

Background: The BTR complex, consisting of the BLM helicase, topoisomerase IIIα, RMI1, and RMI2, dissolves the double Holliday junction (dHJ) to yield non-crossover products exclusively.
Results: RPA physically interacts with RMI1 and stimulates dHJ dissolution.
Conclusion: RPA-RMI1 interaction is required for efficient dHJ dissolution.
Significance: RPA, through an interaction with RMI1, is essential for the functional integrity of the BTR dHJ dissolvasome.
Keywords: Cancer Biology, DNA Binding Protein, DNA Helicase, DNA Repair, Homologous Recombination, Double Holliday Junction, Single-stranded DNA Binding Protein RPA
Abstract
The conserved BTR complex, composed of the Bloom's syndrome helicase (BLM), topoisomerase IIIα, RMI1, and RMI2, regulates homologous recombination in favor of non-crossover formation via the dissolution of the double Holliday Junction (dHJ). Here we show enhancement of the BTR-mediated dHJ dissolution reaction by the heterotrimeric single-stranded DNA binding protein replication protein A (RPA). Our results suggest that RPA acts by sequestering a single-stranded DNA intermediate during dHJ dissolution. We provide evidence that RPA physically interacts with RMI1. The RPA interaction domain in RMI1 has been mapped, and RMI1 mutants impaired for RPA interaction have been generated. Examination of these mutants ascertains the significance of the RMI1-RPA interaction in dHJ dissolution. Our results thus implicate RPA as a cofactor of the BTR complex in dHJ dissolution.
Introduction
Bloom syndrome is an autosomal recessive disorder that shows a strong cancer predisposition (1). The hallmark of cells from Bloom syndrome patients is a dramatic increase in the frequency of sister chromatid exchanges, which is thought to arise from an elevated frequency of chromatid arm crossover formation during homologous recombination (HR)5 (2, 3). Bloom syndrome is caused by mutations in the DNA helicase BLM, which is one of the five RecQ-like DNA helicases in mammals (1, 4).
BLM is associated with topoisomerase IIIα (Topo IIIα, a type IIIa topoisomerase), RMI1, and RMI2 in a stable ensemble called the BTR complex (5, 6). Importantly, BLM functions with Topo IIIα to process the double Holliday Junction (dHJ), a DNA intermediate formed during HR, by convergent DNA branch migration of the two Holliday junctions in the structure and DNA strand decatenation, to yield non-crossover recombinants exclusively (5–9). The dHJ dissolution activity of BLM-Topo IIIα is enhanced strongly by the heterodimeric RMI1-RMI2 complex (10, 11). That the BTR complex promotes non-crossover HR provides a satisfactory explanation for the sister chromatid exchange phenotype of Bloom syndrome cells and of cells that are genetically ablated for other components of this complex (10–13).
The dHJ dissolution activity of the BTR complex is evolutionarily conserved, as is its counterpart (the STR complex) in the budding yeast Saccharomyces cerevisiae, comprising the Sgs1 helicase (orthologous to BLM), Top3 (orthologous to Topo IIIα), and Rmi1, dissolves the dHJ in an analogous reaction (14, 15). Abundant genetic data have implicated the STR complex in HR regulation in favor of non-crossover formation. It should be noted that, in HR events that are triggered by DNA double-strand breaks, the STR complex cooperates with the Dna2 nuclease in 5′ strand resection from the DNA ends to generate 3′ single-stranded DNA tails for the assembly of the HR machinery (15–17). A function of the BLM helicase in 5′ end resection has also been revealed in cell-based and biochemical studies (18–20).
By coimmunoprecipitation, the heterotrimeric single-stranded DNA binding protein RPA has been found to associate with the BTR complex in cell extracts (11, 13). In this study, we have asked whether RPA influences the BTR-mediated dHJ dissolution reaction and examined possible physical interactions with the BTR subunits. Using a DNA substrate that permits examination of the late stages of dHJ dissolution, we have found that RPA up-regulates the dHJ dissolution activity of the BTR complex in a species-specific manner. Importantly, we present results from biochemical mapping and mutant analyses to show that the specific interaction of RPA with RMI1 is indispensable for the stimulation of dHJ dissolution. Our study thus identifies RPA as an important cofactor of the BTR complex in the dHJ dissolution reaction, and it also suggests that RPA plays an intimate role in the suppression of crossover HR.
EXPERIMENTAL PROCEDURES
Construction and Purification of RMI1 Fragments
The pGEX-based (GE Healthcare) plasmids that express GST-tagged RMI11–211, RMI1212–424, and RMI1425–625 have been described (9). GST-tagged RMI1212–300 and RMI1212–334 were generated using QuikChange mutagenesis (Stratagene) to insert stop codons into the RMI1 coding region of pGEX-RMI1212–424. The DNA fragment encoding RMI1301–424 was amplified from pGEX-RMI1212–424 by PCR and introduced into the EcoRI and XhoI sites of the pGEX-6P1 vector (GE Healthcare) to add a GST tag to the N-terminal end of the RMI1 fragment. The purification of the GST-tagged RMI1 fragments followed our published procedure (9).
Constructions and Purification of the RMI1Δ301–337-RMI2 and RMI14EA -RMI2 Complexes
The pMAL-p2X (New England Biolabs) plasmid with the cDNA coding for MBP-RMI1-(His)6 has been described (10). The RMI1 truncation mutant lacking residues 301–337 (designated as RMI1Δ301–337) and the RMI1-E312A/E313A/E317A/E318A mutant (designated as RMI14EA) were generated in this plasmid by QuikChange mutagenesis. The oligonucleotides used in the mutagenesis procedure are listed in supplemental Table S1. The mutant RMI1 proteins were coexpressed in Escherichia coli with FLAG-tagged RMI2 (10), and the purification of the tagged mutant RMI complexes followed our published procedure (10).
Purification of Other Proteins
The MBP-RMI1-(His)6-FLAG-RMI2 and MBP-RMI1425–625-FLAG-RMI2 complexes were expressed in E. coli and purified to near homogeneity as described (10). (His)6-tagged BLM and (His)6-tagged Topo IIIα were expressed in yeast and E. coli, respectively, and purified following our published procedures (9, 21). MBP-RMI1-(His)6 was expressed in E. coli and purified as described (10). Human and yeast RPA proteins were expressed in E. coli and purified as described (22). E. coli SSB was purchased from New England Biolabs. The hRPAFWAA mutant (23) was expressed in E. coli and purified as for wild-type hRPA.
Pull-down Assays
For the affinity pull-down of (His)6-tagged proteins, hRPA (5 μg) was incubated with or without 5 μg of (His)6-tagged BLM, 5 μg of Topo IIIα, or 5 μg of RMI1 in 30 μl of buffer K (20 mm KH2PO4 (pH 7.4), 10% glycerol, 0.5 mm EDTA, 0.01% Igepal, 1 mm DTT) containing 150 mm KCl and 10 mm imidazole for 30 min at 4 °C. The reactions were mixed gently with 15 μl of Ni-NTA resin (Qiagen) for 30 min at 4 °C to capture the (His)6-tagged protein and associated hRPA. After washing the resin three times with 200 μl of the same buffer, bound proteins were eluted with 20 μl of 2% SDS. The supernatant, wash, and SDS eluate, 10 μl of each, were analyzed by 7.5% SDS-PAGE and Coomassie Blue staining.
For the affinity pull-down of MBP-tagged proteins, 5 μg of hRPA was incubated with MBP-tagged RMI1425–625-RMI2, RMI1-RMI2, RMI1Δ301–337-RMI2, RMI14EA-RMI2, or MBP (5 μg) and mixed with amylose resin (New England Biolabs) to capture the MBP-tagged protein and associated RPA, as above. The reactions were processed and analyzed as above.
For the affinity pull-down of GST-tagged proteins, 5 μg of hRPA was incubated with 5 μg of GST-tagged RMI1 fragments or 5 μg of GST and mixed with glutathione resin (GE Healthcare) as above. The reactions were processed and analyzed as above.
dHJ Substrate
The dHJ substrate was prepared by hybridizing and ligating two oligonucleotides as described (7). The substrate was purified by polyacrylamide gel electrophoresis and elution from gel slices (5).
dHJ Dissolution Assay
Reaction mixtures containing combinations of BLM (10 nm), Topo IIIα (10 nm), RMI1-RMI2 (7.5 nm), and the indicated amount of hRPA, yRPA, or SSB were assembled and incubated on ice for 10 min in 11.5 μl of reaction buffer (50 mm Tris-HCl (pH 7.8), 1 mm DTT, 2.4 mm MgCl2, 200 μg/ml BSA, 2 mm ATP, 140 mm KCl, and an ATP-regenerating system consisting of 10 mm creatine phosphate and 50 μg/ml creatine kinase). After the addition of the dHJ substrate (10 nm or as indicated) in 1 μl of water, the reaction was incubated at 37 °C for 12 min (or time as indicated). In Fig. 5D, hRPA and yRPA were preincubated with 200 nm oligo dT20 before testing. The reaction mixtures were processed and analyzed as before (5).
FIGURE 5.

Role of the hRPA DNA binding function in dHJ dissolution. A, purified hRPA and hRPAFWAA were analyzed by SDS-PAGE. B, mixtures of MBP-tagged RMI1 or MBP with hRPA or hRPAFWAA were incubated with amylose resin and then analyzed as in Fig. 2. S, supernatant; W, wash; E, SDS eluate. C, effect of the hRPAFWAA mutation on dHJ dissolution mediated by the BTR complex. D, hRPA or yRPA was preincubated with oligo dT20 before being tested in the BTR-mediated dHJ dissolution reaction. The error bars in the graph in C and D represent mean ± S.D. from three independent experiments. E, schematic representation of the protein interaction domains in RMI1. The numbers refer to amino acid residues in RMI1.
RESULTS
Enhancement of dHJ Dissolution by hRPA
Because RPA coimmunoprecipitates with the BTR complex from human cell extracts (11, 13), we asked whether it might stimulate the dHJ dissolution activity of the latter. A radiolabeled dHJ substrate (Fig. 1A), constructed as described (5, 7, 24), was incubated with the combination of BLM, Topo IIIα, RMI1-RMI2, and different amounts of hRPA, and the dissolution products were analyzed as before (5, 6). The results showed that, as expected (5, 9–11), the BLM-Topo IIIα pair is capable of dHJ dissolution, and its activity is enhanced by RMI1-RMI2 (Fig. 1B, lanes 2 and 4). Interestingly, we found that hRPA stimulates, in a concentration-dependent manner, dHJ dissolution catalyzed by the BTR ensemble (Fig. 1B, lanes 5–9). At the highest concentration of hRPA (160 nm), an ∼5-fold enhancement of the dissolution reaction was seen. We next tested two heterologous single-stranded binding proteins, RPA from the budding yeast S. cerevisiae (yRPA) and E. coli SSB, to see whether they would similarly stimulate BTR-mediated dHJ dissolution. The results revealed that the heterologous single-stranded binding proteins are significantly less capable of reaction enhancement. At 20 and 40 nm, although hRPA elevated the dHJ dissolution efficiency by more than 2- and 4-fold, respectively, after 12 min, only a slight enhancement was seen for either of the heterologous proteins (Fig. 1B). A time course experiment to examine the effects of hRPA and yRPA in the dHJ dissolution reaction revealed that the rate of dissolution with hRPA is 2.5-fold of that with either of the heterologous single-stranded binding proteins (Fig. 4B). These and other observations presented below helped establish that a species-specific interaction of hRPA with RMI1 is indispensable for maximal up-regulation of the dHJ activity of the BTR ensemble.
FIGURE 1.

RMI1-RMI2-dependent enhancement of dHJ dissolution by RPA. A, schematic of dHJ dissolution. The asterisk denotes the 32P labeled strand. B, dHJ dissolution by the BTR complex was examined with and without hRPA, yRPA, or E. coli SSB. C, effect of hRPA, yRPA, and SSB on dHJ dissolution in the absence of RMI1-RMI2. The dHJ substrate concentration was reduced from 10 to 1.5 nm. The error bars in the graphs in B and C represent mean ± S.D. from three independent experiments.
FIGURE 4.

Stimulation of dHJ dissolution by hRPA is dependent on hRPA-RMI1 interaction. A, examination of RMI1-RMI2, RMI1Δ301–337-RMI2, and RMI14EA-RMI2 in the dHJ dissolution reaction with and without hRPA. B, time course of dHJ dissolution with RMI1-RMI2, RMI1Δ301–337-RMI2, or RMI14EA-RMI2 and hRPA or yRPA. The concentration of hRPA and yRPA was 80 nm. The error bars in the graph in B and C represent mean ± S.D. from three independent experiments.
Stimulation of dHJ Dissolution by RPA Is Dependent on RMI1-RMI2
We asked whether stimulation of dHJ dissolution would still occur if RMI1-RMI2 were omitted. Because the BLM-Topo IIIα pair is less active than the BTR complex in the dissolution reaction, we lowered the concentration of the dHJ substrate (from 10 to 1.5 nm) in this experiment to obtain a reasonable signal. Importantly, in the absence of RMI1-RMI2, hRPA, in fact, exerted a significant inhibition, whereas the heterologous proteins had little or no effect on the reaction efficiency (Fig. 1C). The results thus indicate that stimulation of dHJ dissolution by a single-stranded binding protein requires the presence of RMI1-RMI2.
Species-specific Interaction of hRPA with RMI1
Taking advantage of the affinity tag on our purified BTR components, we applied biochemical pull-down to examine a possible interaction of untagged hRPA with these purified components. When Ni-NTA resin was used to pull down RMI1 through its (His)6 tag, hRPA was also retained on the affinity matrix (Fig. 2A, lane 9). Importantly, neither (His)6-tagged BLM nor Topo IIIα could retain hRPA on the affinity resin (Fig. 2A, lanes 3 and 6). As expected, hRPA alone did not associate with the affinity resin in the absence of the BTR components (Fig. 2A, lane 12).
FIGURE 2.
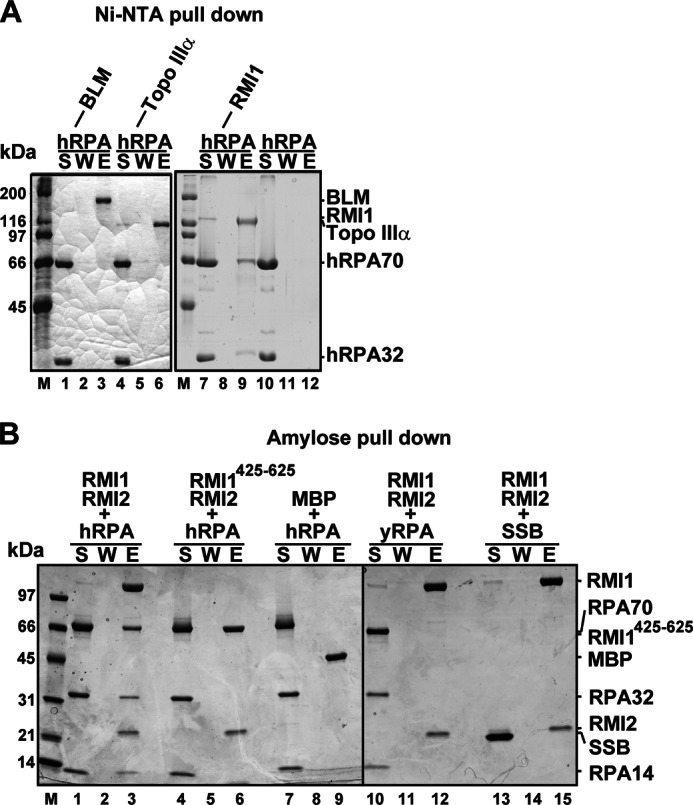
Interaction of hRPA with RMI1. A, mixtures of BLM, Topo IIIα, and RMI1 with hRPA were incubated with Ni-NTA resin, which was washed and then treated with SDS to elute bound proteins. hRPA alone was also incubated with the resin. The supernatant (S), wash (W) and SDS eluate (E) from these pull-down reactions were analyzed by SDS-PAGE. B, mixtures of MBP-tagged RMI1-RMI2, RMI1425–625-RMI2, or MBP with hRPA, yRPA, or SSB were incubated with amylose resin and then analyzed as in A.
Because of its strong tendency to aggregate (10), RMI2 alone could not be tested for hRPA interaction. However, RMI2 could be purified in complex with full-length RMI1 or RMI1 fragments that harbor the C-terminal residues 425–625 (RMI1425–625) in which the RMI2 interaction domain resides (10, 11). We therefore expressed and purified a MBP-tagged form of RMI1 or RMI1425–625 bound to RMI2 and subjected the protein complexes to the pull-down assay using amylose resin (specific for the MBP tag) to capture any complex with hRPA. Importantly, although, as expected, RMI1-RMI2 associated with hRPA avidly, little or no hRPA was retained on the affinity resin when RMI1425–625-RMI2 was used (Fig. 2B, lanes 1–9). We note that the interaction of RPA with RMI1 is species-specific, as RMI1-RMI2 has little or no affinity for either yRPA or E. coli SSB (Fig. 2B, lanes 12 and 15). Taken together, these results reveal that RMI1 is the only BTR complex component that has a high affinity for hRPA, and they further suggest that the C-terminal portion of RMI1 (harboring residues 425–625) is not responsible for RPA interaction.
An Acidic Region in RMI1 Helps Mediate hRPA Interaction
The results in Fig. 2B suggest that the hRPA-binding domain likely resides in a region outside of the C terminus of RMI1. To further define the hRPA interaction domain in RMI1, we expressed and purified three overlapping RMI1 fragments spanning its entire length, namely, residues 1–211, 212–424, and 425–625 (Fig. 3A), as fusions to GST to allow for affinity pull-down using glutathione resin (specific for the GST tag). The affinity pull-down analysis showed that RMI1212–424, but not RMI11–211 or RMI1425–625, interacts with hRPA (supplemental Fig. S1). On the basis of this information, we made additional GST fusions harboring RMI1 residues 212–300, 212–334, and 301–424. Pull-down analysis employing these RMI1 fragments enabled us to deduce that the hRPA interaction domain resides within residues 212–334 (supplemental Fig. S1).
FIGURE 3.

Biochemical mapping of the hRPA binding domain in RMI1 and mutants of RMI1 impaired for hRPA interaction. A, schematic of the RMI1 fragments and deletion mutants (left panel) and a summary of their ability to interact with hRPA (right panel). B, mixtures of MBP-tagged RMI1301–334 or MBP with hRPA were incubated with amylose resin and then analyzed as above. The black arrow points to MBP or the MBP-tagged RMI1 fragment. C, sequence alignment of the acidic region in RMI1 orthologs predicted to contribute to hRPA interaction. D, purified RMI1-RMI2, RMI1Δ301–337-RMI2, and RMI14EA-RMI2 were analyzed by SDS-PAGE. E, mixtures of MBP-tagged RMI1-RMI2, RMI1Δ301–337-RMI2, or RMI14EA-RMI2 with hRPA were incubated with amylose resin and then analyzed as in Fig. 2. S, supernatant; W, wash; E, SDS eluate.
Knowing that various RPA interacting proteins such as yeast Rad52, the tumor suppressor p53, and the nuclease MRE11 (25–27) often employ an acidic domain to mediate RPA binding, we sought to find a similar domain in RMI1. Alignment of the deduced hRPA-interacting fragment of RMI1 against the equivalent region of RMI1 orthologs revealed a clustering of acidic amino acids within residues 301–334 (Fig. 3C), which resembles the hallmark characteristic of the RPA interaction domain in the aforementioned protein factors. To directly test the validity of this premise, we expressed the acidic portion in the predicted hRPA binding region of RMI1 spanning residues 301–334 as a MBP fusion protein and purified it. Importantly, by affinity pull-down, we found that the MBP-tagged RMI1301–334 binds hRPA (Fig. 3B), whereas, as expected, it has little or no affinity for either yRPA or E. coli SSB (data not shown).
Mutants of RMI1 Impaired for hRPA Interaction
To further verify that the acidic region in RMI1 (Fig. 3C) is important for hRPA interaction, we constructed two RMI1 mutants, one lacking residues 301–337 and the other harboring the change of four conserved acidic residues (E312A, E313A, E317A, and E318A) within this region to alanine (the 4EA mutant). These RMI1 mutants were N-terminally tagged with MBP, coexpressed with RMI2, and then the resulting protein complexes were purified (Fig. 3D) for testing in the pull-down assay (this section) and for analysis in the dHJ dissolution reaction (see later). As shown in Fig. 3E, hRPA interaction is impaired by both RMI1 mutations (Fig. 3E, lanes 6 and 9). In contrast, both mutant RMI1-RMI2 complexes bind BLM and Topo IIIα just as avidly as the wild-type counterpart (supplemental Fig. S2).
Taken together, these results strongly suggest that the conserved acidic region in RMI1 helps mediate hRPA interaction. The fact that the RMI1-RMI2 mutant complexes retain the ability to associate with BLM and Topo IIIα normally also indicates that the RMI1 mutations do not induce any gross protein misfolding.
Relevance of RMI1-hRPA Interaction in dHJ Dissolution
The availability of RMI1 mutants impaired for hRPA interaction allowed us to determine whether the RMI1-hRPA complex is important for dHJ dissolution. For this, dHJ dissolution assays were conducted with BLM; Topo IIIα; and RMI1-RMI2, RMI1Δ301–337-RMI2, or RMI14EA-RMI2 with and without increasing amounts of hRPA. The results revealed that the two mutant RMI1-RMI2 complexes are just as adept as the wild-type counterpart in the enhancement of dHJ dissolution when hRPA is absent (Fig. 4A, compare lanes 3, 7, and 11 with lane 2). However, neither of the mutant RMI1-RMI2 complexes is nearly as effective as the wild-type counterpart when hRPA is present. For instance, in the presence of wild-type RMI1-RMI2, although hRPA, at 20 nm, enhanced dissolution more than 2-fold after 12 min, much less stimulation occurred with either of the mutant RMI1-RMI2 complexes. In time course experiments done with a higher hRPA concentration (80 nm), the reaction with wild-type RMI1-RMI2 proceeded at a faster pace than that harbored by either of the mutant complexes (kcat of 0.083 min−1 for wild-type RMI1-RMI2 versus 0.040 min−1 for the two mutant RMI1-RMI2 complexes, Fig. 4B). We note that the diminished level of dHJ dissolution seen with the mutant RMI1-RMI2 complexes in conjunction with hRPA resembles that observed with the wild-type RMI complex and yRPA or SSB (Figs. 1B and 4A). Importantly, we also verified that the two mutant RMI complexes are just as adept as the wild-type counterpart in dHJ dissolution with yRPA (supplemental Fig. S3). Altogether, these results allow us to conclude that the species-specific stimulation of dHJ dissolution by hRPA is, to a large degree, reliant on its interaction with RMI1.
The DNA Binding Function of hRPA Is Needed for dHJ Dissolution Enhancement
We next asked whether the DNA binding function of RPA is needed for dHJ dissolution enhancement. To address this question, we purified and tested a hRPA variant that harbors the FWAA mutation (with phenylalanine 238 and tryptophan 361 of the hRPA70 subunit having been changed to alanine) that was shown previously to strongly impair DNA binding (23) (Fig. 5A). Importantly, even though hRPAFWAA retains the ability to physically interact with RMI1 (Fig. 5B), it is completely devoid of stimulatory activity in the dHJ dissolution reaction (C, lanes 6 and 7). The results thus indicate that the DNA binding function of RPA is essential for dHJ dissolution enhancement. Consistent with this deduction, we found that preincubation of wild-type hRPA (or yRPA) with a DNA oligonucleotide (dT20) ablates its ability to enhance the dHJ dissolution reaction (Fig. 5D) without changing its ability to interact with RMI1 (supplemental Fig. S4).
DISCUSSION
Prompted by the observation that RPA associates with the BTR complex in human cell extracts (11, 13), we sought to define its role in the dHJ dissolution reaction and to identify the subunit of the BTR complex that physically interacts with it. Using a well characterized model DNA substrate that resembles a late intermediate of dHJ dissolution, we showed that RPA elevates the reaction efficiency rather substantially. By biochemical analyses involving the use of heterologous single-stranded binding proteins, we have presented evidence that hRPA sequesters a single-stranded DNA intermediate during dHJ dissolution and that a specific interaction of hRPA with RMI1 is necessary for optimal reaction efficiency. In support of these conclusions, mutants of RMI1 that fail to stably associate with RPA and a DNA binding mutant of RPA are impaired for the enhancement of dHJ dissolution. Overall, the published results (8, 11, 13, 14, 28) and the findings presented here help implicate RPA in dHJ dissolution in human cells via a specific interaction with the RMI1 subunit. Our results showing species specificity of RPA in dHJ dissolution enhancement also suggest that targeting of RPA to HR sites to regulate dHJ dissolution is dependent on its interaction with RMI1.
In human cells, the BTR complex interacts with the DNA branch migration protein FANCM via physical contacts with RMI1 and Topo IIIα, and the higher-order BTR-FANCM complex has been implicated in the repair of injured DNA replication forks by catalyzing the regression of the injured fork to allow replication restart or lesion bypass (29). We have summarized, in Fig. 5E, the various protein interaction domains that RMI1 possesses. Future studies will determine whether RPA also plays a role in targeting the BTR-FANCM complex to single-stranded DNA associated with injured replication forks. In these regards, the RMI1 mutants impaired for RPA interaction (this work) could prove to be a valuable tool for investigating the role of the RMI1-RPA complex in HR regulation and replication fork repair.
We note that S. cerevisiae possesses an ortholog of RMI1 but is apparently devoid of a RMI2 equivalent. The S. cerevisiae Rmi1 protein is much smaller than the human ortholog (241 residues versus 625 residues), as it lacks the middle and C-terminal regions of hRMI1 that harbor the RPA binding domain (this study) and the RMI2 interaction domain (30), respectively. Consistent with this, yeast Rmi1 and RPA do not appear to physically interact (data not shown). In studies conducted with purified Sgs1 and Top3 (14, 28), Rmi1 was found to specifically enhance the late, decatenation step of dHJ dissolution. Interestingly, in the yeast system (14, 28), RPA or E. coli SSB is equally effective in up-regulating the efficiency of dHJ dissolution, which led to the proposal that the primary role of RPA is to bind and stabilize single-stranded DNA generated as a result of DNA strand separation by Sgs1 (14). Such an action of yeast RPA in the STR-mediated dHJ dissolution reaction is congruent with our conclusion that sequestration of a single-stranded intermediate, a task that can be fulfilled by a single-strand binding protein without species specificity, in the late stage of dHJ dissolution helps maximize the reaction efficiency (this study). Interestingly, the fruit fly Drosophila melanogaster possesses neither a RMI1 nor RMI2 ortholog. Thus, dHJ dissolution in that organism may be mediated by the Blm-Topo IIIα complex (8, 31). Consistent with this premise, it has been shown that RPA enhances dHJ dissolution catalyzed by the D. melanogaster Blm-Topo IIIα pair.
As defined in genetic studies in S. cerevisiae, in preparation for double-strand break repair by HR, the 5′ strand of the DNA break ends is resected by partially redundant nucleases, including the Mre11 nuclease, the 5′ to 3′ exonuclease Exo1, and the endonuclease Dna2 (17, 32–35). In S. cerevisiae cells, the STR complex functions with Dna2 in a major pathway of long range DNA end resection, and the activity of Dna2 is further regulated by a direct interaction with RPA. In human cells, there is a good amount of preliminary evidence that the BTR complex is also needed for 5′ strand resection of double-strand breaks to initiate lesion removal via HR (18–20). Interestingly, in this regard, BLM not only functionally synergizes with the DNA2 protein, but it also interacts with and up-regulates the activity of EXO1 (18–20). It will be of considerable interest to examine whether the RMI1-RPA complex we have documented herein is relevant for double-strand break end resection in human cells.
Acknowledgments
We thank Marc Wold (University of Iowa) for providing the hRPAFWAA expression plasmid and members of our laboratory for stimulating discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 ES015632, RO1 ES07061, and RO1 GM57814.

This article contains supplemental Figs. S1–S4 and Table S1.
- HR
- homologous recombination
- Topo IIIα
- topoisomerase IIIα
- dHJ
- double Holliday junction
- RPA
- replication protein A
- SSB
- E. coli single-stranded DNA binding protein
- hRPA
- human replication protein A
- NTA
- nickel-nitrilotriacetic acid
- yRPA
- S. cerevisiae replication protein A
- BTR
- BLM-Topo IIIα-RMI
- STR
- Sgs1-Top3-Rmi1
- MBP
- maltose binding protein
- FANCM
- Fanconi anemia complementation group M gene product.
REFERENCES
- 1. Ellis N. A., Groden J., Ye T. Z., Straughen J., Lennon D. J., Ciocci S., Proytcheva M., German J. (1995) The Bloom's syndrome gene product is homologous to RecQ helicases. Cell 83, 655–666 [DOI] [PubMed] [Google Scholar]
- 2. Chaganti R. S., Schonberg S., German J. (1974) A manyfold increase in sister chromatid exchanges in Bloom's syndrome lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 71, 4508–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kuhn E. M., Therman E. (1986) Cytogenetics of Bloom's syndrome. Cancer Genet. Cytogenet. 22, 1–18 [DOI] [PubMed] [Google Scholar]
- 4. Cheok C. F., Bachrati C. Z., Chan K. L., Ralf C., Wu L., Hickson I. D. (2005) Roles of the Bloom's syndrome helicase in the maintenance of genome stability. Biochem. Soc. Trans. 33, 1456–1459 [DOI] [PubMed] [Google Scholar]
- 5. Raynard S., Bussen W., Sung P. (2006) A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIα, and BLAP75. J. Biol. Chem. 281, 13861–13864 [DOI] [PubMed] [Google Scholar]
- 6. Wu L., Bachrati C. Z., Ou J., Xu C., Yin J., Chang M., Wang W., Li L., Brown G. W., Hickson I. D. (2006) BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc. Natl. Acad. Sci. U.S.A. 103, 4068–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu L., Hickson I. D. (2003) The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426, 870–874 [DOI] [PubMed] [Google Scholar]
- 8. Plank J. L., Wu J., Hsieh T. S. (2006) Topoisomerase IIIα and Bloom's helicase can resolve a mobile double Holliday junction substrate through convergent branch migration. Proc. Natl. Acad. Sci. U.S.A. 103, 11118–11123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raynard S., Zhao W., Bussen W., Lu L., Ding Y. Y., Busygina V., Meetei A. R., Sung P. (2008) Functional role of BLAP75 in BLM-topoisomerase IIIα-dependent Holliday junction processing. J. Biol. Chem. 283, 15701–15708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh T. R., Ali A. M., Busygina V., Raynard S., Fan Q., Du C. H., Andreassen P. R., Sung P., Meetei A. R. (2008) BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev. 22, 2856–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu D., Guo R., Sobeck A., Bachrati C. Z., Yang J., Enomoto T., Brown G. W., Hoatlin M. E., Hickson I. D., Wang W. (2008) RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev. 22, 2843–2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sung P., Klein H. (2006) Mechanism of homologous recombination. Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 7, 739–750 [DOI] [PubMed] [Google Scholar]
- 13. Yin J., Sobeck A., Xu C., Meetei A. R., Hoatlin M., Li L., Wang W. (2005) BLAP75, an essential component of Bloom's syndrome protein complexes that maintain genome integrity. EMBO J. 24, 1465–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cejka P., Plank J. L., Bachrati C. Z., Hickson I. D., Kowalczykowski S. C. (2010) Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-Top3. Nat. Struct. Mol. Biol. 17, 1377–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Niu H., Chung W. H., Zhu Z., Kwon Y., Zhao W., Chi P., Prakash R., Seong C., Liu D., Lu L., Ira G., Sung P. (2010) Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 467, 108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cejka P., Cannavo E., Polaczek P., Masuda-Sasa T., Pokharel S., Campbell J. L., Kowalczykowski S. C. (2010) DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467, 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mimitou E. P., Symington L. S. (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gravel S., Chapman J. R., Magill C., Jackson S. P. (2008) DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22, 2767–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nimonkar A. V., Genschel J., Kinoshita E., Polaczek P., Campbell J. L., Wyman C., Modrich P., Kowalczykowski S. C. (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nimonkar A. V., Ozsoy A. Z., Genschel J., Modrich P., Kowalczykowski S. C. (2008) Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. U.S.A. 105, 16906–16911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bussen W., Raynard S., Busygina V., Singh A. K., Sung P. (2007) Holliday junction processing activity of the BLM-Topo IIIα-BLAP75 complex. J. Biol. Chem. 282, 31484–31492 [DOI] [PubMed] [Google Scholar]
- 22. Van Komen S., Macris M., Sehorn M. G., Sung P. (2006) Purification and assays of Saccharomyces cerevisiae homologous recombination proteins. Methods Enzymol. 408, 445–463 [DOI] [PubMed] [Google Scholar]
- 23. Walther A. P., Gomes X. V., Lao Y., Lee C. G., Wold M. S. (1999) Replication protein A interactions with DNA. 1. Functions of the DNA-binding and zinc-finger domains of the 70-kDa subunit. Biochemistry 38, 3963–3973 [DOI] [PubMed] [Google Scholar]
- 24. Fu T. J., Tse-Dinh Y. C., Seeman N. C. (1994) Holliday junction crossover topology. J. Mol. Biol. 236, 91–105 [DOI] [PubMed] [Google Scholar]
- 25. Plate I., Hallwyl S. C., Shi I., Krejci L., Müller C., Albertsen L., Sung P., Mortensen U. H. (2008) Interaction with RPA is necessary for Rad52 repair center formation and for its mediator activity. J. Biol. Chem. 283, 29077–29085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bochkareva E., Kaustov L., Ayed A., Yi G. S., Lu Y., Pineda-Lucena A., Liao J. C., Okorokov A. L., Milner J., Arrowsmith C. H., Bochkarev A. (2005) Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. U.S.A. 102, 15412–15417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu X., Vaithiyalingam S., Glick G. G., Mordes D. A., Chazin W. J., Cortez D. (2008) The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Mol. Cell Biol. 28, 7345–7353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cejka P., Plank J. L., Dombrowski C. C., Kowalczykowski S. C. (2012) Decatenation of DNA by the S. cerevisiae Sgs1-Top3-Rmi1 and RPA complex. A mechanism for disentangling chromosomes. Mol. Cell 47, 886–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deans A. J., West S. C. (2009) FANCM connects the genome instability disorders Bloom's Syndrome and Fanconi anemia. Mol. Cell 36, 943–953 [DOI] [PubMed] [Google Scholar]
- 30. Mullen J. R., Nallaseth F. S., Lan Y. Q., Slagle C. E., Brill S. J. (2005) Yeast Rmi1/Nce4 controls genome stability as a subunit of the Sgs1-Top3 complex. Mol. Cell Biol. 25, 4476–4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen S. H., Wu C. H., Plank J. L., Hsieh T. S. (2012) Essential functions of C terminus of Drosophila topoisomerase IIIα in double Holliday junction dissolution. J. Biol. Chem. 287, 19346–19353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu Z., Chung W. H., Shim E. Y., Lee S. E., Ira G. (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134, 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jazayeri A., Falck J., Lukas C., Bartek J., Smith G. C., Lukas J., Jackson S. P. (2006) ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 8, 37–45 [DOI] [PubMed] [Google Scholar]
- 34. Llorente B., Symington L. S. (2004) The Mre11 nuclease is not required for 5′ to 3′ resection at multiple HO-induced double-strand breaks. Mol. Cell Biol. 24, 9682–9694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsubouchi H., Ogawa H. (2000) Exo1 roles for repair of DNA double-strand breaks and meiotic crossing over in Saccharomyces cerevisiae. Mol. Biol. Cell 11, 2221–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]