

To gain new insights into p53 biology, Kenzelmann Broz et al. used high-throughput sequencing to analyze global p53 transcriptional networks in primary mouse embryo fibroblasts in response to DNA damage. This approach identified autophagy genes as direct p53 target genes. p53-induced autophagy was important for both p53-dependent apoptosis and transformation suppression by p53. These data highlight an intimate connection between p53 and autophagy and suggest that autophagy contributes to p53-dependent apoptosis and cancer suppression.
Keywords: p53, ChIP-seq, RNA-seq, tumor suppression, autophagy
Abstract
The mechanisms by which the p53 tumor suppressor acts remain incompletely understood. To gain new insights into p53 biology, we used high-throughput sequencing to analyze global p53 transcriptional networks in primary mouse embryo fibroblasts in response to DNA damage. Chromatin immunoprecipitation sequencing reveals 4785 p53-bound sites in the genome located near 3193 genes involved in diverse biological processes. RNA sequencing analysis shows that only a subset of p53-bound genes is transcriptionally regulated, yielding a list of 432 p53-bound and regulated genes. Interestingly, we identify a host of autophagy genes as direct p53 target genes. While the autophagy program is regulated predominantly by p53, the p53 family members p63 and p73 contribute to activation of this autophagy gene network. Induction of autophagy genes in response to p53 activation is associated with enhanced autophagy in diverse settings and depends on p53 transcriptional activity. While p53-induced autophagy does not affect cell cycle arrest in response to DNA damage, it is important for both robust p53-dependent apoptosis triggered by DNA damage and transformation suppression by p53. Together, our data highlight an intimate connection between p53 and autophagy through a vast transcriptional network and indicate that autophagy contributes to p53-dependent apoptosis and cancer suppression.
p53 is a critical tumor suppressor, as evidenced by both sporadic and inherited human cancers with p53 mutations and the completely penetrant tumor predisposition of p53-null mice (Kenzelmann Broz and Attardi 2010; Olivier et al. 2010). p53's capacity to suppress tumor development is thought to relate to its function as a cellular stress sentinel, in which it responds to diverse stress signals—including DNA damage, oncogene expression, and nutrient deprivation—by promoting cell cycle arrest or apoptosis to prevent the propagation of damaged or compromised cells (Vousden and Lu 2002; Vousden and Prives 2009). At the molecular level, p53 acts primarily as a transcription factor, inducing a network of genes that contributes to its biological responses (Bieging and Attardi 2012).
The precise cellular response triggered by p53 is cell type-dependent but also relates to the intensity and type of activating signal (Vousden and Lu 2002; Vousden and Prives 2009). For example, cells sustaining extensive DNA damage may undergo either a p53-driven permanent cell cycle arrest known as senescence or p53-dependent apoptosis, both of which will lead to the functional elimination of damaged cells from tissues. Alternatively, cells sustaining limited DNA damage may undergo a temporary p53-mediated cell cycle arrest, affording cells an opportunity to repair damaged DNA, and in such a capacity, p53 can act as a survival factor. p53's ability to promote cell survival is not limited to genotoxic stresses, as wild-type cells subjected to glucose starvation engage a metabolic checkpoint and arrest, while p53-deficient cells fail to arrest upon such nutrient deprivation and undergo apoptosis (Jones et al. 2005). Hence, in some settings, p53 can preserve viability in response to stress signals.
The ability of p53 to respond to DNA damage is thought to be an ancient function, since it is shared by all members of the p53 family, comprising p53 and the related transcription factors p63 and p73. In primitive organisms, a p63/p73 ancestor protects germline integrity by inducing apoptosis in response to DNA damage, and this role is conserved in flies, worms, and mice, where p63 is the key driver of oocyte death upon irradiation (Belyi et al. 2010; Rutkowski et al. 2010). Although each family member ultimately specialized, with p53 action being central for tumor suppression and p63 and p73 for promoting stratified epithelial development and nervous system function, respectively, the presence of a p53 family member in single-cell eukaryotes such as choanoflagellates suggests that there are important functions of this family beneficial to unicellular organisms. The ancestral functions of the p53 superfamily, however, remain incompletely understood.
Genome-wide studies of p53 transcriptional responses have been used successfully to gain novel insights into p53 biology. For example, an early study using chromatin immunoprecipitation (ChIP) paired end ditag (ChIP-PET) technology defined a list of novel p53 target genes involved in a variety of functions and whose expression correlated with clinical outcome in human cancer patients (Wei et al. 2006). More recently, a p53 ChIP array study in mouse embryonic stem cells (ESCs) uncovered a role for p53 in regulating differentiation through the induction of Wnt pathway genes (Lee et al. 2010). In addition, global genomic profiling of mouse ESCs was used to elaborate a dual mechanism for p53 regulation of self-renewal and pluripotency by showing that p53 binding in promoter sites correlates with activation of differentiation gene expression, while p53 binding at more distant sites interferes with distal enhancer activity to repress expression of stem cell-related genes (Li et al. 2012).
Here, to gain new insights into p53 function, we leveraged the state-of-the-art genomic techniques of ChIP sequencing (ChIP-seq) and RNA sequencing (RNA-seq) to characterize p53 transcriptional responses to DNA damage signals in a global and unbiased manner. We focused on the response of primary fibroblasts to DNA damage to best understand how p53 behaves in differentiated normal cells. Interestingly, we identified a host of autophagy genes induced by p53 in conjunction with its family members. The induction of the autophagy transcriptional program in response to p53 activation correlates with increased levels of autophagy in a variety of settings, and we show that autophagy is important for p53-dependent apoptosis and transformation suppression. Collectively, these studies suggest that transcriptional activation of this extensive autophagy network is a fundamental component of p53 biological function.
Results
ChIP-seq in DNA damage-treated primary mouse embryo fibroblasts (MEFs) reveals myriad p53-bound sites
MEFs represent an important primary cell model system and have been widely used to discover many aspects of p53 function, including its roles in apoptosis and cell cycle arrest as well as the molecular pathways through which p53 acts in these contexts (Kastan et al. 1992; Debbas and White 1993; Lowe and Ruley 1993). To gain new insights into p53 biology, we aimed to obtain a genome-wide profile of genes bound and regulated by p53 in MEFs using high-throughput ChIP-seq and RNA-seq techniques (Fig. 1A). Beyond being primary cells in which the biology of p53 responses is very well characterized, MEFs afford the possibility of cleanly assessing the p53 dependence of gene expression and biological responses through the comparison of wild-type and p53-deficient MEFs derived from littermate wild-type and p53−/− embryos. While wild-type MEFs treated with the DNA double-strand break inducer doxorubicin undergo permanent G1 arrest and senescence, p53−/− MEFs fail to do so and ultimately die, likely from mitotic catastrophe (Brown and Attardi 2005). Importantly, the availability of robust polyclonal antibodies directed against full-length mouse p53 provided a means to ensure isolation of all p53 isoforms bound to DNA.
Figure 1.

Genome-wide analysis of p53 binding in response to DNA damage. (A) Experimental outline. Wild-type and p53−/− MEFs were derived from embryonic day 13.5 (E13.5) embryos and either left untreated or treated with doxorubicin to induce DNA damage. RNA extracted from these cells was used for expression profiling by RNA-seq (3SEQ), and chromatin was used for genome-wide analysis of p53 binding to DNA by ChIP-seq. The overlap in genes identified from these analyses will reveal direct p53 target genes. (B) Genomic location of the p53 ChIP-seq peaks with respect to annotated genes. (C) p53 consensus motif derived from MEME de novo motif analysis compared with the TRANSFAC p53 matrix. Our motif shows a strong preference for the “core” C and G but more variability in the A and T residues in the “CATG” core as well as more degeneracy in the flanking nucleotides. (D) Distribution of spacer lengths between p53 response element half-sites, suggesting that many p53-binding sites have spacers >3 nt. (E) Biological process GO terms enriched in our p53-bound gene data set, with P-values as calculated by DAVID using a modified Fisher exact test. Percentages of genes bound by p63 are indicated, as determined by analysis of a published p63 ChIP-seq data set (Kouwenhoven et al. 2010).
To derive a global profile of genes directly activated and repressed by p53, we chose to map p53-binding sites at relatively early time points after p53 activation after 6 h of doxorubicin treatment, when p53 was stabilized and target gene induction was observed (Supplemental Fig. 1A,B). Our p53 ChIP-seq analysis of wild-type MEFs, with p53−/− MEFs as a negative control, identified 4785 p53-bound sites at a false discovery rate (FDR) of 0.02% (Supplemental Table 1). By associating these sites with specific RefSeq genes if the binding site was within 10 kb of the gene, we identified 3193 genes with p53-binding sites, which we term “p53-bound genes” (Supplemental Table 2). In addition, we found a significant number of p53-bound sites >10 kb from the annotated genes. Our study, whose comprehensive nature is underscored by the identification of a high percentage of classical p53 target genes involved in a wide array of functions, thus identifies a large set of p53-bound genes (Supplemental Fig. 1C,D; Riley et al. 2008). A replicate experiment produced similar results. Quantitative PCR (qPCR) analyses of randomly selected individual binding sites, with enrichments spanning from just above the enrichment cutoff to the most enriched peaks, validated virtually all of these (34 out of 35) (Supplemental Fig. 1E).
The majority of p53-bound sites are localized near or within genes (Fig. 1B). Most of these are either within 10 kb upstream of the transcription start site (13.8%) or within introns (41.7%), consistent with previous studies showing that p53-binding sites are commonly found in introns of target genes (Wei et al. 2006; Smeenk et al. 2011). In addition, a significant percentage (31.1%) of p53-bound sites lie >10 kb from annotated genes, likely representing distal regulatory sites for known genes or more local regulatory elements for novel intergenic genes such as noncoding RNAs (ncRNAs). Analysis of conservation in vertebrates shows that genomic DNA sequences located within both gene-proximal and intergenic p53-bound sites are more conserved than flanking sequences (Supplemental Fig. 2A), indicating the potential functional significance of these sites.
To determine whether our peaks contain p53 response elements, we performed de novo motif analysis using MEME with the 200 bases flanking the peak coordinates and found that our binding sites are indeed enriched for p53 response elements. Furthermore, using MEME with sequences from the 1000 most highly enriched peaks, we generated an “ideal” p53 consensus motif, which we found resembled the known p53 response element in the Transfac database comprising two half-sites of 10 nucleotides (nt) (Fig. 1C). Using our p53 matrix, we then scanned all ChIP-seq peaks for p53-binding sites, accounting for the spacer of 1–13 nt by separating the two half-sites and scanning them individually. Clear p53 response elements were present in 3152, or 65.9%, of our ChIP-seq peaks using a P-value of 0.05, with little difference in frequency between gene-associated and intergenic peaks (Supplemental Fig. 2B). In addition, the total of 6461 matrix scan hits in all peaks indicates that many peaks contain multiple p53 response elements. While the majority of p53-bound sites contain p53 response elements recognized by the p53 matrix, the remaining p53-bound sites still contain less highly conserved p53 motifs, as indicated by MEME analysis. Interestingly, although the majority of p53 response elements have either no spacer (35%) or a spacer of 1 base pair (bp) (9.5%), longer spacers of 3–13 nt are detected in a significant fraction of ChIP-seq peaks, which together represent a large fraction of p53 response elements (Fig. 1D). Moreover, the variability both within the sequence of the p53 response element and in spacer length poses challenges to the computational prediction of p53-binding sites and highlights the need for biological binding data to define p53-binding sites.
To obtain a global overview of the biological functions of proteins encoded by p53-bound genes, we performed a functional annotation using DAVID (Database for Annotation, Visualization, and Integrated Discovery) analysis for Kyoto Encyclopedia of Genes and Genomics (KEGG) pathways and biological processes gene ontology (GO) terms (Fig. 1E; Supplemental Fig. 2C; Supplemental Tables 9,10). Beyond the pathways and biological processes previously associated with p53, such as “regulation of apoptosis” and “blood vessel development,” we observed enrichment for genes involved in pathways and biological processes not clearly related to p53 function, including “regulation of small GTPase-mediated signal transduction,” “regulation of cell size,” and “vesicle-mediated transport” (Fig. 1E; Teodoro et al. 2006). Interestingly, we noted many functions relevant to the developmental roles of the p53 family members p63 and p73, such as “cell adhesion” and “neuron differentiation,” respectively. Indeed, we found that 61.8%–82.3% of p53-bound genes were also bound by p63 in a p63 ChIP-seq data set of human keratinocytes, suggesting a considerable overlap in genes bound by p53 family members (Fig. 1E; Kouwenhoven et al. 2010).
Combined ChIP-seq and RNA-seq analysis to identify direct, p53-regulated target genes
To determine which p53-bound genes are also transcriptionally regulated by p53, we performed RNA-seq of wild-type and p53−/− MEFs either left untreated or treated with doxorubicin for 6 h (Fig. 1A). Using the R library DEseq, we identified 1323 doxorubicin-regulated, p53-dependent genes (Supplemental Table 3), of which 432 (32.65%) are bound by p53, thus likely representing direct p53 targets (Fig. 2A; Supplemental Tables 4,6; Anders and Huber 2010). Strikingly, of the 3193 p53-bound genes, only 13% appear regulated by p53 in response to DNA damage. While some of these p53-bound genes may not clearly be regulated at such an early time point, in other cases, p53 binding alone may be insufficient for transcriptional regulation. Therefore, genes may be poised for p53-dependent regulation pending the recruitment of cell type-specific or stress response-specific cofactors to induce their expression. Of the p53-dependent, DNA damage-regulated genes, 69.39% are induced and 30.61% are repressed (Fig. 2A), while of the 432 p53-bound and p53-dependent, DNA damage-regulated genes, most (84.49%) are induced and only 15.55% are repressed. This finding indicates that either direct repression is not a major component of the global p53 transcriptional program or fewer repressed genes were identified because of the biological variability that we observed with repression. Beyond verifying that our list of 432 genes is highly enriched for direct p53 target genes such as Cdkn1a and Bbc3/Puma, we identified a set of new p53 target genes, including those that we described previously as novel p53 tumor suppression-associated target genes, such as Abhd4 and Crip2 (Supplemental Fig. 3A,B; Brady et al. 2011). Other p53 target genes, such as Apaf1 and Zmat3/Wig1, did not pass our statistical criteria, indicating that some bona fide p53 targets may be missed in this analysis due to the early time point or biological variation.
Figure 2.

p53-bound and regulated genes in response to DNA damage. (A, left) Overlap between all p53-bound genes and genes differentially expressed in a p53-dependent manner in response to DNA damage (DD), with the overlap defining a set of 432 p53-bound and regulated, direct p53 target genes. (Right) Summary of percentages of induced and repressed p53-dependent, DNA damage-regulated genes and p53-bound and p53-dependent DNA damage-regulated genes. (B) Biological process GO terms enriched in our p53-bound and regulated gene data set, with P-values as calculated by DAVID using a modified Fisher exact test. (C) Lists of genes within the categories shown in B, with fold changes in wild-type MEFs in response to DNA damage (FC DD) and adjusted P-values (pval adj) derived from DEseq analysis. Repressed genes are highlighted in gray. Inf denotes infinitely induced genes, reflecting lack of expression in untreated cells.
To identify novel biological processes regulated by p53, we subjected the list of 432 bound and regulated genes to DAVID analysis (Fig. 2B,C; Supplemental Tables 11,12). Our gene set was enriched for several gene ontology (GO) terms related to cell cycle arrest and programed cell death, classical p53 responses, thus validating our list. Intriguingly, we found that Ppp1r13l/iAspp, which encodes a protein that binds p53 and inhibits its apoptotic function, is a direct p53 target gene (Supplemental Fig. 3C; Sullivan and Lu 2007). Moreover, we found that p53 binds and up-regulates another of its important negative regulators, Mdm4, which was only recently reported to be a direct p53 target gene (Li et al. 2010). These observations suggest novel aspects of p53 negative feedback regulation. In addition, Dffb, encoding an endonuclease responsible for DNA fragmentation and chromatin condensation during apoptosis, is p53-bound and induced in response to DNA damage, indicating that p53 regulates genes at all levels of the apoptotic pathway, from induction of apoptosis to DNA fragmentation. Thus, this data set implicates new players in various aspects of p53 function and regulation.
Interestingly, there are multiple enriched GO terms related to the negative regulation of growth or cell size, a relatively unexplored function for p53 that would integrate well with known p53 functions (Supplemental Tables 10, 12; Figs. 1E, 2B,C). For example, while cells are undergoing cell cycle arrest, there is no need for cell growth, and p53 might help to ensure that resources are used for survival or DNA repair. The identification of Ulk1, which encodes a key initiator of macroautophagy (referred to as autophagy hereafter)—a major pathway for cellular quality control and energy production through the degradation of long-lived proteins and organelles—suggested that p53 might directly activate genes encoding autophagy core machinery components. In addition, this category includes the known p53 target genes Ei24 and Bbx/Puma, which have been associated with autophagy (Yee et al. 2009; Tian et al. 2010). Together, these findings suggested the possibility that the direct activation of central autophagy pathway genes is part of the transcriptional program contributing to p53 function.
In addition to p53-dependent DNA damage-regulated genes, we also derived a list of 1269 genes differentially regulated between unstressed wild-type and p53−/− cells, termed basal p53-regulated genes (Supplemental Table 7). Again, of the 3193 p53-bound genes, only a subset—253, or 7.9%—are regulated by p53 at basal levels (Supplemental Fig. 4A). Percentages of genes induced or repressed in wild-type MEFs relative to p53−/− MEFs are similar for all basal p53-regulated genes (44.92% and 55.08%, respectively), while in the p53-bound group, a higher percentage of genes is induced in the presence of p53 (62.45%) than repressed (37.55%), suggesting a more prominent role for p53-dependent activation of gene expression in unstressed cells, as during the DNA damage response. To identify biological processes regulated by p53 in unstressed cells, we performed DAVID analysis (Supplemental Fig. 4B; Supplemental Tables 13,14). The p53-bound and basal p53-regulated gene set was enriched for KEGG pathways and GO terms related to apoptosis, proliferation, cell growth, and blood vessel development similar to the p53-bound and p53-dependent DNA damage-regulated genes. In addition, we also identified KEGG pathways and GO terms related to cell migration, such as “focal adhesion,” “cell projection organization,” and “cytoskeleton organization,” suggesting that regulation of these processes by p53 might be more prominent in unstressed cells than in DNA damage-treated cells.
p53 binds to a host of autophagy genes
Autophagy is a complex process in which cytosolic contents, including proteins and organelles, become engulfed by nascent vesicles called autophagosomes and degraded after fusion with lysosomes (Kroemer et al. 2010). The activation and execution of autophagy thus require the coordinated action of myriad proteins. To further explore the idea that p53 may regulate the autophagy machinery, we closely inspected our p53-bound gene list for other key autophagy genes. In fact, we discovered p53-bound genes encoding proteins involved in multiple steps of autophagy, including upstream regulators of the pathway, core machinery components, and lysosomal proteins (Fig. 3A,B; Supplemental Fig. 5A,B). Upstream of autophagy, both the mTor and Ampk pathways comprise components encoded by previously known p53 target genes, including Tsc2, Ddit4/Redd1, Foxo3a, and the β1 and β2 subunits of Ampk (Ellisen et al. 2002; Feng et al. 2007). In our list of p53-bound genes, we identified three additional genes encoding components of the Ampk or PI3K pathways: Stk11/Lkb1, the upstream activator of Ampk and a tumor suppressor; Prkag2, the γ-subunit of Ampk; and Pik3r3, a phosphoinositide-3-kinase regulatory subunit in the PI3K pathway. Interestingly, in addition to Ulk1, we also identified a set of autophagy core machinery-encoding genes, including Atg2b, Atg4a, Atg4c, Atg7, Atg10, Tmem49/Vmp1, Ulk2, and Uvrag, as p53-bound. Furthermore, we found that several lysosomal protein-encoding genes—including the established p53 target gene Ctsd and the genes Laptm4a, Tpp1, and Vmp4—are bound by p53 (Wu et al. 1998). As most of these p53-bound autophagy genes did not initially appear in our list of p53-bound and regulated targets, we tested whether they might be regulated at a later time point than our RNA-seq analysis by performing qRT–PCR analyses after 24 h of doxorubicin treatment. We found that genes for all autophagy components are indeed induced by doxorubicin in wild-type cells, similarly to p21 (Fig. 3C; Supplemental Fig. 5C). Interestingly, we found that the induction of a subset of these genes, such as Atg2b and Ulk1, was partially compromised in p53−/− MEFs, while others were induced normally, suggesting that their expression is not strictly p53-dependent.
Figure 3.
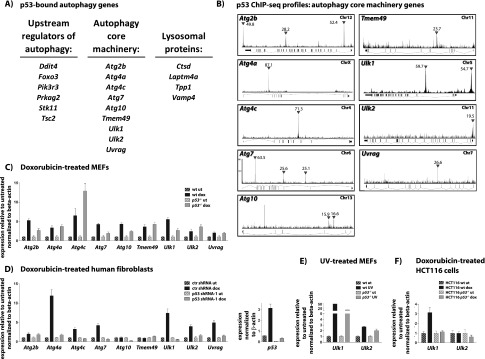
p53 regulates a program of autophagy genes. (A) Lists of p53-bound genes encoding proteins involved in the upstream regulation of autophagy, autophagy core machinery function, and lysosomal function. Peak enrichment for Atg10 and Ulk2 is slightly below our threshold for peak calling at 20, but they were still judged reliable upon inspection of the ChIP-seq binding profile and were therefore included in our list. (B) p53 ChIP-seq profiles showing p53 binding to autophagy core machinery component-encoding genes. The schematics show the p53 ChIP-seq profile and the gene organization, with the direction of transcription indicated by the orientation of the exon connectors: upward for the 5′ end of gene to the left, and downward for the 5′ end of gene to the right. Inverted triangles mark “called” peaks, and the numbers indicate ChIP enrichment. (C) qRT–PCR analysis of wild-type (wt) and p53−/− MEFs shows induction of p53-bound core autophagy genes after 24 h of doxorubicin treatment, with varying extents of p53-dependent contribution. Expression levels represent the average ± SD of technical triplicates after normalization to expression in untreated cells and to β-actin. (D, left) qRT–PCR analysis of human fibroblasts transduced with control (ctr) or p53 shRNAs shows p53-dependent induction of p53-bound core autophagy genes after 24 h of doxorubicin treatment. Expression levels represent the average ± SD of technical triplicates after normalization to expression in untreated cells and to β-actin. (Right) Analysis of average p53 expression levels ± SD of technical triplicates after normalization to β-actin confirms efficient p53 knockdown. (E) qRT–PCR analysis of Ulk1 and Ulk2 expression in UVC-treated wild-type and p53−/− MEFs after 24 h. Expression levels represent the average ± SD of technical triplicates after normalization to expression in untreated cells and to β-actin. (F) qRT–PCR analysis of Ulk1 and Ulk2 expression in doxorubicin-treated wild-type and p53-deficient HCT116 human colon carcinoma cells after 24 h. Expression levels represent the average ± SD of technical triplicates after normalization to expression in untreated cells and to β-actin.
We further evaluated the role of p53 in activating these autophagy targets by investigating their induction by p53 in other contexts. First, we assessed whether DNA damage also induces autophagy core genes in human cells using primary human fibroblasts transduced with either a control or a p53 shRNA-expressing construct. Most of these genes—including Atg4a, Atg7, Atg4c, Ulk1, Ulk2, and Uvrag—show clear p53-dependent induction in response to treatment with doxorubicin, like p21, suggesting that the activation of autophagy genes by p53 occurs in human cells (Fig. 3D; Supplemental Fig. 5D). These results were confirmed with a second p53 shRNA (Supplemental Fig. 5E). A small subset, including Atg10 and Tmem49, was not clearly induced by doxorubicin at this time point or had a p53-independent contribution to induction. Next, we examined the induction of Ulk1 and Ulk2, both of which are mammalian Atg1 homologs, as representative autophagy target genes in response to p53 activation by a different DNA damaging agent, UVC radiation, which induces apoptosis in MEFs. In addition, we tested p53-dependent induction of autophagy target genes in epithelial cancer cells using HCT116 wild-type or p53−/− colorectal cancer cells. In these contexts, we found that Ulk1 exhibited either complete or partial dependence on p53 for induction by DNA damage (Fig. 3E, F; Supplemental Fig. 5F,G). In contrast, we found that Ulk2 showed induction by UVC in MEFs but with minimal p53-dependence and that Ulk2 was not induced by doxorubicin in HCT116 cells, suggesting a cell type specificity for the induction of some autophagy genes by DNA damage. Collectively, these findings indicate that many p53-bound autophagy genes are inducible by DNA damage in both mouse and human cells, but the dependency on p53 displays both some gene-specificity and some cell type specificity.
p53 family members contribute to the regulation of the autophagy program
The fact that some p53 autophagy target genes maintained partial or complete DNA damage-dependent up-regulation in p53−/− MEFs suggested the possibility that other factors might compensate for the loss of p53. We considered the p53 family members p63 and p73, which are also activated in response to DNA damage and have a DNA-binding specificity similar to that of p53. We compared our p53 ChIP-seq data set with published genome-wide DNA-binding data sets for the p53 family members, including a p63 ChIP-seq data set in primary human keratinocytes (Kouwenhoven et al. 2010) and a p73 ChIP–Chip data set in human rhabdomyosarcoma cells (Rosenbluth et al. 2011). Indeed, many of the autophagy genes, including numerous core autophagy genes, were bound by p63 when these data were analyzed using the same parameters as for our ChIP-seq data analysis, despite differences in cell type, treatment, and species (Fig. 4A). Moreover, by comparing our list of p53-bound genes with the 3071 genes described in the p73 ChIP–Chip study, we found that a number of p53 autophagy targets are also bound by p73 in human cancer cells, including various core autophagy genes. These comparisons indicate that binding to autophagy genes is a shared function of the three p53 family members and occurs in different cell types and species. To test whether p53 family members contribute to the regulation of p53-bound autophagy genes, we used RNAi to attenuate p63 and p73 expression in wild-type and p53−/− MEFs and analyzed expression of autophagy genes and p21 after DNA damage by qRT–PCR. With some autophagy genes—including Atg4a, Atg7, and Atg10—as well as with p21, we noted some compromised induction by DNA damage in wild-type MEFs with knockdown of both p63 and p73 (Fig. 4B; Supplemental Fig. 6). Strikingly, the down-regulation of these p53 family members in p53−/− cells prevented efficient induction of all autophagy genes by doxorubicin. Collectively, these findings indicate that p53 and its family members can induce expression of a host of autophagy target genes in response to DNA damage. In the setting of p53-null MEFs, p63 and p73 appear to compensate for chronic p53 deficiency. In wild-type cells, p53 plays a more dominant role in autophagy gene induction, as suggested by the acute p53 knockdown experiments in human fibroblasts, but p53 family members can still contribute to the activation of autophagy genes by DNA damage.
Figure 4.
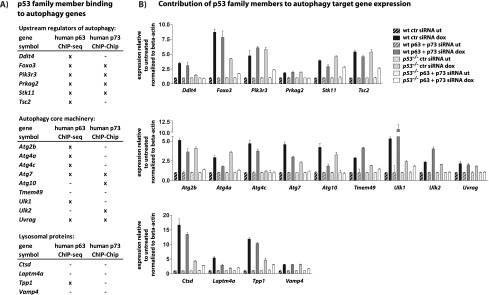
p53 family members contribute to regulation of the autophagy program. (A) Table showing which of the p53-bound autophagy genes are bound by p63, based on a ChIP-seq study in primary human keratinocytes (Kouwenhoven et al. 2010), and by p73, based on a ChIP–Chip study in human rhabdomyosarcoma cells (Rosenbluth et al. 2011). (B) qRT–PCR analysis of autophagy gene expression in untreated (ut) or doxorubicin-treated (dox) wild-type and p53−/− MEFs transfected with either control (ctr) or p63 and p73 siRNAs. Expression levels represent the average ± SD of technical triplicates after normalization to expression in untreated cells and to β-actin.
p53 induces autophagy in diverse contexts and in a transactivation-dependent manner
To determine whether the p53-driven induction of autophagy genes translates into increased levels of autophagy, we analyzed autophagy in response to p53 activation in diverse settings. First, we examined autophagy in wild-type MEFs upon p53 activation by DNA damage, verifying p53 activity by induction of the p53 target genes p21 and Ulk1 (Supplemental Fig. 7A). We measured levels of autophagy by immunoblotting for modified LC3 (LC3-II), which is incorporated into autophagic vesicles and thus correlates with levels of autophagy. To confirm that any enhanced LC3-II signal represents increased autophagic flux rather than a block in autophagy resulting from accumulation of autophagosomes, we also analyzed cells treated for 1 h with BafilomyinA1 (BafA1), an inhibitor of autophagic flux. We observed increased LC3-II levels upon DNA damage treatment, which further increased with BafA1 treatment, indicating that autophagic flux is up-regulated in response to DNA damage (Fig. 5A). Accordingly, accumulation of LC3 in discrete puncta was detected by immunofluorescence staining for endogenous LC3 in doxorubicin-treated wild-type MEFs (Fig. 5B). To further confirm the induction of autophagy by p53, we measured autophagy in response to genotoxic stress-independent activation of p53 by either treatment of wild-type MEFs with the Mdm2–p53 interaction antagonist Nutlin-3a or genetic activation of p53 expression in MEFs carrying conditional p53 alleles in which p53 expression is induced by Cre-mediated recombination of an upstream transcriptional STOP cassette (Olive et al. 2004; Brady et al. 2011). First, we observed that Nutlin-3a treatment enhanced p53 transcriptional activity, LC3-II levels by Western blot, and autophagosome formation by immunofluorescence, indicating that activation of p53 alone is sufficient to drive autophagy (Fig. 5A,B; Supplemental Fig. 7A). Second, using the genetic activation system, we infected p53LSL-wt/LSL-wt MEFs with either empty adenoviruses (Ad-empty), leaving the cells p53-null, or adenoviruses expressing Cre (Ad-Cre), to allow p53 expression. Western blot analysis and immunofluorescence confirmed efficient and widespread expression of p53 throughout the population (Fig. 5C; Supplemental Fig. 7B). We observed increased LC3-II with Ad-Cre infection relative to Ad-empty infection (Fig. 5C, lanes 1–4). Finally, to determine whether p53-dependent regulation of autophagy genes may be relevant in another cell type and in response to oncogenic signals, we examined autophagy levels in response to p53 activation in cells derived from lung tumors from KRasG12D-expressing mice carrying the inducible p53 allele. In this model, restored p53 induces cell cycle arrest but no significant apoptosis (Feldser et al. 2010). As with the MEFs, transduction of KRasG12D;p53LSL-wt/LSL-wt lung cancer cell lines with Ad-Cre to induce p53 expression enhanced autophagy relative to Ad-empty-infected cells (Fig. 5D,E; Supplemental Fig,. 7B). Induction of p21 and autophagy genes in response to p53 expression activated by Ad-Cre treatment was verified by qRT–PCR (Supplemental Fig. 7C). Taken together, our findings indicate that p53 activation promotes autophagy in response to a variety of stimuli, including DNA damage, oncogenic signaling, direct pharmacological activation by Nutlin-3a, and genetic activation. Thus, autophagy appears to be a core p53 response.
Figure 5.

Activation of p53 promotes autophagy in a transactivation-dependent manner. (A, top) Western blot analysis of p53-induced autophagy in wild-type MEFs in response to Nutlin-3a and DNA damage (dox) at 24 h using modified LC3 (LC3-II) levels as a marker for autophagic vesicles and BafA1 treatment to determine autophagic flux. One representative Western blot from at least two independent experiments using at least two independently derived primary MEF lines is shown. p53 is shown as a control, and β-actin (actb) serves as a loading control. (Bottom) Quantification of LC3-II levels normalized to β-actin levels. (B) Immunostaining for endogenous LC3 in wild-type MEFs treated with Nutlin-3a or DNA damage (dox), where the formation of autophagic vesicles is indicated by LC3 puncta. DAPI marks cell nuclei. (C, top) Western blot analysis of MEFs homozygous for conditional p53 alleles. Ad-Cre-induced excision of a transcriptional STOP cassette activated p53 expression, and Ad-empty was used as control. Autophagy induction in response to activation of wild-type p53 (lanes 1–4), a transactivation-dead p53 mutant (p5325,26,53,54; lanes 5–8), and a human cancer-derived p53 mutant (p53R270H; lanes 9–12) was assessed by LC3-II blotting 48 h after Ad-Cre infection. β-Actin (actb) serves as a loading control, and p53 expression in Ad-Cre-infected cells was verified by Western blot. One representative Western blot from three independent experiments is shown. (Bottom) Quantification of LC3-II levels normalized to β-actin levels. (D, top) Western blot analysis of KRasG12D lung cancer cells homozygous for conditional p53 alleles. Ad-Cre-induced excision of a transcriptional STOP cassette activated p53 expression, and Ad-empty was used as control. Autophagy induction in response to activation of wild-type p53 in two independent KRasG12D;p53LSL-wt/LSL-wt cell lines (lanes 1–8) and one p53−/− lung cancer cell line (lanes 9–12) was assessed by LC3-II blotting 48 h after Ad-Cre infection. β-Actin (actb) serves as a loading control, and p53 expression in Ad-Cre-infected cells was verified by Western blot. Shown is one representative Western blot from three independent experiments. (Bottom) Quantification of LC3-II levels normalized to β-actin levels. (E) Immunostaining for endogenous LC3 in lung cancer cells with (Cre) or without (empty) activation of a conditional p53 allele. (F) Heat map showing expression of p53-bound autophagy genes using microarray data generated from HrasV12;p53+/+ MEFs undergoing senescence or HrasV12 MEFs either lacking p53 (p53−/−) or homozygous for a transactivation-dead p53 mutant allele (p5325,26,53,54/25,26,53,54), both of which fail to undergo senescence. Induced genes appear red on the heat map, and repressed genes appear blue.
To determine whether the induction of autophagy is dependent on p53 transcriptional activity, as suggested by the p53-activated transcriptional program we uncovered, we used MEFs homozygous for a conditional knock-in allele encoding a p53 mutant with alterations in both transcriptional activation domains, p5325,26,53,54, which we previously showed to be transactivation-dead on a genome-wide level (Brady et al. 2011). Indeed, transcriptomic analyses in MEFs showed that while the p53-bound autophagy target genes are induced in MEFs with wild-type p53 compared with p53-null MEFs, the p5325,26,53,54 mutant fails to activate these genes (Fig. 5F). Accordingly, we found that, unlike wild-type p53, p5325,26,53,54 could not induce autophagy, suggesting that transcriptional activation is important for p53 to activate autophagy (Fig. 5C, lanes 5–8; Supplemental Fig. 7B). Consistent with this notion, the tumor-derived p53 mutant p53R270H, which has a mutation in the DNA-binding domain that abrogates binding to p53 response elements in DNA and thus prevents transactivation of p53 target genes, also fails to induce autophagy (Fig. 5C, lanes 9–12; Supplemental Fig. 7B). Collectively, these results highlight the importance of p53 transcriptional activity for the induction of autophagy, consistent with the requirement of p53-mediated activation of autophagy genes to drive the autophagy program.
Autophagy deficiency does not compromise DNA damage-induced cell cycle arrest and survival but impairs p53-dependent apoptosis
To determine the role of autophagy in the p53 pathway, we queried how autophagy contributes to p53 DNA damage responses in primary cells using MEFs conditionally deficient for the essential autophagy gene Atg5 (Hara et al. 2006). We confirmed efficient recombination of the Atg5fl/fl alleles by PCR (Supplemental Fig. 8A) and immunoblotting, where we noted a dramatic decrease in Atg5 protein levels (Fig. 6A; Supplemental Fig. 8B). Importantly, Atg5 deletion inhibited autophagy, as established by LC3-II Western blot analysis.
Figure 6.

Autophagy deficiency does not compromise DNA damage-induced cell cycle arrest and survival but impairs p53-dependent apoptosis and suppression of transformation. All experiments were performed with two independent Atg5fl/fl MEF cell lines and at least in duplicate. (A, top) Western blot analysis confirming decreased Atg5 protein levels and inhibition of autophagy as assessed by LC3-II 48 h after Ad-Cre (+Cre) or Ad-empty (−Cre) infection. β-Actin serves as a loading control. (Bottom) Quantification of Atg5 and LC3-II is shown relative to β-actin. (B) Graph showing the average S-phase ratios of γ-irradiated/untreated MEFs for each genotype. The P-value was calculated by the Student's t-test. (C) p53-dependent cell survival in primary MEFs upon exposure to DNA damage (dox) for 72 h. Shown are the average percentages ± SD of technical replicates of AnnexinV-negative cells from a representative experiment. (D) p53-dependent apoptosis in E1A;HrasV12 MEFs of different genotypes after 18 h of DNA damage treatment (dox). Shown are the average percentages ± SD of AnnexinV-positive cells from a representative experiment using two MEF lines per genotype. The P-value was calculated by the Student's t-test. (E) p53-dependent apoptosis in E1A;HrasV12;wild-type and E1A;HrasV12;Ulk1−/− MEFs transduced with negative control GFP shRNAs or Ulk2 shRNAs after 18 h of DNA damage treatment (dox). Shown are the average percentages ± SD of AnnexinV-positive cells from one representative of more than three independent experiments, with error bars indicating technical replicates, and P-values calculated by the Student's t-test. (F) Soft agar assay for p53-dependent suppression of transformation in E1A;HrasV12 MEFs. Quantification shows average colony number ± SD. The P-values were calculated by the Student's t-test. Shown are representative wells of 3-wk Giemsa-stained colonies from one experiment of four, each with technical triplicates.
In response to DNA damage, p53 can drive G1 arrest to allow cells to repair DNA damage before proceeding through the cell cycle, thereby limiting the propagation of oncogenic mutations. Importantly, the p53-dependent checkpoint response to DNA damage is also critical for cell survival, since upon extended exposure to DNA damage, wild-type cells arrest but remain viable, while p53−/− MEFs continue to proliferate, accumulate DNA damage, and ultimately die due to mitotic catastrophe (Brown and Attardi 2005). To determine whether autophagy might be important for this arrest and survival function of p53, we assessed G1 checkpoint function in the absence of autophagy by treating wild-type and Atg5-deficient MEFs with 5 Gy of γ-irradiation and analyzing cell cycle distribution by flow cytometry. Like wild-type MEFs, Atg5-deficient MEFs displayed an intact G1 checkpoint response, as determined by the ratio of the S-phase fraction in irradiated cells relative to untreated cells, contrasting with p53−/− MEFs, which failed to arrest (Fig. 6B; Supplemental Fig. 8B,C). To test whether autophagy contributes to cell survival in the face of prolonged DNA damage, we treated control and Atg5-deficient MEFs with doxorubicin for 72 h. We found that the Atg5-deficient MEFs are able to survive as well as the wild-type MEFs, unlike p53−/− MEFs (Fig. 6C). Together, these observations suggest that the role of autophagy downstream from p53 in this primary cell setting is not to promote survival upon genotoxic stress.
The other major p53 response to DNA damage is to stimulate apoptosis to cull damaged cells. To determine whether autophagy is involved in promoting p53-dependent apoptosis, we introduced the oncogenes E1A and HrasV12 into MEFs, which sensitize MEFs to undergo apoptosis rather than cell cycle arrest in response to DNA damage. Interestingly, we found that upon doxorubicin treatment, apoptosis in E1A;HrasV12;Atg5-deficient MEFs was significantly attenuated relative to E1A;HrasV12;wild-type MEFs, although not to the extent of E1A;HrasV12;p53−/− MEFs, which failed to undergo apoptosis (Fig. 6D; Supplemental Fig. 8D,E). Importantly, it is likely that we are underestimating the degree of apoptosis compromise with Atg5 deficiency, as we do not completely abolish Atg5 expression or autophagy (Supplemental Fig. 8D). These findings indicate that autophagy is required for robust p53-dependent apoptosis. Since autophagy-independent functions have been described for several Atg proteins, we confirmed our result with an independent genetic model of autophagy deficiency, cells depleted of Ulk1 and Ulk2, mammalian homologs of Atg1, and direct targets of p53 in our studies. We generated E1A;HrasV12;wild-type or E1A;HrasV12;Ulk1−/− MEFs expressing either GFP or Ulk2 shRNAs and verified reduced expression of Ulk1 and Ulk2 by qRT–PCR (Supplemental Fig. 8F). Indeed, we observed decreased doxorubicin-triggered, p53-dependent apoptosis in cells deficient for both Ulk1 and Ulk2, whereas either inactivation of Ulk1 or depletion of Ulk2 alone was insufficient to attenuate apoptosis (Fig. 6E). Collectively, these data indicate that in response to DNA damage, p53-induced autophagy does not mediate p53-dependent cell cycle arrest or survival but contributes to efficient p53-dependent apoptosis.
Autophagy deficiency compromises transformation suppression
The ability of p53 to induce apoptosis is critical for tumor suppression in some settings (Symonds et al. 1994; Schmitt et al. 2002). For example, oncogene-expressing MEFs with wild-type p53 are prone to apoptosis and grow poorly in transformation assays (Lowe et al. 1994; Soengas et al. 1999). Deficiency for either p53 or apoptosis genes relieves this block and enables efficient growth of oncogene-expressing MEFs in soft agar transformation assays and as tumors in immunocompromised mice. To determine whether autophagy contributes to the tumor suppressor function of p53 in this context, we tested whether autophagy deficiency promotes transformation in a well-established p53-dependent transformation suppression assay using MEFs expressing the oncogenes E1A and HrasV12. We compared E1A;HrasV12;wild-type and E1A;HrasV12;Atg5-deficient MEFs for growth in a soft agar assay, which tests for p53-dependent inhibition of anchorage-independent growth and serves as a surrogate of in vivo tumor growth. As expected, E1A;HrasV12;p53−/− MEFs yielded significantly more colonies than E1A;HrasV12;wild-type MEFs (Fig. 6F). Interestingly, we found that E1A;HrasV12;Atg5-deficient MEFs also produced more colonies than their wild-type counterparts, indicating that autophagy deficiency indeed promotes transformation. Notably, the colonies forming from autophagy-deficient cells do not appear as numerous and/or as large as those observed with p53 deficiency, consistent with the notion that p53 regulates processes other than autophagy that are compromised upon p53 loss, thus further enhancing colony formation. These findings demonstrate that autophagy contributes to p53-dependent suppression of transformation, underscoring an important novel mechanism by which p53 may generally suppress tumorigenesis.
Discussion
Here, to gain novel insights into p53 function, we used combined ChIP-seq and RNA-seq analyses to describe the genome-wide binding and direct regulation of genes by p53 in primary MEFs in response to DNA damage. Interestingly, our studies revealed numerous novel downstream components of p53 as well as a vast autophagy network activated by p53 and its family members. We showed that p53 induces autophagy in various contexts, including in response to DNA damage and direct activation by Nutlin-3a and after genetic activation, as well as in different cell types, including primary MEFs and lung cancer cell lines. The induction of autophagy relies on p53 transcriptional activity, consistent with a requirement for p53-mediated induction of the autophagy gene network we identified. Finally, we found that in the context of p53 activation, autophagy does not promote survival but instead contributes to efficient p53-dependent apoptosis and suppression of transformation.
Genome-wide profiling of p53 binding and transcriptional activity has been performed previously and led to several general insights (Wei et al. 2006; Smeenk et al. 2008, 2011; Lee et al. 2010; Botcheva et al. 2011; Li et al. 2012; Nikulenkov et al. 2012). First, p53 binds genes representing a wide variety of functional categories, including cell adhesion and axon guidance, and many of these can also be bound by the p53 family members p63 and p73 (Smeenk et al. 2008). Second, p53 ChIP-seq studies have shown that in cancer cells treated with different agents to induce either apoptosis or cell cycle arrest, total p53-binding profiles are remarkably similar despite transcriptional differences (Smeenk et al. 2011; Nikulenkov et al. 2012). This observation suggests that other factors, such as cofactors recruited in a cell type-specific and stress response-specific manner, are required for inducing changes in gene expression (Wei et al. 2006; Smeenk et al. 2011; Nikulenkov et al. 2012). Third, studies have found p53 response elements in p53-bound sites, indicating direct binding of p53, as well as several motifs for other transcription factors such as AP-1, Sp1, and Ets2 that may cooperate with p53 (Smeenk et al. 2008). Our study in the MEF primary cell system also indicates that p53 binds to genes involved in a vast array of biological functions, many of which are also bound by the p53 family members. While only a subset of the p53-bound genes is regulated in response to DNA damage in MEFs, others may be poised for regulation in a different cell type or in response to a different stress. We found p53 response elements in a majority of p53-bound sites, although searching for the half-sites separately allowed us to identify a large fraction of p53 response elements with spacers >1 nt, indicating that there is more variability in spacer length than previously anticipated and that binding data, rather than bioinformatic prediction, are important for defining p53-binding sites in the genome. In addition, our study significantly extends the previous studies by unveiling a vast autophagy program regulated by p53 (Fig. 7A).
Figure 7.

Summary and model. (A) Schematic drawing of the autophagic process, showing proteins involved in different steps of autophagy. Autophagy core proteins encoded by genes that we identified as p53-bound are highlighted in red. Induction of autophagy is regulated by mTor and Ampk, which phopshorylate the Ulk1 complex to regulate autophagy. Subsequently, a Beclin-1 complex induces formation of the phagophore by recruiting the LC3 conjugation machinery. Two ubiquitin-like conjugation systems mediate the attachment of a phosphatidylethanolamine moiety (PE) to LC3 for recruitment to the autophagic membrane. The growing autophagosome engulfs cytosolic contents, such as damaged proteins and organelles. Finally, the mature autophagosome fuses with a lysosome, resulting in the degradation of its contents and the autophagic membrane. (B) Model showing the contribution of autophagy to p53 responses. p53, in collaboration with p63 and p73, regulates apoptosis target genes to induce apoptosis, which contributes to tumor suppression. Similarly, p53 and its family members regulate the newly identified autophagy program (e.g., Atg2b, Atg4a, Atg4c, Atg7, Atg10, Tmem49, Ulk1, Ulk2, and Uvrag) to induce autophagy, which contributes to tumor suppression through apoptosis. Autophagy may also contribute directly to tumor suppression through apoptosis-independent mechanisms as indicated by the question mark next to the arrow from autophagy to tumor suppression.
Interestingly, our studies have revealed that the p53 family members p63 and p73 participate in regulating the p53-bound autophagy genes that we identified, as indicated by the binding of p63 and p73 to these genes in published human ChIP-seq and ChIP–Chip experiments and by our genetic analyses showing that p63 and p73 contribute to the induction of autophagy target genes by DNA damage (Kouwenhoven et al. 2010; Rosenbluth et al. 2011). The idea that p53 family members can participate in activating p53 target genes is not unprecedented, as p63 and p73 contribute to the induction of proapoptotic genes during apoptosis in E1A MEFs (Flores et al. 2002). The finding that p53 family members all contribute to regulating autophagy genes suggests that the induction of autophagy may represent an ancestral function of the family. As the DNA damage-triggered apoptotic response is thought to be a critical ancient function of the p53 family for protection of the germline, the induction of autophagy by p53 family members could have evolved to promote this response. It is also plausible that p53 family member-regulated autophagy evolved originally as a general stress response to stresses such as nutrient deprivation, a function that is compatible with the presence of p53 family members in unicellular organisms.
p53 has been implicated previously in autophagy through a couple of mechanisms. The primary link has been through transcriptional induction of a small set of target genes by p53, although basal levels of p53 can also inhibit autophagy in the cytoplasm (Tasdemir et al. 2008; Maiuri et al. 2010). The most central p53-activated target gene involved in autophagy is Dram1, which encodes a lysosomal protein involved in late stages of autophagy (Crighton et al. 2006). In addition, p53 can contribute to autophagy through activation of its apoptotic targets, including Bax and Puma, which participate in the cross-talk between apoptosis and autophagy (Maiuri et al. 2007). Furthermore, p53 transcriptionally activates components of pathways upstream of autophagy, including Tsc2 and Ampk, which inhibit mTor, resulting in the activation of autophagy, as shown, for example, in response to DNA damage (Feng et al. 2005, 2007). Recently, Ulk1 and the autophagy modulator ISG20L1 were also identified as direct p53 targets in human cells (Eby et al. 2010; Gao et al. 2011), and several autophagy genes were found to be bound by p73 in human cells (Rosenbluth and Pietenpol 2009). p53 has also been proposed to be a regulator of autophagic homeostasis by post-transcriptional modulation of LC3 levels, which ensures autophagic flux and promotes survival in response to chronic starvation (Scherz-Shouval et al. 2010). Here, we expand the direct role of p53 in regulating autophagy by defining an entire program of autophagy genes directly activated by p53 and its family members, including genes involved in each step of the autophagic process (Fig. 7A).
Autophagy has a context-dependent role in cancer development. Several autophagy-deficient mouse strains are susceptible to tumorigenesis, including Becn1+/− mice, Atg4c−/−mice, and mice lacking Atg5 or Atg7 in the liver, which develop spontaneous tumors, chemical carcinogen-induced tumors, and benign liver tumors, respectively (Morgan and Kastan 1997; Yue et al. 2003; Takamura et al. 2011). This tumor suppressor role is consistent with autophagy enabling apoptosis or autophagy promoting cellular integrity through the destruction of damaged proteins and organelles, limiting reactive oxygen species (ROS), and preventing genomic instability. However, in other contexts, autophagy can promote the adaptation to environmental stresses and increased metabolic demands, thereby enhancing the survival of cancer cells in unfavorable conditions. This notion has been supported by models where loss of autophagy impedes progression to malignancy, as in mosaic Atg5 knockout mice or liver-specific Atg7 knockout mice, which develop benign liver carcinomas that do not progress to a more malignant state (Takamura et al. 2011). Additionally, the growth of Hras transformed xenograft tumors and pancreatic tumors in xenograft and autochthonous cancer models was impaired by autophagy inhibition (Guo et al. 2011; Kim et al. 2011; Yang et al. 2011). These seemingly distinct roles for autophagy can be reconciled by proposing a dual role for autophagy in cancer development depending on the stage. Specifically, while initially preventing tumor growth by maintaining cellular and genomic integrity, autophagy is exploited at later stages to promote cancer cell growth when cells face increased metabolic demands or external stresses. Moreover, as p53 is frequently mutated or inactivated in cancer cells, regulation of autophagy in this setting will likely be p53-independent. Interestingly, our observation that transformed autophagy-deficient cells produce more colonies than wild-type controls suggests a tumor suppressor role for autophagy downstream from p53 in this context. Furthermore, because the transformation suppression triggered by p53 in E1A;HrasV12 MEFs is through induction of apoptosis, our findings suggest that autophagy may promote tumor suppression in this setting through enhancement of p53-dependent apoptosis (Fig. 7B).
In response to cancer therapy, roles for autophagy in promoting survival as well as in enhancing cell death have been described, although the prevailing view is that therapy-induced autophagy is a survival and resistance mechanism and that inhibiting autophagy is therefore a therapeutic strategy (Rubinsztein et al. 2012). However, since cancer cells generally lack a functional p53 pathway, these experiments most likely address p53-independent cell death, in which autophagy may have a different role and promote survival. Certainly, a more detailed understanding of the settings in which autophagy promotes apoptosis versus survival is important to maximize the benefits of modulating autophagy in cancer treatment. Our findings showing that autophagy promotes p53-dependent apoptosis in a genetically defined setting of E1A;HrasV12 transformed MEFs with wild-type p53 suggests that genotoxic cancer therapies could promote regression of tumors with wild-type p53 in part through autophagy. This idea is supported by studies showing that the p53 autophagy target gene Dram contributes to efficient p53-triggered apoptosis (Crighton et al. 2006).
Our genome-wide studies have elaborated an extensive connection between p53 and autophagy. p53 regulates autophagy directly by induction of core autophagy genes, which helps promote p53 responses. How, exactly, autophagy contributes to p53-dependent apoptosis and transformation suppression remains an open question. It will be very interesting to further dissect the mechanisms by which autophagy contributes to p53-dependent tumor suppression in the future.
Materials and methods
Cell culture
MEFs, lung cancer cells, and HCT116 cells were cultured in DMEM with 10% FCS. Human fibroblasts were obtained from Coriell Cell Depository and maintained in DMEM with 15% FCS. Doxorubicin (Sigma) was used at 0.2 μg/mL, and Nutlin-3a (Sigma-Aldrich) was used at 0.5 mM. UVC treatment was at 20 J/m2. For autophagy experiments, cells were treated with BafA1 for 1 h prior to harvesting. Pulses for cell cycle experiments were 3 μg/mL BrdU or 5 μg/mL EdU for 4 h. Lentiviral and adenoviral infections were carried out as described (Brady et al. 2011). siRNA transfection was performed according to the manufacturer's instructions. Briefly, siRNAs were reverse-transfected at a final concentration of 25 nM using Dharmafect 4 (Dharmacon) and 300,000 MEFs per 35-mm well. Previously published p63 and p73 siRNAs (Lang et al. 2004) and siGENOME nontargeting siRNA pools (D-001206-14, Dharmacon) were used for p53 family member knockdown experiments.
ChIP-seq experiments
MEFs were seeded at 4 × 106 cells per 15-cm dish and treated with 0.2 μg/mL doxorubicin for 6 h on the following day. Chromatin from 100 × 106 cells was used for each ChIP-seq experiment. ChIP was carried out essentially as described (Johnson et al. 2007) using p53 polyclonal antibodies (CM-5, Vector Laboratories) with a few modifications: Washes were performed two times with low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl at pH 8.1, 150 mM NaCl), three times with high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl at pH 8.1, 500 mM NaCl), and four times with LiCl wash buffer (0.25 M LiCl, 1% IGEPAL CA630, 1% deoxycholic acid [sodium salt], 1 mM EDTA, 10 mM Tris at pH 8.1). Chromatin-immunoprecipitated DNA was quantified by qPCR using SYBR Green (SA-Biosciences) and a 7900HT Fast Real-Time PCR machine (Applied Biosystems). Libraries were generated according to the manufacturer's instructions (Illumina), and quality was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies). Thirty-five–base-pair reads were generated on an Illumina Genome Analyzer II, and raw data were uploaded to the high-throughput sequencing data analysis platform DNAnexus and mapped to the 2007 release of the mouse genome (mm9). The quality of the ChIP-seq libraries was confirmed by the number and percentage of mapped and confidently mapped reads. All ChIP-seq data are available in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/gds) under the accession number GSE46240.
RNA-seq experiments
3SEQ libraries were prepared from purified mRNA from untreated and doxorubicin-treated wild-type or p53−/− cells as described (Beck et al. 2010) using at least two independently derived MEF lines per genotype. Raw sequencing data generated on the Illumina Genome Analyzer II were uploaded to the high-throughput sequencing data analysis platform DNAnexus and mapped to the 2007 release of the mouse genome (mm9). The quality of the 3SEQ library was confirmed by the number and percentage of mapped and confidently mapped reads. All RNA-seq data are available in the GEO database (http://www.ncbi.nlm.nih.gov/gds) under the accession number GSE46240.
ChIP-seq and RNA-seq data analysis
Peak calling was performed using DNAnexus with settings for peak enrichment >20, peak-to-background enrichment >3, a minimum ratio between uniquely and repetitively mapped reads of 3:1, a kernel bandwith of 30.0, and enabling FDR calculation. MEME was used for de novo motif analysis of the genomic DNA sequences flanking ChIP-seq peaks (Bailey and Elkan 1994). The percentage of peaks containing a p53 consensus motif was determined with RSA-tools matrix scan (Turatsinze et al. 2008). DAVID Bioinformatics Resources 6.7 was used to analyze the gene lists derived from ChIP-seq data or ChIP-seq data combined with 3SEQ data to find enriched pathways and biological functions (Huang et al. 2009a,b).
3SEQ raw data were uploaded to DNAnexus, and quantification was performed using transcriptome-based quantification using sense reads and RefSeq gene annotation. Read counts were further analyzed using R environment and the DEseq package according to the suggestions in DEseq's vignette to derive a list of differentially regulated genes. In summary, the size factor was first calculated to normalize the samples for the size of the library. After estimation of variation, a method based on the negative binomial distribution was used to call differentially expressed genes between DNA damage-treated and untreated conditions for wild-type and p53−/− MEFs or between untreated wild-type and p53−/− MEFs. Genes with an adjusted P-value ≤0.1 were then used for further analysis. To derive p53-dependently DNA damage-regulated genes, we excluded genes that were significantly regulated by DNA damage in p53−/− MEFs with >70% of the fold change observed in wild-type MEFs.
qPCR analysis
For qRT–PCR, RNA was isolated using Trizol (Invitrogen) and reverse-transcribed using MMLV reverse transcriptase (Invitrogen) and random primers. PCR was performed in triplicate using SYBR Green (SA-Biosciences) and a 7900HT Fast Real-Time PCR machine (Applied Biosystems), and the results were computed relative to a standard curve made with cDNA pooled from all samples. Primer sequences for qRT–PCR and ChIP-qPCR are listed in the Supplemental Material.
Western blotting
Western blotting was performed according to standard protocols using p53 antibodies (1:1000; CM5, Vector Laboratories), LC3 antibodies (1:1000; NB600-1384, Novus), and HRP-conjugated β-actin antibodies (1:1000; Santa Cruz Biotechnology). Protein lysates were prepared in RIPA buffer (150 mM NaCl, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris at pH 8.0) with freshly added protease inhibitor cocktail (Roche). Western blots were quantified with ImageJ.
Immunofluorescence
Immunofluorescence was performed as described using anti-mouse p53 (1:150; rabbit CM5, Vector Laboratories) and anti-LC3 (1:200; rabbit CST no. 3868, Cell Signaling Technology). For LC3 staining, cells were permeabilized with methanol and blocked with DAKO blocking buffer (X0909, Dako).
FACS analysis
Apoptosis assays were performed as described (Johnson et al. 2005). Per sample, 0.2 × 106 to 0.3 × 106 cells were seeded per 35-mm well, and cells were analyzed 18 h (for apoptosis) or 72 h (for survival) after treatment with doxorubicin. For cell cycle analysis, 0.5 × 106 cells were seeded per 60-mm dish and irradiated with 5 Gy of γ-radiation. Click-IT EdU labeling and SYTOX staining for DNA content (both Invitrogen) were performed according to the manufacturer's protocols 18 h post-treatment.
Soft agar assays
E1A;HrasV12 MEFs (3 × 104) were resuspended in 1.5 mL of 0.3% top agar in complete DMEM growth medium and placed on a layer of 2 mL of 0.5% bottom agar in a 35-mm dish. Each cell line was seeded in triplicate, and cells were fed every 3 d. After 2 and 3 wk, colonies were stained with Giemsa, and photographs were taken. Colony number was quantified using ImageJ. Briefly, images of Giemsa-stained colonies were processed using the color deconvolution plugin and then converted to binary (black and white) images. The “analyze particles” function was used to determine the number of particles (colonies).
Acknowledgments
We thank Ziming Weng, Joe Foley, Eric Van Nostrand, Stuart Kim, and Gill Bejerano for technical assistance. We thank Karla Kirkegaard, Michel Brahic, and Noboru Mizushima for providing us with Atg5fl/fl mice, and Dan Egan and Reuben Shaw for providing us with Ulk1−/−MEFs and Ulk2 shRNA constructs. We also thank Julien Sage, Kay Macleod, and Heiyoun Jung for critical reading of the manuscript. This work was supported by a Swiss National Science Foundation Fellowship to D.K.B., a C.A.P.E.S Fellowship to S.S.M., an American Cancer Society Post-doctoral Fellowship to K.T.B., and funding from the National Institutes of Health (CA140875), American Cancer Society, and the Leukemia and Lymphoma Society to L.D.A.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.212282.112.
References
- Anders S, Huber W 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Beck AH, Weng Z, Witten DM, Zhu S, Foley JW, Lacroute P, Smith CL, Tibshirani R, van de Rijn M, Sidow A, et al. 2010. 3′-End sequencing for expression quantification (3SEQ) from archival tumor samples. PLoS ONE 5: e8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio-Kuter A, Levine AJ 2010. The origins and evolution of the p53 family of genes. Cold Spring Harb Perspect Biol 2: a001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging KT, Attardi LD 2012. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol 22: 97–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botcheva K, McCorkle SR, McCombie WR, Dunn JJ, Anderson CW 2011. Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle 10: 4237–4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. 2011. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145: 571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Attardi LD 2005. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer 5: 231–237 [DOI] [PubMed] [Google Scholar]
- Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM 2006. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126: 121–134 [DOI] [PubMed] [Google Scholar]
- Debbas M, White E 1993. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev 7: 546–554 [DOI] [PubMed] [Google Scholar]
- Eby KG, Rosenbluth JM, Mays DJ, Marshall CB, Barton CE, Sinha S, Johnson KN, Tang L, Pietenpol JA 2010. ISG20L1 is a p53 family target gene that modulates genotoxic stress-induced autophagy. Mol Cancer 9: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, Oliner JD, McKeon F, Haber DA 2002. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell 10: 995–1005 [DOI] [PubMed] [Google Scholar]
- Feldser DM, Kostova KK, Winslow MM, Taylor SE, Cashman C, Whittaker CA, Sanchez-Rivera FJ, Resnick R, Bronson R, Hemann MT, et al. 2010. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 468: 572–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S 2005. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci 102: 8204–8209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ 2007. The regulation of AMPKβ1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1–AKT–mTOR pathways. Cancer Res 67: 3043–3053 [DOI] [PubMed] [Google Scholar]
- Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T 2002. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416: 560–564 [DOI] [PubMed] [Google Scholar]
- Gao W, Shen Z, Shang L, Wang X 2011. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ 18: 1598–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. 2011. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25: 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889 [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA 2009a. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Johnson TM, Hammond EM, Giaccia A, Attardi LD 2005. The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat Genet 37: 145–152 [DOI] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502 [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB 2005. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 18: 283–293 [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ Jr 1992. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71: 587–597 [DOI] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Attardi LD 2010. In vivo analysis of p53 tumor suppressor function using genetically engineered mouse models. Carcinogenesis 31: 1311–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, Hwang SG, Yoon G, Lee SJ 2011. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem 286: 12924–12932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouwenhoven EN, van Heeringen SJ, Tena JJ, Oti M, Dutilh BE, Alonso ME, de la Calle-Mustienes E, Smeenk L, Rinne T, Parsaulian L, et al. 2010. Genome-wide profiling of p63 DNA-binding sites identifies an element that regulates gene expression during limb development in the 7q21 SHFM1 locus. PLoS Genet 6: e1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B 2010. Autophagy and the integrated stress response. Mol Cell 40: 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. 2004. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119: 861–872 [DOI] [PubMed] [Google Scholar]
- Lee KH, Li M, Michalowski AM, Zhang X, Liao H, Chen L, Xu Y, Wu X, Huang J 2010. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc Natl Acad Sci 107: 69–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Cheng Q, Li Z, Chen J 2010. p53 inactivation by MDM2 and MDMX negative feedback loops in testicular germ cell tumors. Cell Cycle 9: 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, He Y, Dubois W, Wu X, Shi J, Huang J 2012. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol Cell 46: 30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Ruley HE 1993. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev 7: 535–545 [DOI] [PubMed] [Google Scholar]
- Lowe SW, Jacks T, Housman DE, Ruley HE 1994. Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc Natl Acad Sci 91: 2026–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G 2007. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy 3: 374–376 [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G 2010. Autophagy regulation by p53. Curr Opin Cell Biol 22: 181–185 [DOI] [PubMed] [Google Scholar]
- Morgan SE, Kastan MB 1997. p53 and ATM: Cell cycle, cell death, and cancer. Adv Cancer Res 71: 1–25 [DOI] [PubMed] [Google Scholar]
- Nikulenkov F, Spinnler C, Li H, Tonelli C, Shi Y, Turunen M, Kivioja T, Ignatiev I, Kel A, Taipale J, et al. 2012. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ 19: 1992–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T 2004. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119: 847–860 [DOI] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, Hainaut P 2010. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2: a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A 2008. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9: 402–412 [DOI] [PubMed] [Google Scholar]
- Rosenbluth JM, Pietenpol JA 2009. mTOR regulates autophagy-associated genes downstream of p73. Autophagy 5: 114–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth JM, Mays DJ, Jiang A, Shyr Y, Pietenpol JA 2011. Differential regulation of the p73 cistrome by mammalian target of rapamycin reveals transcriptional programs of mesenchymal differentiation and tumorigenesis. Proc Natl Acad Sci 108: 2076–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B 2012. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11: 709–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski R, Hofmann K, Gartner A 2010. Phylogeny and function of the invertebrate p53 superfamily. Cold Spring Harb Perspect Biol 2: a001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M 2010. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci 107: 18511–18516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW 2002. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 1: 289–298 [DOI] [PubMed] [Google Scholar]
- Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, Denissov S, Stunnenberg HG, Lohrum M 2008. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res 36: 3639–3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk L, van Heeringen SJ, Koeppel M, Gilbert B, Janssen-Megens E, Stunnenberg HG, Lohrum M 2011. Role of p53 serine 46 in p53 target gene regulation. PLoS ONE 6: e17574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW 1999. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science 284: 156–159 [DOI] [PubMed] [Google Scholar]
- Sullivan A, Lu X 2007. ASPP: A new family of oncogenes and tumour suppressor genes. Br J Cancer 96: 196–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symonds H, Krall L, Remington L, Saenz-Robles M, Lowe S, Jacks T, Van Dyke T 1994. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell 78: 703–711 [DOI] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N 2011. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25: 795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al. 2008. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10: 676–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodoro JG, Parker AE, Zhu X, Green MR 2006. p53-mediated inhibition of angiogenesis through up-regulation of a collagen prolyl hydroxylase. Science 313: 968–971 [DOI] [PubMed] [Google Scholar]
- Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, et al. 2010. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 141: 1042–1055 [DOI] [PubMed] [Google Scholar]
- Turatsinze JV, Thomas-Chollier M, Defrance M, van Helden J 2008. Using RSAT to scan genome sequences for transcription factor binding sites and cis-regulatory modules. Nat Protoc 3: 1578–1588 [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X 2002. Live or let die: The cell's response to p53. Nat Rev Cancer 2: 594–604 [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C 2009. Blinded by the light: The growing complexity of p53. Cell 137: 413–431 [DOI] [PubMed] [Google Scholar]
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. 2006. A global map of p53 transcription-factor binding sites in the human genome. Cell 124: 207–219 [DOI] [PubMed] [Google Scholar]
- Wu GS, Saftig P, Peters C, El-Deiry WS 1998. Potential role for cathepsin D in p53-dependent tumor suppression and chemosensitivity. Oncogene 16: 2177–2183 [DOI] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. 2011. Pancreatic cancers require autophagy for tumor growth. Genes Dev 25: 717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH 2009. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ 16: 1135–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N 2003. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci 100: 15077–15082 [DOI] [PMC free article] [PubMed] [Google Scholar]