

Background: RNA polymerase-binding protein A (RbpA) plays an unknown essential role in Mycobacterium tuberculosis.
Results: The structure of RbpA was solved using NMR.
Conclusion: RbpA binds sigma factors A and B via its conserved N and C termini.
Significance: The identified interactions shed light on the function of RbpA in regulating transcription.
Keywords: Mycobacterium tuberculosis, NMR, Protein Structure, RNA Polymerase, Transcription, Tuberculosis, RbpA, Sigma Factor
Abstract
RNA polymerase-binding protein A (RbpA), encoded by Rv2050, is specific to the actinomycetes, where it is highly conserved. In the pathogen Mycobacterium tuberculosis, RbpA is essential for growth and survival. RbpA binds to the β subunit of the RNA polymerase where it activates transcription by unknown mechanisms, and it may also influence the response of M. tuberculosis to the current frontline anti-tuberculosis drug rifampicin. Here we report the solution structure of RbpA and identify the principle sigma factor σA and the stress-induced σB as interaction partners. The protein has a central ordered domain with a conserved hydrophobic surface that may be a potential protein interaction site. The N and C termini are highly dynamic and are involved in the interaction with the sigma factors. RbpA forms a tight complex with the N-terminal domain of σB via its N- and C-terminal regions. The interaction with sigma factors may explain how RbpA stabilizes sigma subunit binding to the core RNA polymerase and thereby promotes initiation complex formation. RbpA could therefore influence the competition between principal and alternative sigma factors and hence the transcription profile of the cell.
Introduction
Tuberculosis (TB)5 is an infectious disease that has plagued humanity for thousands of years, indeed, molecular evidence of TB infection has been found in a 5,400-year-old Egyptian mummy (1). Through the long years of coexistence between TB and humankind Mycobacterium tuberculosis, the main etiological agent of human TB, has evolved to become one of the most successful human pathogens. In 2011, M. tuberculosis claimed 1.4 million lives and caused an estimated 8.7 million incident cases of TB (2).
Transcription by bacterial RNA polymerase (RNAP) is a key point of control of gene expression and an important target for antimicrobial chemotherapy, including the frontline anti-tuberculosis drug rifampicin. RNAP is formed by a core of five subunits (α2ββ′ω), which is competent for transcription elongation, and by a sixth σ-subunit (or σ factor) that is essential for promoter recognition and transcription initiation. All bacterial genomes encode one primary σ-subunit that is responsible for the transcription of housekeeping genes and is essential for the growth of the organism (3). However, the total number of σ-subunits encoded can vary from 1 in Mycoplasma pneumoniae (4) to 65 in Streptomyces coelicolor A3(2) (5). Each σ-subunit has different promoter specificity, so the use of different σ-subunits, which is often linked to a specific stimulus such as stress response, represents an efficient mechanism of gene expression control.
M. tuberculosis encodes 13 distinct σ-subunits (σA–σM) (6): σA is the primary σ-subunit, σB is a primary-like σ-subunit that shares a high degree of sequence homology with the last 300 residues of σA, and σC–σM are alternative σ-subunits (3). Interestingly, M. tuberculosis has the highest ratio of alternative σ-subunits compared with genome size of any obligate pathogen (7), possibly reflecting the complex stages of infection that require tight regulation.
Switching between different σ-subunits to reprogram transcription is a common strategy to control gene expression. However, other mechanisms that control the transcription activity of the RNAP include transcription factors, other RNAP-binding proteins, non-protein ligands, and the folded bacterial chromosome structure (8, 9). The expression of a particular gene is thus the result of the interaction of different players in a complex and dynamic network that, for M. tuberculosis, is still not completely understood.
A novel small RNAP-binding protein RbpA was identified in S. coelicolor and is highly conserved in the actinomycetes (10). RbpA of M. tuberculosis contains 111 amino acids, is encoded by Rv2050, and is thought to be part of the gene expression control network (11). Hu et al. (11) have shown that RbpA activates transcription by stabilizing the formation of the RNAP holoenzyme containing σA but does not activate σF-dependent transcription. RbpA and its homologue in Mycobacterium smegmatis have been reported to bind to the β-subunit of RNAP, but the basis of sigma factor specificity is unknown (11, 12). One hypothesis is that RbpA can modify the RNAP core structure to specifically increase the affinity for σA (11).
Further evidence for the role of RbpA in transcription comes from its role in reducing inhibition of the RNAP by rifampicin, during in vitro transcription and cell-based assays in M. tuberculosis (11), S. coelicolor (13), and M. smegmatis (14). Rifampicin binds to the β-subunit of the RNAP. The binding site of rifampicin does not overlap with the predicted binding site of RbpA, and the role of RbpA in cell sensitivity to rifampicin remains unclear (11, 14). RbpA expression is up-regulated in several stress conditions: starvation, hypoxia, in mouse macrophages, and in the presence of rifampicin and vancomycin (13, 15, 16). Furthermore, RbpA was predicted to be essential for normal growth of M. tuberculosis (17). This early prediction was confirmed using an RbpA conditional knockdown mutant strain in M. tuberculosis (18). However, it is not clear yet what essential function(s) RbpA performs in M. tuberculosis.
The essential role of RbpA in the regulation of gene expression and its role in rifampicin tolerance make this protein an important subject for study. Here we present evidence that RbpA can directly interact with both σA and σB, thereby providing insights into the mechanism(s) used by RbpA to modify the transcription activity of the RNAP. Furthermore, we report the high resolution structure of the N-terminal domain of RbpA (residues 1–79, RbpA1–79), which reveals the presence of a well structured core with long and extremely flexible, N and C termini. We investigated the interaction between RbpA and the σ-subunit and found that the structured central domain of RbpA is not involved in the formation of the complex with the σ-subunit, but both ends of the construct are involved in the interaction, although only the C terminus is essential.
EXPERIMENTAL PROCEDURES
Preparation of Expression Vectors
The genes encoding σA (Rv2703), σB (Rv2710), RbpA (Rv2050), and truncated versions of these genes σB1–228, RbpA1–79, RbpA1–92, and RbpA24–111 were amplified by PCR from M. tuberculosis H37Rv genomic DNA and cloned by a ligase-independent method (BD In-Fusion® PCR cloning kit; Clontech) into an expression vector derived from pET-43.1a(+) (Novagen) encoding a cleavable His6 tag at the N terminus.
Expression and Purification of RbpA Constructs
Escherichia coli BL21(DE3) cells, transformed with the relevant expression vector, were grown in LB medium containing 100 μg/ml ampicillin at 37 °C with shaking to an absorbance at 600 nm of ∼0.7. Protein expression was then induced by the addition of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, and the cell growth was continued at 30 °C for 4 h. The cells were lysed in a buffer containing 50 mm Tris-HCl, pH 8.0, and 0.1 mm EDTA plus 1 mm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF) and 0.1 mg/ml lysozyme. The cell lysate was then centrifuged (15,000 × g for 30 min), and the supernatant was loaded on a nickel-Sepharose 5-ml column (GE Healthcare) pre-equilibrated with a buffer containing 25 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 30 mm imidazole. Subsequently the His6-tagged protein was eluted in the same buffer with a linear imidazole gradient (30–500 mm). Fractions containing the His6-tagged protein were subjected to size exclusion chromatography using a Superdex 75 16/60 (GE Healthcare) column and a buffer containing 25 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 2 mm DTT.
To uniformly label RbpA with 15N, 13C, or 15N/13C, E. coli BL21(DE3) cells transformed with the relevant expression vector were grown in modified Spizizen's minimal medium (19) containing 100 μg/ml ampicillin, where the sole source of nitrogen and carbon were 1 g/liter of [15N]ammonium sulfate and 2 g/liter of 13C-d-(+)-glucose as appropriate. The same expression protocol was applied to uniformly label RbpA1–79, except for 15N/13C-labeled RbpA1–79, which was prepared using nonisotopically labeled aromatic amino acids (His, Tyr, Trp, and Phe at 50 mg/liter) in the minimal medium. The preparation of uniformly labeled 15N/13C/2H RbpA was achieved following a protocol for high density expression of labeled protein (20). Purification of the labeled proteins were performed as for isotopically unlabeled RbpA.
Expression and Purification of σA, σB, and σB1–228
E. coli BL21(DE3) cells, transformed with the relevant expression vector, were grown in LB medium (containing 100 μg/ml ampicillin) at 37 °C with shaking to an absorbance at 600 nm of ∼0.7. Protein expression was induced by the addition of isopropyl 1-thio-β-d-galactopyranoside at 0.05 mm, and the cell growth continued at 16 °C overnight. The cells were lysed in a buffer containing 50 mm Tris-HCl, pH 7.9, 500 mm NaCl, and 5% glycerol plus 1 mm AEBSF, 0.1 mg/ml lysozyme, 0.1 mg/ml deoxyribonuclease I, and 5 mm MgCl2. The cell lysate was then centrifuged (15,000 × g for 30 min), and the supernatant was loaded on a 5-ml nickel-nitrilotriacetic acid column (Qiagen) pre-equilibrated with the lysis buffer containing 20 mm imidazole. The His6-tagged protein was eluted in the same buffer with a linear imidazole gradient (20–500 mm). Fractions containing the His6-tagged protein were subjected to size exclusion chromatography using a Superdex 75 16/60 (GE Healthcare) column and a buffer containing 50 mm Tris-HCl, pH 7.9, 500 mm NaCl, 5% glycerol, 0.1 mm EDTA, and 0.5 mm DTT.
His6 Tag Cleavage
The His6 tag was removed from the His6-tagged proteins by enzymatic cleavage using His6-TEV protease (provided by PROTEX, University of Leicester). The reaction results in complete removal of the tag leaving a single serine residue. His6-TEV protease and the His6 tag were removed from the reaction mixture using a 5-ml nickel-nitrilotriacetic acid column (Qiagen).
NMR Spectroscopy
NMR spectra were acquired from 0.35-ml samples of RbpA (∼0.2 mm) and RbpA1–79 (∼0.35 mm) dissolved in a buffer containing 25 mm K2HPO4, pH 6.5, 100 mm KCl, 0.5 mm DTT, 0.5 mm EDTA, 0.02% (w/v) NaN3, 0.2 mm AEBSF, and 0.5 mm Tris(2-carboxyethyl) phosphine with 10% D2O with 90% H2O or 100% D2O as appropriate. All of the NMR data were recorded at 35 °C using either 600 MHz Bruker Avance/DRX systems or an 800 MHz Bruker Avance II spectrometer fitted with cryogenically cooled probe heads.
The two- and three-dimensional spectra recorded to obtain sequence specific assignments for RbpA were: 1H-1H TOCSY (21) with mixing times of 35 and 55 ms, 1H-1H NOESY (22) with an NOE mixing time of 80 ms, 15N/1H TROSY (23), 15N/1H NOESY-TROSY with an NOE mixing time of 50 ms (24), 13C/1H HC(C)H-TOCSY and (H)CCH-TOCSY (25) with mixing times of 18 ms, 15N/13C/1H HBHA(CO)NH (26), TROSY-HNCO (27), TROSY-HNCA (27), TROSY-HN(CO)CA (28), CBCA(CO)NH (29), and TROSY-HNCACB (30). A uniformly 15N/13C/2H-labeled sample of RbpA was used to acquire the 15N/1H TROSY and 15N/13C/1H TROSY-HNCACB spectra. Typical acquisition times in the two-dimensional experiments were: 60 ms (15N) and 19 ms (1H) in F1 and 65–72 ms (1H) in F2. Typical acquisition times in the three-dimensional experiments were: 4–9 ms (13C), 9–18 ms (15N) in F1 and F2, and 65–83 ms in F3 (1H).
The two- and three-dimensional spectra recorded to obtain sequence specific assignments and 1H-1H distance constraints for RbpA1–79 were: 1H-1H TOCSY (21) with mixing times of 35 and 55 ms, 15N/1H HSQC (23), TOCSY-HSQC (31) and 15N/13C/1H HNCACB (30) with a mixing time of 60 ms, 15N/13C/1H HNCO (27), HNCACB (30), and 1H-1H NOESY (22), 13C/1H NOESY-HSQC (32), and 15N/1H NOESY-HSQC (24) with NOE mixing times of 200 ms. An 15N/13C-labeled sample of RbpA1–79 with nonisotopically labeled aromatic amino acids (His, Tyr, Trp, and Phe) was used to acquire the 15N/13C/1H HNCACB, HNCO, and 13C/1H NOESY-HSQC spectra. Typical acquisition times in the two-dimensional experiments were: 60 ms (15N) and 35–39 ms (1H) in F1 and 80–120 ms (1H) in F2. Typical acquisition times in the three-dimensional experiments were: 9–12 ms (13C), 19–34 ms (15N), and 15–18 ms (1H) in F1 and F2 and 70–100 ms (1H) in F3.
To identify changes in the RbpA signals induced by His6-σB1–228 binding, NMR spectra were acquired from 0.35-ml samples of 15N/13C/2H RbpA (0.19 mm) and of 15N/13C/2H RbpA (0.1 mm) bound to unlabeled His6-σB1–228 (1:1 molar ratio) dissolved in a buffer containing 25 mm K2HPO4, pH 7.5, 100 mm KCl, 0.5 mm DTT, 0.5 mm EDTA, 0.02% (w/v) NaN3, 0.2 mm AEBSF, and 0.5 mm Tris(2-carboxyethyl) phosphine with 10% D2O with 90% H2O. The two- and three-dimensional spectra recorded were: 15N/1H TROSY and 15N/13C/1H TROSY-HNCO. Acquisition times in the two-dimensional experiments were: 26 ms in F1 (15N) and 65 ms in F2 (1H). Acquisition times in the three-dimensional experiments were: 12 ms in F1 (13N), 10 ms F2 (15N), and 65 ms in F3 (1H). The two-dimensional NMR spectra were acquired typically for ∼0.5 h, and the three-dimensional spectra were acquired typically for ∼65 h.
The WATERGATE method (33) was used to suppress the water signal when required. The NMR data collected were processed using the program Topspin (Bruker Biospin Ltd.) with linear prediction used to extend the effective acquisition time up to 2-fold in F1 and F2. The spectra were analyzed with the Sparky package (T. D. Goddard and D. G. Kneller, University of California, San Francisco).
Sequence-specific Assignment
Sequence-specific backbone assignments were obtained for RbpA from the identification of intra- and inter-residue connectivities in a series of double and triple resonance spectra: 15N/1H TROSY (23), NOESY TROSY (24), 15N/13C/1H HBHA(CO)NH (26), TROSY-HNCO (27), TROSY-HNCA (27), TROSY-HN(CO)CA (28), CBCA(CO)NH (29), and TROSY-HNCACB (30). Assignments were then extended to the side chains using correlations observed in: 1H-1H TOCSY (21), NOESY (22), 13C/1H HC(C)H-TOCSY, (H)CCH-TOCSY (25), and NOESY-TROSY (24). Initially, tentative assignments for RbpA1–79 were obtained by transposing the assignment from RbpA onto the 1H-1H TOCSY (21), 1H-1H NOESY (22), 15N/1H HSQC (23), and TOCSY-HSQC (31) spectra. The inter-residue amide nitrogen/proton to Cα and Cβ connectivities observed in the HNCACB (30) and the NH to NH and CH to CH NOEs identified in the 15N- and 13C-edited NOESY spectra (24, 32) were used to confirm the assignment.
Structure Calculation
The program Cyana (34) was used to calculate the family of converged structures of RbpA1–79 in a two-stage process. Initially, the combined automated NOE assignment and structure determination protocol (CANDID) was used to automatically assign NOE cross-peaks and to generate a preliminary family of converged structures. The inputs used for this first step were: 15N, 13C, and 1H resonance assignments, a set of 72 backbone torsion angle constraints, determined by the protein backbone dihedral angle prediction program TALOS+ (35), and three lists of manually picked NOE cross-peaks that were identified in a two-dimensional NOESY spectrum (399 peaks) and in a three-dimensional 15N- and a 13C-edited NOESY spectra (881 and 1049 peaks, respectively). The CANDID calculation was carried out using the standard parameters of Cyana but with a chemical shift tolerance set at 0.03 ppm for 1H and at 0.4 ppm for 15N and 13C. In the second stage, the final family of converged RbpA1–79 structures was produced through several cycles of simulated annealing combined with redundant dihedral angle constraints (36), resulting in 98 structures with no distance violations greater than 0.5 Å and no dihedral angle violations greater than 5 °. The structures obtained were then refined in a generalized Born solvent model (37) using the AMBER 10 package (38) as described previously (39), but with one cycle of restrained molecular dynamics simulated annealing. The 35 structures with the lowest AMBER energy were selected and analyzed using MOLMOL (40) and CING (41).
Sequence Alignment and Conservation Score Assignment
RbpA homologue sequences were identified using the BLAST algorithm (42) with the default settings leading to the identification of 416 sequences. The alignment was performed using the software COBALT (43) with sequences from Actinobacteria. When multiple sequences were identified from the same genus, only the sequence with the highest similarity score was selected. The resulting alignment was used to assign a conservation score to each RbpA residue, calculated using the analysis of multiply aligned sequences method (44) implemented in Jalview (45).
Construction of a Genomic M. tuberculosis Yeast Two-hybrid Library
The total DNA of M. tuberculosis H37Rv (kindly provided by Roland Brosch, Pasteur Institute, Paris) was partially digested with AciI and HpaII. The fragments were subsequently cloned into the single ClaI site of plasmid pGADT7 (Clontech) to generate a genomic M. tuberculosis library for yeast two-hybrid analyses. To avoid religation of the digested vector, the ligation mix was treated with ClaI before transformation into E. coli. The obtained library consists of ∼3 × 105 independent clones with an insertion frequency of nearly 100% and an average insert size of ∼500 base pairs.
Two-hybrid Screening for σB-interacting Proteins
The entire coding sequence of σB was amplified by PCR with M. tuberculosis H37Rv DNA as a template and fused in-frame to the DNA of the yeast Gal4 DNA-binding domain using the restriction sites EcoRI and PstI of plasmid pGBT9 (Clontech). Subsequently, this construct was used to screen for σB-interacting proteins. The “bait” and “prey” containing plasmids were co-transformed by the method of Klebe et al. (46) into the yeast strain Y190 (MATa, ura3–52, his3–200, ade2–101, lys2–801, trp1–901, leu2–3, −112, gal4Δ, gal80Δ, cyhr2, LYS2∷GAL1UAS-HIS3TATA-HIS3, URA3∷GAL1UAS-GAL1TATA-lacZ; Clontech). Yeast transformants were selected and cultivated on SD synthetic medium (2% glucose and 0.67% yeast nitrogen base without amino acids) supplemented with essential amino acids and nucleotides. Potential interaction partners of σB were monitored using the β-galactosidase as a reporter system by a filter lift assay (47, 48).
Interaction Assay by Split-Trp
Protein-protein interactions were tested in E. coli using the Split-Trp method (49, 50). The genes encoding σB and RbpA were each cloned into PL184 and PL185 for expression as fusions with Ntrp and Ctrp. Pairs of plasmids were transformed in E. coli ΔtrpF, and strains were grown in LB then plated on VB minimal agar containing 0 or 60 μg/ml tryptophan and incubated at 25 °C. Photographs were taken on the sixth day.
Analytical Size Exclusion Chromatography
The interaction between RbpA and the sigma factors was assessed by size exclusion chromatography using a Superdex 75 10/300 GL column (GE Healthcare) or a Superdex 200 10/300 GL column (GE Healthcare) and a buffer containing 50 mm Tris-HCl, pH 7.9, 500 mm NaCl, 5% glycerol, 0.1 mm EDTA, and 0.5 mm DTT. The oligomeric organization of RbpA was assessed by size exclusion chromatography using a Superdex 75 10/300 GL column (GE Healthcare) and a buffer containing 25 mm K2HPO4 pH 6.5, 100 mm KCl, 0.5 mm DTT, 0.5 mm EDTA, and 0.02% (w/v) NaN3.
Mapping the σB Binding Site on RbpA
Changes in the positions of RbpA NMR signals resulting from binding to His6-σB1–228 were analyzed using the minimal shift approach (51–53). The combined HN, N, and CO chemical shift differences (Δδ) between the peaks detected in HNCO spectra recorded on free 15N/13C/2H RbpA and on 15N/13C/2H RbpA bound to unlabeled His6-σB1–228 were calculated using the following equation: Δδ = √((ΔδHN)2 + (ΔδN*0.2)2 + (ΔδCO*0.35)2), where ΔδHN, ΔδN, and ΔδCO correspond to the differences in 1H, 15N, and 13C chemical shifts between pairs of HNCO peaks, and 0.2 and 0.35 are scaling factors required to account for differences in the range of amide proton, amide nitrogen, and carbonyl chemical shifts. For each peak detected in the HNCO spectra recorded on free 15N/13C/2H RbpA, the minimal shift induced by the binding with His6-σB1–228 was taken as the lowest calculated combined shift value (Δδ).
Complementation of Rv2050 Mutant
The construction of a conditional mutant strain of M. tuberculosis H37Rv in which the gene Rv2050 is under the control of a pristinamycin-inducible promoter has been described previously (18). The coding region of the gene Rv2050 with the 304-base pair promoter region was amplified with specific oligonucleotides and cloned into plasmid pMV306 to obtain the plasmid pMV306-RbpA. pMV306 is an integrative plasmid in which the expression cassette of pMV361 is replaced by a multiple cloning site (54). A truncated version, pMV306-RbpA1–79, missing 32 residues at the C terminus was constructed in an identical manner. Each complementing plasmid, and, as a control, pMV306, was electroporated into the conditional mutant. Colonies were obtained on Middlebrook 7H10 agar supplemented with 10% ADN (5% albumin, 2% dextrose, 145 mm NaCl), kanamycin (40 μg/ml), hygromycin (100 μg/ml), and pristinamycin (0.5 μg/ml). A few clones for each transformation were grown in Middlebrook 7H9 medium supplemented with ADN, Tween 80 (0.05%), kanamycin, hygromycin, and pristinamycin for 10 days and then serially diluted and spotted in parallel onto 7H10 agar plates with and without pristinamycin. Photographs were taken after 24 days of incubation at 37 °C.
RESULTS
The Role of the C-terminal Region of RbpA in Oligomerization
The protocol adopted for the purification of RbpA involves metal affinity chromatography followed by size exclusion chromatography. In the latter step, it was noticed that RbpA eluted from the column earlier than is expected for a protein with a molecular mass of 13 kDa. The oligomerization status of RbpA was investigated by performing a number of size exclusion chromatography experiments on RbpA samples at different concentrations (Fig. 1). The resulting chromatograms reveal asymmetric peaks with the elution volume strongly dependent on the concentration of the sample. These observations suggest that there is a dynamic and concentration-dependent equilibrium between different oligomeric species of RbpA.
FIGURE 1.

Overlay of the size exclusion chromatography elution profiles of RbpA samples loaded at different concentrations. Samples of RbpA were loaded at different concentrations onto a size exclusion chromatography column (Superdex 75 10/300). The elution volumes of protein molecular mass standards (conalbumin, ovalbumin, carbonic anhydrase, and ribonuclease) are indicated by dashed lines.
Initial NMR experiments aimed at determining the solution structure of RbpA were recorded and analyzed. For residues 1–76, the extent of the assignment is 94.1% of all the aliphatic 13C resonances and 88.1 and 91.5% of all the 15N and 1H resonances, respectively. However, approximately half of the residues 77–111 do not generate observable NMR signals. This prevented unequivocal assignment of the last 35 residues of RbpA. The lack of backbone NMR signals for this region implies that it samples a number of discrete structural states on a second to millisecond time scale.
The secondary structure prediction for RbpA (Fig. 2A) reveals that two distinct α-helices are likely to be formed in the C-terminal region (residues 80–111). Interestingly, the second predicted helix (residues 91–108) has a marked amphipathic character (Fig. 2B). The hydrophobic side of this helix could potentially interact with the equivalent face on a second RbpA molecule, leading to homo-oligomerization. The elongated shape of the peak obtained during the size exclusion chromatography analysis of RbpA (Fig. 2C, blue trace) indicates that there is a constant interchange between different oligomerization species. Conformational exchange in the C-terminal region of RbpA could account for the poor quality of the NMR signals generated by the last 35 residues: some signals are weak, some are very broad, and some are missing.
FIGURE 2.
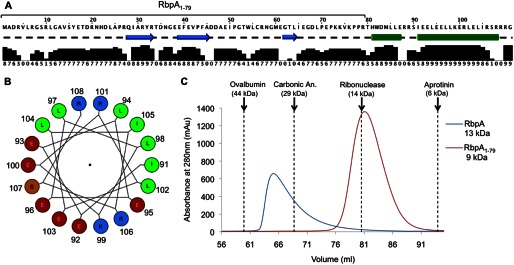
The role of the C-terminal region of RbpA in oligomerization. A, the sequence of RbpA and its secondary structure prediction, performed using JPred 3 (69), with the relative prediction confidence underneath (levels 0–9). Cyan arrows represent β-strands, green cylinders represent α-helices, and a dashed line indicates a lack of secondary structure. B, the helical wheel projection of RbpA residues 91–108 (IEELEELLKERLELIRSR). The following color code is used to highlight differently charged residues: red for acidic residues, blue for basic residues, green for nonpolar residues, and orange for polar, uncharged residues. C, the overlay of the size exclusion chromatography elution profiles of RbpA at 85 μm and RbpA1–79 at 330 μm (Superdex 75 16/60). The elution volumes of protein molecular mass standards are indicated by dashed lines.
To investigate the role of the C-terminal region in oligomerization, we expressed and purified a shorter version of RbpA, termed RbpA1–79, which consists of residues 1–79 and therefore lacks the predicted amphipathic helix. The elution profile of this truncated protein in size exclusion chromatography is consistent with the predicted molecular mass of the monomer (9 kDa; Fig. 2C) and is independent of the protein concentration (not shown). The difference in retention between RbpA and RbpA1–79 clearly indicates that the C-terminal region is essential for oligomerization.
Solution Structure Calculation of RbpA1–79
There was a significant improvement in the quality of the NMR data obtained for RbpA1–79 as compared with full-length RbpA, which is evident from both the increased signal to noise ratio and decreased line widths observed for RbpA1–79 as shown in the spectra reported in Fig. 3 and supplemental Fig. S1. Comparison of the backbone chemical shifts of RbpA and RbpA1–79 revealed only minor changes (Fig. 3), indicating that the structure of the first 79 residues of RbpA is maintained in the shorter RbpA1–79 construct. Therefore, RbpA1–79 was chosen for further structural characterization.
FIGURE 3.

Overlay of HSQC spectra recorded on RbpA and RbpA1–79. HSQC spectra were recorded for 15N RbpA (210 μm), shown in black, and 15N RbpA1–79 (350 μm), shown in red.
The quality of the NMR data acquired for RbpA1–79 allowed us to obtain comprehensive backbone and side chain assignments. For example, nearly complete assignments of backbone resonances (N, NH, Hα, Hβ, Cα, and Cβ) were made for all residues except: Met1 (N, NH, Hα, Hβ2, Hβ3, and Cβ), Gly8 (Hα2 and Hα3), Tyr16 (Cα and Cβ), His22, Tyr32 (Cα and Cβ), Phe40 (Cα and Cβ), Phe44 (Cα and Cβ), Arg57 (N and NH), Pro70 (Hα, Hβ2, and Hβ3), Pro77, and Pro78 (Hα, Hβ2, and Hβ3). The extent of the assignment is 78.5% of all the aliphatic 13C resonances, 86.4 and 91% of all the 15N and 1H resonances, respectively. The CANDID protocol (34) was used to automatically assign the NOE cross-peaks identified in two-dimensional 1H-1H NOESY and three-dimensional 15N- and 13C-edited NOESY spectra. Unique assignments were obtained for 87% of the NOE cross-peaks identified. The structure of RbpA1–79 was calculated with the program Cyana (34) using the constraints listed in Table 1. In the final round of structure calculation, 98 satisfactorily converged RbpA1–79 structures were obtained from 100 initial random structures. They had no distance or van der Waals violations greater than 0.5 Å, had no dihedral angle violations greater than 5°, and had an average value for the Cyana target function of 1.17 Å2. The structures were refined using the AMBER 10 package (38), and the 35 structures with the lowest AMBER energy were selected. The final family of converged structures has no distance constraint violations greater than 0.37 Å and no dihedral angle violations.
TABLE 1.
NMR constraints and structural statistics for the RbpA1–79 structure
The table shows a summary of the NMR constraints used for the calculation of the RbpA1–79 structure and of the structural statistics for the final family of 35 RbpA1–79 converged structures.
| NMR constraints | |
| No. of upper distance limits | 1276 |
| Short range |i − j| ≤ 1 | 618 |
| Medium range 1 ≤ |i − j| ≤ 5 | 165 |
| Long range |i − j| ≥ 5 | 493 |
| No. of backbone torsion angle constraints | 72 |
| Violations | |
| Maximum distance violation | 0.37 Å |
| Maximum dihedral angle violation | 0° |
| Energies | |
| Mean AMBER energy | −3696.46 kcal mol−1 |
| Mean NOE energy | 11.51 kcal mol−1 |
| Deviation for idealized geometry | |
| Bond lengths | 0.0108 ± 1.09 × 10−4 Å |
| Bond angles | 2.139 ± 0.036° |
| Root mean square deviation for the structured core (residues 26–66) | |
| Backbone | 0.32 ± 0.11 Å |
| Heavy atoms | 1.08 ± 0.16 Å |
| Ramachandran plot (residues 26–66) | |
| Residues in most favored regions | 97.6% |
| Residues in additionally allowed regions | 2.4% |
| Residues in generously allowed regions | 0% |
| Residues in disallowed regions | 0% |
Superposition of the final family of converged structures (Fig. 4A) reveals the presence of a well defined central domain (residues 26–66), whereas the N- and C-terminal regions of the protein are both disordered and highly flexible. The structure of the central domain was determined to high precision, as is clearly evident from the overlay shown in Fig. 4B, and is reflected in low root mean square deviation values to the mean structure for both the backbone and all the heavy atoms of 0.32 and 1.08 Å, respectively (a summary of the NMR constraints and structural statistics is reported in Table 1). The central domain is primarily composed of four distinct β-strands: β_1 (residues 27–33), β_2 (residues 39–45), β_3 (residues 53–55), and β_4 (residues 61–65), which fold to form two antiparallel β-sheets linked by turns and loops (Fig. 4C). The two β-sheets lie perpendicular to each other forming a β-sandwich-like structure, which is stabilized by a cluster of aromatic and nonpolar residues (Tyr32, Val42, Phe44, and Trp54). Analysis of the electrostatic surface of the central domain (Fig. 5) revealed the presence of a cluster of conserved residues (Arg27, Val42, Phe44, Ala45, Asp47, Ala48, Glu49, and Trp54) that form a surface patch of 742.2 Å2 with a hydrophobic center surrounded by charged residues. This patch is the most conserved region on the surface of the central domain (Fig. 5B) and may form a potential protein interaction site.
FIGURE 4.

Solution structure of RbpA1–79. A, a best fit superposition of the final family of 35 converged structures obtained for RbpA1–79. B, a best fit superposition of the structured core (residues 26–66) of the final family of converged structures of RbpA1–79. The backbone is shown in blue, the side chains are colored in red, except for the aromatic side chains, which are colored cyan. The orientation is identical to that shown in A. C, a ribbon representation of the RbpA1–79 structure, from residue 21 to residue 71, with an orientation identical to that shown in A and B.
FIGURE 5.

Surface view of the structured core of RbpA (residues 26–66). A, contact surface views of the central domain colored according to electrostatic potential. Neutral areas are shown in white, and areas of significant charge are shown in red (negative) and blue (positive). A dashed line is used in the left panel to locate the conserved patch. B, the same views are colored according to amino acid conservation among homologues with the least conserved residues in blue, scaling to completely conserved residues in orange, and invariant residues in red. Conservation scores were taken from Jalview: blue, 0–3; cyan, 4–6, yellow, 7–9; orange, 10; and red, 11).
Comparison of the structure of the RbpA central domain (residues 26–66) with the known folds in the Protein Data Bank using Dali (55) identified 14 structures (supplemental Table S1) that share structural homology (z score > 2). However, the structural homology of each with RbpA central domain is relatively low (z score < 3.5), and no specific function has been assigned to this fold. The highest scoring structural homologue (Protein Data Bank code 3UXQ, chain 6) is a ribosomal protein that has a Cα root mean square deviation to the RbpA central domain of 2.8 Å (supplemental Table S1) but no sequence homology (2% amino acid identity).
Identification of RbpA as a Binding Partner of σB
M. tuberculosis is unusual, but not unique, in having a second principle-like sigma factor σB that is up-regulated during stress. As part of an investigation into the function of σB, we performed a yeast two-hybrid screen to identify potential binding partners encoded within the genome of M. tuberculosis. Of ∼75,000 yeast transformants analyzed, one clone was identified, which scored positive after several rounds of verification. Sequencing revealed that the gene Rv2050 was fused in-frame to the activation domain of Gal4.
Because this interaction had not been observed previously, we additionally verified that RbpA binds to σB in bacterial cells using a protein fragment complementation assay in E. coli (49, 50). Strains carrying Ntrp-σB and RbpA-Ctrp grew on minimal medium, suggesting that interaction of σB with RbpA reconstituted the Trp1p enzyme (Fig. 6). The strain carrying Ntrp-σB and σB-Ctrp formed a negative control, as σB is not thought to form homo-oligomers.
FIGURE 6.
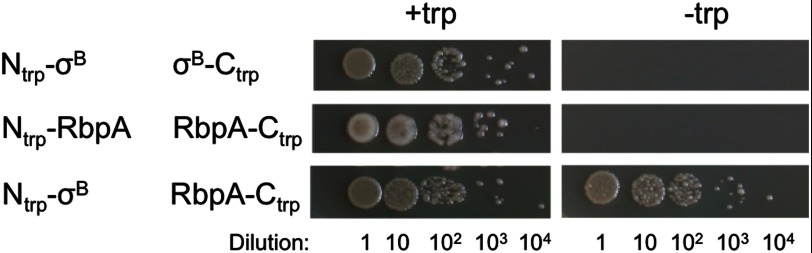
Evidence for RbpA interaction with σB from the Split-Trp protein fragment complementation assay. Tryptophan auxotrophic E. coli were transformed with pairs of plasmids, serially diluted, and plated on minimal medium in the presence or absence of tryptophan. The interaction between RbpA and σB led to reconstitution of the Trp1p enzyme and hence the ability of the strain to grow on medium lacking tryptophan. The strain expressing Ntrp-σB plus σB-Ctrp is a negative control because σB is not thought to form dimers or multimers. Tryptophan-containing medium is a control to demonstrate that all strains are viable.
Characterization of the Interaction of RbpA with Sigma Factors
To further characterize the identified interaction in vitro and to determine the domains responsible for interaction, we expressed and purified σB (full-length, residues 1–323) and a truncated version, σB1–228 (residues 1–228), which contains conserved motifs 1.2–3.1 (56). The site of σB truncation was chosen using multiple sequence alignment and structural modeling with Phyre (57) based on the structure of the Thermus thermophilus principal σ subunit, Protein Data Bank code 1IW7 (45% of sequence identity) (58). Both versions were tested for the ability to interact with RbpA during size exclusion chromatography (below) and affinity chromatography (not shown). RbpA was found to bind σB1–228 (Fig. 7) or full-length σB (data not shown). Co-purification demonstrates that the interaction is tight (KD < 10−6 m) and specific and shows that the first 228 residues of σB are sufficient to form the complex with RbpA.
FIGURE 7.

His6-RbpA binds and forms a tight complex with His6-σB1–228. A, elution profiles from a size exclusion column (Superdex 75 10/300) of His6-RbpA or His6-σB1–228 individually and the complex formed between them (1:1 molar ratio). The elution volumes of the protein molecular mass standards are indicated by dashed lines. B, samples from the fractions collected during the different size exclusion chromatography runs were analyzed by SDS-PAGE. Lane MW shows molecular mass markers, lane L shows the column load, and lanes 8–12 are labeled according to the elution volume (ml).
To investigate the stoichiometry of the complex, further size exclusion chromatography experiments were performed using different molar ratios: 2:1 and 1:2 of RbpA:σB1–228 (supplemental Fig. S2). In each case a 1:1 complex was eluted from the column, with the excess material eluting as expected for the free protein. The elution volume for the His6-RbpA·His6-σB1–228 complex is lower (earlier) than that predicted by the molecular mass of the complex, 43.9 kDa but is nevertheless closer to the prediction for a 1:1 complex than a 2:2 complex (Fig. 7). Hence we suggest that the His6-RbpA·σB1–228 complex has a 1:1 stoichiometry and that the slightly earlier elution may be due to the extended conformation that is known for sigma factors (57).
Sigma factor A (σA), the primary sigma factor of M. tuberculosis, shares high homology with σB including 64% amino acid identity in residues 1–228 of σB. We hypothesized that σA could also interact with RbpA. To test this hypothesis, we expressed and purified σA and assessed the binding with RbpA by size exclusion chromatography experiments, confirming that σA can also bind and form a tight complex with RbpA (data not shown).
Insights on the Interaction between RbpA and σB
To better understand the binding mode of RbpA with the sigma subunit, we took advantage of the RbpA NMR assignment to investigate which residues of RbpA are involved in the interaction with σB1–228 by NMR minimal shifts. To this purpose, a sample of triply labeled 15N/13C/2H RbpA was mixed in an equimolar ratio with unlabeled His6-σB1–228, and the resulting complex was purified by size exclusion chromatography. TROSY and TROSY-HNCO spectra were acquired on the complex sample and on a triply labeled sample of 15N/13C/2H RbpA in the free form. The differences observed in the peak positions between the complex and free RbpA are reported in Fig. 8. Comparison between the TROSY spectra recorded on RbpA free and on the RbpA·His6-σB1–228 complex (Fig. 8A) show evident perturbations in both assigned and unassigned resonances. In total, at least 19 RbpA backbone amide groups are clearly affected by the binding with His6-σB1–228, and 10 of these are unassigned resonances (supplemental Fig. S3). All the unassigned RbpA backbone amide groups—except Met1, Gly8, and Arg10—lie in the last 35 residues of the protein. Therefore, the relatively high number of perturbations observed in the unassigned peaks show that the C terminus of RbpA is affected by the binding with His6-σB1–228. Furthermore, the only resonance that we were able to assign in the region 77–111, the indole group of the tryptophan in position 82, is clearly affected by the binding with His6-σB1–228 (Fig. 8A). Among the assigned resonances that are affected by the binding with His6-σB1–228, there is a clear cluster between residues 11 and 20 (Fig. 8C).
FIGURE 8.
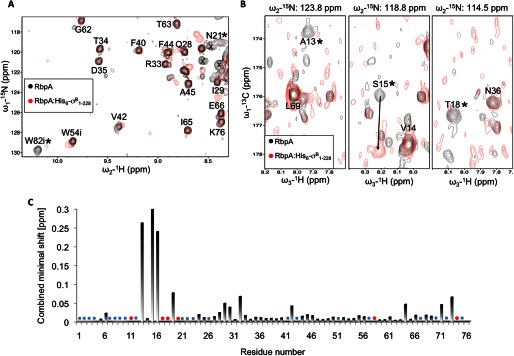
RbpA chemical shifts changes induced by binding of His6-σB1–228. A, an overlay of a selected region from TROSY spectra recorded on 15N/13C/2H RbpA (190 μm) (shown in black) and on a complex of 15N/13C/2H RbpA with His6-σB1–228 (100 μm) (shown in red). The letter i indicates a peak generated by indole groups, asterisks indicate assigned peaks that are affected by His6-σB1–228 binding, and the letter X indicates unassigned peaks that are affected by His6-σB1–228 binding. B, an overlay of selected regions from TROSY-HNCO spectra recorded on 15N/13C/2H RbpA (190 μm) (shown in black) and on a complex of 15N/13C/2H RbpA with unlabeled His6-σB1–228 (100 μm) (shown in red). Asterisks indicate peaks that are affected by His6-σB1–228 binding, and an arrow shows the shift for the Ser15 peak. C, combined minimal shift changes observed comparing the TROSY-HNCO spectra described. Assigned resonances for RbpA that cannot be detected in the complex are marked with a red circle (●), whereas RbpA residues for which the assignment in the TROSY-HNCO spectrum is missing are indicated with a blue square (■).
This NMR analysis of the RbpA·His6-σB1–228 complex led us to hypothesize that both the N- and C-terminal regions of RbpA are involved in the binding with His6-σB1–228. To support this hypothesis, we tested three truncated versions of RbpA: RbpA1–79, RbpA1–92 (residues 1–92), and RbpA24–111 (residues 24–111) for interaction with the σ factor by size exclusion chromatography experiments (data not shown). The truncation site at Glu92 was chosen to remove the predicted amphipathic C-terminal helix but retain residues 79–89 that are almost invariant among RbpA homologues and include Trp82, which is thought to be involved in σB binding based on the minimal shift assay. The results obtained are summarized in Fig. 9. From this set of experiments, it is evident that residues 1–24 of RbpA are not essential for the interaction with His6-σB1–228, but the whole C-terminal region is required for the formation of a stable complex with His6-σB1–228.
FIGURE 9.

Summary of the size exclusion chromatography experiments performed to investigate binding between RbpA and the sigma factors. A check indicates that the proteins formed a complex, a cross indicates that no interaction was detected, and a dash indicates that the combination was not tested.
Complementation of an RbpA Conditional Mutant Strain in M. tuberculosis
The gene encoding RbpA was predicted to be essential for the growth of M. tuberculosis (17), and essentiality was confirmed recently by constructing a strain where the expression of RbpA is under the control of the antibiotic pristinamycin I (PI) (18). This strain exhibits a conditional lethal phenotype when PI is withdrawn (18). To confirm that the lethal phenotype is due to the lack of RbpA expression and to investigate the domains of RbpA, we complemented this mutant strain with RbpA or RbpA1–79 expressed from the pMV306 plasmid vector. Ectopic expression of full-length RbpA led to complementation, as measured by growth on agar lacking PI (Fig. 10). By contrast, expression of RbpA1–79 did not complement the conditional mutant, suggesting that residues 80–111 are required for the physiological function of RbpA.
FIGURE 10.

RbpA, but not RbpA1–79, complements the conditional lethal phenotype of the conditional RbpA mutant strain of M. tuberculosis. Liquid cultures of the conditional mutant strain transformed with pMV306, pMV306-RbpA, or pMV306-RbpA1–79 were diluted (from 10- to 106-fold) and grown on 7H10 agar with or without PI. The plates were placed on a white light box for photography. Plasmid pMV306-RbpA, but not pMV306-RbpA1–79, complemented the conditional lethal phenotype, allowing growth in the absence of PI. The control strain carrying unmodified plasmid pMV306 did not grow in the absence of PI. All strains grew in the presence of PI.
DISCUSSION
The essential functions of RbpA are important in understanding the control of bacterial transcription, characterizing the target of a frontline anti-tuberculosis drug, and identifying new vulnerabilities that could be exploited for future drug development. Although the interaction between RbpA and RNAP was reported in 2001 (10) and the influence of RbpA expression levels on rifampicin sensitivity was reported in 2006 (13), the mechanisms by which RbpA influences transcription and rifampicin sensitivity are still unclear. Our results showing that RbpA can bind directly to σA supports the proposal by Hu et al. (11), that RbpA may promote open complex formation and influence the competition between sigma subunits for binding to RNAP. The fact that RbpA can bind to both RNAP and σA provides the simplest mechanism by which RbpA could stabilize the holo-RNAP. We have shown that RbpA can form a tight complex with σ subunits alone, whereas previous studies have shown that RbpA can bind apo-RNAP (11, 14), but evidence is lacking over the order of binding. Previous studies, using RbpA labeled with a chemical cross-linker (12, 14) or a chemical protease (11), have shown that RbpA binds to the β subunit of RNAP but disagree about the likely binding site. Given the structural flexibility of the termini of RbpA and the fact that σB interaction occurs through these termini, it is not possible to propose which, if any, of the previously proposed binding sites might be compatible with RbpA binding to the β and σ subunits simultaneously.
Although Hu et al. (11) postulate that RbpA could bind RNAP as a dimer, there is conflicting data on the likely oligomerization state of RbpA and its homologues. The retention during size exclusion chromatography was the reason that RbpA (11) and its homologue from S. coelicolor (13) were proposed to be homodimers, whereas the same method led Dey et al. (14) to propose that the homologue from M. smegmatis is monomeric. These homologues share high amino acid identity with RbpA (55 and 92%), and each is also predicted to have an amphipathic helix at the C terminus (not shown). We suggest that the apparent disagreement in molecular size could be due to relatively low affinity homo-oligomerization, with a KD close to the range of protein concentrations we used (3–200 μm) so that the oligomerization state depends on the concentration (Fig. 1). Given the homo-oligomerization observed for RbpA in vitro, a positive result might have been expected for the protein fragment complementation assay with E. coli expressing Ntrp-RbpA and RbpA-Ctrp (Fig. 6). There are many possible reasons for the observed negative result, such as the low expression levels of fusion proteins in this system or the influence of fusion to Trp1p fragments.
The results of size exclusion chromatography of the complex between RbpA and σB1–228 indicate that it is likely to have a 1:1 stoichiometry. Together with the established 1:1 stoichiometry of sigma subunit binding to RNAP, this leads us to suggest that a single RbpA binds per RNAP. Whether oligomerization of RbpA occurs in vivo would depend on its concentration, as well as the local environment, and competition with other (non-self) binding partners. In our hands, there was evidence of oligomerization at all concentrations tested (≥3 μm). The intracellular concentration of RNAP in bacteria depends on growth phase and has been estimated at ∼2.5 μm (59). Therefore, we cannot rule out a role for RbpA multimerization in binding RNAP or indeed for other functions, for example protection from nonspecific interactions, as reported for the anti-sigma factor AsiA (60).
The direct interaction between RbpA and σA not only provides a mechanism for stabilizing the holo-RNAP, but may also explain the observed sigma factor/promoter selectivity of RbpA transcription activation. The identification of a direct interaction with σB potentially broadens the function of RbpA to the σB regulon, which includes transcription factors and genes related to cell envelope stress. It is attractive to postulate a role for RbpA in activating σB-driven transcription, because both RbpA and σB are up-regulated under multiple stress conditions. Here we established that RbpA binds to both σA and σB, which are highly homologous in the binding region (σB residues 1–228; 64% amino acid identity). Of the remaining sigma factors in M. tuberculosis, the stress sigma factor σF shares the most similarity with the binding region (31% amino acid identity). However, σF-driven transcription is not activated by RbpA (11), and this would suggest that RbpA binding is restricted to σA and σB and that its activity is restricted to σA- and σB-driven transcription.
We have recorded and analyzed NMR experiments for RbpA1–79 and full-length RbpA, concluding that there are only minor spectral changes between the two forms (Fig. 3) despite the change in oligomerization state. Because chemical shifts are extremely sensitive to the chemical environment, the similarity between spectra indicates that the structure of the first 79 residues of RbpA is independent of the C-terminal region. We suggest that RbpA consists of three regions: residues 1–26, which are a flexible random coil; residues 26–67, which form a stably folded central domain that is connected by an unstructured linker to the predicted helical region, and residues 80–111. Residues 1–79 are not involved in the oligomerization of RbpA, because the chemical shifts for these residues in oligomeric RbpA are essentially unchanged in monomeric RbpA1–79.
Hoping to characterize the binding site on RbpA for sigma factors, we compared the spectra of RbpA with those of the RbpA·σB1–228 complex. This analysis, summarized in Fig. 8, shows that the central domain of RbpA is not directly involved in the formation of the complex with σB1–228, but the N and C termini are clearly affected by binding. Because it was not possible to assign the NMR signals for the C-terminal region of RbpA, we cannot map the precise residues involved. It is not unusual for conformational averaging caused by protein flexibility to occur at high affinity protein-protein interaction sites; indeed there are many documented examples (52, 61–64). The N and C termini are on the same side of the structured core (Fig. 4A), suggesting that they are in close proximity. Therefore, both ends could take part in the interaction with the σ-subunit. A series of deletion experiments (Fig. 9) show that the whole C-terminal region is essential for a tight interaction with the sigma factor. Somewhat surprisingly, given the shifts in signals from residues in the N terminus, the first 23 amino acids are not required for RbpA to bind σB but may stabilize the complex. It is also possible that the conserved N terminus may play other roles in the function of RbpA.
Although the structural core of RbpA does not appear to form part of the interaction surface with σB, there is a significant hydrophobic patch on the surface, bordered by conserved charged residues (Fig. 5), which could form a putative interaction surface, perhaps for RNAP. Notably, the residues that form this patch are highly conserved in all homologues, including Trp54, which is invariant in RbpA homologues, but not conserved throughout the structural homologues.
With the work of Hu et al. (11), RbpA became recognized as part of a repertoire of small RNAP-binding proteins that modulate transcription. Here we establish that RbpA binds not only to RNAP, but also to the sigma factors σA and σB. In E. coli, Crl is also thought to be able to bind to RNAP and to a sigma factor (65–67). Crl is thought to perform a chaperone-like function, influence competition between sigma factors, and promote open complex formation. Potentially there may be similarities with the function of RbpA, but there are significant differences. First, RbpA binds to the principle sigma factor and principle-like sigma factor, whereas Crl binds to the alternative stress responsive sigma factor σS (equivalent to σF of M. tuberculosis). Second, Crl binds to σS and RNAP with low affinity, and it is not able to compete for interaction of σS with other binding partners, such as RssB, which targets σS to the proteasome (66). Stoichiometric co-purification of RbpA with σB indicates a tight complex (KD < 10−6 m), and so it is possible that RbpA may affect sigma factor stability in cells or promote/prevent interactions with additional ancillary proteins. Lastly, Crl is nonessential, whereas a loss of RbpA leads rapidly to loss of growth and viability of M. tuberculosis (18), although the essential nature of RbpA function has not yet been identified clearly. σB is dispensable for growth in vitro and in infection models (68), so the likely essential function could be related to the activation of transcription driven by essential σA. Our results with the conditional mutant of M. tuberculosis (Fig. 10) support the importance of direct interaction with sigma subunits as part of the essential function of RbpA because truncated RbpA1–79 that is unable to bind sigma subunits is also unable to complement the conditional mutant.
An interesting feature of RbpA is that it is conserved only in actinomycetes. This information, together with its essentiality in M. tuberculosis, makes RbpA a potential anti-tuberculosis drug target (11, 15). In the last few years, significant progress has been made toward characterization of RbpA, and this work contributes a structure and new interaction partners to shed light on the function and mechanism of action.
Acknowledgments
We thank Julie Morrissey for critical reading of the manuscript, Gisbert Schumann for generation of the two-hybrid library, and the Centre for Core Biotechnology Services at the University of Leicester for support with containment level 3 experiments
This work was supported by a College of Medicine, Biological Sciences and Psychology, University of Leicester college studentship (to A. B.), a Research Council UK fellowship (to H. M. O.), Biotechnology and Biological Sciences Research Council Grant BB/H007865/1 (to H. M. O.), UCB support (to L. C. W. and P. W. A.), and NM4TB EC_VI Framework Contract LSHP-CT-2005-018923 (to D. G., F. F., and H. M. O.).

This article contains supplemental Table S1 and Figs. S1–S3.
The atomic coordinates and structure factors (code 2M4V) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TB
- tuberculosis
- RNAP
- RNA polymerase
- PI
- pristinamycin I
- NOESY
- NOE spectroscopy
- AEBSF
- 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride
- TOCSY
- total correlation spectroscopy
- HSQC
- heteronuclear single quantum coherence
- TROSY
- transverse relaxation optimized spectroscopy.
REFERENCES
- 1. Crubézy E., Ludes B., Poveda J. D., Clayton J., Crouau-Roy B., Montagnon D. (1998) Identification of Mycobacterium DNA in an Egyptian Pott's disease of 5,400 years old. C. R. Acad. Sci. III 321, 941–951 [DOI] [PubMed] [Google Scholar]
- 2. World Health Organization (2012) Global Tuberculosis Report 2012 (in IRIS), World Health Organization, Geneva [Google Scholar]
- 3. Manganelli R., Provvedi R., Rodrigue S., Beaucher J., Gaudreau L., Smith I. (2004) Sigma factors and global gene regulation in Mycobacterium tuberculosis. J. Bacteriol. 186, 895–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Himmelreich R., Hilbert H., Plagens H., Pirkl E., Li B. C., Herrmann R. (1996) Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 24, 4420–4449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bentley S. D., Chater K. F., Cerdeño-Tárraga A. M., Challis G. L., Thomson N. R., James K. D., Harris D. E., Quail M. A., Kieser H., Harper D., Bateman A., Brown S., Chandra G., Chen C. W., Collins M., Cronin A., Fraser A., Goble A., Hidalgo J., Hornsby T., Howarth S., Huang C. H., Kieser T., Larke L., Murphy L., Oliver K., O'Neil S., Rabbinowitsch E., Rajandream M. A., Rutherford K., Rutter S., Seeger K., Saunders D., Sharp S., Squares R., Squares S., Taylor K., Warren T., Wietzorrek A., Woodward J., Barrell B. G., Parkhill J., Hopwood D. A. (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147 [DOI] [PubMed] [Google Scholar]
- 6. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 7. Rodrigue S., Provvedi R., Jacques P. E., Gaudreau L., Manganelli R. (2006) The sigma factors of Mycobacterium tuberculosis. FEMS Microbiol Rev 30, 926–941 [DOI] [PubMed] [Google Scholar]
- 8. Browning D. F., Busby S. J. (2004) The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2, 57–65 [DOI] [PubMed] [Google Scholar]
- 9. Haugen S. P., Ross W., Gourse R. L. (2008) Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat. Rev. Microbiol. 6, 507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paget M. S., Molle V., Cohen G., Aharonowitz Y., Buttner M. J. (2001) Defining the disulphide stress response in Streptomyces coelicolor A3(2). Identification of the sigmaR regulon. Mol Microbiol 42, 1007–1020 [DOI] [PubMed] [Google Scholar]
- 11. Hu Y., Morichaud Z., Chen S., Leonetti J. P., Brodolin K. (2012) Mycobacterium tuberculosis RbpA protein is a new type of transcriptional activator that stabilizes the sigma A-containing RNA polymerase holoenzyme. Nucleic Acids Res. 40, 6547–6557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dey A., Verma A. K., Chatterji D. (2010) Role of an RNA polymerase interacting protein, MsRbpA, from Mycobacterium smegmatis in phenotypic tolerance to rifampicin. Microbiology 156, 873–883 [DOI] [PubMed] [Google Scholar]
- 13. Newell K. V., Thomas D. P., Brekasis D., Paget M. S. (2006) The RNA polymerase-binding protein RbpA confers basal levels of rifampicin resistance on Streptomyces coelicolor. Mol. Microbiol. 60, 687–696 [DOI] [PubMed] [Google Scholar]
- 14. Dey A., Verma A. K., Chatterji D. (2011) Molecular insights into the mechanism of phenotypic tolerance to rifampicin conferred on mycobacterial RNA polymerase by MsRbpA. Microbiology 157, 2056–2071 [DOI] [PubMed] [Google Scholar]
- 15. Provvedi R., Boldrin F., Falciani F., Palù G., Manganelli R. (2009) Global transcriptional response to vancomycin in Mycobacterium tuberculosis. Microbiology 155, 1093–1102 [DOI] [PubMed] [Google Scholar]
- 16. Murphy D. J., Brown J. R. (2007) Identification of gene targets against dormant phase Mycobacterium tuberculosis infections. BMC Infect. Dis. 7, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sassetti C. M., Boyd D. H., Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 [DOI] [PubMed] [Google Scholar]
- 18. Forti F., Mauri V., Dehò G., Ghisotti D. (2011) Isolation of conditional expression mutants in Mycobacterium tuberculosis by transposon mutagenesis. Tuberculosis 91, 569–578 [DOI] [PubMed] [Google Scholar]
- 19. Karunairatnam M. C., Spizizen J., Gest H. (1958) Preparation and properties of protoplasts of Rhodospirillum rubrum. Biochim. Biophys. Acta 29, 649–650 [DOI] [PubMed] [Google Scholar]
- 20. Sivashanmugam A., Murray V., Cui C., Zhang Y., Wang J., Li Q. (2009) Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein Sci. 18, 936–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Braunschweiler L., Ernst R. (1983) Coherence transfer by isotropic mixing. Application to proton correlation spectroscopy. J. Magn. Reson. 53, 521–528 [Google Scholar]
- 22. Macura S., Ernst R. R. (1980) Elucidation of cross relaxation in liquids by two-dimensional n.m.r. spectroscopy. Mol. Phys. 41, 95–117 [Google Scholar]
- 23. Bodenhausen G., Ruben D. J. (1980) Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 69, 185–189 [Google Scholar]
- 24. Marion D., Kay L. E., Sparks S. W., Torchia D. A., Bax A. (1989) Three-dimensional heteronuclear NMR of nitrogen-15 labeled proteins. J. Am. Chem. Soc. 111, 1515–1517 [Google Scholar]
- 25. Kay L. E., Xu G. Y., Singer A. U., Muhandiram D. R., Formankay J. D. (1993) A gradient-enhanced HCCH-TOCSY experiment for recording side-chain 1H and 13C correlations in H2O samples of proteins. J. Magn. Reson. 101, 333–337 [Google Scholar]
- 26. Grzesiek S., Bax A. (1993) Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J. Biomol. NMR 3, 185–204 [DOI] [PubMed] [Google Scholar]
- 27. Kay L. E., Ikura M., Tschudin R., Bax A. (1990) Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. J. Magn. Reson. 89, 496–514 [DOI] [PubMed] [Google Scholar]
- 28. Bax A., Ikura M. (1991) An efficient 3D NMR technique for correlating the proton and 15N backbone amide resonances with the α-carbon of the preceding residue in uniformly 15N/13C enriched proteins. J. Biomol. NMR 1, 99–104 [DOI] [PubMed] [Google Scholar]
- 29. Grzesiek S., Bax A. (1992) Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114, 6291–6293 [Google Scholar]
- 30. Grzesiek S., Bax A. (1992) An efficient experiment for sequential backbone assignment of medium-sized isotopically enriched proteins. J. Magn. Reson. 99, 201–207 [Google Scholar]
- 31. Marion D., Driscoll P. C., Kay L. E., Wingfield P. T., Bax A., Gronenborn A. M., Clore G. M. (1989) Overcoming the overlap problem in the assignment of 1H NMR spectra of larger proteins by use of three-dimensional heteronuclear 1H-15N Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy. Application to interleukin 1β. Biochemistry 28, 6150–6156 [DOI] [PubMed] [Google Scholar]
- 32. Muhandiram D. R., Farrow N. A., Xu G. Y., Smallcombe S. H., Kay L. E. (1993) A gradient 13C NOESY-HSQC experiment for recording NOESY spectra of 13C-labeled proteins dissolved in H2O. J. Magn. Reson. Ser. 102, 317–321 [Google Scholar]
- 33. Sklenar V., Piotto M., Leppik R., Saudek V. (1993) Gradient-tailored water suppression for 1H-15N HSQC experiments optimized to retain full sensitivity. J. Magn. Reson. Ser. 102, 241–245 [Google Scholar]
- 34. Herrmann T., Güntert P., Wüthrich K. (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 [DOI] [PubMed] [Google Scholar]
- 35. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+. A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Güntert P., Wüthrich K. (1991) Improved efficiency of protein structure calculations from NMR data using the program DIANA with redundant dihedral angle constraints. J. Biomol. NMR 1, 447–456 [DOI] [PubMed] [Google Scholar]
- 37. Tsui V., Case D. A. (2000) Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 56, 275–291 [DOI] [PubMed] [Google Scholar]
- 38. Case D. A., Cheatham T. E., 3rd, Darden T., Gohlke H., Luo R., Merz K. M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Muskett F. W., Thouta S., Thomson S. J., Bowen A., Stansfeld P. J., Mitcheson J. S. (2011) Mechanistic insight into human ether-a-go-go-related gene (hERG) K+ channel deactivation gating from the solution structure of the EAG domain. J. Biol. Chem. 286, 6184–6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koradi R., Billeter M., Wuthrich K. (1996) MOLMOL. A program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55, 29–32 [DOI] [PubMed] [Google Scholar]
- 41. Doreleijers J. F., Sousa da Silva A. W., Krieger E., Nabuurs S. B., Spronk C. A., Stevens T. J., Vranken W. F., Vriend G., Vuister G. W. (2012) CING. An integrated residue-based structure validation program suite. J. Biomol. NMR 54, 267–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 43. Papadopoulos J. S., Agarwala R. (2007) COBALT. Constraint-based alignment tool for multiple protein sequences. Bioinformatics 23, 1073–1079 [DOI] [PubMed] [Google Scholar]
- 44. Livingstone C. D., Barton G. J. (1993) Protein sequence alignments. A strategy for the hierarchical analysis of residue conservation. Comput. Appl. Biosci. 9, 745–756 [DOI] [PubMed] [Google Scholar]
- 45. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., Barton G. J. (2009) Jalview Version 2. A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Klebe R. J., Harriss J. V., Sharp Z. D., Douglas M. G. (1983) A general method for polyethylene-glycol-induced genetic transformation of bacteria and yeast. Gene 25, 333–341 [DOI] [PubMed] [Google Scholar]
- 47. Teutschbein J., Schumann G., Möllmann U., Grabley S., Cole S. T., Munder T. (2009) A protein linkage map of the ESAT-6 secretion system 1 (ESX-1) of Mycobacterium tuberculosis. Microbiol. Res. 164, 253–259 [DOI] [PubMed] [Google Scholar]
- 48. Mihatsch K., Nestler M., Saluz H. P., Henke A., Munder T. (2009) Proapoptotic protein Siva binds to the muscle protein telethonin in cardiomyocytes during coxsackieviral infection. Cardiovasc. Res. 81, 108–115 [DOI] [PubMed] [Google Scholar]
- 49. Tafelmeyer P., Johnsson N., Johnsson K. (2004) Transforming a (β/α)8-barrel enzyme into a split-protein sensor through directed evolution. Chem. Biol. 11, 681–689 [DOI] [PubMed] [Google Scholar]
- 50. O'Hare H., Juillerat A., Dianisková P., Johnsson K. (2008) A split-protein sensor for studying protein-protein interaction in mycobacteria. J. Microbiol. Methods 73, 79–84 [DOI] [PubMed] [Google Scholar]
- 51. Farmer B. T., 2nd, Constantine K. L., Goldfarb V., Friedrichs M. S., Wittekind M., Yanchunas J., Jr., Robertson J. G., Mueller L. (1996) Localizing the NADP+ binding site on the MurB enzyme by NMR. Nat. Struct. Biol. 3, 995–997 [DOI] [PubMed] [Google Scholar]
- 52. Williamson R. A., Carr M. D., Frenkiel T. A., Feeney J., Freedman R. B. (1997) Mapping the binding site for matrix metalloproteinase on the N-terminal domain of the tissue inhibitor of metalloproteinases-2 by NMR chemical shift perturbation. Biochemistry 36, 13882–13889 [DOI] [PubMed] [Google Scholar]
- 53. Muskett F. W., Frenkiel T. A., Feeney J., Freedman R. B., Carr M. D., Williamson R. A. (1998) High resolution structure of the N-terminal domain of tissue inhibitor of metalloproteinases-2 and characterization of its interaction site with matrix metalloproteinase-3. J. Biol. Chem. 273, 21736–21743 [DOI] [PubMed] [Google Scholar]
- 54. Stover C. K., de la Cruz V. F., Fuerst T. R., Burlein J. E., Benson L. A., Bennett L. T., Bansal G. P., Young J. F., Lee M. H., Hatfull G. F. (1991) New use of BCG for recombinant vaccines. Nature 351, 456–460 [DOI] [PubMed] [Google Scholar]
- 55. Holm L., Rosenström P. (2010) Dali server. Conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lonetto M., Gribskov M., Gross C. A. (1992) The sigma 70 family. Sequence conservation and evolutionary relationships. J. Bacteriol. 174, 3843–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vassylyev D. G., Sekine S., Laptenko O., Lee J., Vassylyeva M. N., Borukhov S., Yokoyama S. (2002) Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature 417, 712–719 [DOI] [PubMed] [Google Scholar]
- 58. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the Web. A case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 59. Shepherd N., Dennis P., Bremer H. (2001) Cytoplasmic RNA polymerase in Escherichia coli. J. Bacteriol. 183, 2527–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gilmore J. M., Bieber Urbauer R. J., Minakhin L., Akoyev V., Zolkiewski M., Severinov K., Urbauer J. L. (2010) Determinants of affinity and activity of the anti-sigma factor AsiA. Biochemistry 49, 6143–6154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Waters L. C., Veverka V., Böhm M., Schmedt T., Choong P. T., Muskett F. W., Klempnauer K. H., Carr M. D. (2007) Structure of the C-terminal MA-3 domain of the tumour suppressor protein Pdcd4 and characterization of its interaction with eIF4A. Oncogene 26, 4941–4950 [DOI] [PubMed] [Google Scholar]
- 62. Veverka V., Henry A. J., Slocombe P. M., Ventom A., Mulloy B., Muskett F. W., Muzylak M., Greenslade K., Moore A., Zhang L., Gong J., Qian X., Paszty C., Taylor R. J., Robinson M. K., Carr M. D. (2009) Characterization of the structural features and interactions of sclerostin. Molecular insight into a key regulator of Wnt-mediated bone formation. J. Biol. Chem. 284, 10890–10900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Williamson R. A., Muskett F. W., Howard M. J., Freedman R. B., Carr M. D. (1999) The effect of matrix metalloproteinase complex formation on the conformational mobility of tissue inhibitor of metalloproteinases-2 (TIMP-2). J. Biol. Chem. 274, 37226–37232 [DOI] [PubMed] [Google Scholar]
- 64. Gao G., Semenchenko V., Arumugam S., Van Doren S. R. (2000) Tissue inhibitor of metalloproteinases-1 undergoes microsecond to millisecond motions at sites of matrix metalloproteinase-induced fit. J. Mol. Biol. 301, 537–552 [DOI] [PubMed] [Google Scholar]
- 65. England P., Westblade L. F., Karimova G., Robbe-Saule V., Norel F., Kolb A. (2008) Binding of the unorthodox transcription activator, Crl, to the components of the transcription machinery. J. Biol. Chem. 283, 33455–33464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Typas A., Barembruch C., Possling A., Hengge R. (2007) Stationary phase reorganisation of the Escherichia coli transcription machinery by Crl protein, a fine-tuner of sigmas activity and levels. EMBO J. 26, 1569–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bougdour A., Lelong C., Geiselmann J. (2004) Crl, a low temperature-induced protein in Escherichia coli that binds directly to the stationary phase sigma subunit of RNA polymerase. J. Biol. Chem. 279, 19540–19550 [DOI] [PubMed] [Google Scholar]
- 68. Fontán P. A., Voskuil M. I., Gomez M., Tan D., Pardini M., Manganelli R., Fattorini L., Schoolnik G. K., Smith I. (2009) The Mycobacterium tuberculosis sigma factor σB is required for full response to cell envelope stress and hypoxia in vitro, but it is dispensable for in vivo growth. J. Bacteriol. 191, 5628–5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cole C., Barber J. D., Barton G. J. (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]