrometry revealed a close association between Psb27 and Loop E of CP43 (10). Using a combination of genetic and biochemical techniques, other research groups independently reached similar conclusions (11). The binding interface between CP43 and Psb27, however, remains unknown, as does the mechanism that triggers dissociation of Psb27 from CP43.
Mass spectrometry-based protein footprinting methodology is developing rapidly, in part because it takes advantage of the tools of analytical proteomics (12). Footprinting methods are becoming capable of probing protein folding and unfolding dynamics and providing new perspectives on the biophysics of protein-ligand interactions. We report here that carboxyl-group footprinting, specifically with glycine ethyl ester (GEE) labeling, can be used to map the Psb27-CP43 binding interface in the PSII complex from the cyanobacterium Synechocystis sp. strain PCC 6803 (hereafter called Synechocystis 6803). To map the CP43-Psb27 interface, we compared PSII complexes from a strain enriched in Psb27-containing complexes (HT3ΔctpA) and a Psb27-deletion strain (HT3ΔctpAΔ27), both of which accumulate complexes in which pD1 has not been processed. In the next step, we sought to identify changes to the CP43-Psb27 interface upon processing of pD1 to D1. We accomplished this by comparing the modification extent of a second pair of PSII assembly intermediates isolated from HT3ΔctpA and His27 cells. Complexes from the former strain reveal the Psb27-CP43 interface before pD1 processing, and complexes from the latter reveal the interface after pD1 processing. Our data indicate a novel function for the Loop E of CP43 as a hub of PSII donor-side assembly during PSII biogenesis, in addition to its well-established role providing coordination for the Mn4CaO5 cluster during steady-state oxygen evolution.
MATERIALS AND METHODS
Mutant Construction
Generation of the His-27 strain was previously reported (5). The HT3 strain (13), which was genetically modified to encode a C-terminal His6 tag on the CP47 protein, was a kind gift from Dr. Terry Bricker. The HT3ΔctpAΔ27 mutant was generated by transforming the HT3ΔctpA strain by using the Δpsb27 construct reported in Ref. 4. The tagged PSII complexes were purified essentially as in Refs. 13 and 14. Protein electrophoresis and immunodetection were performed as in Refs. 15 and 16, unless otherwise indicated.
Carboxyl Group Modification
PSII preparations were resuspended at 0.3 mg/ml chlorophyll a (Chl a) in 25% glycerol, 10 mm MgCl2, 5 mm CaCl2, 50 mm MES buffer, pH 6.5. The modification reaction was carried out for 1 h at 23 °C in the dark with 0.3 m GEE (Sigma) and 50 mm EDC (Pierce), using freshly prepared 1.5 m GEE and 0.5 m EDC stock solutions. The reaction was quenched by adding an equal volume of 1 m sodium acetate, and the sample was desalted and buffer-exchanged using a ZebaTM column (Thermo Scientific, Rockford, IL) according to the manufacturer's protocol.
Absorption Spectroscopy
PSII samples with and without GEE modification were diluted to 0.5 μg/ml of Chla and analyzed on a DW 2000 conversion spectrophotometer (On-Line Instrument Systems, Bogart, GA).
LC-MS/MS
The modified PSII samples were digested by using sequencing-grade trypsin (Sigma) following the manufacturer's instructions. An aliquot (5 μl) of sample after 1:5 dilution was loaded onto a custom-built silica capillary column (0.075 mm x 150 mm) packed with C18 reversed-phase material (Magic, 5 μm 300 Å, Michrom, Auburn, CA). The gradient was from 2% solvent B (97% acetonitrile, 3% water, 0.1% formic acid) and 98% solvent A (97% water, 3% acetonitrile, 0.1% formic acid) to 60% solvent B over 60 min, then to 85% solvent B for 10 min at a flow rate of 260 nL/min followed by a 10 min re-equilibration step. The solution was sprayed directly from the column into an LTQ-Orbitrap mass spectrometer (Thermo Fisher, Waltham, MA) by using a PicoView PV-500 nanospray source (New Objective, Woburn, MA). A full mass spectrum of eluting peptides was recorded at high mass resolving power (100,000 for ions of m/z 400) with the FT mass spectrometer component while product-ion (MS/MS) spectra were obtained for the six most abundant ions from peptides eluting at that time. The normalized collision energy was 35%, with a 2 Da isolation width and wide-band activation. Ions submitted to MS/MS were placed in a dynamic exclusion list for 8 s. Identification of peptides and sites of aspartic acid (D) and glutamic acid (E) modification was performed by taking advantage of accurate-mass measurements for the peptides and the product-ion mass spectra by searching against the bacterial entries in the NCBI database with the Mascot search engine (Matrix Science Inc, Boston, MA). The extracted ion chromatograms were used to quantify the chemical-modification percentage of each peptide from three PSII complexes, as described in “Results.” Integration of peak areas was performed using Qual Browser (Xcalibur: Thermo-Scientific, San Jose, CA).
RESULTS
PSII complexes from three genetically modified strains were prepared, and their D/E residues were subjected to protein footprinting by chemical modification with GEE (17, 18). A schematic diagram of the experimental design and procedure is shown in Fig. 1. We performed the experiments under physiological conditions by using the zero-length cross-linker, 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (EDC). It should be noted that under our experimental conditions (0.3 m GEE, see “Materials and Methods”), the EDC-triggered GEE modification reactions dominate EDC-mediated cross-linking reactions (19, 20), essentially eliminating formation of lysine (K)-D/E cross-links.
FIGURE 1.

Scheme showing the experimental design. Solvent-exposed amino acid residues of isolated PSII complexes were covalently modified by GEE. The labeling sites were located by LC/MS/MS after quenching, purification, and trypsin digestion. The modification extent of different peptides were compared by determining the interaction interface. Psb27 (black rectangle), PsbO (blue rectangle), Psb28 (gray oval). For clarity, some parallel procedures are omitted in the figure.
The PSII complexes we compared are grouped into two pairs. Pair A consists of HT3ΔctpAΔ27PSII and HT3ΔctpAPSII, and Pair B consists of HT3ΔctpAPSII and His27PSII (Fig. 1). Pair A was used to map the CP43-Psb27 binding interface before pD1 processing, since the only difference between these two complexes is that HT3ΔctpAΔ27PSII does not contain Psb27 and HT3ΔctpAPSII does contain Psb27. Pair B was used to probe any structural perturbations to the CP43-Psb27 interface when pD1 is processed to D1 (Fig. 1).
To establish that the three PSII preparations are of sufficient quality for footprinting, we analyzed them by SDS-PAGE (Fig. 2A). Note the essential differences of protein composition between His27PSII and the other two PSII preparations. However, because of the identical chlorophyll-binding protein compositions in the three preparations (i.e. CP47 and CP43), the three PSII preparations showed consistent characteristic absorption spectra of chlorophyll a in aqueous solution (Fig. 2, B–D), specifically strong Qy and Soret bands. Peaks from carotenoid absorption in the 400–500 nm regions were recognizable in the absorption spectra of the three PSII samples. Notably, all samples showed identical absorption spectra before and after GEE modification (Fig. 2, B–D), indicating that the footprinting caused no significant conformational changes that could lead to misleading modification of formerly protected side chains.
FIGURE 2.

A, polypeptide profiles of isolated His27PSII (lane 2), HT3ΔctpAΔ27PSII (lane 3), and HT3ΔctpAPSII (lane 4) separated on SDS-PAGE and visualized by Coomassie Brilliant Blue R250 staining. Lane 1, molecular weight standards. The positions of major PSII protein components are indicated on the left. On the right, a (CP47, untagged), b (PsbO), c (D1, processed), d (Psb27, His6-tagged), e (Psb28). Characteristic absorption spectra of three PSII complexes, B, absorption spectra of HT3ΔctpAΔ27PSII complex unmodified (black) and GEE-modified complex (red). C, HT3ΔctpAPSII. D, His27PSII.
Once the samples were modified, the reaction was quenched. Quenching reagent and other non-protein chemicals were removed as described in “Materials and Methods,” and the samples were subjected to trypsin digestion followed by LC-MS/MS analysis (Fig. 1; an example of experimental output is shown in Fig. 3). The analytical approach employed is one that is typically used in proteomics studies. Quantification of modified and unmodified peptides was accomplished by obtaining extracted ion chromatograms (EICs) and integrating areas under relevant peaks. The modification extent of a peptide was computed to be the area under a peak representing the modified peptide divided by the sum of the peak areas of all forms of that peptide that could be detected by MS (Fig. 1): (AreaC + AreaD)/(AreaB + AreaC + AreaD), as in Fig. 3 for HT3ΔctpAΔ27PSII. In this manner, the modification extent of the same peptides from all three PSII complexes were quantified.
FIGURE 3.
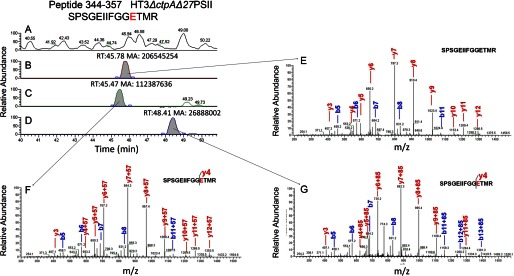
Representative chromatograms from LC-MS/MS showing peptide from CP43 with D/E modifications. A, LC chromatogram of the trypsin-digested HT3ΔctpAΔ27 protein complex. B, EIC of the unmodified peptide 344–357 of CP43 protein of m/z 740.85. C, EIC of the modified peptide 344–357 of m/z 769.36 with E modification (+57.05 Da). D, EIC of the modified peptide 344–357 of m/z 783.38 with E modification (+85.05 Da). The D/E modification sites were determined by the product ions (E, F, G). The retention times of the peptides and the area of EIC chromatograms were labeled, and the mass accuracy was better than 10 ppm. The p values for all the modified and unmodified peptides are below 10−5.
Chemical modification of D/E sites by GEE shifts the tryptic peptide mass by +85.0528 Da (C4H7NO) (Fig. 3N) or +57.0515 (C2H3NO) Da (Fig. 3O) (17); the latter is a hydrolysis product formed under the acidic conditions used in the LC separation. Both modified and unmodified peptides are identifiable with high certainty in the LC-MS/MS analysis by tracking the accurate masses of the modified peptides. For example, the +85.0528 Da peptide generally elutes ∼1–3 min later than the unmodified peptide, whereas the +57.0515 Da peptide elutes at approximately the same time or a little earlier than the unmodified peptide (17). The same peptides from all three PSII complexes were analyzed and compared separately.
The CP43 protein from Synechocystis 6803 has 460 amino acid (AA) residues and is highly conserved in cyanobacteria and higher plants (21). Our sequence coverage of CP43 by LC-MS/MS analysis is 41%, which is comparable or even superior to that observed for this protein by other investigators (32 and 26% from Nakamura et al. (22) and Frankel et al. (23), respectively). Low coverage is likely due to the difficulty of detecting large tryptic peptides in our analysis. If there are no missed cleavages in the trypsin digestion, CP43 is expected to produce 19 different peptides containing at least 6 AA (supplemental Table S1). Four peptides (36–122, 212–248, 250–307, and 379–410), however, have more than 30 AA and are technically difficult to identify by LC-MS/MS. Signals for peptide 166–184 (supplemental Table S1) on Loop C are barely above the noise level and only could be found occasionally. Hence, these peptides were not considered further. It should be noted that most missing peptide fragments are parts of transmembrane helices and analyzing them is not informative for locating a protein-protein interface.
Of the remaining 15 peptides, 6 peptides (supplemental Table S1), most of which were detected, do not have any D/E residues. Of the remaining nine peptides, which do contain D/E residues, four peptides (14–28, 123–139, 437–444, 449–460) are either buried in the hydrophobic membrane (see “Discussion”) and are not accessible for modification, or are exposed to the cytosolic side of PSII and undergo modification reactions. The modification extent of the D/E residues on the cytosolic side of PSII is not expected to change among the three PSII complexes under investigation because the Psb27 binding site and the pD1 processing site are both present on the lumenal side of PSII.
Various authors have used different numbering systems for PSII proteins from a variety of different organisms. Note that peptide sequence numbering used in this study hereafter is based on the PSII structure in Ref. 1 (PDB ID: 3ARC) for literature consistency. A protein sequence alignment of CP43 based on this PSII crystal structure (PDB ID: 3ARC) and the corresponding Synechocystis 6803 sequence was performed for comparison (data not shown).
We detected tryptic peptides from CP43 in the digests of all three PSII complexes and classified them into different groups. For Pair A (Fig. 1), our results demonstrate that peptide 344–357, located in the loop E of CP43 on the lumenal side of PSII (Fig. 4A and supplemental Table S2) was modified to approximately an identical level in both the absence and presence of Psb27 (compare the red bars with the green bars in Fig. 4B). It should be noted that the modification extent of peptide 450–457 showed essentially no statistical difference between the two complexes in Pair A (Fig. 4B, red and green bars), indicating that solvent accessibility of E456 on the PSII cytosolic side is not dependent on the presence of Psb27, which is bound on the lumenal side of PSII. We view this outcome as a negative control to assure us that our approach produces reliable results.
FIGURE 4.

Modification extent of key CP43 peptides and their location in the PSII crystal structure (A) Location of four CP43 lumenal peptides from Fig. 4B in the PSII structure (PDB ID: 3ARC). PsbA (blue), PsbB (salmon), PsbC (wheat), PsbD (violet), all other PSII subunits (light green). B, two peptides have similar modification extent (peptide 344–357 and 450–457), and three peptides (363–370, 371–379, and 382–390) show different modification levels (*: p = not significant, **: p < 0.05, ***: p < 0.01, Student's t test, n = 3, error bars indicate S.D.). The inset shows the theoretical computation (Macrodox) of exposed area (Å2) of side-chain carboxyl oxygen in CP43's loop E determined from five PSII crystal structures (PDB ID: 3ARC, 3AOB, 3BZ1, 3PRQ, and 2AXT). C, magnified view of the model shown in B. E367, D376, D378, and D383 are represented as sticks. Note N378 of 3ARC is represented as D378 (Synechocystis 6803). The Psb27 binding site is indicated (dashed oval). D, modification extent of Psb27, peptides 24–35, 36–43, 44–52, 57–63, and 95–104. The inset shows the theoretical computation (Macrodox) of exposed area (Å2) of side-chain carboxyl oxygen of Psb27 (PDB ID: 2KND, n = 20, states from NMR). E, location of four peptides of Psb27 in NMR structure (PDB ID: 2KND). Peptide 24–43 (purple), peptide 44–52, 57–63, and 95–104 (orange). D/E are represented as sticks.
In contrast, the D/E modification extent of three other peptides, 363–370, 371–379, 382–390, showed statistically significant decreases in the presence of Psb27 in Pair A than in its absence (Fig. 4B, red and green bars). This indicates that the regions corresponding to these peptides are shielded when Psb27 binds in HT3ΔctpAPSII. Theoretically, even structurally shielded domains can be modified by EDC-facilitated GEE labeling. The modification extent depends on the solvent accessibility of the site and on the duration of the modification reaction. What is important, therefore, is whether the modification extent changes between the various biological states. Such changes can indicate, for example, binding of a ligand or a change in conformation. To draw reliable conclusions, modification reactions must be conducted under consistent, controlled physiological conditions, and multiple biological replicates must demonstrate statistically significant results. Our data fit these criteria.
The core subunits of PSII are highly conserved (21). For example, the protein sequences of the Loop E of CP43 from T. elongatus, T. vulcanus, and Synechocystis 6803 are more than 90% identical (data not shown). Building on a foundation from earlier studies, we took the crystal structures of PSII from the first two thermophiles as representative of the latter. Using the solvent exposures of every atom on Loop E of CP43 (based on PDB ID:3ARC and four other published PSII structures: 3AOB, 3BZ1, 3PRQ, and 2AXT), we used MacroDox (v 4.5.3. S. H. Northrup, Tennessee Technological University) to compute and plot the surface exposure in Å2 for the carboxyl oxygen of each D/E residue of Loop E (Fig. 4B, insert). The experimental GEE modification extent consistently paralleled the calculated solvent accessibilities of the D/E carboxyl groups taken from the five PSII crystal structures. Thus, a change in modification extent for a given peptide can be interpreted confidently as a change in solvent accessibility. To our knowledge, this is the first attempt to establish a relationship between surface exposure of a carboxyl group and its experimental modification extent by GEE. It should be noted that data from the PSII structure (PDB ID:3ARC) (1), with the highest atomic resolution (1.9 Å resolution), does not give the best fit with our observed experimental data (Fig. 4B). Instead, the PSII structure 3BZ1 (2.9 Å resolution) gives the best fit and is more consistent with the other three PSII structures.
For Pair B, the D/E modification extents are similar for peptides 344–357 and 450–457 (Fig. 4B, green and blue bars). The D/E modification extent of peptides 363–370 and 371–379 of CP43, however, are significantly increased (Fig. 4B, green and blue bars and supplemental Table S2). This indicates that upon pD1 processing in His27PSII, the solvent accessibility of the regions represented by these two peptides increases significantly. For peptide 382–390, there is no statistically significant difference between the modification extent of the two Pair B complexes (∼10–15% modification in both cases).
Trypsin digestion yielded 68% sequence coverage over the 110 AA of Psb27 from the two complexes in Pair B as determined by LC-MS/MS analysis. The missing regions correspond to either small tryptic peptides that contain less than six AA or two large peptides that contain more than 20 AA (data not shown). Nevertheless, there are several informative features from the peptides that were detected (Fig. 4, D and E). First, peptides 24–35 and 36–43 display high modification extents. This trend is consistent with the calculated carboxyl group exposures of the D and E residues in Psb27 as computed using MacroDox and the NMR structure of Psb27 (2KND (7) and 2KMF (24) data not shown). The Fig. 4D insert shows the calculated result using 2KND. Second, for both of these peptides, there is no statistically significant modification difference between the two Pair B complexes. This shows that the solvent exposure of these regions is independent of the processing of pD1 to D1. Third, the modification extent of peptides 44–52 and 95–104 are low (less than 10%). The general trend of these modifications matches more closely the theoretical computations for 2KND than for 2KMF (data not shown). Finally, a comparison of the modification level of these regions in Pair B suggests that upon processing of pD1 to D1, solvent accessibility increases. The modification extent of peptide 57–63 also showed a significant difference in Pair B. However, this fragment showed unusual chromatographic behavior in our experiment, eluting early and over a long chromatographic time. It is possible that this phenomenon arose from a trypsin digestion bias due to the presence of an RRK sequence on the N-terminal side of this fragment. Overall, modification-extent comparison of these three peptides indicated that binding of Psb27 to CP43 after D1 processing in His27PSII was not as tight as it was in HT3ΔctpAPSII, before D1 processing.
DISCUSSION
CP43-Psb27 Binding Interface
Both CP43 and CP47, core antenna proteins of PSII, contain six trans-membrane helices and a large extrinsic domain, 132 and 191 amino acids long, respectively, that joins the lumenal ends of helices V and VI. The six hydrophobic trans-membrane domains are conserved and bind 13 and 16 chlorophyll a molecules in CP43 and CP47, respectively. As might be expected, charged amino acid residues, including the trypsin cleavage sites of lysine and arginine, rarely occur in transmembrane helices, and this trend holds true for CP43 (data not shown) and CP47.
Based on the significantly different modification extent shown by three peptides between HT3ΔctpAΔ27PSII and HT3ΔctpAPSII (Pair A), we were able to locate the CP43-Psb27 binding interface to be on the distal surface of Loop E of CP43 (Fig. 5A). In our Pair A experiment, CP43 E367, D376, D378, and D383 are protected by the presence of Psb27 in HT3ΔctpAPSII (Figs. 4A and 5B). These results indicate that binding of any other extrinsic proteins in this region will be decreased or even abolished because of steric interference. This explains the absence of PsbV and PsbO in the Psb27-containing Pair A PSII complexes (HT3ΔctpA (25) and His27ΔctpA (5)).
FIGURE 5.

A, location of Psb27 on loop E of CP43, a model based on D/E footprinting. B, Psb27 and PsbO in one complex, His27PSII. Charge-charge interactions of the N-terminal domain of PsbO and the Loop E of CP43 (D8- R362, sphere) are affected but not abolished by the presence of Psb27 (dashed oval). PsbO (yellow), CP43 (wheat). C, steric conflicts of Psb27 and PsbV. CP43 (wheat), PsbO (yellow), PsbV (ruby), E90:R390, represented as sticks (red). Peptide 64–94 of Psb27 is represented as a backbone (cyan) for clarity. D, relative solvent exposure of CP43 peptides in His27PSII by treating the modification extent in HT3ΔctpAΔ27PSII as full exposure and the modification extent in HT3ΔctpAPSII as full protection. Peptide fragments are labeled as peptide numbers in Fig. 4. All structure figures were prepared using PyMol software (36).
In the PSII crystal structures, it appears that CP43 interacts with both PsbO and PsbV through its Loop E domain. In particular, two residues (R362 and K381) in loop E are predicted to interact with two residues (D8 and D99) of PsbO through charge-charge interaction over a distance of less than 3 Å (26). Additionally, the presence of Psb27 may also contribute to decreased binding of PsbO to PSII by shielding the N-terminal binding site of PsbO to PSII and abolishing the cation-π interaction Y7-R370 (PsbO-CP43). Another residue (R390) in the Loop E of CP43 is predicted to participate in a charge-charge interaction with E90 (within 4.8 Å) of PsbV. All three of these residues on the Loop E of CP43 participate in the CP43-Psb27 binding interface as observed in this study.
Our results show that Psb27 prevents PsbO and PsbV from binding to loop E by at least partially blocking their binding sites (Fig. 5C). The absence of PsbU in the Pair A complexes should result from the absence of PsbO and PsbV, as found in previous biochemical data (27, 28). Even though our footprinting results indicate that the lumenal domain of CP43 (Loop E) provides structural accommodation for the binding site for Psb27 (Fig. 5A), interactions between Psb27 and the pD1 protein (29) cannot be excluded at this stage. However, peptide 324–334 of pD1/D1 did not show a significant difference in modification extent among the three PSII complexes studied here (data not shown).
Another approach to determining binding interfaces is hydrogen/deuterium amide exchange (HDX). Comparisons of the outcomes of HDX and GEE modification experiments in other systems demonstrated that the two methods are comparable and give consistent results (30). In principle, HDX experiments can also be performed to track the conformational change of PSII subunits' lumenal domains. However, we chose not to use HDX because there are technical difficulties in isolating and digesting the proteins in complexes while minimizing back-exchange. GEE modification of large protein complexes is less problematic than HDX experiments because of the irreversible nature of GEE modification.
Processing of pD1 to D1 Alters the Interaction between CP43 and Psb27
The in vivo proteolytic removal of the C-terminal extension in pD1 is essential for the assembly of the Mn4CaO5 cluster and, hence, for oxygen evolution activity. Our Pair B complexes provided insights into conformational changes that occur upon pD1 processing by allowing us to compare solvent accessibility of putative domains involved in the protein-protein interactions. For peptide 363–370, a significant increase in solvent accessibility occurs (Fig. 4B. green and blue bars). Even in the presence of Psb27 in the HT3ΔctpAPSII complex, the solvent accessibility of this domain increases to a level of 80% of a Psb27-deletion PSII complex (i.e. HT3ΔctpAΔ27PSII) (Fig. 4B, compare the blue to red bar). The modification extent of peptide 371–379 in His27PSII is in between those of HT3ΔctpAΔ27PSII and HT3ΔctpAPSII, but it is closer to that of the latter than the former. We derived a relative solvent-exposure plot of peptides 363–370, 371–379, and 382–390 in His27PSII by setting HT3ΔctpAΔ27PSII as complete exposure, when Psb27 is absent in the complex, and setting HT3ΔctpA as complete protection, when Psb27 is present (Fig. 1). These three domains of His27PSII (Fig. 5D) show an uneven decreasing relative exposure; specifically, peptide 363–370 is exposed much more than peptides 371–380 and 382–390 in His27PSII. These data indicate that the partial dissociation of Psb27 from CP43 in His27PSII does not occur evenly across all three peptide domains involved in the association with Psb27. Peptide 363–370 moves away from Psb27 far more than do both peptides 371–379 and 382–390. We interpret that this uneven loosening of CP43 from Psb27 may expose peptide 363–370 and nearby domains to be more accessible to the N-terminal domain of PsbO (Fig. 5B). Notably, we observed an increase in the modification extent of the Psb27 peptide 57–63 (Fig. 4D). The increased modification extent of peptide 363–370 of CP43 parallels the elevated modification extent of peptide 57–63 of Psb27 and is consistent with our model deduced from chemical cross-linking experiments (10).
Our results shed light on another unanswered question. It is known that in the presence of Psb27, the binding stoichiometry of PsbO in PSII is less than that in active PSII (5, 31). The partial dissociation of Psb27 in His27PSII may create the environment for partial PsbO binding. It is known that CP47, CP43, D1, and D2 possess residues that contribute to the binding of PsbO (26). No reports demonstrated yet that CP47 and D2 dynamics can affect PsbO binding to PSII. On the other hand, early experiments indicated that PsbO binds to the PSII docking site and then undergoes folding that enables the protein to be functionally assembled into PSII (32–34). FTIR experiments further showed that PsbO increases in β-sheet content at the expense of random-coil content (35) upon binding to PSII. In line with these observations, we speculate that the Loop E of CP43 plays an important role in modulating the binding affinity of PsbO and its subsequent folding process.
PsbV was not observed in the Psb27-containing PSII complex even after pD1 processing (5). This may result from the high protection of peptide 382–390 owing to Psb27 binding, as shown in our model of His27PSII (Fig. 5C). One of the charge-charge interactions, PsbV:CP43 (E90:R390), is still shielded by the presence of Psb27 (Fig. 4B, peptide 382–390, green and blue bars). Our data indicate that such steric conflicts can be resolved only after Psb27 is completely removed from the PSII assembly intermediate, and PsbV can then be recruited to the PSII lumenal side.
These protein footprinting studies suggest that pD1 to D1 processing plays an essential role in regulating the conformational dynamics of CP43 to allow different binding partners to be recruited sequentially. It should be noted that in the crystal structures, Loop E of CP43 is in its post-pD1 processing and post-Psb27-removal conformation. As our results show, the conformation of this loop changes throughout the PSII assembly process.
In summary, we have shown that Psb27 binds to CP43's loop E surface, which is located on the distal side of the pseudo-symmetrical D1-D2 axis. Our results demonstrate that processing of pD1 to D1 weakens CP43-Psb27 interactions, allowing PsbO and, ultimately after the dissociation of Psb27, the remaining three extrinsic proteins PsbU, PsbV, and PsbQ, to be recruited to PSII. These results not only demonstrate that the Loop E of CP43 plays a key role in PSII assembly, but are experimental evidence for the long-standing idea that conformational rearrangement of PSII intermediates helps regulate the intricate life cycle of PSII. Processing of pD1 to D1 by CtpA not only exposes the C-terminal carboxylate group of A344 for the coordination of the Mn4CaO5 cluster, but also triggers the sequential recruitment of extrinsic proteins that stabilize the final photo-activated Mn4CaO5 cluster.
Acknowledgments
We thank Dr. Terry M. Bricker for the HT3 strain and other members of the Pakrasi and Gross laboratories and members of the R.E. Blankenship group for collegial discussions, especially Drs. Hao Zhang and Jianzhong Wen.
This work was supported, in whole or in part, by National Institutes of Health Grant P41 GM103422 (to M. L. G.) and by NSF-MCB0745611 (to H. B. P.).

This article contains supplemental Tables S1 and S2.
- PSII
- Photosystem II
- GEE
- glycine ethyl ester
- EDC
- 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride
- EIC
- extracted ion chromatograms
- HDX
- hydrogen/deuterium amide exchange.
REFERENCES
- 1. Umena Y., Kawakami K., Shen J. R., Kamiya N. (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473, 55–60 [DOI] [PubMed] [Google Scholar]
- 2. Nowaczyk M. M., Hebeler R., Schlodder E., Meyer H. E., Warscheid B., Rögner M. (2006) Psb27, a cyanobacterial lipoprotein, is involved in the repair cycle of photosystem II. Plant Cell 18, 3121–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bentley F. K., Luo H., Dilbeck P., Burnap R. L., Eaton-Rye J. J. (2008) Effects of inactivating psbM and psbT on photodamage and assembly of photosystem II in Synechocystis sp. PCC 6803. Biochemistry 47, 11637–11646 [DOI] [PubMed] [Google Scholar]
- 4. Roose J. L., Pakrasi H. B. (2008) The Psb27 protein facilitates manganese cluster assembly in photosystem II. J. Biol. Chem. 283, 4044–4050 [DOI] [PubMed] [Google Scholar]
- 5. Liu H., Roose J. L., Cameron J. C., Pakrasi H. B. (2011) A genetically tagged Psb27 protein allows purification of two consecutive photosystem II (PSII) assembly intermediates in Synechocystis 6803, a cyanobacterium. J. Biol. Chem. 286, 24865–24871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hwang H. J., Nagarajan A., McLain A., Burnap R. L. (2008) Assembly and disassembly of the photosystem II manganese cluster reversibly alters the coupling of the reaction center with the light-harvesting phycobilisome. Biochemistry 47, 9747–9755 [DOI] [PubMed] [Google Scholar]
- 7. Cormann K. U., Bangert J. A., Ikeuchi M., Rögner M., Stoll R., Nowaczyk M. M. (2009) Structure of Psb27 in solution: implications for transient binding to photosystem II during biogenesis and repair. Biochemistry 48, 8768–8770 [DOI] [PubMed] [Google Scholar]
- 8. Fagerlund R. D., Eaton-Rye J. J. (2011) The lipoproteins of cyanobacterial photosystem II. J. Photochem. Photobiol. B. 104, 191–203 [DOI] [PubMed] [Google Scholar]
- 9. Michoux F., Takasaka K., Boehm M., Komenda J., Nixon P. J., Murray J. W. (2012) Crystal structure of the Psb27 assembly factor at 1.6Å: implications for binding to Photosystem II. Photosynth. Res. 110, 169–175 [DOI] [PubMed] [Google Scholar]
- 10. Liu H., Huang R. Y., Chen J., Gross M. L., Pakrasi H. B. (2011) Psb27, a transiently associated protein, binds to the chlorophyll binding protein CP43 in photosystem II assembly intermediates. Proc. Natl. Acad. Sci. U.S.A. 108, 18536–18541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Komenda J., Knoppová J., Kopečná J., Sobotka R., Halada P., Yu J., Nickelsen J., Boehm M., Nixon P. J. (2012) The Psb27 assembly factor binds to the CP43 complex of photosystem II in the cyanobacterium Synechocystis sp. PCC 6803. Plant Physiol. 158, 476–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mendoza V. L., Vachet R. W. (2009) Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom. Rev. 28, 785–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bricker T. M., Morvant J., Masri N., Sutton H. M., Frankel L. K. (1998) Isolation of a highly active photosystem II preparation from Synechocystis 6803 using a histidine-tagged mutant of CP 47. Biochim. Biophys. Acta 1409, 50–57 [DOI] [PubMed] [Google Scholar]
- 14. Kashino Y., Lauber W. M., Carroll J. A., Wang Q. J., Whitmarsh J., Satoh K., Pakrasi H. B. (2002) Proteomic analysis of a highly active photosystem II preparation from the cyanobacterium Synechocystis sp. PCC 6803 reveals the presence of novel polypeptides. Biochemistry 41, 8004–8012 [DOI] [PubMed] [Google Scholar]
- 15. Kashino Y., Koike H., Yoshio M., Egashira H., Ikeuchi M., Pakrasi H. B., Satoh K. (2002) Low-molecular-mass polypeptide components of a photosystem II preparation from the thermophilic cyanobacterium Thermosynechococcus vulcanus. Plant Cell Physiol. 43, 1366–1373 [DOI] [PubMed] [Google Scholar]
- 16. Kashino Y., Koike H., Satoh K. (2001) An improved sodium dodecyl sulfate-polyacrylamide gel electrophoresis system for the analysis of membrane protein complexes. Electrophoresis 22, 1004–1007 [DOI] [PubMed] [Google Scholar]
- 17. Wen J., Zhang H., Gross M. L., Blankenship R. E. (2009) Membrane orientation of the FMO antenna protein from Chlorobaculum tepidum as determined by mass spectrometry-based footprinting. Proc. Natl. Acad. Sci. U.S.A. 106, 6134–6139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang H., Shen W., Rempel D., Monsey J., Vidavsky I., Gross M. L., Bose R. (2011) Molecular Cell Proteomics 10(6):M110.005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Swaisgood H., Natake M. (1973) Effect of carboxyl group modification on some of the enzymatic properties of l-glutamate dehydrogenase. J. Biochem. 74, 77–86 [DOI] [PubMed] [Google Scholar]
- 20. Hoare D. G., Koshland D. E., Jr. (1967) A method for the quantitative modification and estimation of carboxylic acid groups in proteins. J. Biol. Chem. 242, 2447–2453 [PubMed] [Google Scholar]
- 21. Eaton-Rye J. J., Putnam-Evans C. (2005) in Photosystem II: The Water/Plastoquinone Oxidoreductase of Photosynthesis (Wydrzynski T., Satoh K., eds), pp. 95–120, Springer, Dordrecht, Netherlands [Google Scholar]
- 22. Nakamura T., Dohmae N., Takio K. (2004) Characterization of a digested protein complex with quantitative aspects: an approach based on accurate mass chromatographic analysis with Fourier transform-ion cyclotron resonance mass spectrometry. Proteomics 4, 2558–2566 [DOI] [PubMed] [Google Scholar]
- 23. Frankel L. K., Sallans L., Limbach P. A., Bricker T. M. (2012) Identification of oxidized amino acid residues in the vicinity of the Mn(4)CaO(5) cluster of Photosystem II: implications for the identification of oxygen channels within the Photosystem. Biochemistry 51, 6371–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mabbitt P. D., Rautureau G. J., Day C. L., Wilbanks S. M., Eaton-Rye J. J., Hinds M. G. (2009) Solution structure of Psb27 from cyanobacterial photosystem II. Biochemistry 48, 8771–8773 [DOI] [PubMed] [Google Scholar]
- 25. Roose J. L., Pakrasi H. B. (2004) Evidence that D1 processing is required for manganese binding and extrinsic protein assembly into photosystem II. J. Biol. Chem. 279, 45417–45422 [DOI] [PubMed] [Google Scholar]
- 26. Bricker T. M., Roose J. L., Fagerlund R. D., Frankel L. K., Eaton-Rye J. J. (2012) The extrinsic proteins of Photosystem II. Biochim. Biophys. Acta 1817, 121–142 [DOI] [PubMed] [Google Scholar]
- 27. Shen J. R., Ikeuchi M., Inoue Y. (1992) Stoichiometric association of extrinsic cytochrome c550 and 12 kDa protein with a highly purified oxygen-evolving photosystem II core complex from Synechococcus vulcanus. FEBS Lett. 301, 145–149 [DOI] [PubMed] [Google Scholar]
- 28. Shen J. R., Inoue Y. (1993) Binding and functional properties of two new extrinsic components, cytochrome c-550 and a 12-kDa protein, in cyanobacterial photosystem II. Biochemistry 32, 1825–1832 [DOI] [PubMed] [Google Scholar]
- 29. Wei L., Guo J., Ouyang M., Sun X., Ma J., Chi W., Lu C., Zhang L. (2010) LPA19, a Psb27 homolog in Arabidopsis thaliana, facilitates D1 protein precursor processing during PSII biogenesis. J. Biol. Chem. 285, 21391–21398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang H., Wen J., Huang R. Y., Blankenship R. E., Gross M. L. (2012) Mass spectrometry-based carboxyl footprinting of proteins: method evaluation. Int. J. Mass Spectrom. 312, 78–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nowaczyk M. M., Krause K., Mieseler M., Sczibilanski A., Ikeuchi M., Rögner M. (2012) Deletion of psbJ leads to accumulation of Psb27-Psb28 photosystem II complexes in Thermosynechococcus elongatus. Biochim. Biophys. Acta 1817, 1339–1345 [DOI] [PubMed] [Google Scholar]
- 32. Popelkova H., Yocum C. F. (2011) PsbO, the manganese-stabilizing protein: analysis of the structure-function relations that provide insights into its role in photosystem II. J. Photochem. Photobiol. B. 104, 179–190 [DOI] [PubMed] [Google Scholar]
- 33. Popelkova H., Im M. M., Yocum C. F. (2003) Binding of manganese stabilizing protein to photosystem II: identification of essential N-terminal threonine residues and domains that prevent nonspecific binding. Biochemistry 42, 6193–6200 [DOI] [PubMed] [Google Scholar]
- 34. Lydakis-Simantiris N., Betts S. D., Yocum C. F. (1999) Leucine 245 is a critical residue for folding and function of the manganese stabilizing protein of photosystem II. Biochemistry 38, 15528–15535 [DOI] [PubMed] [Google Scholar]
- 35. Hutchison R. S., Betts S. D., Yocum C. F., Barry B. A. (1998) Conformational changes in the extrinsic manganese stabilizing protein can occur upon binding to the photosystem II reaction center: an isotope editing and FT-IR study. Biochemistry 37, 5643–5653 [DOI] [PubMed] [Google Scholar]
- 36. Delano W. L. (2002) The PyMol Molecular Graphics System, Delano Scientific, Palo Alto, CA [Google Scholar]