

Background: The NF-κB transcription factor mediates the inflammatory response through canonical and non-canonical pathways.
Results: TRAF-1 binds and stabilizes NIK by disrupting its association with TRAF2·cIAP2 complex.
Conclusion: These integrated computational-experimental studies identify TRAF1·NIK as a feed-forward complex coupling canonical and non-canonical pathways.
Significance: These findings provide insight into how cells decode inflammatory signals into distinct genomic responses.
Keywords: Inflammation, Mathematical Modeling, NF-κB Transcription Factor, TRAF, Tumor Necrosis Factor (TNF), NIK
Abstract
The NF-κB transcription factor mediates the inflammatory response through distinct (canonical and non-canonical) signaling pathways. The mechanisms controlling utilization of either of these pathways are largely unknown. Here we observe that TNF stimulation induces delayed NF-κB2/p100 processing and investigate the coupling mechanism. TNF stimulation induces TNF-associated factor-1 (TRAF-1) that directly binds NF-κB-inducing kinase (NIK) and stabilizes it from degradation by disrupting its interaction with TRAF2·cIAP2 ubiquitin ligase complex. We show that TRAF1 depletion prevents TNF-induced NIK stabilization and reduces p52 production. To further examine the interactions of TRAF1 and NIK with NF-κB2/p100 processing, we mathematically modeled TRAF1·NIK as a coupling signaling complex and validated computational inference by siRNA knockdown to show non-canonical pathway activation is dependent not only on TRAF1 induction but also NIK stabilization by forming TRAF1·NIK complex. Thus, these integrated computational-experimental studies of TNF-induced TRAF1 expression identified TRAF1·NIK as a central complex linking canonical and non-canonical pathways by disrupting the TRAF2-cIAP2 ubiquitin ligase complex. This feed-forward kinase pathway is essential for the activation of non-canonical pathway.
Introduction
NF-κB is a tightly regulated transcription factor family essential for regulating a broad range of biological processes including inflammation, immune response, cell growth, and survival (1). Inappropriate NF-κB activation by exogenous ligands or activating somatic mutations has been linked to diabetes, vascular disease, solid- and hematopoietic cancers, and accelerated aging (2, 3). Two distinct NF-κB activation pathways have been described that are referred to as the canonical and non-canonical pathways. The canonical pathway is activated by monokines (tumor necrosis factor (TNF), interleukin-1 (IL-1)), bacterial patterns (lipopolysaccharide), and genotoxic agents (1, 4). The rate-limiting step in the canonical pathway is mediated by IκB kinase complex-mediated phosphorylation and subsequent proteosomal degradation of IκB (primarily IκBα) that normally sequesters RelA·p50 heterodimers in the cytoplasm. After IκBα degradation, RelA·p50 can translocate to the nucleus where it interacts in hyperdynamic exchange with binding sites in regulatory sequences of inducible genes, affecting their expression through a process involving transcriptional elongation (5–7). Genes regulated by the canonical pathway are primarily associated with innate immunity, anti-apoptosis, and inflammation (8).
By contrast, the non-canonical pathway is primarily activated by receptor activator of nuclear factor κB ligand, B cell activating factor, dsRNA, and CD40 ligands (9). The rate-limiting step in non-canonical NF-κB pathway activation is mediated by MAP3K14/NF-κB-inducing kinase (NIK),2 an upstream MAP kinase responsible for phosphorylating and activating IκB kinase α primarily (10). Activated IκB kinase α in turn phosphorylates the 100-kDa NFKB2 gene product (p100) in its carboxyl terminus, leading to proteosome-mediated partial degradation to form a 52-kDa DNA binding subunit (p52) that translocates to the nucleus as heterodimers with RelA or RelB and binds to NF-κB sites in the promoter region of certain NF-κB-dependent genes to activate transcription (11, 12). The non-canonical dependent genes are under cell type-specific control, for example Naf-1 in epithelial cells and SCF/BLC/ELC lymphokines in dendritic cells.
Presently we understand that NIK is maintained at a low steady state level in resting cells as a result of active protein turnover mediated by TNF receptor-associated factors (TRAFs). TRAF3 binding NIK recruits a complex of TRAF2 and cellular inhibitor of apoptosis (cIAP-1) and -2 ubiquitin ligases. In this complex, NIK undergoes Lys-48-linked polyubiquitination by cIAP1/2 resulting in rapid proteosomal degradation (13, 14). By contrast, in response to non-canonical stimuli, cIAP1/2 ubiquitinates TRAF3, promoting its degradation. NIK is thereby released from negative regulation leading to its stabilization and intracellular accumulation. An increased level of NIK causes its activation, presumably by concentration-dependent oligomerization and autophosphorylation (15). However, the role of TRAF1 in regulating non-canonical pathway is not well described.
In the current study, we investigated the mechanism by which the non-canonical pathway is coupled to the canonical pathway after TNF stimulation. Here we demonstrate that tonic TNF stimulation induces TRAF1 expression and delayed p52 formation. TRAF1 binds to NIK with high avidity and stabilizes it by preventing its interaction with the TRAF2-cIAP2 complex. siRNA-mediated silencing of TRAF1 or NIK demonstrates the requirement for both factors in non-canonical gene expression. A deterministic mathematical model of the canonical-non-canonical pathway is developed and experimentally verified. Together our results provide evidence that inducible TRAF1 expression is coupled to a stimulus-dependent feed-forward pathway to NIK stabilization and non-canonical pathway activation.
EXPERIMENTAL PROCEDURES
Cell Culture
Human A549 pulmonary epithelial cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) at 37 °C in a 5% CO2 incubator. A FLAG-tagged full-length NIK expression vector (FLAG-EGPF-NIK) was constructed as described earlier (16). CloneticsTM primary normal human bronchial epithelial (NHBE) cells were purchased from Lonza and cultured in special BEGM BulletKit media as per the manufacturer's protocol.
Preparation of Subcellular Extracts
Cells were harvested in phosphate-buffered saline and centrifuged, and the pellets were resuspended sequentially in low salt, sucrose, and high salt solutions to obtain cytosolic and highly purified nuclear extracts as previously described (17).
Immunoprecipitation and Immunoblotting
Protein extracts were precleared by incubating for 45 min with washed protein-A beads (Sigma) followed by incubation with specific antibody. Bound proteins subjected to WB.
Quantitative Real Time PCR (Q-RT-PCR)
2 μg of TRIzol-extracted total RNA was reverse-transcribed using the SuperScript III First-Strand Synthesis System from Invitrogen and amplified in a 20-μl reaction system containing 10 μl of iQ SYBR Green Supermix (Bio-Rad) and 400 nm primer mixtures. Relevant primers were purchased from SA Bioscience (Frederick, MD). Results were analyzed by the iQ5 program (Bio-Rad), and the data were analyzed using the ΔCT method in reference to GAPDH (18).
Mathematical Modeling Reactions and Parameters
The new mathematical model includes reactions and parameters for both canonical and non-canonical arms of NF-κB pathway. All proteins in the system are present in large numbers; therefore, the effect of stochasticity on the time evolution of individual molecules is negligible. In the model, whereas RelA is synthesized as a mature product for canonical gene expression, the second transcription factor p52 required for non-canonical pathway undergoes proteolytic cleavage from a large precursor p100. This poly-ubiquitination-mediated proteolytic processing of p100 is facilitated by TRAF1·NIK complex. However, synthesis of p100 is itself NF-κB-dependent and requires stimulation. Below are the protein-specific descriptions.
TRAF1/TRAF2
The TRAF1 kinetics is directed by our laboratory experimental data as well as coefficients for similar processes published earlier. Importantly, the TRAF1 mRNA half-life is estimated to be at least 2 h (to fit with our time-profiles too) from the study by Hao and Baltimore (19) on temporal order of induced gene expression, resulting in its rate constant as 9.62e−05 s−1. The rate parameters of TRAF2 are assumed to be same as that of A20 because kinetics of both proteins is nearly same.
For TRAF1 protein dynamics,
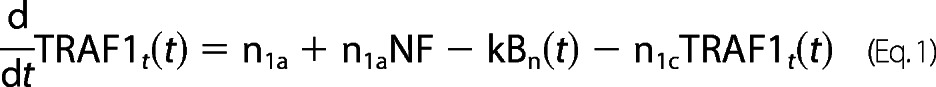 |
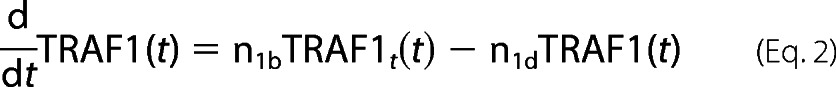 |
For TRAF2 protein dynamics,
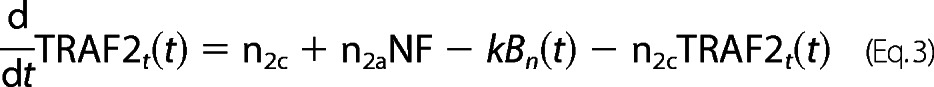 |
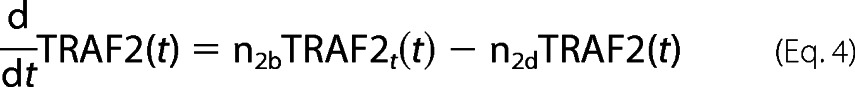 |
NIK
The role of TRAFs and cIAPs in NIK degradation and stabilization involves complicated and unresolved kinetics. It lead us to simplify the model by approximating NIK gene expression under an unknown (UNK) promoter by both de novo synthesis having a zero order constant production term as well as an first-order inducible term. The rate constants describing the association of NIK with TRAF2 (b1) to catalyze its degradation (b2), TRAF1 association with TRAF2·NIK complex (b3) to form a TRAF1·NIK complex (b4) are either fitted to experimental data or assumed, under experimentally known physiological constraints. The time-evolution of all these reactions to reach to a certain threshold activity levels take nearly 3 h. This correlates with the fact that the process of instant degradation of NIK by TRAF2 is extended to almost 3 h once TRAF1 is synthesized and initiates the NIK rescuing process.
For NIK protein dynamics,
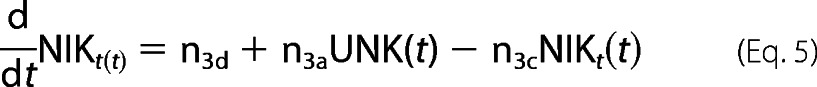 |
 |
For bi- and tri-molecular complexes,
 |
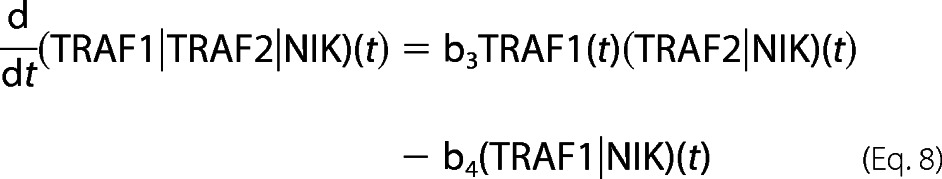 |
p100/p52
Most of reaction parameters involving p100/p52 are fitted in accordance with our laboratory experimental observations as well as the Basak et al. (11) model. For example, the p100 mRNA half-life is estimated to be nearly 6–7 h. There is a time delay of 90 min in p100 transcription, resulting in delay in its translation too (which we see in the time-course profile of p100 in our Western blots). However, p100 processing by TRAF1·NIK complex to generate p52 (nc5) involves fast kinetics of less than 5′. This is evident from our laboratory Western blot profiles. The fast kinetics emphasizes the possibility of co-translational processing mechanism (20). Thus, our model includes p52 generation from newly synthesized p100 and then its import into nucleus.
For P100 protein dynamics,
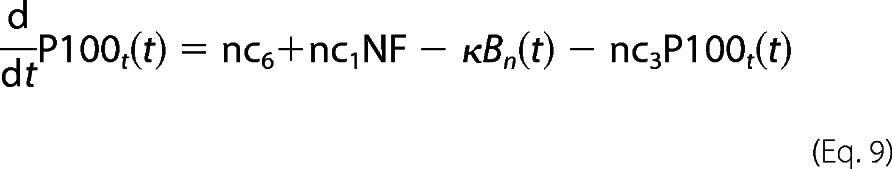 |
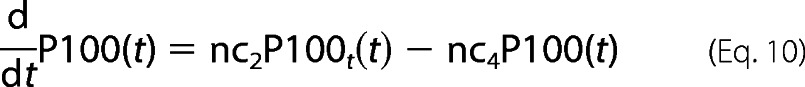 |
 |
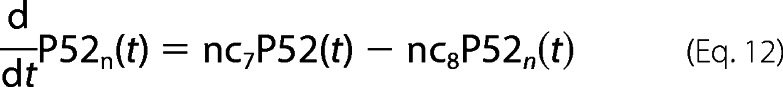 |
Naf1
The reaction coefficients pertaining to Naf1 kinetics is assumed similar to A20/IκBα except for the fact that Naf1 expression is under the p52 promoter. All the complex cascading signaling and associated inherent delays with p52 generation seem to be a simple explanation about gene expression of genes such as Naf1 here.
For Naf1 protein dynamics,
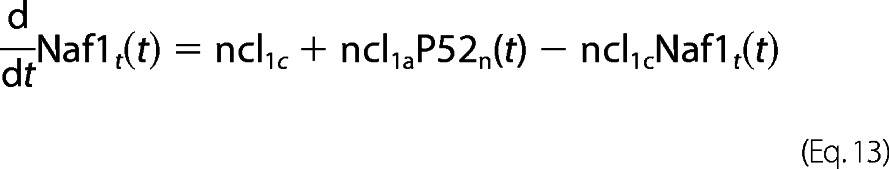 |
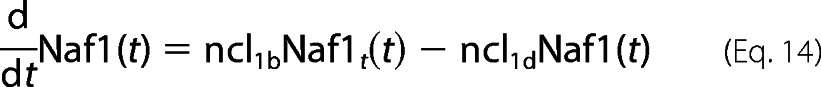 |
Selected Reaction Monitoring (SRM) Data Analysis
All SRM data were processed using Xcalibur® 2.1. All data were manually inspected to ensure peak detection and accurate integration. The chromatographic retention time and the relative product ion intensities of the analyte peptides were compared with those of the stable isotopically labeled peptide standard (SIS) peptides. Signature peptides that stoichiometrically represent the protein candidate were selected on the following criteria: tryptic peptides must be unique to the target protein; length of 8–25 residues; absence of missed tryptic cleavages, chemically active amino acid residues (such as cysteine or methionine), and no basic amino acids residues (such as cysteine or methionine) and no basic amino acids on either cleavage site of the peptide sequence (21). The tryptic peptides that met the above criteria were considered as potential signature peptides. Pilot LC-MS-MS experiments were performed using tryptic digests of recombinant NIK and TRAF1 to identify the highest MS responding peptides for each protein. The extracted ion chromatograms based on the monoisotopic peak for all charge states and modifications detected from sequence-identified peptides were compared, and the peptides with the highest MS response were selected as the high-responding signature peptide for each target proteins. These signature peptides are listed in Table 1.
TABLE 1.
Parameters of SRM assays of NIK, TRAF1, and p52/p100
m/z values listed are for the natural forms of the peptides.
| Gene name | UniProt accession no. | Peptide sequence | Peptide molecular mass | Precursor ion (m/z) | Product ion (m/z) | Ion type | Collision energy |
|---|---|---|---|---|---|---|---|
| Da | V | ||||||
| TRAF1 | Q13077 | VVELQQTLAQK | 1255.721 | 628.864 | 688.398 | y6 | 25 |
| 628.864 | 816.457 | y7 | 25 | ||||
| 628.864 | 929.541 | y8 | 25 | ||||
| 628.864 | 1058.583 | y9 | 25 | ||||
| NIK | Q99558 | IASEPPPVR | 964.541 | 483.274 | 468.292 | y4 | 20 |
| 483.274 | 565.345 | y5 | 20 | ||||
| 483.274 | 694.388 | y6 | 20 | ||||
| 483.274 | 781.420 | y7 | 20 | ||||
| p52/p100 | Q00653 | DSGEEAAEPSAPSR | 415–428 | 701.808 | 359.203 | y3 | 27 |
| 701.808 | 614.325 | y6 | 27 | ||||
| 701.808 | 743.368 | y7 | 27 | ||||
| 701.808 | 814.405 | y8 | 27 | ||||
| 701.808 | 885.442 | y9 | 27 | ||||
| 701.808 | 1014.484 | y10 | 27 |
For each high-responding signature peptides, four y-ions were selected. The y-ions were preferred over b-ions because b-ions were usually less prominent or absent in the triple quadrupole mass spectrometer (QQQ-MS) MS/MS spectra; only the y-ions, whose m/z values exceed precursor ion m/z values, were chosen. The selection of fragment ions were on the basis of MS/MS spectra of the peptide from SRM-triggered MS/MS experiments, the highest intensity fragment ions in MS/MS were selected to maximize the sensitivity of detection. The collision energy for each signature peptide was predicted by the generic formula collision energy = 0.034 × (precursor ion m/z) + 3.314. The S-lens voltage for each signature peptide was derived from S-lens table generated during MS calibration. The selected Q1/Q3 transitions and their collision energy are tabulated in Table 1.
Synthesis of Native and SIS
Native tryptic peptides were synthesized using N-(9-fluorenyl) methoxycarbonyl chemistry. SIS peptides were commercially synthesized incorporating isotopically labeled [13C6,15N4]arginine to a 98% isotopic enrichment (Sigma). Both native and SIS peptides were HPLC-purified to >98% purity. The molecular weights were measured with electrospray mass spectrometry and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. The specific peptide concentration was determined by amino acid analysis. Individual SIS peptide stocks of 20 pmol/μl were made in 80% acetonitrile.
Trypsin Digestion of Affinity-purified NIK
The beads from the NIK pulldown experiment were washed 3 times using 50 mm ammonium bicarbonate (NH4HCO3) buffer (pH 8). The beads were resuspended with 200 μl of NH4HCO3 buffer. Then 500 fmol of NIK and TRAF1 stable isotope-labeled standard peptides and 2 μg of trypsin (Promega, Madison, WI) were added to each sample. The samples were incubated at 37 °C for 16 h with gentle vortex. Digestion was stopped by adding 2 μl of 10% trifluoroacetic acid. The beads and the supernatant were separated by using an empty TopTip micro spin column (Columbia, MD), and the flow-through was collected. The beads were washed with 100 μl of 50% acetonitrile three times. The flow-through and the eluates were combined and dried in a SpeedVac. The dried peptide samples were then redissolved in 30 μl of 5% formic acid, 0.01% trifluoacetic acid. 10 μl of the redissolved samples were injected and analyzed by LC-SRM-MS.
LC-SRM-MS Analysis
LC-SRM-MS analysis was performed with a TSQ Vantage triple quadrupole mass spectrometer equipped with nanospray source (Thermo Finnigan, San Jose, CA). The online desalting and chromatography were performed using an Eksigent NanoLC-2D HPLC system (AB SCIEX, Dublin, CA). An aliquot of 10 μl of each of the tryptic digests was injected on a C18 peptide trap (Agilent, Santa Clara, CA) desalted with 0.1% formic acid at a flow rate of 2 μl/min for 45 min. Peptides were eluted from the trap and separated on a reverse-phase nano-HPLC column (PicoFritTM, 75 μm × 10 cm; tip ID 15 μm) packed in-house using Zorbax SB-C18 (5-μm diameter particles, Agilent). Separations were performed using a flow rate of 500 nl/min with a 20-min linear gradient from 2 to 40% mobile phase B (0.1% formic acid, 90% acetonitrile) in mobile phase A (0.1% formic acid) followed by a 0.1-min gradient from 40 to 90% mobile phase B and 5 min of 90% mobile phase B. The TSQ Vantage was operated in high resolution SRM mode with Q1 and Q3 set to 0.2 and 0.7-Da full width half maximum (FWHM). All acquisition methods used the following parameters: 1800 V ion spray voltage, a 275 °C ion transferring tube temperature, a collision-activated dissociation pressure at 1.5 mTorr, and the S-lens voltage used the values in the S-lens table generated during MS calibration. Each sample was analyzed by LC-MS twice.
SRM Quantification of NIK
To more quantitatively ascertain the positive role of TRAF1 in regulating NIK stability, we developed NIK- and TRAF1-specific SRM assays. The assay dynamic range was assessed by the method of standard addition (21–23). In this method, serial dilutions of target proteins with a fixed amount of SIS peptide are spiked into the similar matrix to the neat specimen, and a response calibration curve is generated for each 12C analyte peptide. We diluted the tryptic digest to generate a range of analyte concentrations spanning a 10,000-fold concentration range (from 50 amol to 5 pmol on the column). These analyte concentrations were then combined with a constant amount of 13C-analyte peptide internal standard. Linear regression analysis was performed on the observed peak area ratios (native: heavy) versus concentration ratios to generate calibration curves (data not shown). The SRM assay yielded linear responses over a >20,000-fold concentration range with a strong linear correlations (r2 > 0.9991). The error bars indicate S.D. of the measurements.
RESULTS AND DISCUSSION
Tonic TNF Stimulation Produces Delayed Activation of the Non-canonical Pathway
Earlier studies from our laboratory have shown that a brief pulse of TNF stimulation produces a monotonic activation of the RelA translocation, whereas tonic TNF stimulation produces a prolonged NF-κB translocation profile (8). Although TNF is thought to primarily stimulate the canonical NF-κB pathway, we determined whether the non-canonical pathway was also being activated. For this purpose, A549 cells were stimulated with TNF and fractionated into nuclear and cytoplasmic fractions (9), and nuclear localization of RelA, RelB, and NF-κB2 was assayed by Western blot (WB). We observed rapid 2–4-fold nuclear translocation of RelA and cytoplasmic IκBα degradation within 30 min of TNF treatment (Fig. 1A). Live cell imaging data at a single cell resolution clearly demonstrate a robust increase in the nuclear RelA translocation after TNF stimulation measured by confocal microscopy (24). By contrast, p52 translocation was significantly increased after 3 h of stimulation (Fig. 1B). RelB translocation was biphasic with the initial translocation seen at 30 min followed by its nuclear depletion followed by a second translocation coincident with p52 translocation, 3 h after stimulation. Together, we interpret these data to suggest that TNF stimulation induces a delayed response of non-canonical pathway activation.
FIGURE 1.

TNF-induced TRAF1 expression coincides with late non-canonical pathway activation. A, A549 cells were treated with TNF (25 ng/ml) for the indicated times. Shown is the WB analysis of nuclear NF-κB proteins. NE: nuclear extracts, CE: cytoplasmic extracts. B, WBs show a time course of TRAF1 and TRAF2 expression in cytosolic fraction of TNF-treated A549 cells. β-Actin served as a loading control in both these figures. C, TRAF1 expression requires canonical pathway activation. Q-RT-PCR shows TRAF1 expression in HeLa cells expressing nondegradable IκBα (−Dox; light bars) is completely suppressed as compared with cells expressing normal IκBα proteins (+Dox; dark bars). Data represent the mean ± S.D. of three independent experiments and were analyzed by two-way ANOVA with multiple comparison (time and doxycycline treatment) and Tukey's post hoc test for significance between time intervals and the treatment groups. Significantly different from TNF (0 h)-treated samples: *, p < 0.01; significantly different from doxycycline-treated samples: †, p < 0.001.
TRAF1 mRNA is rapidly induced by tonic TNF stimulation, at times before p100 processing (8). This finding, in conjunction with a recent report suggests a positive role of TRAF1 in p100 processing (25), prompted us to examine the kinetics of TRAF1 protein expression. A WB of TNF-stimulated cytoplasmic extracts showed that TRAF1 is detectable at 3 h and increases by 6 h of stimulation, whereas the steady state abundance of TRAF2 is not significantly affected (Fig. 1C). To determine whether TRAF1 is exclusively NF-κB-dependent, we used HeLatTA/FLAG-IκBαMut, a clonal cell line expressing a tetracycline-regulated NF-κB dominant-negative inhibitor (8, 26). These cultured cells in the absence of doxycycline (Dox) express the non-degradable S32A/S36A IκBα mutant that blocks canonical pathway activation. In the presence of Dox, TNF induces a 7-fold increase in TRAF1 mRNA, peaking to 30-fold at 6 h (Fig. 1D). However, in the absence of Dox, we note that TRAF1 expression is completely inhibited. The profile of TRAF1 mRNA induction is similar to that of TRAF1 protein. To further confirm the activation of the non-canonical pathway after TNF stimulation, NHBE cells were treated with TNF for 0–9 h, and TRAF1 expression kinetic and p52 nuclear translocation was assayed by Western blot. We observed a delayed increase in p52 nuclear accumulation peaking at 3 h, similar to A549 cells. Similarly, TRAF1 expression peaks at 3 h of TNF treatment in these cells (Fig. 2, A and B). The delayed activation of non-canonical pathway coincides with the expression of Naf1, a typical late responsive gene (Fig. 2D) compared with early response gene A20 (Fig. 2C) (8). Taken together, these findings suggest that TNF-induced TRAF1 expression is exclusively canonical NF-κB pathway-dependent and temporally precedes non-canonical pathway activation.
FIGURE 2.

TNF-induced non-canonical pathway activation in NHBE cells. A, shown are cells treated with TNF (25 ng/ml) for the indicated times. Shown is the WB analysis of nuclear p52. B, a WB shows a time course of TRAF1 expression in the cytosolic fraction of TNF-treated NHBE cells. C and D, NHBE cells were stimulated with TNF (25 ng/ml) for the indicated times. A20 and ABIN1/Naf1 expression was measured by Q-RT-PCR. Data expressed as -fold change was compared with untreated cells after normalizing to internal controls, GAPDH. Data were analyzed by one-way ANOVA with multiple comparisons. Significantly different from untreated samples: *, p < 0.01.
TRAF1 Associates with and Stabilizes NIK
Our findings that TNF administration induces p52 processing suggested that TNF stimulation induces NIK activity. Currently NIK activation is thought to be primarily mediated by its post-translational stabilization, oligomerization, and autophosphorylation (15). Thus we investigated if TRAF1 associates with NIK and/or if TRAF1 is involved in NIK stability. HEK 293 cells were co-transfected with a constant concentration of myc epitope-tagged NIK and increasing concentrations of FLAG epitope-tagged TRAF1 expression plasmids. Fig. 3A shows a dose-dependent increase in NIK binding with overexpressed TRAF1. It was remarkable to observe NIK and TRAF1 interaction even at a very low concentration of TRAF1 expression (0.1 μg/ml) where the TRAF1 expression in cellular lysates is undetectable. The lower panel shows increased accumulation of NIK at higher concentrations of TRAF1. However, TRAF1 expression alone did not alter NIK expression at mRNA level (data not shown), suggesting that TRAF1-mediated NIK stabilization is a post-translational event. These findings suggest that TRAF1 not only binds to NIK with high affinity, but increased abundance of cellular TRAF1 facilitates increased steady state levels of NIK.
FIGURE 3.

Interaction with TRAF1 stabilizes NIK protein. A, HEK 293 cells were co-transfected with myc-NIK (0.4 μg/ml) and FLAG-TRAF1 (0.05, 0.2, 0.4, and 1.0 μg/ml). Whole cell extracts were prepared in radioimmune precipitation assay buffer, and overexpressed TRAF1 was immunoprecipitated (IP) by FLAG antibodies. Shown is the WB depicting increased binding of NIK with increasing TRAF1 expression. The lower panel shows the overexpression of these proteins and effect of these protein expressions on p100 processing in the lysate. ns: non-specific band. B, increased expression and association of TRAF1 with NIK disrupts TRAF2 and cIAP2 interaction. HEK 293 cells were transfected as described above, and proteins were immunoprecipitated with either myc or TRAF2 antibodies. Interacting TRAFs and cIAP2 was measured by WB. The lower panel shows the expression levels of proteins involved in NIK degradation in the lysates. C, TNF-induced TRAF1 binds to endogenous NIK and stabilizes it by displacing TRAF2 from the complex. A549 cells stably transfected with FLAG-NIK were treated with TNF (25 ng/ml) for the specified time intervals. Whole cell extracts were fractionated on 10% gel. WBs show differential binding of NIK with TRAF1 and TRAF2 after TNF treatment as a function of time.
NIK interaction with TRAF3 and subsequent degradation is critical to maintain its low cellular steady state levels; we asked if TRAF1 interaction with NIK alters this interaction and thus protects it from degradation. HEK 293 cells were transfected with Myc-NIK alone or in the presence of a constant concentration of FLAG-TRAF3 and increasing concentrations of FLAG-TRAF1 expression vectors. In the absence of TRAF3 expression, NIK was precipitated, but no FLAG-TRAF3 was detected (Fig. 3B). However, in the presence of TRAF3 expression, a strong association of TRAF1 with NIK is seen. Importantly, the abundance of TRAF3 immunoprecipitated with NIK remained the same in the presence or absence of TRAF1. Even at a higher TRAF1 concentration, the ability of NIK to interact with TRAF3 remained unaltered, suggesting that TRAF1 and TRAF3 binding to NIK are independent events. TRAF1 competition with TRAF3 for NIK binding does not explain the phenomenon of NIK stabilization by TRAF1.
We next examined the possibility that increased TRAF1 expression could interfere with TRAF2·cIAP2 complex. Consistent with Fig. 3A, a dose-dependent increase in TRAF1 binding with NIK was observed with increasing TRAF1 expression. Surprisingly, cIAP2 binding to TRAF2 was significantly reduced in the presence of TRAF3 and TRAF1 at all the concentrations used. cIAP2 binding with TRAF2 was undetectable at higher TRAF1 expression. Additionally, loss of cIAP2 from the TRAF2 coincides with the expression of FLAG-TRAF3. This could be because TRAF2 interacts with TRAF3 as well as cIAP2 through the same domain. TRAF-C domain of TRAF3 interacts with the TRAF-N domain (amino acids 264–344) and zinc (fingers 4 and 5 of TRAF2 (27). The region between amino acids 267 and 328 of TRAF2 is also required for its interaction with cIAP2 (28). Thus, TRAF3 overexpression in this experiment could potentially compete off cIAP2 interaction with TRAF2. Therefore, we observe a decline in TRAF2·cIAP2 interaction after TRAF3 expression even in absence of TRAF1 overexpression (compare lanes 1 and 3, Fig. 3B). Nevertheless, overexpression of TRAF1 alone is sufficient to reduce TRAF2·cIAP2 interaction even in the absence of TRAF3 overexpression (compare lanes 1 and 2, Fig. 3B). These findings suggest that TRAF1 accumulation disrupts the TRAF2 complex with cIAP2 without affecting NIK binding with TRAF3.
NIK Interaction with TRAF2 Is Reduced by a Concomitant Increase in TRAF1 Binding
To understand if TNF-induced TRAF1 expression disrupts endogenous NIK association with its TRAF2·cIAP2 degradation complex, A549 cells stably expressing FLAG-EGFP-NIK were treated with TNF for various time intervals. TRAF2-NIK interaction was then determined by non-denaturing co-immunoprecipitation using anti-TRAF2 antibody. As expected, NIK strongly associates with TRAF2 in the absence of TNF treatment. However, TRAF2 interaction with NIK is dramatically reduced for up to 6 h of TNF treatment (Fig. 3C). The reduction in TRAF2 binding at 6 h was associated with increased TRAF1 binding. An earlier report suggests that TNF-induced non-canonical pathway is mainly regulated by the RIP1 kinase, a kinase that not only potentiates TNF-induced canonical pathway activation but also inhibits activation of non-canonical pathway (29). In this model the loss of RIP1 leads to increased TRAF2 degradation leading to NIK stabilization and subsequent non-canonical pathway activation in fibroblast cells. However, we do not observe TNF inducible TRAF2 degradation (Fig. 1C). Our data, therefore, suggest an alternative mechanism where TNF-induced NIK stabilization and non-canonical pathway activation are associated with increased association of NIK with TRAF1 with simultaneous loss of TRAF2 from the complex. Because TRAF3 is the main adaptor protein that links NIK to TRAF2-cIAP1/2 ubiquitin ligase complex, potential mechanisms for NIK stabilization could be 1) that increased levels of TRAF1 alter the specificity of the associated ubiquitin ligase activity toward TRAF3, leading to its degradation and relieving NIK from its negative regulation or 2) TRAF1 could compete TRAF3 from binding NIK, and thus. TRAF1-NIK is not a potent substrate for the TRAF2-cIAP1/2 ubiquitin ligase. However, our findings show that increasing TRAF1 expression binds and stabilizes NIK without affecting TRAF3 protein expression levels or its interaction with NIK. Because the amount of NIK being bound to TRAF3 remains constant at various levels of TRAF1 expression, we suggest that TRAF-1 and -3 interact independently with NIK.
Another possibility for NIK stabilization could be that increasing the concentration of TRAF1 could alter the constituents of NIK ubiquitin ligase complex. In this context, Zheng et al. (28) show that TRAF1 alone interacts weakly with cIAP2 in crystal as well solution; however, TRAF1 preferentially makes a heterotrimer, TRAF1·(TRAF2)2, and significantly increases TRAF2 interaction with cIAP2 compared TRAF2 alone. Thus, these findings suggest that up-regulation of TRAF1 by various stimuli could potentially modulate the interaction of TRAF2 and cIAP2, two critical components of ubiquitin ligase responsible for NIK degradation. However, these studies do not predict how these interactions will be altered in the presence of NIK. Here we have clearly shown that TRAF1 overexpression in the presence of NIK disrupts interaction of TRAF2 and cIAP2. Also, in response to TNF treatment, we observed increased binding of TRAF1 with NIK and a corresponding loss of TRAF2 from this complex. Therefore, our data suggest that although TRAF2 and TRAF3 bind and target NIK for degradation, increasing TRAF1 expression competes for NIK binding with TRAF2. These data are further supported by the findings that TRAF1 and TRAF2 interaction domains map to the same region of NIK between amino acids 624 and 947 (30, 31).
Formulation of an NF-κB Feed-forward Model
Our current data suggest that TNF-induced TRAF1·NIK complex could couple the canonical and non-canonical NF-κB pathways. Therefore, we expanded our recently published deterministic model of the canonical NF-κB pathway under negative feedback control (24) by incorporating TRAF1·NIK as a central, rate-limiting signaling complex for non-canonical pathway activation. The current non-canonical model consists of 14 ordinary differential equations and 31 reaction parameters (Table 2). The overall two-compartmental kinetic model describing the NF-κB signaling system consists of 28 ordinary differential equations and 58 reaction parameters, where IκB kinase and NF-κB are activator proteins, TNFAIP3/A20 and IκBα act as inhibitory proteins mediating the negative feedback loops, and TRAF1·NIK are feed-forward signaling proteins linking canonical to non-canonical pathway activation (Fig. 4). The essential assumptions in model development were that TRAF1 mRNA expression is under canonical NF-κB control (Fig. 1D) and NF-κB2/p100 processing is co-translational-dependent (20).
TABLE 2.
Rate parameters for the Feed-Forward NF-κB non-canonical ODE model
All other parameters pertaining to canonical NF-κB model are reported in Kalita et al. (24) after being fitted to the single-cell dynamical data in the study. All the new parameters for the feed-forward model are described in the table below. The comment column indicates the origin of the nominal values for this model.
| Symbol | Values | Units | Description | Comments |
|---|---|---|---|---|
| n1a | 5 × 10−7 | s−1 | TRAF1-inducible mRNA synthesis | Assumption |
| n1b | 0.5 | s−1 | TRAF1 translation rate | Fitted |
| n1c | 9.62 × 10−5 | s−1 | TRAF1 mRNA degradation | Fitted, Hao and Baltimore (19) |
| n1d | 0.0003 | s−1 | TRAF1 degradation rate | Fitted |
| n1e | 0.0 | μms−1 | TRAF1 constitutive mRNA synthesis | Assumption |
| n2a | 5 × 10−7 | s−1 | TRAF2-inducible mRNA synthesis | Assumption |
| n2b | 0.5 | s−1 | TRAF2 translation rate | Fitted |
| n2c | 0.0004 | s−1 | TRAF2 mRNA degradation | Fitted |
| n2d | 0.0003 | s−1 | TRAF2 degradation rate | Fitted |
| n2e | 0.0 | μms−1 | TRAF2 constitutive mRNA synthesis | Assumption |
| n3a | 2.5 × 10−9 | s−1 | NIK-inducible mRNA synthesis (NF-κB-independent) | Assumption |
| n3b | 0.5 | s−1 | NIK translation rate | Fitted |
| n3c | 0.0004 | s−1 | NIK mRNA degradation | Fitted |
| n3d | 0.0 | μms−1 | NIK-constitutive mRNA synthesis | Assumption |
| b1 | 1.0 | s−1 | TRAF2-NIK association | Any large |
| b2 | 6.42 × 10−5 | s−1 | NIK degradation from TRAF2-NIK complex | Fitted |
| b3 | 0.5 | s−1 | TRAF1 association with TRAF2-NIK complex | Assumption |
| b4 | 0.25 | s−1 | formation of TRAF1-NIK complex by displacing TRAF2 from TRAF2-NIK complex | Fitted |
| nc1 | 2.5 × 10−8 | s−1 | P100-inducible mRNA synthesis | Basak et al. (11) |
| nc2 | 0.5 | s−1 | P100 translation rate | Fitted |
| nc3 | 3.2 × 10−5 | s−1 | P100 mRNA degradation | Basak et al. (11) |
| nc4 | 0.0004 | s−1 | P100 degradation rate | Fitted |
| nc5 | 0.002 | s−1 | TRAF1-NIK and p100 association | Fitted |
| nc6 | 0.0 | μms−1 | p100-constitutive mRNA synthesis | Assumption |
| nc7 | 7.5 × 10−4 | s−1 | p52 nuclear import | Basak et al. (11) |
| nc8 | 0.0002 | s−1 | p52 nuclear export | Basak et al. (11) |
| ncl1a | 5 × 10−7 | s−1 | Naf1-inducible mRNA synthesis | Assumption |
| ncl1b | 0.5 | s−1 | Naf1 translation rate | Fitted |
| ncl1c | 0.0004 | s−1 | Naf1 mRNA degradation | Fitted |
| ncl1d | 0.0003 | s−1 | Naf1 degradation rate | Fitted |
| ncl1e | 0.0 | μms−1 | Naf1-constitutive mRNA synthesis | Assumption |
FIGURE 4.

Numerical solution of a two-compartment-based TNF-stimulated feed-forward NF-κB model. A, the schematic model represents both canonical and non-canonical pathways coupled by TRAF1 dynamics. n represents nuclear protein. Each reaction is represented by a kinetic parameter (Table 2). UNK: unknown NIK promoter. B, shown are kinetic time profiles of NF-κB-dependent TRAF1 and TRAF2 proteins in the canonical pathway. C, shown are kinetic time profiles of NF-κB-dependent p100 and its processing to p52; a time evolution of newly synthesized p100 is shown here that increases with time. The proteolytic processing of p100 leads to the generation of p52 (cytoplasmic), which then translocates into nucleus (p52n) using an import-export mechanism as mentioned in Table 2. D, shown are kinetic time profiles of the activation complex of TRAF1·NIK and its involvement in non-canonical activation; the inducible TRAF1-mediated NIK stabilization activates the p100 processing mechanism to produce p52. The nuclear p52 then binds to promoters of non-canonical genes such as Naf1 to initiate its transcription.
Consistent with our earlier model, the addition of new parameters did not alter the canonical NF-κB dynamics (24, 32). Notably, simulated kinetic profiles of NF-κB-induced expression of TRAF1 and TRAF2 proteins and the delay in p52 processing from newly synthesized p100 (Fig. 4, B and C) provided an in silico confirmation of our WB analysis (Fig. 1). The activation kinetics of non-canonical product Naf1 due to in silico formation of functional TRAF1·NIK complex by TRAF1 mediated NIK stabilization and subsequent p100 precursor processing mechanism (Fig. 4D) correlates with our experimentally observed data (Fig. 3). Thus, combining computational methods and biochemical assays has further developed the existing canonical model to capture TRAF1 dependent non-canonical NF-κB signaling dynamics. Sufficient correlation between experimental observables and modeling parameters demonstrated the mechanism of non-canonical gene expression in response to tonic TNF stimulation. This newly developed model allowed us to explore the signaling cross-talk between two NF-κB pathways.
TRAF1 and NIK Are Required for Non-canonical Gene Expression
To further establish the significance of TRAF1-mediated NIK stabilization on TNF-induced p52 formation and NF-κB-dependent gene expression, we first performed in silico knock-out (iKO) simulations of TRAF1 or NIK genes, individually. The iKO of TRAF1 showed that p100 processing to p52 is completely abrogated, although p100 synthesis itself remains unaffected (Fig. 5A). Likewise, NIK stabilization is also reduced, resulting in undetectable levels of the TRAF1·NIK complex. As a result, TRAF1 iKO suppressed non-canonical pathway activation with no effect on canonical pathway. To confirm these in silico predictions, TRAF1 was experimentally knocked down by siRNA transfection. No detectable TRAF1 was observed at any time points as compared with control siRNA-transfected cells (Fig. 5B). To investigate if TRAF1 association with NIK affects its turnover rate, TRAF1 siRNA transfected cells were TNF-pre-treated for 3 h to induce TRAF1 expression followed by inhibition of protein synthesis using cyclohexamide (50 ng/ml) treatment for various times before cellular harvest. Blocking of new protein biosynthesis allowed us to measure the effects of TRAF1 depletion on NIK stability. A 3-h TNF stimulation time was chosen based on data derived from Fig. 1, C and D, to allow sufficient time for TRAF1 expression. A549 cells transfected with control siRNA served as a control. To overcome the challenge of low cellular abundance of NIK, it was immunoprecipitated from these cell lysates and quantitated by SRM. The SRM assay yields linear responses over a >20,000-fold concentration range with a strong linear correlations (r2 > 0.9991) (data not shown). SRM analysis showed a 10-fold lower abundance of basal NIK levels in TRAF1-depleted cells as compared with controls (Fig. 5C). However, after 80 min of cyclohexamide exposure, NIK levels were undetectable in TRAF1-silenced cells, whereas NIK protein expression levels remained unchanged in response to cyclohexamide treatment. These results indicate that TRAF1 is required for stabilizing NIK.
FIGURE 5.

TRAF1 is important for the activation of TNF-induced Naf1 and NF-κB2. A, shown is in silico iKO simulation of TRAF1. A simulation of p52 processing and subsequent non-canonical signaling was performed by in silico knockdown of transcription of TRAF1 by making n1a = 0.0. B, WB analysis shows TRAF1 knockdown in A549 cells treated with TNF for different times. C, TRAF1 knockdown increases NIK turnover. A549 cells stably expressing NIK were transfected with control or TRAF1-specific siRNA (100 nm) for 72 h followed by TNF (25 ng/ml) for 3 h before cyclohexamide (50 ng/ml) addition for various times as indicated. Immunoprecipitated NIK from these cells was analyzed for abundance of NIK by SRM and expressed as native/SIS ratio as described under “Experimental Procedures.” D, TRAF1 expression is essential for p100 processing. Nuclear extracts from control and TRAF1 knockdown cells were subjected SRM analysis to quantify p52. Values were normalized β-actin and are expressed as % -fold change as compared with cells with no TNF treatment. E–H, A549 cells transfected with control (dark bars) or TRAF1 specific siRNA (light bars) were treated with TNF (25 ng/ml) for various time intervals. RNA isolated thereafter was used to quantify expression of A20, IL8, Naf1, and NF-κB 2 by Q-RT-PCR using specific primers. Data are expressed as -fold change as compared with untransfected cells after normalizing to internal controls, GAPDH. Data represent the mean ± S.D. of three independent experiments. Data were analyzed by two-way ANOVA with multiple comparison (time and siRNA treatment) and Tukey's post hoc test for significance between time intervals and the treatment groups. Significantly different from TNF (0 h)-treated samples: *, p < 0.001; significantly different from control siRNA-transfected samples: †, p < 0.05; ††, p < 0.001.
The critical step in non-canonical pathway activation is the partial carboxyl-terminal degradation of p100 to produce p52. Thus, to examine if TRAF1 requirement is critical for TNF-induced p100 processing, p52 abundance in nuclear extracts from control or TRAF1-silenced A549 cells were determined by SRM assay. The high-responding signature peptide for each target protein is listed in Table 1. Because the basal NIK levels varies in control and TRAF1 knockdown cells, p52 abundance measurements were expressed as -fold change from the respective starting p52 levels to reflect the component of inducible p100 processing. Consistent with Fig. 1B, 3 h of TNF treatment resulted in an 8-fold increase in p52 protein abundance in control cells as compared with cells without TNF exposure (Fig. 5D). However, in TRAF1-silenced cells, TNF stimulation did not result in p52 production. These results clearly suggest that TRAF1-mediated NIK stabilization is essential for TNF-induced p100 processing. The positive role of TRAF1 in regulating the non-canonical pathway was further emphasized by the fact that knocking down TRAF1 blocks p100 processing in Hodgkin disease-derived cells (25). Thus, TRAF1 could be a negative regulator of the canonical pathway, whereas it positively regulates non-canonical pathway activation via NIK stabilization.
We and others have reported that TNF-induced NF-κB-dependent genes have at least three different expression profiles in epithelial cells termed early, middle, and late response genes, peaking 1, 3, and 6 h after stimulation, respectively (8, 19, 33). Although the earlier group was responsive to pulsatile TNF stimulation, the later was not and required tonic TNF stimulation (8). Thus, the effect of TRAF1 knockdown on the expression of TNF-induced genes with early and late kinetics was measured. Q-RT-PCR was used to measure expression of TNF-induced canonical (TNFAIP3/A20, CXCL8/IL8) and non-canonical (ABIN1/Naf1, NF-κB2) genes. We observed that TRAF1 knockdown had no effect on TNF-induced expression of A20 or IL8 (Fig. 5, E and F). By contrast, the 10-fold induction of Naf1 was significantly reduced to 2.5-fold in the absence of TRAF1 after 6 h of TNF treatment (Fig. 5G). A similar effect was observed on NF-κB2, where the 25-fold induction of NF-κB2 gene expression was reduced to 5-fold in the absence of TRAF1 (Fig. 5H). Furthermore, we investigated the requirement for NIK on TNF-induced NF-κB-dependent gene expression. Similar to TRAF1 iKO, NIK iKO also abolishes non-canonical gene expression without effecting canonical signaling events (Fig. 6A). These simulations were further validated by siRNA-mediated NIK knockdown in A549 cells. Control or NIK siRNA-transfected cells were stimulated with TNF, and steady state NIK levels were quantified using Western immunoblot. Relative to scrambled siRNA controls, transfection with 50 and 100 nm NIK-specific siRNA reduced NIK protein expression by ∼90% after 72 h in Western immunoblot (Fig. 6B). The expression of TNFAIP3/A20 and CXCL8/IL8 was not detectably affected (Fig. 6, C and D). Importantly, NIK silencing produced a dramatic reduction in TNF-inducible ABIN1/Naf1 and NF-κB2 gene expression, most significantly at 6 h of stimulation, reducing Naf1 expression from 14- to 4-fold and NF-κB2 from 17- to 7-fold (Fig. 6, E and F). These data suggest that TNF-induced expression of Naf1 and NF-κB2 genes are TRAF1- and NIK-dependent. By contrast, both TRAF1 and NIK are dispensable for the canonical pathway-controlled A20 and IL8.
FIGURE 6.

NIK is essential for optimum induction of a set of TNF-induced late responsive gene expression. A, iKO simulation of NIK is shown. The n3a parameter represents the NIK transcription rate in the system, which plays an important role in feed-forward signaling. A simulation of p52 processing and subsequent non-canonical signaling was performed by in silico knockdown of transcription of NIK by making n3a = 0.0. B, á Western blot shows NIK knockdown in A549 cells by 50 and 100 nm NIK-specific siRNA as compared with the cells transfected with nonspecific control siRNA. β-Actin in the lower panel serves as a loading control. C–F, A549 cells transfected with control (dark bars) or NIK specific siRNA (light bars) were treated with TNF (25 ng/ml) for various time intervals. RNA isolated thereafter was used to quantify expression of A20, IL8, Naf1, and NF-κB2 by Q-RT-PCR using specific primers. Data are expressed as -fold change as compared with untransfected cells after normalizing to internal controls, GAPDH. Data represent the mean ± S.D. of three independent experiments and analyzed by two-way ANOVA with multiple comparison (time and siRNA treatment) and Tukey's post hoc test for significance between time intervals and the siRNA- treatment groups. Significantly different from TNF (0 h)-treated samples: *, p < 0.05; **, p < 0.001; significantly different from control siRNA-transfected samples: †, p < 0.01.
One of the proposed mechanisms for the differences in the kinetics of early and late NF-κB-responsive genes suggests differences in chromatin accessibility of RNA polymerase II on these promoters (19, 34); however, this is not consistent with our data. Using quantitative chromatin immunoprecipitation, we demonstrated that the early and late genes are accessed by NF-κB/RelA with similar kinetics (6).
Our data presented here indicate that TNF-induced delayed activation of the non-canonical NF-κB pathway depends upon a feed-forward mechanism activated by canonical NF-κB pathway-induced TRAF1 expression. Here we have shown how TNF-induced TRAF1 expression directly binds to NIK and disrupts TRAF2- and TRAF3-mediated degradation. In this way, the TRAF1·NIK complex couples the canonical and non-canonical pathways and activates the complete array of NF-κB-dependent genes with early and late expression kinetics. These data provide insight into how cells decode inflammatory signals into distinct genomic responses.
This work was supported, in whole or in part, by National Institutes of Health Grants AI062885 (to A. R. B.), HHSN268201000037C (NHLBI; to A. R. B.), 1U54RR02614 (University of Texas Medical Branch Clinical and Translational Science Award; to A. R. B.), and National Institute of Diabetes and Digestive and Kidney Diseases DK079053 (to S. C.). This work was also supported by American Heart Association Grant 0630100N (to S. C.).
- NIK
- NF-κB-inducing kinase
- TRAF
- TNF receptor-associated factor
- cIAP
- cellular inhibitor of apoptosis
- NHBE
- normal human bronchial epithelial
- Q-RT-PCR
- quantitative real time PCR
- SRM
- selected reaction monitoring
- WB
- Western blot
- SIS
- stable isotopically labeled peptide standards
- Dox
- doxycycline
- iKO
- in silico knock-out.
REFERENCES
- 1. Hayden M. S., Ghosh S. (2004) Signaling to NF-κB. Genes Dev. 18, 2195–2224 [DOI] [PubMed] [Google Scholar]
- 2. Donath M. Y., Shoelson S. E. (2011) Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11, 98–107 [DOI] [PubMed] [Google Scholar]
- 3. Aloisi F., Pujol-Borrell R. (2006) Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 6, 205–217 [DOI] [PubMed] [Google Scholar]
- 4. Bonizzi G., Karin M. (2004) The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 25, 280–288 [DOI] [PubMed] [Google Scholar]
- 5. Brasier A. R., Tian B., Jamaluddin M., Kalita M. K., Garofalo R. P., Lu M. (2011) RelA Ser-276 phosphorylation-coupled Lys-310 acetylation controls transcriptional elongation of inflammatory cytokines in respiratory syncytial virus infection. J. Virol. 85, 11752–11769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nowak D. E., Tian B., Jamaluddin M., Boldogh I., Vergara L. A., Choudhary S., Brasier A. R. (2008) RelA Ser-276 phosphorylation is required for activation of a subset of NF-κB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol. Cell. Biol. 28, 3623–3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vallabhapurapu S., Karin M. (2009) Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733 [DOI] [PubMed] [Google Scholar]
- 8. Tian B., Nowak D. E., Brasier A. R. (2005) A TNF-induced gene expression program under oscillatory NF-κB control. BMC Genomics 6, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choudhary S., Boldogh S., Garofalo R., Jamaluddin M., Brasier A. R. (2005) Respiratory syncytial virus influences NF-κB-dependent gene expression through a novel pathway involving MAP3K14/NIK Expression and nuclear complex formation with NF-κB2. J. Virol. 79, 8948–8959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Woronicz J. D., Gao X., Cao Z., Rothe M., Goeddel D. V. (1997) IκB kinase-β. NF-κB activation and complex formation with IκB kinase-α and NIK. Science 278, 866–869 [DOI] [PubMed] [Google Scholar]
- 11. Basak S., Kim H., Kearns J. D., Tergaonkar V., O'Dea E., Werner S. L., Benedict C. A., Ware C. F., Ghosh G., Verma I. M., Hoffmann A. (2007) A fourth IκB Protein within the NF-κB signaling module. Cell 128, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qing G., Xiao G. (2005) Essential role of IκB kinase α in the constitutive processing of NF-κB2 p100. J. Biol. Chem. 280, 9765–9768 [DOI] [PubMed] [Google Scholar]
- 13. Vallabhapurapu S., Matsuzawa A., Zhang W., Tseng P. H., Keats J. J., Wang H., Vignali D. A., Bergsagel P. L., Karin M. (2008) Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat. Immunol. 9, 1364–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zarnegar B. J., Wang Y., Mahoney D. J., Dempsey P. W., Cheung H. H., He J., Shiba T., Yang X., Yeh W. C., Mak T. W., Korneluk R. G., Cheng G. (2008) Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2, and TRAF3 and the kinase NIK. Nat. Immunol. 9, 1371–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin X., Mu Y., Cunningham E. T., Jr., Marcu K. B., Geleziunas R., Greene W. C. (1998) Molecular determinants of NF-κB-inducing kinase action. Mol. Cell. Biol. 18, 5899–5907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu P., Li K., Garofalo R. P., Brasier A. R. (2008) Respiratory syncytial virus induces RelA release from cytoplasmic 100-kDa NF-κB2 complexes via a novel retinoic acid-inducible gene-I·NF-κB-inducing kinase signaling pathway. J. Biol. Chem. 283, 23169–23178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Starkey J. M., Haidacher S. J., LeJeune W. S., Zhang X., Tieu B. C., Choudhary S., Brasier A. R., Denner L. A., Tilton R. G. (2006) Diabetes-induced activation of canonical and noncanonical nuclear factor-κB pathways in renal cortex. Diabetes 55, 1252–1259 [DOI] [PubMed] [Google Scholar]
- 18. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. METHODS 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 19. Hao S., Baltimore D. (2009) The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 10, 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mordmüller B., Krappmann D., Esen M., Wegener E., Scheidereit C. (2003) Lymphotoxin and lipopolysaccharide induce NF-κB-p52 generation by a co-translational mechanism. EMBO Rep. 4, 82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao Y., Widen S. G., Jamaluddin M., Tian B., Wood T. G., Edeh C. B., Brasier A. R. (2011) Quantification of activated NFκB/RelA complexes using ssDNA aptamer affinity-stable isotope dilution-selected reaction monitoring-mass spectrometry. Mol. Cell. Proteomics 10, M111.008771–M111.0087716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Keshishian H., Addona T., Burgess M., Mani D. R., Shi X., Kuhn E., Sabatine M. S., Gerszten R. E., Carr S. A. (2009) Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 8, 2339–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kalita M. K., Sargsyan K., Tian B., Paulucci-Holthauzen A., Najm H. N., Debusschere B. J., Brasier A. R. (2011) Sources of cell-to-cell variability in canonical nuclear factor-κB (NF-κB) signaling pathway inferred from single cell dynamic Images. J. Biol. Chem. 286, 37741–37757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lavorgna A., De Filippi R., Formisano S., Leonardi A. (2009) TNF receptor-associated factor 1 is a positive regulator of the NF-κB alternative pathway. Mol. Immunol. 46, 3278–3282 [DOI] [PubMed] [Google Scholar]
- 26. Tian B., Zhang Y., Luxon B. A., Garofalo R. P., Casola A., Sinha M., Brasier A. R. (2002) Identification of NF-κB-dependent gene networks in respiratory syncytial virus-infected cells. J. Virol. 76, 6800–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. He L., Grammer A. C., Wu X., Lipsky P. E. (2004) TRAF3 forms heterotrimers with TRAF2 and modulates its ability to mediate NF-κB activation. J. Biol. Chem. 279, 55855–55865 [DOI] [PubMed] [Google Scholar]
- 28. Zheng C., Kabaleeswaran V., Wang Y., Cheng G., Wu H. (2010) Crystal structures of the TRAF2·cIAP2 and the TRAF1·TRAF2·cIAP2 complexes. Affinity, specificity, and regulation. Mol. Cell 38, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim J. Y., Morgan M., Kim D. G., Lee J. Y., Bai L., Lin Y., Liu Z. G., Kim Y. S. (2011) TNFα-induced noncanonical NF-κB activation is attenuated by RIP1 through stabilization of TRAF2. J. Cell Sci. 124, 647–656 [DOI] [PubMed] [Google Scholar]
- 30. Song H. Y., Régnier C. H., Kirschning C. J., Goeddel D. V., Rothe M. (1997) Tumor necrosis factor (TNF)-mediated kinase cascades. Bifurcation of nuclear factor-κB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc. Natl. Acad. Sci. U.S.A. 94, 9792–9796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luftig M. A., Cahir-McFarland E., Mosialos G., Kieff E. (2001) Effects of the NIK aly Mutation on NF-+ B Activation by the Epstein-Barr Virus Latent Infection Membrane Protein, Lymphotoxin + Receptor, and CD40. J. Biol. Chem. 276, 14602–14606 [DOI] [PubMed] [Google Scholar]
- 32. Lipniacki T., Paszek P., Brasier A. R., Luxon B., Kimmel M. (2004) Mathematical model of NF-κB regulatory module. J Theor. Biol. 228, 195–215 [DOI] [PubMed] [Google Scholar]
- 33. Tian B., Nowak D. E., Jamaluddin M., Wang S., Brasier A. R. (2005) Identification of direct genomic targets downstream of the nuclear factor-κB transcription factor mediating tumor necrosis factor signaling. J. Biol. Chem. 280, 17435–17448 [DOI] [PubMed] [Google Scholar]
- 34. Hargreaves D. C., Horng T., Medzhitov R. (2009) Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138, 129–145 [DOI] [PMC free article] [PubMed] [Google Scholar]