

Background: Development of liver cancer involves alterations of multiple pathways of gene expression.
Results: Translational activation of C/EBPβ-HDAC1 complexes represses p53, SIRT1, and PGC1α, leading to liver cancer.
Conclusion: Modulation of levels of C/EBPβ-HDAC1 complexes at different stages of cancer is involved in liver cancer.
Significance: Understanding the mechanisms of liver cancer is a critical step for the development of therapeutic approaches for cancer.
Keywords: Cancer, Cancer Tumor Promoter, Cyclin-dependent Kinase (CDK), Cell Proliferation, Chromatin Immunoprecipitation (ChIP), C/EBP, Cancer, Epigenetic, Liver, Translation
Abstract
Cancer changes biological processes in the liver by altering gene expression at the levels of transcription, translation, and protein modification. The RNA binding protein CUGBP1 is a key regulator of translation of CCAAT enhancer binding protein β and histone deacetylase 1 (HDAC1). These proteins form complexes that are involved in the regulation of liver biology. Here we show a critical role of the translational activation of CCAAT/enhancer binding protein β-HDAC1 complexes in the development of liver cancer mediated by diethylnitrosamine. We found that diethylnitrosamine increases the levels of CUGBP1 and activates CUGBP1 by phosphorylation, leading to the formation of the CUGBP1-eIF2 complex, which is an activator of translation of CUGBP1-dependent mRNAs. The elevation of the CUGBP1-eIF2 complex increases translation of C/EBPβ and HDAC1, resulting in an increase of C/EBPβ-HDAC1 complexes at later stages of liver cancer. We found that C/EBPβ-HDAC1 complexes repress promoters of three key regulators of liver functions: p53, SIRT1, and PGC1α. As the result of this suppression, the p53-, SIRT1-, and PGC1α-dependent downstream pathways are reduced, leading to increased liver proliferation. We also found that the proper regulation of C/EBPβ-HDAC1 complexes is required for the maintenance of biological levels of p53, SIRT1, and PGC1α in quiescent livers and at early stages of liver cancer. Taken together, these studies showed that the development of liver cancer includes a tight regulation of levels of C/EBPβ-HDAC1 complexes on the levels of transcription, translation, and posttranslational modifications.
Introduction
Epigenetic control is involved in the regulation of many liver functions, including protection of the liver from cancer development (1, 2). Modifications of histones are one of the major pathways in the epigenetic control of gene expression. The main modifications include methylation, acetylation, and phosphorylation (3). The acetylation of histones is recognized as one of the critical steps of epigenetic control of gene expression. A family of histone deacetylases (HDACs)2 includes several members, which are expressed in the liver. Among these members, HDAC1 has been shown to be involved in the control of liver proliferation after partial hepatectomy because the inhibition of HDAC1 activity by siRNA leads to impaired liver regeneration (4). Several recent studies suggest that HDAC1 is involved in the development of liver cancer. Wang et al. (5) showed that HDAC1 up-regulates microRNA-244 in human hepatocellular carcinoma, which is involved in the development of liver cancer. In agreement with these findings, Buurman et al. (6) showed that HDAC1 activates hepatocyte growth hormone signaling by repression of microRNA 449 in human hepatocellular carcinoma cells. It has also been shown that inactivation of HDAC1 in human hepatocellular carcinoma leads to activation of p21 and p27 and subsequent suppression of proliferation (7).
Although HDAC1 deacetylates histones on DNA, it does not bind to DNA directly and needs to be delivered to certain regions of the chromatin by transcription factors. Data from our laboratory and from other groups showed that two members of the C/EBP family of transcription factors, C/EBPα and C/EBPβ, interact with HDAC1 and that these interactions are involved in the regulation of gene expression in several biological situations (8). C/EBPα interacts with HDAC1 in livers of old mice, leading to the formation of a complex that also contains E2F4 and the chromatin remodeling protein Brm (9, 10). Generation of C/EBPα-S193D knockin mice, which express age-like isoforms of C/EBPα, confirmed that the C/EBPα-HDAC1 complexes are involved in development of aging phenotypes in liver (11). It has also been shown that the HDAC1 forms complexes with another member of C/EBP family, C/EBPβ, and that these complexes are increased in livers of old mice, leading to repression of certain promoters such as those of the SIRT1 and GSK3β genes (12, 13). Although the C/EBPβ-HDAC1 complexes are less abundant in the livers of young mice, these complexes are involved in a proper regulation of transcription during liver regeneration after partial hepatectomy (4). We have shown previously that expression of C/EBPβ and HDAC1 and the formation of C/EBPβ-HDAC1 complexes are regulated by RNA binding protein CUGBP1 on the level of translation. CUGBP1 binds to the 5′ regions of the C/EBPβ and HDAC1 mRNAs and increases translation of these proteins (4, 14). In this work we present data showing that the CUGBP1-mediated elevation of C/EBPβ-HDAC1 complexes at the late stages of liver cancer eliminates p53-dependent inhibition of liver proliferation and reduces the levels of SIRT1 and PGC1α.
EXPERIMENTAL PROCEDURES
Experiments with Mice and DEN-mediated Cancer
All experiments using mice were approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine (protocol AN-1439). Liver tumors were induced in mice by the diethylnitrosamine (DEN) tumor liver induction protocol as described (15, 16). 5 μg or 25 μg of DEN/g body weight was injected into mice. Two sets of experiments were performed. We have found previously that the higher dose of DEN (25 μg/g body) causes a stronger inhibition of C/EBPβ and C/EBPβ-HDAC1 complexes (16). Therefore, one set examined the expression of proteins at very early time points after 25 μg of DEN/g body weight injection. For these studies, animals were sacrificed at 24, 48, and 72 h. We also sacrificed mice at 6, 10, 20, 25, 30, and 35 weeks after injection of 5 μg of DEN/g body weight. Livers were harvested and analyzed. Eight animals were used per time point after DEN injection.
BrdU Uptake
DNA replication was examined by BrdU uptake. BrdU was injected 2 h before the animals were sacrificed. Livers were harvested and stained with antibodies to BrdU as described previously (15, 16).
Identification of Consensuses for FXR and C/EBP and Construction of Plasmids for the Reporter Luciferase Assay
We screened the 5′ upstream regions of the mouse C/EBPβ, p53, SIRT1, and PGC1α genes for potential FXR (FXREs) and C/EBP sites using a matrix-based computational approach (NUBIScan). One DR6 (direct repeat 6) element (GGGCCAgggctgGGGTCA) located at −1014 relative to the transcription start site and one DR9 element (GGGTCAatgggtcggGGGTCA) at −1002 were identified. Interestingly, these two elements shared one hexanucleotide repeat: GGGTCA. Subsequently, a 1608-bp fragment (from nucleotides −1501 to +107) containing these two putative FXREs were amplified and cloned onto the pGL 4.10 luciferase reporter vector. The sequences of the primers were 5′-CGTTAGGTACCCAGGTGAGATGGGCTTTCAGGTCAGG-3′ (sense) and 5′-TATTATCTCGAGGAACGCGGGGCCCGCGGGCT-3′ (antisense). Two mutants with point mutations in these two elements were constructed using a site-directed mutagenesis approach. The sequences of the primers used to construct mutant 1 were 5′-CGGGGCCAGGGCTGGTTTCAATGTATCGGGAATCAGCCCCTGACAGG-3′ (sense) and 5′-CCTGTCAGGGGCTGATTCCCGATACATTGAAACCAGCCCTGGCCCCG-3′ (antisense). The sequences of the primers used to construct mutant 2 were 5′-GCCGGGGCCAGGGCTGATTACAATGGGTCGGGGGTC-3′ (sense) and 5′-GACCCCCGACCCATTGTAATCAGCCCTGGCCCCGGC-3′ (antisense).
Regarding C/EBP sites within the p53 and PGC1α promoters, the identified consensuses are shown in Figs. 4A and 5A. The C/EBP binding sites within the SIRT1 promoter were identified and characterized in our previous work (13).
FIGURE 4.

The C/EBPβ-HDAC1 complex represses the p53 promoter. A, location of C/EBP sites within the p53 promoter and diagram of the mutant constructs. luc, luciferase. B, EMSA with the probe covering C/EBP binding site 1 within the p53 promoter. The probe was incubated with nuclear extracts (NE) from livers of DEN-treated mice. Antibodies (shown at the top) were added to the binding reactions. Note that Abs to C/EBPβ neutralize the binding. SS, supershift; free, free probe; NS, nonspecific band. C, mutation of C/EBP sites reduces the basal activity of the p53 promoter and the ability of C/EBPβ to activate the p53 promoter. The luciferase constructs (shown in A) were cotransfected with C/EBPβ into Hep3B2 cells, and the luciferase activity was calculated. V, vector; A–E, constructs shown in A. *, p < 0.05. D, the C/EBPβ-HDAC1 complex inhibits the p53 promoter. The luciferase construct containing the 1.6-kb p53 promoter was cotransfected with different amounts of C/EBPβ and HDAC1. Luciferase activity was determined (bar graphs). The top panel shows levels of C/EBPβ in experimental cells determined by Western blotting. E, knockdown of endogenous HDAC1 activates the p53 promoter and increases the ability of C/EBPβ to activate the p53 promoter. Endogenous HDAC1 was inhibited by siRNA in cells transfected with the p53-luc constructs and in cells transfected with the p53-luc construct and C/EBPβ. The bottom panel shows levels of HDAC1 after inhibition by siRNA. F, knockdown of endogenous C/EBPβ reduces the ability of HDAC1 to repress the p53 promoter. The experiments were performed as described above. The bottom panel shows inhibition of C/EBPβ by siRNA. G, C/EBPβ-HDAC1 complexes repress the p53 promoter in DEN-treated mice. A ChIP assay was performed with chromatin solutions from control (DEN 0) and DEN-treated mice (DEN 20 and DEN 35). Ag, negative control; AcK9 and MeK9 immunoprecipitations with Abs to histone H3 acetylated at Lys-9 and trimethylated at Lys-9, respectively. H, diagram summarizing the effects of CUGBP1 and the C/EBPβ-HDAC1 complex on the p53 promoter.
FIGURE 5.
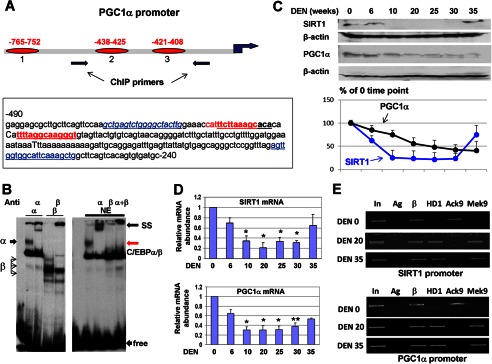
The C/EBPβ-HDAC1 complex down-regulates SIRT1 and PGC1α at later stages of liver cancer. A, top panel, location of three C/EBP sites within the PGC1α promoter. The bottom panel shows the nucleotide sequence of the PGC1α promoter and the C/EBP consensus sites. B, EMSA with the probe covering PGC1α site 3. The left panel shows the EMSA with overexpressed C/EBPα and C/EBPβ. The right panel shows the EMSA with liver nuclear extracts (NE). Positions of C/EBPα/β complexes, supershift (SS), and free probe are shown. C, the protein levels of SIRT1 and PGC1α are reduced at later stages of liver cancer. Western blotting was performed with the antibodies shown at the left. The bottom panel shows levels of SIRT1 and PGC1α as ratios to β-actin. D, levels SIRT1 and PGC1α mRNA are reduced at later stages of liver cancer. Quantitative RT-PCR was performed. Bar graphs show the results of analyses of three to four animals of each group. *, p < 0.05; **, p < 0.01, DEN treatment group versus control group. E, the SIRT1 and PGC1α promoters are repressed by C/EBPβ-HDAC1 complexes at later stages of liver cancer. A ChIP assay was performed with chromatin solutions as described in the legend to Fig. 4G. In, input; Ag, agarose beads; β, C/EBPβ.
EMSA Assay
Conditions for the EMSA assay were described in our papers (12, 13). For EMSA with C/EBPβ promoters, two 32P-labeled oligomers, b9 and b10, were used as the probes for the FXR binding analysis. The sequences of oligomer b9 were 5′AGGTGCCGGGGCCAGGGCTGGGGTCAATGG3′ (top strand) and 5′TGGCCATTGACCCCAGCCCTGGCCCCGGCA3′ (bottom strand), containing a putative FXRE (DR6 element, 5′GGGCCAgggctgGGGTCA). The sequences of b10 probe were 5′AGGCTGGGGTCAATGGGTCGGGGGTCAGCCT3′ (top strand) and 5′TGGAGGCTGACCCCCGACCCATTGACCCCAG3′ (bottom strand), containing another putative FXRE (DR9 element, 5′GGGTCAatgggtcggGGGTCA3′). A polyclonal anti-FXR antibody (catalog no. sc-1204X, Santa Cruz Biotechnology, Inc.) was used for the supershift assay to confirm binding specificity.
EMSA with C/EBP sites within the p53 and PGC1α promoters were performed with the probes shown in Figs. 4 and 5. For these experiments we used nuclear extracts from mouse livers and C/EBPα and C/EBPβ overexpressed in Hep3B2 cells. Antibodies to C/EBPα and C/EBPβ were added to the binding reactions. DNA-protein complexes were separated by non-denaturing 6% polyacrylamide gel electrophoresis in 0.5× Tris borate EDTA buffer. After electrophoresis, the gel was dried and exposed to x-ray film.
RT-PCR
Total RNA from livers was extracted with an RNeasy mini kit (Qiagen, Germantown, MD) according to the instructions of the manufacturer. cDNA was synthesized using SuperScript III first-strand (Invitrogen) and random primer hexamers. cDNA was subsequently amplified using a GoTaq Green master kit (Promega) or Brilliant II SYBR Green quantitative PCR master mix kit (Agilent Technologies). The sequences of PCR primers were as follows: mouse p53, 5′-TCCTCCCCTCAATAAGCTATTCTG-3′ (forward) and 5′-TGGCGCTGACCCACAAC-3′ (reverse); mouse p21, 5′-TCTGAGCGGCCTGAAGATTC-3′ (forward) and 5′-TTCAGGGTTTTCTCTTGCAGAAG-3′ (reverse); mouse SIRT1, 5′-ACTGCAGAAACTTTTAGCCTTTCAA-3′ (forward) and 5′- GGCAATGTTCCAAAGAAGTCTGT-3′ (reverse); mouse PGC1α, 5′-AACCACACCCACAGGATCAGA-3′ (forward) and 5′-TCTTCGCTTTATTGCTCCATGA-3′ (reverse); and mouse β-actin, 5′-AAGTGCTTCTAGGCGGACTGTT-3′ (forward) and 5′- TTTTCTGCGCAAGTTAGGTTTTG-3′ (reverse).
Chromatin Immunoprecipitation Assay
The ChIP assay was performed as described in our previous articles (10, 13) using the Chip-It kit. The sequences of primers used in the work were as follows: mouse p53, 5′-TTAAACACGGTGGTGCGATA-3′ (forward) and 5′-GTTGAGGGCAAGAAATGGAG-3′ (reverse); mouse PGC1α, 5′-GCTGAGTCTGGGGCTACTTG-3′ (forward) and 5′-AGCTTTGAATGCCACCAACT-3′ (reverse); mouse SIRT1, 5′-TCCTGGCCCTGTCATTTTAG-3′ (forward) and 5′-GACCCACCAGGTCCTCTGTA-3′ (reverse); and mouse C/EBPβ, 5′-AAGGGCACAGGGAGATGTCATT-3′ (forward) and 5′-TGCTCAACCTTCGGTGTTATCTGC-3′ (reverse). The final volume of PCRs was 50 μl. We usually loaded 5 μl of the PCR reaction on the agarose or polyacrylamide gel.
Antibodies and Reagents
Antibodies to FXR (Cys-20 and His-130), C/EBPβ (Cys-19), C/EBPα (144A), Cdk4, cyclin D3, elF2, calreticulin, CUGBP1, p53, and HDAC1 (H-51) were obtained from Santa Cruz Biotechnology, Inc. Antibodies to PGC1α, p53, and p21 were purchased from Abcam. Monoclonal anti-β-actin antibody was from Sigma. The BrdU uptake assay kit was from Invitrogen. Coimmunoprecipitation studies were performed using TrueBlot reagents as described in our previous papers (13, 15).
Cotransfection Studies
C/EBPβ and p53 promoter constructs were cotransfected with an FXR-expressing plasmid or with C/EBPβ- and HDAC1-expressing plasmids into Hep3B2 cells as described (13, 15, 16). For the studies of the C/EBPβ promoter, 24 h after transfection, the cells were treated with the FXR ligands CDCA or GW 4064. For the studies of the p53 promoter, endogenous C/EBPβ and HDAC1 were inhibited by siRNA as described in our previous papers (4, 13). The luciferase assay was performed using the dual luciferase reporter assay system (Promega). Luciferase activity was normalized by the Renilla internal control.
Protein Isolation and Western Blotting
Cytoplasmic and nuclear extracts were isolated from livers, and Western blotting was performed as described previously (13, 15). The Western blotting images show representative pictures for each experiment. The summaries (bar graphs) of the Western blotting data represent the results of studies with three to four mice/time point after DEN injections.
Statistical Analysis
Data are expressed as the mean ± S.D. unless specified. Overall significant differences involving multiple groups were determined by analysis of variance. Two-group statistical comparisons were determined by Student's t tests, and differences were considered significant at p < 0.05 and p < 0.01.
RESULTS
Translational Activity of CUGBP1 Is Increased during DEN-mediated Carcinogenesis
Our previous studies of samples from patients with liver cancer showed that CUGBP1 is activated in these patients and that this activation correlates with the elevation of C/EBPβ-HDAC1 complexes (4). Because the studies of human liver samples are limited because of the small size of the available tissues and because they provide only correlative observations, we have initiated examination of the role of CUGBP1 in cancer using mouse models of liver cancer. It was also important to determine targets of C/EBPβ-HDAC1 complexes at different stages of liver cancer. Therefore, we applied a DEN protocol to initiate liver cancer as described in our previous papers (15, 16) and examined the CUGBP1 pathway and its targets during development of liver cancer. Initial experiments were performed with mice sacrificed at 6, 10, 20, 25, 30, and 35 weeks after DEN injection. Under these conditions, mice develop liver cancer at 33–35 weeks. Fig. 1A shows a typical picture of liver cancer observed in our experimental animals. More details for these DEN-induced liver cancers are described in our previous papers (15, 16). Because translational activity of CUGBP1 is activated by cyclin D3-Cdk4, leading to the increased interactions of CUGBP1 with the eukaryotic translation initiation factor α (eIF2α), we first examined expression of cyclin D3, Cdk4, eIF2α, and CUGBP1 in the cytoplasm of the liver cells at different time points after DEN injection. We found that cyclin D3 and Cdk4 are increased in cytoplasmic extracts of livers 6 weeks after DEN treatments, whereas CUGBP1 is elevated at 20 weeks (Fig. 1B). Co-IP studies showed that the interactions of Cdk4 with CUGBP1 are increased starting at 6 weeks after DEN injections (Fig. 1C). We next determined the phosphorylation status of CUGBP1 using a two-dimensional Western approach and found that a portion of CUGBP1 is hyperphosphorylated in DEN-treated livers to a much higher degree than in control livers (Fig. 1D). It has been shown that hyperphosphorylation of CUGBP1 increases formation of the CUGBP1-eIF2α complex which, in turn, binds to and activates translation of certain mRNAs (8, 9). To determine the amounts of the CUGBP1-eIF2α complexes, we used two approaches: co-IP and HPLC-based size exclusion chromatography. Co-IP studies showed that amounts of the CUGBP1-eIF2α complexes are increased at 6–25 weeks after DEN injection (Fig. 1E). Examination of CUGBP1-eIF2 complexes by size exclusion chromatography showed that cytoplasmic extracts of the liver from DEN-treated mice contain the CUGBP1-eIF2 complexes that are located in the regions of very high molecular weight complexes (Fig. 1F) and perhaps contain additional proteins. Thus, these studies show that DEN-mediated liver cancer activated cyclin D3-Cdk4 in cytoplasm, leading to phosphorylation of CUGBP1 and to subsequent elevation of the CUGBP1-eIF2 complexes (Fig. 1G).
FIGURE 1.

Activation of translational functions of CUGBP1 during development of liver cancer after DEN injections. A, upper panel, a typical picture of liver cancer at 35 weeks after DEN injection. The arrows show large tumor nodules. B, cyclin D3, Cdk4, and CUGBP1 are elevated in the cytoplasm after DEN injection. Cytoplasmic extracts were isolated from livers at different time points after DEN injection (weeks (wks) shown on top) and examined by Western blotting with the Abs shown on the right. Calreticulin (CRT) served as a loading control for cytoplasmic proteins. C, association of Cdk4 with CUGBP1 is increased in DEN-treated mice. CUGBP1 was immunoprecipitated (IP), and the immunoprecipitates were probed with Abs to Cdk4. Positions of Cdk4 and heavy and light chains of IgGs are shown. D, CUGBP1 is hyperphosphorylated in DEN-treated mice. Cytoplasmic extracts from control livers (0 time point) and from livers at 20 weeks after DEN injection were separated by two-dimensional technique and probed with Abs to CUGBP1. Additional phosphorylated isoforms of CUGBP1 are indicated by red arrows. E, amounts of CUGBP1-eIF2 complexes are increased in livers of DEN-injected mice. The CUGBP1 immunoprecipitates (filter in Fig. 1C) were reprobed with Abs to eIF2α. F, examination of CUGBP1-eIF2 complexes by size exclusion chromatography. Cytoplasmic proteins from control mice and from livers of mice 20 weeks after DEN treatments were separated on a SEC400 column, and the fractions were analyzed by Western blotting with Abs to CUGBP1, cyclin D3, Cdk4, and eIF2α. CUGBP1-IP, CUGBP1 was immunoprecipitated from each fraction, and the immunoprecipitates were probed with Abs to eIF2α. Positions of eIF2α and heavy chains of IgG are indicated by arrows. G, diagram summarizing DEN induction of CUGBP1-eIF2 complexes and their association with Cdk4/cyclin D3 up-regulation.
Amounts of HDAC1, C/EBPβ, and C/EBPβ-HDAC1 Complexes Are Increased in DEN-treated Mice
Previous studies showed that the CUGBP1-eIF2 complex binds to the 5′ regions of C/EBPβ and HDAC1 mRNAs and activates translation of the corresponding proteins (4, 14). Given the elevation of the CUGBP1-eIF2 complex in DEN-treated mice, we examined whether these downstream targets of the complex are also elevated. Western blotting analyses of nuclear extracts showed that C/EBPβ and HDAC1 are increased up to 3- to 4-fold, starting at 10 weeks after DEN injections, and then stay at high levels (Fig. 2A). Examination of mRNA levels using quantitative RT-PCR showed that C/EBPβ and HDAC1 mRNAs are not changed significantly (Fig. 2B). This suggests that the elevation of the HDAC1 and C/EBPβ proteins is mediated by translational mechanisms. We next applied a co-IP approach and found that amounts of the C/EBPβ-HDAC1 complexes are elevated in nuclei of DEN-treated mice, with maximum levels between 6 and 25 weeks after DEN injection (Fig. 2C). In the course of these studies, we found that amounts of the C/EBPβ-HDAC1 complex are increased 6 weeks after DEN injections. However, protein levels of C/EBPβ and HDAC1 are increased at later time points. Searching for possible mechanisms of activation of these complexes at 6 weeks, we found that migration of HDAC1 in the gel electrophoresis is different in samples isolated from 6- and 10-week DEN-treated mice (Fig. 2A, red arrow). This suggested that HDAC1 might undergo posttranslational modifications and that these modifications might accelerate formation of the C/EBPβ-HDAC1 complex. Therefore, we examined HDAC1 isoforms in control livers and in livers of mice treated with DEN for 6 weeks using the two-dimensional Western approach. We found that DEN-treated mice contained two additional isoforms of HDAC1 located in the acidic region of the two-dimensional gel (Fig. 2D). These new isoforms are likely to be phosphorylated forms because treatments of protein extracts with alkaline phosphatase (CIP) eliminated these additional forms (Fig. 2D). We noted that several other isoforms of HDAC1 were moved to the alkaline region after CIP treatments. To determine whether this phosphorylation of HDAC1 is involved in the formation of the C/EBPβ-HDAC1 complexes, we examined these complexes after CIP treatment of the nuclear extracts. As can be seen in Fig. 2E, the elimination of phosphorylated forms of HDAC1 significantly reduced C/EBPβ-HDAC1 complexes 6 and 10 weeks after DEN injections. On the basis of these data, we suggest that the phosphorylation of HDAC1 6–10 weeks after DEN injection might serve as a triggering signal for the formation of C/EBPβ-HDAC1 complexes.
FIGURE 2.

Accumulation of complexes of C/EBPβ and HDAC1 in DEN-treated mice. A, levels of C/EBPβ, HDAC1, and C/EBPβ-HDAC1 complexes are increased in DEN-treated mice. Western blotting was performed with nuclear extracts isolated at different time points after DEN injection. Wks, weeks. B, levels of C/EBPβ and HDAC1 mRNAs are not changed during development of liver cancer. Quantitative RT-PCR was performed with RNA isolated from livers at different time points after DEN injection. C, C/EBPβ-HDAC1 complexes are elevated 6–25 weeks after DEN injection. HDAC1 was immunoprecipitated from nuclear extracts, and C/EBPβ was examined after immunoblotting of these immunoprecipitates. D, examination of HDAC1 by two-dimensional gel electrophoresis. Nuclear extracts from control livers (Con) and livers from DEN-treated mice were separated by a two-dimensional technique and probes with Abs to HDAC1. DEN + CIP, proteins were treated with alkaline phosphatase prior to two-dimensional separation. E, dephosphorylation of HDAC1 reduces formation of C/EBPβ-HDAC1 complexes. Protein extracts from 6 and 10 weeks of DEN-treated mice were incubated with CIP and used for co-IP and Western blot analyses. C/EBPβ was immunoprecipitated, and HDAC1 was examined in these immunoprecipitates. Data for two animals for each time point are shown.
The p53-p21 and p53-E2F7 Pathways of Inhibition of Liver Proliferation Are Reduced in DEN-treated Mice
Previous reports showed that C/EBPβ binds to and regulates the p53 promoter (17, 18). Given the elevation of C/EBPβ-HDAC1 complexes during development of liver cancer, we hypothesized that these complexes might be involved in the repression of p53 and subsequent development of cancer. To test this hypothesis, we initially examined the levels of p53 mRNA and protein. We found that both p53 mRNA and protein are dramatically reduced in DEN-treated mice, with the maximum reduction 6–25 weeks after DEN injection (Fig. 3A). The reduction of p53 correlates with the elevation of C/EBPβ-HDAC1 complexes. Interestingly, the levels of p53 mRNA are increased slightly in the end of DEN-mediated carcinogenesis but are still below the original levels.
FIGURE 3.

The p53-p21 and p57-E2F7 pathways are reduced in DEN-treated mice. A, protein and RNA levels of p53 are reduced in DEN-treated mice. Western blotting was performed with nuclear extracts from livers at different time points after DEN injections. The filter was reprobed with Abs to β-actin, and the levels of p53 were calculated as ratios to β-actin and then as percentages of the 0 time point (center panel). The bottom panel shows quantitative RT-PCR with mRNA isolated from livers at different time points after DEN treatments. Bar graphs show the results of analyses of three animals of each group. *, p < 0.05; **, p < 0.01; DEN treatment group versus control group. B, protein and RNA levels of p21 and E2F7 are reduced in DEN-treated mice. The experiment was performed as described above for p53. Bar graphs show the results of analyses of three animals of each group. *, p < 0.05, DEN treatment group versus control group; #, p < 0.05, DEN treatment group versus control group. C, liver proliferation is increased in DEN-treated mice. BrdU uptake was performed with livers isolated from mice at 20, 25, 30, and 35 weeks after DEN injections. The top panels show typical pictures of BrdU staining. The bottom panel shows percentages of BrdU-positive hepatocytes as an average of data with five animals for each time point.
To examine the consequences of down-regulation of p53, we have determined the expression of two targets of p53, p21 and E2F7. These two targets were selected for the studies because they control a major drive of cell cycle: E2F-dependent activation of the cell cycle. E2F7 is a member of E2F family, which does not heterodimerize with a DP partner and does not have an Rb-binding domain (19). Therefore, it operates as a dominant negative molecule by binding to DNA and inhibiting activities of other members of E2F family, leading to inhibition of cell proliferation. It has been shown recently that p53 up-regulates transcription of E2F7 and that this activation contributes to p53-dependent growth arrest (20). Quantitative RT-PCR and Western blotting showed that both mRNA and protein levels of p21 and E2F7 are reduced at 10 and 30 weeks after DEN treatments (Fig. 3B). This reduction of p21 and E2F7 highly correlated with that of the activation of CUGBP1 (Fig. 1), with elevation of CUGBP1-eIF2 complexes and with following up-regulation of C/EBPβ-HDAC1 complexes. We next asked whether the reduction of the p53-p21 and p53-E2F7 pathways might lead to the increased proliferation. Examination of BrdU uptake showed that DNA replication is increased in livers of DEN-injected mice starting from 20 weeks (Fig. 3C), which correlated with the down-regulation of the p53-p21 pathway. Thus, these studies demonstrated that the p53-p21 pathway of inhibition of liver cancer is dramatically reduced during development of liver cancer mediated by DEN. We suggest that this inhibition is caused by CUGBP1-mediated activation of C/EBPβ-HDAC1 complexes.
Elevation of the CUGBP1-C/EBPβ-HDAC1 Pathway in DEN-treated Mice Represses the p53 Promoter
Given the correlation between elevation of C/EBPβ-HDAC1 complexes and the reduction of p53 mRNA, we examined the hypothesis that these complexes repress the p53 promoter during development of liver cancer. Examination of the p53 promoter found two strong consensuses for C/EBP proteins (Fig. 4A). EMSA analysis of liver nuclear extracts has shown that C/EBPα and C/EBPβ bind to the consensus sites (Fig. 4B, data for site 2 are not shown). It appears that C/EBP proteins bind to the p53 promoter as heterodimers because antibodies to each individual protein supershifted/neutralized all specific complexes. To examine whether these C/EBP sites are involved in the regulation of the p53 promoter, we generated a WT p53-promoter luciferase reporter construct and constructs that contained mutations in a single site and in both sites (Fig. 4A). These constructs were transfected in Hep3B2 cells alone or cotransfected with C/EBPβ. Examination of the basal activity of these constructs showed that the C/EBP sites are positive regulators of the p53 promoter and that mutation of each site reduces the basal activity of the promoter (Fig. 4C). Examination of the effects of C/EBPβ on the p53 promoter showed that C/EBPβ alone activates the WT p53 promoter and, to a lesser degree, promoters with a single mutation. However, C/EBPβ is not able to activate the p53 promoter, which contains mutations in both C/EBP sites (Fig. 4C).
We next examined whether C/EBPβ-HDAC1 complexes might down-regulate the p53 promoter using three approaches. First, we cotransfected the WT p53 promoter with C/EBPβ and C/EBPβ+HDAC1 and examined its activity. We found that C/EBPβ alone activates the promoter but that the simultaneous transfection of HDAC1 and C/EBPβ blocks C/EBPβ-dependent activation of the promoter (Fig. 4D). Second, we used siRNA-mediated down-regulation of endogenous HDAC1 and C/EBPβ. We found that the inhibition of endogenous HDAC1 activated the p53 promoter and increased the ability of C/EBPβ to activate the promoter (Fig. 4E). In agreement with these findings, the inhibition of endogenous C/EBPβ abolished the ability of HDAC1 to repress the p53 promoter (Fig. 4F). Third, we have applied a ChIP assay using chromatin solutions from livers of mice at 0, 20, and 35 weeks after DEN injection. The ChIP assay showed that C/EBPβ-HDAC1 complexes occupy and repress the p53 promoter in DEN-treated mice because histone H3 is trimethylated at Lys-9 (Fig. 4G). Taken together, the studies of the p53 promoter revealed that C/EBPβ-HDAC1 complexes repress the promoter in cultured cells and in livers of mice treated with DEN (Fig. 4H).
C/EBPβ-HDAC1 Complexes Down-regulate SIRT1 and PGC1α
The development of liver cancer is characterized by the reduced ability of the liver to support its functions and overall homeostasis. A member of the surtuins family, SIRT1, and its downstream target PGC1α are important regulators of liver functions. We have shown previously that C/EBPβ-HDAC1 represses SIRT1 in the livers of old mice (13). We suggested that SIRT1 signaling might be also controlled by the C/EBPβ-HDAC1 complex during development of liver cancer. Therefore, we examined the expression of SIRT1 and PGC1α during development of liver cancer. In the course of these studies, we found, surprisingly, that PGC1α is also directly regulated by C/EBPβ-HDAC1 complexes. Examination of the PGC1α promoter showed that the promoter of PGC1α contains three C/EBP sites in close proximity to the start of transcription (Fig. 5A). The first site is located in position −765, whereas two other sites are located in positions −438 and −421 (Fig. 5A). An EMSA approach showed that C/EBP proteins bind to the C/EBP sites located in positions −438 and −421 (Fig. 5B, data for site 2 are not shown). Given the presence of high-affinity sites for C/EBP proteins in the SIRT1 (13) and PGC1α promoters, we next examined whether the CUGBP1-C/EBPβ-HDAC1 pathway down-regulates these proteins during DEN-mediated carcinogenesis through the repression of their promoters. Examination of levels of these proteins and corresponding mRNAs showed that both SIRT1 and PGC1α are reduced in mice treated with DEN and that this reduction correlates with the activation of CUGBP1 and elevation of C/EBPβ-HDAC1 complexes (Fig. 5, C and D). Further analyses of the SIRT1 and PGC1α promoters by ChIP assay revealed that C/EBPβ-HDAC1 complexes bind to and repress these promoters because histone H3 is trimethylated on the promoters after DEN treatments (Fig. 5E).
Biologically Relevant Levels of C/EBPβ-HDAC1 Complexes Are Supported in the Liver by FXR-mediated Regulation of C/EBPβ
C/EBPβ-HDAC1 complexes are detectable in quiescent liver, and they are required for proper liver function (4, 8). Our previous studies showed that farnesoid X receptor, FXR, supports biological levels of C/EBPβ-HDAC1 complexes in quiescent livers through regulation of C/EBPβ (16). However, the mechanisms of this regulation are not known. Therefore, we determined the mechanism by which FXR controls the C/EBPβ-HDAC1 complexes. Because the previous report from Guo and colleagues has shown that FXR binds to the C/EBPβ promoter (21), we examined the hypothesis that FXR supports high levels of C/EBPβ and levels of C/EBPβ-HDAC1 complexes through activation of the C/EBPβ promoter. We have cloned a 1.6-kb region of the C/EBPβ promoter that contains the region of interaction with FXR into the pGEX-luc reporter vector. The sequence of this region is shown in Fig. 6A. Computer analysis has identified two potential binding sites for FXR, b9 and b10, that are located side-by-side (Fig. 6D, red, and underlined; the consensus sites are overlapping). We first determined whether FXR binds to these consensus sites using EMSA approaches. As shown in Fig. 6B, FXR interacts with both consensus sites, and the addition of FXR Abs to the binding reactions supershifted the complexes. We next asked whether FXR binds to the C/EBPβ promoter in liver and whether this binding is reduced at early time points after DEN treatments when levels of C/EBPβ are reduced (16 and Fig 7A). A ChIP assay with chromatin solutions isolated at 0, 24, 48, and 72 h after DEN injection showed that FXR occupies the C/EBPβ promoter in quiescent livers but that the interactions of FXR with the C/EBPβ promoter are not observed at 24, 48, and 72 h after DEN injection (Fig. 6C).
FIGURE 6.
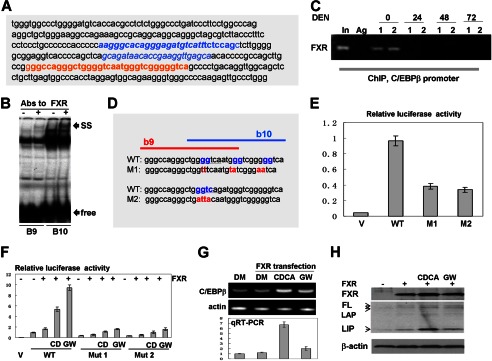
FXR up-regulates C/EBPβ through direct binding to the C/EBPβ promoter. A, nucleotide sequence of the C/EBPβ promoter containing FXR binding sites. Overlapping b9 and b10 FXR consensuses are shown in red. B, EMSA with probes covering FXR binding sites b9 and b10. Abs to FXR were incorporated in the binding reactions. Positions of supershift (SS) and free probes (free) are indicated by arrows. C, FXR binds to the C/EBPβ promoter in quiescent livers but is removed from the promoter in livers at early steps of DEN-mediated cancer. A ChIP assay was performed with chromatin solutions from quiescent livers (0) and livers at 24, 48, and 72 h after DEN injections. Data with two animals for each time point are shown. In, 1/100 input; Ag, agarose beads with IgG from preimmune serum. D, incorporation of mutations in the FXR binding sites within the C/EBPβ promoter. The incorporated mutations are shown in red. E, the mutations of the FXR binding site within the C/EBPβ promoter reduce the activity of the promoter. The Luc-WT and M1 and M2 mutant C/EBPβ promoters were transfected in Hep3B2 cells. The luciferase activity was examined as described above, and it is shown as a ratio to Renilla control. V, vector. F, FXR activates the C/EBPβ promoter through FXR binding sites. CD, CDCA; GW, GW4064. WT and mutant promoters (M1 and M2) were cotransfected with FXR into Hep3B2 cells. Cells were treated with the FXR ligands CDCA and WG4064. DM, dimethyl sulfoxide. G, activation of FXR by ligands increases expression of endogenous C/EBPβ mRNA. The top panel shows a typical picture of a regular RT-PCR assay. β-actin was used as the control. The bottom panel shows the results of quantitative RT-PCR (qRT-PCR) analysis of C/EBPβ mRNA. H, Western blotting shows activation of the endogenous C/EBPβ by FXR. The upper panel shows Western blotting with Abs to FXR. The C/EBPβ filter was reprobed with Abs to β-actin (bottom panel). Positions of the full-length (FL), LAP, and LIP isoforms of C/EBPβ are shown by arrows.
FIGURE 7.

The reduction of C/EBPβ-HDAC1 complexes at early time points after DEN injection leads to the activation of SIRT1, p53, and PGC1α. A, levels of FXR, C/EBPβ, HDAC1, and C/EBPβ-HDAC1 complexes at early time points after DEN injection. Western blotting was performed with Abs shown on the left. The bottom panel (HDAC1-IP:-C/EBPβ Western) shows amounts of C/EBPβ in HDAC1 immunoprecipitates. B, protein levels of SIRT1, PGC1α, and p53 are increased in livers at early time points after DEN injection. Western blotting was performed as described above. Each filter was reprobed with Abs to β-actin. C, levels of C/EBPβ-HDAC1 complexes and p53, SIRT1, and PGC1α proteins were calculated as ratios to β-actin. D, mRNA levels of SIRT1, PGC1α, and p53 are increased in livers at early time points after DEN injection. Bar graphs show the results of three independent repeat analyses of three to four animals of each group. *, p < 0.05, DEN treatment group versus control group.
To further investigate regulation of the C/EBPβ promoter by FXR, we performed a set of experiments in Hep3B2 cells. To show that FXR regulates the C/EBPβ promoter through the b9 and b10 elements, we generated two mutants in which both consensuses were mutated (Fig. 6D, mutants M1 and M2). It is important to note that each individual mutation destroys both consensuses and blocks interactions of FXR with this entire region of DNA. Examination of the basal activity of these constructs showed that the mutation of FXR binding sites significantly reduces activity of the C/EBPβ promoter (Fig. 6E), suggesting that FXR is a positive regulator of the promoter. To test this further, WT and mutant C/EBPβ promoters were cotransfected with a plasmid coding for FXR. To further activate FXR, we treated cells with the ligands CDCA and WG4064. We found that FXR activates the WT C/EBPβ promoter and that the additional activation of FXR by ligands increases this activation (Fig. 6F). Quite different results were obtained with the mutants. FXR-mediated activation of the M1 and M2 mutants is 5 to 6 times lower than that of the WT promoter. To examine whether FXR activates the expression of endogenous C/EBPβ, we determined the expression of endogenous C/EBPβ mRNA and protein after activation of FXR with ligands. RT-PCR analyses revealed that the activation of FXR increases expression of endogenous C/EBPβ mRNA (Fig. 6G). A Western blotting analysis shows that FXR also elevates the levels of all isoforms of endogenous C/EBPβ, full-length FL-C/EBPβ, C/EBPβ-LAP, and C/EBPβ-LIP (Fig. 6H). Taken together, these studies indicate that FXR up-regulates C/EBPβ expression through direct binding to the C/EBPβ promoter.
Early Steps of Liver Cancer Are Characterized by the Reduction of FXR and C/EBPβ-HDAC1 Complexes and by Activation of p53, SIRT1, and PGC1α
To further examine whether the FXR-C/EBPβ-HDAC1 pathway regulates p53, SIRT1, and PGC1α in liver, we performed studies of these proteins at early time points after DEN injection because at those time points FXR is not bound to the C/EBPβ promoter (Fig. 6C). We first examined the expression of FXR, C/EBPβ, HDAC1, and C/EBPβ-HDAC1 complexes at early time points after DEN injection in our experimental animals. Consistent with previous observations (16), Western blotting has shown that FXR and C/EBPβ are reduced significantly and that C/EBPβ-HDAC1 complexes are weak or not detectable at 24–72 h after DEN injection (Fig. 7A). Examination of p53, SIRT1, and PGC1α showed that their protein levels are elevated at 24, 48, and 72 h (Fig. 7, B and C). Quantitative RT-PCR confirmed these data and showed that the mRNAs of p53, SIRT1, and PGC1α are increased at early time points after DEN injection (Fig. 7D). Although there is an elevation of p53, SIRT1, and PGC1α mRNAs after DEN injections, we found that quiescent livers contain relatively high levels of these mRNAs (Fig. 7D). These results are consistent with the hypothesis that the promoters of p53, SIRT1, and PHC1α are partially repressed by C/EBPβ-HDAC1 complexes in quiescent livers. Taken together, these studies showed that there are two opposite events in the regulation of expression of p53, SIRT1, and PGC1α during DEN-mediated liver cancer. An early step includes elevation of these proteins, whereas later stages of liver cancer involve down-regulation of the proteins through changes of different pathways, including phosphorylation of HDAC1 and CUGBP1-mediated elevation of C/EBPβ-HDAC1 complexes.
DISCUSSION
Liver cancer is a multiple-step process that includes alteration of gene expression on different levels within a long duration of cancer development and progression. DEN-mediated liver cancer represents one of the best models in which each step of development of liver cancer might be investigated in detail. In this work, we examined the role of translational and transcriptional regulation of C/EBPβ-HDAC1 complexes in liver cancer and the consequences of the modulation of their levels. Fig. 8 summarizes the data in this work and shows the hypothetical role of C/EBPβ-HDAC1 complexes in the development of liver cancer. The biologically normal levels of C/EBPβ-HDAC1 complexes are supported in quiescent livers by FXR signaling, mainly by direct binding of FXR to the C/EBPβ promoter and subsequent maintenance of high levels of C/EBPβ. At very early stages (within 3 days) of liver cancer mediated by DEN, C/EBPβ-HDAC1 complexes are reduced because of reduction of FXR (16). However, C/EBPβ-HDAC1 complexes are dramatically elevated at later stages, at 6–25 weeks after initiation of the DEN protocol. This elevation of the C/EBPβ-HDAC1 complexes is mediated by at least two mechanisms that operate at different stages of liver cancer. The first mechanism takes place 6–10 weeks after DEN injection and includes phosphorylation of HDAC1 and a subsequent increase of its interaction with C/EBPβ. The second mechanism is activated at later time points of liver cancer (20–35 weeks after DEN injection) and includes the increase of CUGBP1-dependent translation of HDAC1 and C/EBPβ. It has been shown that the phosphorylated CUGBP1 displays its translational functions by interactions with eIF2α (14). In agreement with these observations, the amounts of CUGBP1-eIF2 complexes are increased at later time points after DEN injections. Because FXR regulates C/EBPβ-HDAC1 complexes in quiescent livers, one would expect that FXR would also contribute to the elevation of these complexes at late stages of liver cancer. In fact, we have shown previously that C/EBPβ-HDAC1 complexes are elevated in Little mice that express high levels of FXR at very late stages of liver cancer (35 weeks after DEN injection) (16). However, in WT mice, FXR is reduced during development of cancer, suggesting that the CUGBP1-eIF2-dependent pathway is the major contributor to the elevation of C/EBPβ-HDAC1 complexes at 6–25 weeks after DEN injection.
FIGURE 8.

Hypothetical model showing regulation and the role of C/EBPβ-HDAC1 complexes in the development of liver cancer.
Our previous studies and the data in this work showed that the C/EBPβ-HDAC1 complex binds to and down-regulates promoters of several key regulators of liver biology. These promoters include genes that inhibit liver proliferation and cancer, such as C/EBPα (4) and p53 (Fig. 4). The C/EBPβ-HDAC1 complex also down-regulates the promoter of a small subunit of 26 S-proteasome gankyrin (16), which promotes liver proliferation and liver cancer (10, 16). In addition, C/EBPβ-HDAC1 complexes repress the GSK3β and SIRT1 promoters in livers of old mice (12, 13) and the PGC1α promoter at later stages of liver cancer. The existence of these multiple gene targets for C/EBPβ-HDAC1 complexes with opposite functions in the liver suggests that the levels of the complexes should be tightly regulated. In this work, we show that there are at least three levels of the regulation of C/EBPβ-HDAC1 complexes: FXR-mediated transcriptional control of the complexes, phosphorylation of HDAC1, and translational up-regulation of HDAC1 and C/EBPβ. These multiple pathways allow the liver to control levels of the complexes and expression of the downstream targets at the levels that are required for different biological processes in the liver. These processes include liver proliferation after partial hepatectomy (4, 14), development of aging phenotypes (9, 12, 13), and development of liver cancer (16] and this work). Perhaps C/EBPβ-HDAC1 complexes have larger numbers of targets and might be involved in the control of other liver functions. Theoretically, all genes that contain C/EBP binding sites might be potentially down-regulated by C/EBPβ-HDAC1 complexes under certain settings. In this regard, our recent paper has shown that promoters of five enzymes of triglyceride synthesis contain C/EBP consensus sites and that these promoters are activated by another member of the C/EBP family, C/EBPα, during development of liver steatosis (22). We have recently obtained data that suggest that the biologically relevant levels of enzymes of triglyceride synthesis are controlled by a competition between C/EBPα-p300 complexes (positive regulators) and C/ΕBPβ-HDAC1 complexes (negative regulation) for the binding to their promoters.3
In this work, we show that the levels of C/EBPβ-HDAC1complexes are modulated during development of liver cancer, depending on the stage of liver cancer. We focused our studies on three main targets of the C/EBPβ-HDAC1 complexes: p53, SIRT1, and PGC1α, and on consequences of the down-regulation of p53 at late stages of liver cancer. It has been shown that the p53-p21 pathway inhibits liver proliferation and that it is eliminated during development of liver cancer (23, 24). However, the mechanism of this elimination has not been shown. In this paper, we determined a pathway by which DEN-mediated liver cancer eliminates this pathway. Another target of p53 within the cell cycle is E2F7 (20). We found that CUGBP1-C/EBPβ-HDAC1-mediated repression of p53 also reduces levels of E2F7. Because both p21 and E2F7 are inhibitors of cell proliferation, we suggest that these mechanisms are responsible for the p53-dependent increase of liver proliferation and for development of liver cancer. It is also important to note that two other targets of the CUGBP1-C/EBPβ-HDAC1 pathways, SIRT1 and PGC1α, are involved in the regulation of metabolic processes. Their reduction at later stages of liver cancer is likely to be involved in the metabolic alterations observed in animals with liver cancer.
This work was supported, in whole or in part, by National Institutes of Health Grants GM551888, CA100070, AG039885, AG028865, and CA159942 (to N. A. T.).
J. Jin, P. Iakova, Y. Jiang, K. Lewis, E. Sullivan, N. Jawanmardi, L. Donehower, L. Timchenko, and N. A. Timchenko, unpublished observations.
- HDAC
- histone deacetylase
- C/EBP
- CCAAT/enhancer binding protein
- DEN
- diethylnitrosamine
- FXR
- farnesoid X receptor
- co-IP
- coimmunoprecipitation
- Abs
- antibodies
- CDCA
- chenodeoxycholic acid
- CIP
- alkaline phosphatase.
REFERENCES
- 1. Liu W. R., Shi Y. H., Peng Y. F., Fan J. (2012) Epigenetics of hepatocellular carcinoma. A new horizon. Chin. Med. J. 125, 2349–2360 [PubMed] [Google Scholar]
- 2. Nishida N., Goel A. (2011) Genetic and epigenetic signatures in human hepatocellular carcinoma. A systematic review. Curr. Genomics. 12, 130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mandrekar P. (2011) Epigenetic regulation in alcoholic liver disease. World J. Gastroenterol. 17, 2456–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang G. L., Salisbury E., Shi X., Timchenko L., Medrano E. E., Timchenko N. A. (2008) HDAC1 promotes liver proliferation in young mice via interactions with C/EBPβ. J. Biol. Chem. 283, 26179–26187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y., Lee C. G. (2011) Role of miR-224 in hepatocellular carcinoma. A tool for possible therapeutic intervention? Epigenomics 3, 235–243 [DOI] [PubMed] [Google Scholar]
- 6. Buurman R., Gürlevik E., Schäffer V., Eilers M., Sandbothe M., Kreipe H., Wilkens L., Schlegelberger B., Kühnel F., Skawran B. (2012) Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells. Gastroenterology 143, 811–820 [DOI] [PubMed] [Google Scholar]
- 7. Xie H. J., Noh J. H., Kim J. K., Jung K. H., Eun J. W., Bae H. J., Kim M. G., Chang Y. G., Lee J. Y., Park H., Nam S. W. (2012) HDAC1 inactivation induces mitotic defect and caspase-independent autophagic cell death in liver cancer. PLoS ONE 7, e34265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones K., Timchenko L., Timchenko N. A. (2012) The role of CUGBP1 in age-dependent changes of liver functions. Ageing Res. Rev. 11, 442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang G. L., Salisbury E., Shi X., Timchenko L., Medrano E. E., Timchenko N. A. (2008) HDAC1 cooperates with C/EBPα in the inhibition of liver proliferation in old mice. J. Biol. Chem. 283, 26169–26178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iakova P., Awad S. S., Timchenko N. A. (2003) Aging reduces proliferative capacities of liver by switching pathways of C/EBPα growth arrest. Cell 113, 495–506 [DOI] [PubMed] [Google Scholar]
- 11. Jin J., Wang G. L., Iakova P., Shi X., Haefliger S., Finegold M., Timchenko N. A. (2010) Epigenetic changes play critical role in age-associated dysfunctions of the liver. Aging Cell 9, 895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jin J., Wang G. L., Shi X., Darlington G. J., Timchenko N. A. (2009) The age-associated decline of glycogen synthase kinase 3 β plays a critical role in the inhibition of liver regeneration. Mol. Cell. Biol. 29, 3867–3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jin J., Iakova P., Jiang Y., Medrano E. E., Timchenko N. A. (2011) The reduction of SIRT1 in livers of old mice leads to impaired body homeostasis and to inhibition of liver proliferation. Hepatology 54, 989–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Timchenko L. T., Salisbury E., Wang G. L., Nguyen H., Albrecht J. H., Hershey J. W., Timchenko N. A. (2006) Age-specific CUGBP1-eIF2 complex increases translation of CCAAT/enhancer-binding protein β in old liver. J. Biol. Chem. 281, 32806–32819 [DOI] [PubMed] [Google Scholar]
- 15. Wang G. L., Shi X., Haefliger S., Jin J., Major A., Iakova P., Finegold M., Timchenko N. A. (2010) Elimination of C/EBPα through the ubiquitin-proteasome system promotes the development of liver cancer in mice. J. Clin. Invest. 120, 2549–2562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang Y., Iakova P., Jin J., Sullivan E., Sharin V., Hong I. H., Anakk S., Mayor A., Darlington G., Finegold M., Moore D., Timchenko N. A. (2013) FXR inhibits gankyrin in mouse livers and prevents development of liver cancer. Hepatology 57, 1098–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boggs K., Reisman D. (2007) C/EBPβ participates in regulating transcription of the p53 gene in response to mitogen stimulation. J. Biol. Chem. 282, 7982–7990 [DOI] [PubMed] [Google Scholar]
- 18. Ewing S. J., Zhu S., Zhu F., House J. S., Smart R. C. (2008) C/EBPβ represses p53 to promote cell survival downstream of DNA damage independent of oncogenic Ras and p19(Arf). Cell Death Differ. 15, 1734–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Di Stefano L., Jensen M. R., Helin K. (2003) E2F7, a novel E2F featuring DP-independent repressor of subset of E2F-regulated genes. EMBO J. 22, 6289–6298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carvajal L. A., Hamard P. J., Tonnessen C., Manfredi J. J. (2012) E2F7, a novel target, is up-regulated by p53 and mediates DNA damage-dependent transcriptional repression. Genes Dev. 26, 1533–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thomas A. M., Hart S. N., Kong B., Fang J., Zhong X. B., Guo G. L. (2010) Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 51, 1410–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jin J., Iakova P., Breaux M., Sullivan E., Jawanmardi N., Chen D., Jiang Y., Medrano E. M., Timchenko N. A. (2013) Increased expression of enzymes of triglyceride synthesis is essential for the development of hepatic steatosis. Cell Rep. 3, 831–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pal D., Sur S., Mandal S., Das A., Roy A., Das S., Panda C. K. (2012) Prevention of liver carcinogenesis by amarogentin through modulation of G1/S cell cycle check point and induction of apoptosis. Carcinogenesis 33, 2424–2431 [DOI] [PubMed] [Google Scholar]
- 24. Slee E. A., Benassi B., Goldin R., Zhong S., Ratnayaka I., Blandino G., Lu X. Proc. (2010) Phosphorylation of Ser312 contributes to tumor suppression by p53 in vivo. Natl. Acad. Sci. U.S.A. 107, 19479–19484 [DOI] [PMC free article] [PubMed] [Google Scholar]