

Aiming to identify regulators of glioblastoma multiforme (GBM) maintenance and initiation, Hubert et al. performed genome-wide RNAi screens in patient-derived GBM stem cells (GSCs). This identified the PHD-finger domain protein PHF5A as being required for GSC expansion. PHF5A knockdown in GSCs inhibited splicing of numerous genes, leading to cell cycle arrest and loss of viability. Additionally, PHF5A inhibition compromised GSC tumor formation in vivo and inhibited growth of established GBM patient-derived xenograft tumors. This study highlights the requirement for PHF5A in brain tumor-initiating cells.
Keywords: brain tumors, RNA splicing, RNAi, cancer stem cell
Abstract
To identify key regulators of human brain tumor maintenance and initiation, we performed multiple genome-wide RNAi screens in patient-derived glioblastoma multiforme (GBM) stem cells (GSCs). These screens identified the plant homeodomain (PHD)-finger domain protein PHF5A as differentially required for GSC expansion, as compared with untransformed neural stem cells (NSCs) and fibroblasts. Given PHF5A's known involvement in facilitating interactions between the U2 snRNP complex and ATP-dependent helicases, we examined cancer-specific roles in RNA splicing. We found that in GSCs, but not untransformed controls, PHF5A facilitates recognition of exons with unusual C-rich 3′ splice sites in thousands of essential genes. PHF5A knockdown in GSCs, but not untransformed NSCs, astrocytes, or fibroblasts, inhibited splicing of these genes, leading to cell cycle arrest and loss of viability. Notably, pharmacologic inhibition of U2 snRNP activity phenocopied PHF5A knockdown in GSCs and also in NSCs or fibroblasts overexpressing MYC. Furthermore, PHF5A inhibition compromised GSC tumor formation in vivo and inhibited growth of established GBM patient-derived xenograft tumors. Our results demonstrate a novel viability requirement for PHF5A to maintain proper exon recognition in brain tumor-initiating cells and may provide new inroads for novel anti-GBM therapeutic strategies.
Malignant glioma is the most common and lethal form of brain cancer. Glioblastoma multiforme (GBM) is the most invasive and aggressive grade of glioma and is notoriously drug- and radiation-resistant. There are currently no highly effective therapies against GBM, and with standard of care treatments, including surgery, radiation, and chemotherapy, ∼90% of adult patients die within 2 yr of diagnosis (Latera and Brem 2002; Central Brain Tumor Registry of the United States (CBTRUS), http://www.cbtrus.org), underscoring the need for novel therapeutic targets. The hierarchical organization of adult and pediatric brain tumors suggests a cancer stem cell origin (Hemmati et al. 2003; Singh et al. 2003, 2004; Galli et al. 2004). Consistent with this idea, tumor-initiating GBM stem cells (GSCs) isolated from patients retain the developmental potential and specific genetic alterations found in the original tumor (Hemmati et al. 2003; Singh et al. 2003; Lee et al. 2006; Pollard et al. 2009).
When isolated under serum-free monolayer conditions, GSCs can retain tumor-initiating potential and tumor-specific genetic and epigenetic signatures over extended outgrowth periods (Lee et al. 2006; Pollard et al. 2009). In addition, they have been shown to recreate tumor cellular hierarchies when implanted into the cortex of immunocompromised mice (Lee et al. 2006; Pollard et al. 2009). Furthermore, GSC isolates also retain expression of neural progenitor molecular networks, which may contribute to the aggressive behavior of GBM tumors through enhancing self-renewal or developmental programs (Mangiola et al. 2007; Stiles and Rowitch 2008; Gangemi et al. 2009), DNA repair pathways (Bao et al. 2006a), angiogenesis (Bao et al. 2006b; Cheng et al. 2013), and/or invasiveness (Liu et al. 2009).
Recently, in order to identify new candidate therapeutic targets for GBM, we combined functional genetics and GBM network modeling to identify human kinases required for the expansion of GSCs but dispensable to proliferating neural stem cells (NSCs) (Ding et al. 2013). This approach yielded BUB1B/BUBR1, a critical mitotic spindle kinase, as a GBM-lethal gene. Our studies revealed that certain GBM tumors have an added requirement for a nonessential BUB1B activity to suppress lethal consequences of altered kinetochore function (Ding et al. 2013). Thus, these results demonstrated that patient-derived GSCs can be used to identify cancer- and patient-specific molecular vulnerabilities for GBM that are not observed in tissue-appropriate or other nontransformed control cells (e.g., NSCs and astroyctes).
Here, we expanded our search for GBM-lethal genes by performing genome-wide RNAi screens in multiple GSC patient isolates and NSCs to identify genes differentially required for GSC expansion. These studies revealed that the plant homeodomain (PHD)-finger domain protein PHF5A was differentially required for expansion and viability of multiple GSC isolates. Molecular studies demonstrated that GSCs have a novel requirement for PHF5A activity to facilitate recognition of exons with distinctive 3′ splice sites. PHF5A knockdown resulted in splicing defects in thousands of essential genes, a subset of which are predicted to affect cell division and growth (e.g., CDC20 and RAF1). Cell-based assays revealed that PHF5A/U2snRNP perturbation causes G2/M arrest in GSCs both in vitro and during cell growth in established patient-derived tumors. Importantly, sensitivity to U2snRNP perturbation could be recreated in NSCs and fibroblasts overexpressing MYC. Although MYC is not frequency amplified in GBM, MYC is coordinately activated by mutations in p53 and PTEN, two of the most common mutations in GBM (Zheng et al. 2008b), and is also essential for human GSC self-renewal (Wang et al. 2008). Our results demonstrate an unexpected role for PHF5A in maintaining proper exon recognition in GSCs, which is critical for growth and maintenance of patient-derived tumors.
Results
Functional genetic screens identify PHF5A as differentially required for GSC expansion
To identify genes necessary for the growth and survival of GSCs but likely dispensable to noncancerous neural cells and tissues, we performed functional genetic shRNA screens that targeted 1086 nucleic acid-binding factors in both primary GSC tumor isolates and human fetal NSC-CB660 cells. For these screens, we assayed genes required for GSC and NSC in vitro expansion in serum-free monolayer culture (Fig. 1A; Pollard et al. 2009; Ding et al. 2013).
Figure 1.

Functional genetic screens identify PHF5A as differentially required for GSC expansion. (A) The shRNA screening approach used to initially identify PHF5A as differentially required for GSC expansion. Pooled viruses targeting nucleic acid-binding factors were used in outgrowth screens in human GSC-G166 and NSC-CB660 cells. Candidate genes differentially required for GSC expansion over 21 d of outgrowth were identified through hybridization of viral DNA barcodes to custom microarrays. (B) Average differential between GSCs and NSCs for shRNA clones targeting candidate screen hits by in vitro competition assay. Cells infected with single shRNA clones (GFP+) were mixed with uninfected cells and outgrown for 10 d, and the change in GFP+ cells in each culture was quantified by FACS analysis. (C) Our genome-wide shRNA screening strategy used in NSC-CB660 and three primary GSC isolates. Viral shRNA pools targeting ∼19,000 human genes were infected into cells prior to 21 d of outgrowth in vitro. The change in viral shRNA representation in each cell population was quantified by sequencing. Gene targets statistically underrepresented at the end of the culture period are shown. (D) Viability of NSCs and five GSC isolates infected with three independent shRNA viral clones targeting PHF5A. (M) Mesenchymal subgroup; (PN) proneural subgroup; (*) P-value < 0.002 vs. CB660. (E) Western blot analysis of PHF5A protein expression in NSCs and GSCs after PHF5A knockdown. (F) Images of GSC-0131 cell clones expressing an inducible shRNA construct targeting the endogenous 3′ UTR of PHF5A, with or without rescue by exogenous expression of full length PHF5A. Bar, 50 μm. (G) PHF5A was immunoprecipitated from cellular lysates, and associated binding proteins were analyzed by mass spectrometry. The gene ontology (GO) categories most enriched among PHF5A-bound proteins are presented. See also Supplemental Figure S1.
Cells were infected with pools of shRNAs (Paddison et al. 2004; Luo et al. 2009) and expanded in triplicate screening populations under normal conditions for 21 d. Comparisons of shRNA representation in GSCs or NSCs over time using microarrays or deep sequencing revealed a subset of shRNAs that became significantly under- or overrepresented during expansion. Candidate GSC-specific lethal genes were defined as those shRNAs that were significantly underrepresented in GSC cultures relative to NSC control cultures.
The nucleic acid-binding gene screen, which was performed in a single GBM isolate (G166 cells) along with NSC controls, yielded 27 genes as candidate GBM-lethal hits. Retests of each screen hit were performed using multiple single-shRNA viral clones. Seven genes (26% of candidates) met our validation criteria of two or more single hairpin clones that produced a growth ratio of <0.65 in GSCs cells compared with NSCs with a P-value <0.05 after 7 d of outgrowth (Fig. 1B; Supplemental Fig. 1a). Significantly, knockdown of the gene PHF5A was strongly indicated as the top hit affecting in vitro expansion of GSC-G166 cells (Fig. 1B; Supplemental Fig. 1a). To ensure that the results were applicable to other GBM tumors and that hits would score similarly when comparing the entire genome, we also performed genome-wide shRNA screens in multiple GSC isolates from three different GBM patients that represented two developmental subtypes (G166, mesenchymal; 0131, mesenchymal; 0827, proneural), again in triplicate with NSCs as controls. This approach yielded GSC screen hits that were mainly isolate-specific, likely owing to different tumor backgrounds and heterogeneity in cell populations during expansion (Fig. 1C). However, there were 17 candidate lethal genes shared by each GSC isolate that did not score in NSCs; PHF5A was among these genes (Fig. 1C).
It should be noted that identifying genes that, when inhibited, affect growth of GSCs more than NSCs is unusual. We found that most perturbations in pathways required for cell growth or cell cycle progression (e.g., PI3K pathway, Aurora A and B kinases, heat-shock protein 90, and the microtubule motor protein KIF11/Eg5) (Ding et al. 2013; data not shown) either show no differential effect between NSCs and GSCs or affect NSCs more than GSCs.
PHF5A is a highly conserved PHD-zinc finger domain protein that facilitates interactions between the U2 snRNP complex and DNA/RNA helicases (Rzymski et al. 2008). PHF5A may also bind to chromatin through its PHD domain (Trappe et al. 2002), which, in other PHD family members, can facilitate interactions with specific histone marks on chromatin-bound nucleosomes (Mellor 2006; Musselman and Kutateladze 2009). Consistent with this latter notion, PHF5A protein was found to be an enhancer of estrogen-mediated transcription of the Connexin 43 gene (Oltra et al. 2003). PHF5A has also been characterized as a member of the SF3b component of the U2 snRNP splicing complex (Will et al. 2002). Since PHF5A was a highly reproducible GSC-specific screen hit, yet nothing is known about a cancer-specific role for PHF5A, we further pursued it as a GBM-lethal target.
PHF5A is differentially required for GSC in vitro expansion
To further confirm differential effects of PHF5A knockdown, we performed short-term outgrowth assays in five primary GSC cultures using multiple shRNAs. In each case, PHF5A knockdown showed a strong, GSC-specific loss of viability (Fig. 1D). Next, we examined the effects of PHF5A knockdown on SSEA1+ GSC subpopulations, which are enriched for tumor-initiating cell activity (Son et al. 2009). In the three different GSC isolates examined, PHF5A knockdown compromised outgrowth of SSEA1+ populations over the course of several weeks (Supplemental Fig. S1b). This indicates that PHF5A suppression blocks gross expansion of GSC isolates, including both the bulk cell population and tumor-initiating GSC subpopulations.
GSCs and NSCs express PHF5A at relatively similar levels, and knockdown is equivalently effective in each cell type at both the RNA and protein levels (Fig. 1E; Supplemental Fig. S1c), indicating that the lack of phenotype in NSCs is not due to inefficient knockdown or major differences in expression. Moreover, PHF5A expression levels were similar in GSCs, NSCs, and other tissues, indicating that GSCs do not abnormally overexpress the gene (Supplemental Fig. S1c). We further performed a complementation assay in which a validated, inducible shPHF5A sequence targeting the PHF5A endogenous 3′ untranslated region (UTR) was coexpressed with the PHF5A ORF lacking its endogenous 3′ UTR. Expression of the PHF5A ORF rescued the growth defect observed in PHF5A knockdown GSCs (Fig. 1F; Supplemental Fig. S1d), indicating that the phenotypic effects are PHF5A-specific.
Furthermore, to query what key roles PHF5A might play in our cells, we examined PHF5A-interacting proteins by coimmunoprecipitation (co-IP) mass spectrometry. This yielded a strong enrichment for candidate interacting proteins involved in splicing (GO:0008380 RNA splicing, P = 10−14) as well as gene expression (GO:0010467 gene expression, P = 10−16) (Fig. 1G; Supplemental Table S1).
PHF5A is specifically required for normal exon recognition in GSCs but not NSCs
Because PHF5A has been characterized as both a splicing factor (Will et al. 2002; Rzymski et al. 2008) and a transcriptional regulator (Oltra et al. 2003), we next wished to define which of these activities was most relevant for GBM-specific survival. Recently, Paulsen et al. (2009) found that knockdown of multiple spliceosomal genes in HeLa cells resulted in dsDNA breaks and H2AX phosphorylation. We therefore first examined whether PHF5A knockdown might similarly give rise to DNA damage in GSCs, thereby triggering arrest and growth inhibition. However, upon PHF5A knockdown in GSCs, we did not see an increase in pH2AX levels, phosphorylation of the DNA damage signaling proteins CHK1 and CHK2, or activation of the mitotic spindle checkpoint (Supplemental Fig. S2a). These results suggest that the shPHF5A growth inhibition does not simply arise from a DNA damage response or alterations in the mitotic spindle.
We next hypothesized that PHF5A knockdown might induce GSC-specific aberrant splicing of genes required for cell cycle progression or cell growth. To directly test this hypothesis, we asked whether splicing was globally dysregulated following PHF5A knockdown by performing deep RNA sequencing (RNA-seq) in control NSCs (CB660 cells) and GSCs (G166 and 0827 cells) treated with control or PHF5A-targeting shRNAs. We quantified changes in isoform ratios using only reads that crossed splice sites, an approach that treats all splicing events with equivalent statistical power (Bradley et al. 2012).
This analysis revealed that PHF5A knockdown results in dramatic GSC-specific exon skipping and intron retention events (Figs. 2A; Supplemental Fig. S2b) in hundreds of genes. Other forms of splicing regulation, including selection of competing 5′ and 3′ splice sites and mutually exclusive exon selection, were unaffected (Supplemental Fig. S2b) in both GSCs and NSCs. Most of the resulting GSC-specific splicing changes introduced in-frame stop codons into the mRNAs, strongly suggesting that the splicing changes are aberrant, rather than functionally relevant, splicing (Fig. 2B). Furthermore, the overall expression of GSC mRNAs harboring in-frame stop codons was decreased, consistent with triggering nonsense-mediated mRNA decay (NMD) (Fig. 2B; Amrani et al. 2006).
Figure 2.

PHF5A is globally required by GSCs for proper recognition of an unusual class of exons. (A) In GSCs but not NSCs, PHF5A knockdown causes a dramatic increase in missplicing of constitutive junctions (top row) as well as retention of constitutive introns (bottom row). (B) Many of the splicing changes induced by PHF5A knockdown in GSCs introduce in-frame stop codons, suggesting that the resulting transcripts will be degraded by NMD. Gene expression values were computed with RSEM (Li and Dewey 2011) and normalized with the TMM method (Robinson and Oshlack 2010). Confidence intervals indicate the first and third quartiles of expression. (C) Constitutive junctions that are misspliced following PHF5A knockdown in GSCs (center) have slightly shorter polypyrimidine tracts than do unaffected constitutive junctions (top); in contrast, retained constitutive introns have unusually C-rich polypyrimidine tracts (bottom). (D) Retained constitutive introns are much shorter. Plot illustrates the median intron length, and error bars indicate the standard error estimated by bootstrapping. (E) Retained constitutive introns have branch points that are unusually proximal to the 3′ splice site. Box plots indicate the first and third quartiles of the first upstream AG, a proxy for the branch point location (Gooding et al. 2006). See also Supplemental Figure S2.
Only a relatively small subset of splice junctions was affected by PHF5A depletion in GSCs, indicating that the requirement for PHF5A is not universal across exons. To gain mechanistic insight into the origins of the observed splicing dysregulation, we identified specific features characteristic of 5′ and 3′ splice sites susceptible to PHF5A knockdown in GSCs. We could not detect differences in the 5′ splice site features of affected genes (data not shown). 3′ Splice sites associated with abnormal splicing of constitutive junctions had slightly shorter, but otherwise normal, polypyrimidine tracts relative to unaffected 3′ splice sites. In contrast, 3′ splice sites associated with retained constitutive introns had unusual C-rich tracts (Fig. 2C). The retained constitutive introns were short (Fig. 2D) and had unusually proximal branch points (Fig. 2E). While PHF5A is known as a core component of the spliceosome, it appears to be most important for the recognition of an unusual class of exons with distinctive 3′ splice sites. These data suggested that PHF5A primarily functions to facilitate exon recognition rather than regulate alternative splicing, which is consistent with its characterization as a core component of the spliceosome (Will et al. 2002).
Consistent with the GSC-specific growth defect caused by PHF5A knockdown, we observed severe RNA processing defects in many genes important for cell cycle progression, including CDC16, CDC20, CDC25C, CDC37, CDC45, and RCC2, in GSCs (G166 or 0827 cells) but not NSCs (CB660). For example, the 3′-most constitutive exons of CDC20 (Fig. 3A) and many constitutive exons in RCC2 (Supplemental Fig. S3a) were frequently skipped following PHF5A knockdown in GSCs but not in NSCs. In addition, after PHF5A knockdown, multiple constitutive exons of the well-characterized RTK/Ras signaling effector RAF1 and the cancer-associated deacetylase HDAC6 were skipped in GSCs but not in normal NSCs (Fig. 3B).
Figure 3.

Loss of PHF5A results in splicing defects in GSCs but not NSCs. (A) Select genes important for cell cycle progression, such as CDC20, display broad splicing defects following PHF5A knockdown. Plot illustrates the density of RNA-seq reads crossing splice junctions and was created with IGV (Robinson et al. 2011). Aberrant isoforms lacking constitutive exons appear following knockdown of PHF5A with two distinct shRNAs. cDNA base pair sizes indicate the expected product size of each isoform using the cdc20 primers indicated in C and D. (B) RT–PCR of RNA isoforms of example genes after PHF5A knockdown. PCR products were generated using primers in the indicated exons of each gene. Arrows indicate splicing products specifically induced in GSCs after PHF5A knockdown. (C) RT–PCR as in B using RNA from cells treated for 24 h with 0.5, 1, 2, or 4 μM SudC1. (D) qRT–PCR using primers designed to specifically recognize the splice junctions between consecutive and nonconsecutive exons was performed to determine ψ-values for inclusion of potentially skipped exons in HDAC6 identified above. The change in inclusion rate after PHF5A knockdown is presented. (E) Serial dilutions of GSC lysate with or without PHF5A knockdown were run as Western blots and probed with antibodies specific to example genes with predicted missplicing events. (F) Western blot of GSCs and NSCs with or without PHF5A knockdown probed for levels of the frequently misspliced protein HDAC6. See also Supplemental Figure S3.
To further substantiate these results, we examined the effects of two candidate small molecule inhibitors of the U2 snRNP complex: spliceostatin A (SSA) and sudemycin C1 (SudC1). SSA binds to and inhibits the U2 snRNP subunit SF3b, which contains PHF5A, resulting in a reduction in the fidelity of branch point recognition and a down-regulation of genes important for cell division. (Kaida et al. 2007; Corrionero et al. 2011). SudC1 shares the consensus pharmacophore of SSA and pladienolide (Kotake et al. 2007) and also modulates RNA splicing (Lagisetti et al. 2008, 2009; Fan et al. 2011). We reasoned that if the most relevant GSC-specific function of PHF5A is its function in the splicing activity of the U2 snRNP, then these drugs should show a similar pattern of effects on RNA splicing in GSCs and NSCs. This was indeed the case. Treatment of GSCs with SudC1 resulted in dose-dependent GSC-specific splicing defects (Fig. 3C). Finally, we compared the changes in inclusion of identified skipped exons in the example gene HDAC6 after PHF5A knockdown using quantitative RT–PCR (qRT–PCR) primers designed to span potential consecutive and nonconsecutive exon splice junctions in the mature RNA isoforms. As predicted by the data above, this quantitatively demonstrated greater exclusion of tested exons from mature RNA transcripts in GSCs compared with normal NSCs (Fig. 3D).
If these aberrant mRNAs are translated, they would produce C-terminally truncated proteins. We therefore investigated the effects of PHF5A knockdown on the protein levels of these example genes. As predicted, the protein level of PDGFRA, RAF1, and HDAC6 decreased in knockdown cells (Fig. 3E,F), likely due to effects of NMD (Fig. 2B) and altered protein stability. This severe dysregulation of multiple growth regulatory and essential cell cycle genes in GSCs, but not NSCs, suggests that aberrant splicing in GSCs following PHF5A knockdown may give rise to GBM-specific growth defects and inviability.
Taken together, these results indicate that PHF5A is important for proper recognition of a specific, relatively small class of exons in GSCs. Knockdown of PHF5A causes defective RNA processing of thousands of genes, a subset of which are essential for cell cycle progression. Given the broad splicing dysregulation that we observed, there are likely to be numerous cellular defects induced by PHF5A knockdown that contribute to the observed GSC inviability. This model is consistent with our observation that multiple methods of inhibiting U2 snRNP activity—including knockdown of other spliceosomal genes (below) as well as SudC1 treatment—mimic the effects of PHF5A knockdown even though these distinct perturbations are unlikely to lead to identical defects in RNA processing.
PHF5A-binding partners involved in RNA splicing are also differentially required by GSCs and, when inhibited, trigger GSC-specific G2/M cell cycle arrest
Because PHF5A may play multiple cellular roles (Will et al. 2002; Oltra et al. 2003; Rzymski et al. 2008) and its cellular functions are poorly characterized, we next wished to define whether its splicing role was its critical function in GBM cell survival. To this end, we analyzed knockdown of two PHF5A-binding partners in the spliceosome, U2AF1 and DDX1, which interact with the PHF5A C-terminal and N-terminal domains, respectively (Rzymski et al. 2008). U2AF1 is a key member of the U2 snRNP, which is required for RNA branch point recognition (Kramer 1996; Jurica and Moore 2003), and DDX1 is an ATP-dependent DEAD-box RNA helicase (Fang et al. 2005). Knockdown of either U2AF1 or DDX1 phenocopied PHF5A knockdown in GSCs (Fig. 4A). Moreover, a comprehensive examination of multiple shRNAs against PHF5A and U2AF1 in short-term growth assays showed the same strong trend of requirement of these genes in GSCs but not NSCs (Fig. 4B). These results suggest that the PHF5A function most relevant for GSC-specific viability is associated with its role in splicing and the U2 snRNP complex. Supporting this hypothesis, our examination of PHF5A-interacting proteins by co-IP mass spectrometry yielded a strong enrichment for candidate interacting proteins involved in splicing, especially the U2 snRNP complex, including U2AF1, U2AF2, and multiple DDX/DHX helicase family members (Fig. 1G; Supplemental Table S1).
Figure 4.

PHF5A and its binding partners are differentially required for GSC expansion, and their inhibition triggers G2/M cell cycle arrest in GSCs but not NSCs. (A) Model of PHF5A splicing interactions based on previous research. Knockdown of known PHF5A-binding partners in the spliceosome recapitulates the PHF5A knockdown phenotype, shown. Bar, 50 μm. (B) Cumulative probability plot of multiple shRNAs scoring as cell-lethal versus their GSC to NSC viability ratio. (C) Fluorescence microscopy images of GSCs or NSCs expressing constitutive GFP and PHF5A knockdown or control constructs. (Inset) Immunofluorescent images of phospho-MPM2, indicative of mitotic activity, in GSCs and NSCs depleted for PHF5A. Bar, 100 μm. (D) Quantification of phospho-MPM2 staining in GSCs and NSCs infected with control or shPHF5A virus. (*) P-value < 0.001; (#) insignificant P-value = 0.5. (E) Cellular DNA content as measured by DAPI staining and FACS analysis in GSC, NSC, and IMR90 normal fibroblast cultures with or without PHF5A knockdown. The percentage of cells in each sample with 4n DNA content is shown, indicative of cells that have completed DNA replication in S phase. (F) Viability of GSCs or NSCs treated with increasing doses of SSA. (*) P-value < 0.0003. (G) Immunofluorescent images of phospho-MPM2 in GSCs and NSCs treated with SSA or SudC1. (H) Fluorescent and light images of GSCs expressing Histone 2B-GFP fusion protein, which marks DNA. Cells were pretreated with SSA or SudC1 for 22 h before addition of MG132 for an additional 2 h to arrest dividing cells in metaphase. Circles mark the same cell in corresponding images. See also Supplemental Figure 4 and Supplemental Movies S1–S3.
A striking feature of PHF5A depletion in GSCs was that, preceding widespread GSC cell death, PHF5A knockdown triggered a dramatic cell cycle arrest that resembled the rounded-up phenotype of kinesin motor protein KIF11 knockdown (Sawin et al. 1992), our nonspecific cell-lethal control (Fig. 4C). MPM-2 staining, indicative of CyclinB/CDK activity, dramatically increased in PHF5A knockdown GSCs, confirming mitotic arrest (Fig. 4C [inset], D). Moreover, DNA content analysis showed a pronounced increase in the percentage of G2/M cells in GSCs, but not NSCs or normal fibroblasts, with PHF5A knockdown (Fig. 4E).
Further examination of GSC PHF5A knockdown G2/M-arrested cells showed condensed chromatin and monopolar or multipolar spindles (Supplemental Fig. S4a). Along with high MPM-2 staining and little or no phophosphorylated BubR1, this is consistent with a preanaphase arrest in which the mitotic checkpoint has not been triggered. Consistent with the requirement for U2snRNP activity, treatment of GSCs with SSA or SudC1 resulted in a greater dose-dependent viability loss in GSCs relative to NSCs (Fig. 4F; Supplemental Fig. S4b) and also resulted in the characteristic cell cycle arrest in GSCs but not NSCs at doses within this efficacy window (Fig. 4G).
To better characterize GSC-specific G2/M arrest, we performed metaphase capture assays in H2B-GFP-expressing GSCs treated with the proteasome inhibitor MG132, which arrests mitotic cells at metaphase, blocking APCCdc20-dependent degradation of Cyclin B (Lampson and Kapoor 2005). After overnight exposure to SudC1 or SSA, cells were treated with MG132 for 2 h. Control cells displayed proper enrichment for metaphase cells, with chromosomes aligned along the metaphase plate (Fig. 4H). However, SSA- or SudC1-treated cells were unable to properly arrest, further suggesting a premetaphase arrest (Fig. 4H). Similarly, live-cell imaging of GSC-H2B-GFP cells treated with SudC1 or SSA showed mitotic arrest premetaphase (Supplemental Movies 1–3). We also observed that the viability loss in drug-treated GSC cultures results from the death of previously arrested mitotic cells and not interphase GSCs, identifying the cancer-specific mitotic arrest as a causative event in cancer cell death due to splicing inhibition. A fraction of arrested GSCs were able to survive by progressing through mitosis after arresting, but these cells displayed disorganized, multilobed nuclei and were not observed to successfully divide again (Supplemental Movies 2, 3).
Taken together, the above results establish that PHF5A and U2 snRNP complex activity are differentially required for GSC viability compared with NSCs, and their activity is necessary for GSC but not NSC transit through premetaphase mitosis. Moreover, because treatment of GSCs with SSA or SudC1 did not affect the timing of mitoses for several hours after drug treatment (Supplemental Movies 2, 3), it is unlikely that PHF5A and U2 snRNP activity are directly required for mitotic progression.
Overexpression of MYC recapitulates GSC sensitivity to splicing inhibition
We next wished to determine the possible mechanism by which GSCs become differentially sensitive to inhibition of PHF5A and U2 snRNP activity. One possibility was that the process of cellular immortalization or oncogenic transformation itself resulted in splicing dysregulation. To test this possibility in the context of our normal NSCs, we investigated the expression of multiple human genes known to be involved in cellular transformation (Kendall et al. 2005) and that mimic pathway aberrations frequently found in GBM (Parsons et al. 2008; The Cancer Genome Atlas Research Network 2008). Specifically, we used expression of hTERT, dominant-negative p53DD, CyclinD1, CDK4R24C (p16-resistant), H-RasV12, and MYC either alone or in combination in NSC-CB660 cells and tested the sensitivity of the resulting cell lines to the SF3b inhibitors pladienolide B (Kotake et al. 2007) and SudC1. That the p53 pathway (i.e., p53DD) and the Rb axis (i.e., CyclinD1 and CDK4R24C) were functionally impacted was noted by virtue of the fact that only combined expression of p53DD, CyclinD1, and CDK4R24C was sufficient to bypass RasV12-induced senescence in human NSCs (Supplemental Fig. S5a; data not shown).
Using this platform, we found that expression of MYC alone in NSCs is sufficient to induce sensitivity to U2snRNP perturbation observed in primary GSC cultures (Fig. 5A,B). In each experiment, dramatic cell death was observed at doses not lethal to the parent CB660 cells (Fig. 5C). We further validated this effect in fibroblasts (Supplemental Fig. S5d) and also using two NSC lines immortalized through MYC expression (CX and VM) (Donato et al. 2007) versus two primary NSCs (one embryonic and one adult-derived) without exogenous MYC (Supplemental Fig. S5e). Moreover, we observed that our GSC isolates show higher expression of MYC mRNA than NSCs (Fig. 5D).
Figure 5.
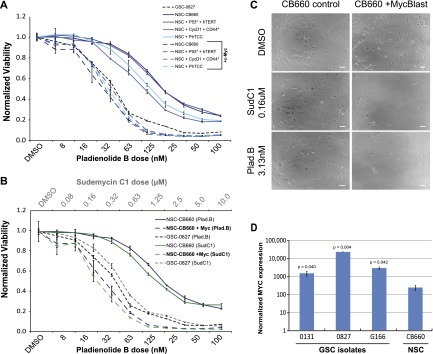
MYC expression in NSCs recapitulates GSC sensitivity to splicing inhibition. (A) Viability of NSCs with or without expression of hTERT, p53DD, CyclinD1, CDK4R24C, and/or MYC after treatment with pladienolide B. (B) Viability of NSCs with or without MYC expression after treatment with SudC1 (top X-axis) or pladienolide B (bottom X-axis). (C) Micrographs of normal CB660 NSCs with or without MYC expression after treatment with the indicated splicing inhibitors. Bar, 64 μm. (D) Log graph of MYC levels in GSCs and NSCs as determined by RNA-seq (fragments per kilobase of exon per million reads mapped [FPKM] normalized; n = 3) (Ding et al. 2013). P-values were determined by Student's t-test. See also Supplemental Figures S5 and S6.
In addition, we found that RasV12 expression alone could also sensitize NSCs, normal human astrocytes, or fibroblasts to PHF5A/U2snRNP perturbation (Supplemental Figs. S5c,e, S6) but did not synergize with MYC expression (Supplemental Fig. S5c). Moreover, the activated MEK allele could partially sensitize cells (Supplemental Fig. S5e). Both results are consistent with the notion that MYC is a downstream target of the Ras pathway by multiple pathways, including ERK and GSK-3 (Sears et al. 2000). Taken together, these results are consistent with recent observations regarding brain tumor-associated MYC activity. For example, concomitant loss of PTEN and p53, two of the most frequently mutated genes in GBM tumors, activates MYC (Zheng et al. 2008a,b), and MYC activity contributes to maintenance of tumor-initiating capacity in mouse and human models of GBM (Wang et al. 2008; Zheng et al. 2008b). The results suggest that inappropriate MYC activity in GBM tumors can give rise to molecular vulnerabilities in PHF5/U2snRNP function. However, future work will need to determine just how MYC function can impact the integrity of 3′ splice site recognition. However, these results raise the possibility that a wide range of MYC- and/or Ras-driven cancer may be vulnerable to PHF5A/U2snRNP inhibition.
Suppression of PHF5A expression compromises GBM tumor formation and maintenance in vivo
Finally, we wished to test whether PHF5A expression was required for GBM tumor formation and maintenance in vivo. To examine tumor formation, we devised an in vivo competition experiment to directly test the proliferative effects of PHF5A suppression in an orthotopic xenograft model of glioblastoma. GSCs were infected with GFP-expressing shPHF5A or shCtrl virus and then mixed with 10% ChFP-expressing control cells. This cell mixture was then either grown in adherent culture or xenografted into the cortex of immunocompromised mice (Supplemental Fig. S7a). Whereas shCtrl cells were able to proliferate and maintain their representation in culture, shPHF5A cells began to exhibit characteristic cell cycle arrest within 2 d of xenograft and were almost completely replaced by ChFP+ control cells within 2 wk (Supplemental Fig. S7b). Likewise, orthotopically xenografted GFP+ shCtrl GSCs were able to proliferate in vivo, whereas GFP+ shPHF5A GSCs were unable to proliferate and meaningfully contribute to in vivo tumor growth (Fig. 6A). The small fraction of coinjected ChFP+ control GSCs were able to engraft and give rise to tumors in every case, and ChFP expression mirrored bulk tumor mass as marked by the Chlorotoxin:Cy5.5 conjugate Tumor Paint (CTX:Cy5.5) (Veiseh et al. 2007). This underscores that expression of PHF5A shRNA was the key determinant in whether GSCs could contribute to tumor growth.
Figure 6.

Suppression of PHF5A expression compromises GBM tumor formation and maintenance in vivo. (A) Fluorescence overlay images of in vivo competition mouse brains 5 wk after xenograft. In addition to GFP and ChFP signal, CTX:Cy5.5 (Tumor Paint) was used to mark total tumor mass. (B,C) Flank xenograft volume over time of GSC-0131 clones expressing Dox-inducible PHF5A shRNA or control (Ctrl) shRNA. Tumors were allowed to progress in the absence of Dox until the tumor volume of each cohort averaged ∼75 mm3. Mice were then randomized onto continuous Dox or vehicle treatment, and tumor volume was monitored over time. (D) Kaplan-Meier analysis of mice bearing brain xenografts of Dox-inducible PHF5A knockdown GSCs. (Inset) At the first sign of symptoms in the first mouse (day 52; CTX:Cy5.5 image) mice were randomized onto continuous Dox or vehicle treatment, and survival was monitored over time. Photographs of representative mice from each cohort are shown. See also Supplemental Figure S7.
We next wished to examine whether PHF5A inhibition in established tumors could compromise tumor maintenance, a key metric in evaluating potential therapeutic avenues. To this end, we generated xenograft mice bearing GSC tumors with doxycycline (Dox)-inducible PHF5A shRNA (Fig. 1F; Supplemental Fig. S1d) or control shRNAs. Tumors were allowed to grow to ∼75 mm3 in size prior to the start of continuous Dox treatment. Whereas control shRNA tumors showed no measurable difference in growth rate upon Dox treatment (Fig. 6B), shPHF5A tumor growth arrested upon Dox administration, and tumors diminished until they were nearly undetectable (Fig. 6C). The onset of this growth arrest corresponded to greatly increased phosphorylation of Histone H3-S10 (Supplemental Fig. S7c,d) indicating a G2/M cell cycle arrest similar to that seen in vitro for shPHF5A-treated GSCs.
Since the above tumor studies were carried out in mouse flanks rather than the brain where GBM arises, we finally asked whether brain-derived, GSC-driven tumors would respond to PHF5A suppression as well. To test this, we xenografted GSCs bearing Dox-inducible PHF5A shRNA into the right cortex of immunocompromised mice. After 52 d, the first mouse showed initial mild symptoms of a brain tumor. CTX:Cy5.5 imaging after sacrifice confirmed a tumor signal in the right cortex (Fig. 6D, inset). We therefore randomized the remaining mice into Dox-treated and vehicle control cohorts and followed their survival over time. Survival was significantly improved by PHF5A suppression in the Dox-treated cohort (P = 0.0006), to the point where, at the conclusion of the study, when all vehicle-treated mice had succumbed to their tumors, 100% of Dox-treated mice were alive and free of symptoms (Fig. 6D). We conclude that PHF5A inhibition compromises both GBM tumor formation and maintenance, suggesting that PHF5A/U2snRNP inhibition may be an effective therapy for GBM.
Discussion
Here, we performed parallel shRNA screens during in vitro expansion of human GSCs and NSCs to identify novel gene activities required for growth and viability of patient-derived GSCs but not normal NSCs. Despite observing a high degree of GSC isolate-specific variation in the screening results, we identified PHF5A as differentially required for expansion of all GSCs examined. PHF5A is a highly conserved PHD-zinc finger domain protein that facilitates interactions between the U2 snRNP complex and ATP-dependent helicases (Rzymski et al. 2008). In vitro assays established that PHF5A activity was required for G2/M progression in GSCs but not NSCs. Consistent with a role in GSC-specific splicing phenomena, knockdown of other U2 snRNP complex members or pharmacological inhibition of U2 snRNP activity both phenocopied PHF5A knockdown. Furthermore, PHF5A knockdown triggered defective splicing of thousands of essential genes, including many important for mitotic progression (e.g., CDC20). Examination of affected splice sites revealed a specific requirement for PHF5A in recognition of 3′ splice sites with C-rich polypyrimidine tracts in GSCs. Moreover, modeling experiments in MYC- and RasV12-expressing NSCs and fibroblasts suggested that oncogenic signaling gives rise to the added requirement for PHF5A and U2 snRNP activity. Finally, in vivo tumor experiments suggested that PHF5A is required for generation and, more importantly, maintenance of GBM tumors. Taken together, our results support a model whereby oncogenic signaling leading to increased MYC activity triggers enhanced reliance on PHF5A/U2snRNP to properly recognize a subclass of 3′ splice sites.
Our results provide new insight into cancer-specific RNA splicing phenomena. Oncogene-associated activities (e.g., MYC, AKT, and Ras) can alter splicing of select genes, including pyruvate kinase (Clower et al. 2010; David et al. 2010), caspases (Shultz et al. 2010), and CD44 (Weg-Remers et al. 2001), to promote tumor formation or growth. Our results provide two important contrasts to these findings. First, while previous examples of cancer-associated splicing events can be largely explained by the activity of canonical splicing regulators on individual substrates, our results indicate that fundamental aspects of 3′ splice site recognition are modulated by transformation. In particular, the GSC-specific reliance on PHF5A suggests that basal spliceosome composition may differ between normal and transformed cells. Therefore, many cancer-associated changes in splicing may be inherently global, rather than specific, phenomena. Second, the many reports of cancer-promoting protein isoforms suggest that splicing can confer tumor robustness. However, our findings suggest a different model, wherein splicing is a source of tumor vulnerability due to “fragile” recognition of specific subclasses of exons.
Future studies are required to define just how this vulnerability might arise in GBM and other transformed cells. One possibility is that oncogenic signaling leads to direct perturbation of U2 snRNP activity by affecting complex assembly, activity, turnover, nuclear localization, and/or coordination with transcription (for review, see Heyd and Lynch 2011). As many splicing factors have complementary or compensatory activity and also participate in feedback loops to maintain cellular homeostasis, one attractive model is that transformation disrupts expression of spliceosomal proteins that normally complement PHF5A's role. A related question is how MYC activity gives rise to PHF5A/U2snRNP sensitivity. MYC is known to affect the expression of specific splicing factors (David et al. 2010; Das et al. 2012), which can alter ratios of spliced isoforms of genes such as pyruvate kinase (David et al. 2010). However, with regard to 3′ splice site recognition and U2snRNP function, it is unclear whether MYC activity has a direct role in causing a perturbation or does so indirectly, for example, by perturbing regulatory pathways that in turn might affect splicing fidelity (e.g., protein turnover/degradation).
Another key question arising from our studies is whether PHF5A and U2 snRNP represent reasonable therapeutic targets for GBM. One notable benefit of targeting their activity is that partial inhibition simultaneously affects the splicing of thousands of essential genes. For the vast majority of affected genes, the resulting isoforms contain in-frame stop codons, resulting in either degradation by NMD or translation of aberrant truncated proteins. Therefore, targeting PHF5A or U2 snRNP leads to partial or complete loss of function for many essential genes, collectively causing loss of viability. For example, the observed arrest phenotype is likely due to simultaneous dysregulation of many genes required for cell cycle progression (CDC16, CDC20, CDC25C, CDC37, CDC45, RCC2, etc.) rather than abrogation of a single “target” gene's activity. In contrast to the yeast cef1-13 example—where the arrest phenotype was rescued by removal of a single misspliced intron in α-tubulin (Burns et al. 2002)—mutations affecting just one of the thousands of dysregulated splice sites are highly unlikely to rescue GSCs. As a result, targeting PHF5A/U2 snRNP may have an advantage over current targeted therapeutic strategies focused on inhibiting the activities of single oncogenic drivers (e.g., EGFR, RAF1, AKT, etc.), which cancer cells can circumvent through mutation or up-regulation of parallel or downstream pathway components. Moreover, classes of synthetic and natural compounds already exist that inhibit U2 snRNP activity (e.g., Pladienolide B, SudC1, and SSA). The natural product splicing modulators were originally identified on the basis of anti-cancer activity in vitro and in vivo (Nakajima et al. 1996; Mizui et al. 2004; Kaida et al. 2007; Kotake et al. 2007), and at least one derivative has entered clinical trials for solid tumors (NCT00499499). Our results suggest that further investigation of this family of compounds may be beneficial for GBM as well as a variety of other MYC- and Ras-driven cancers.
In summary, this study establishes that patient-derived GSCs are vulnerable to perturbation in recognition of a subclass of 3′ splice sites, which results in a reduction in GSC viability and loss of GBM tumor maintenance. Since standard of care therapies are ineffective against GBM, we proffer that targeting PHF5A and/or U2 snRNP activity may offer a new therapeutic inroad for this cancer.
Materials and methods
Pooled shRNA barcode screens and analysis
For both the focused and genome-wide RNAi screens, GSCs or NSCs were infected with pooled GIPZ lentivirus (Open Biosystems) at a multiplicity of infection (MOI) <1 and selected with puromycin (Sigma) to remove uninfected cells. Cells were propagated in culture for an additional 21 d, during which time a minimal representation of 1000-fold per replicate was maintained. For each corresponding sample, shRNA barcodes (for microarray or one-half hairpin for deep shRNA sequencing) were PCR-recovered from genomic DNA samples. The change in the relative abundance of each shRNA in the library over time was measured using the normalized Cy3/Cy5 ratio of its probe signal or sequence counts. Barcode probes depleted in the GSC samples were considered candidate screen hits. Hits from the genome-wide screens were further filtered based on cellular expression as measured by RNA-seq.
Additional methods can be found in the Supplemental Material.
Acknowledgments
We thank Stephen Tapscott, Bruce Clurman, and Valeri Vasioukhin for critical reading of this manuscript; Minoru Yoshida, Howard Fine, Russell Pieper, Do-Hyun Nam, Yoshihiko Kotake, and Austin Smith for providing cell lines and/or reagents; Stacey Hansen, Kyle Pedro, and Sally Ditzler for technical help; Pam Lindberg and Laima Abele for administrative support; and members of the Paddison and Olson laboratories for helpful discussions. This work was supported by Fred Hutchinson Cancer Research Center institutional funds (P.J.P. and R.K.B.) and grants from AACI Translational Cancer Research Fellowship (to Y.D.), Accelerate Brain Cancer Cure (to P.J.P.), Damon Runyon Cancer Research Foundation (DFS 04-12 to R.K.B.), Department of Defense Translational New Investigator Award CA100735 (to P.J.P.), National Cancer Institute/National Institutes of Health (NCI/NIH) (R21CA170722-01 and P30CA15704 to P.J.P.; and R01CA155360 , R01 CA135491, and 5R01 CA114567 to J.M.O.), NIH Interdisciplinary Training in Cancer Research Program (no. T32CA080416 to C.G.H.), the Pew Biomedical Scholars Program (to P.J. P.), a Phi Beta Psi Sorority Cancer Research Grant (to P.J.P.), and U.K.-U.S. Stem Cell Collaboration Development Award (to S.M.P. and P.J.P.).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.212548.112.
References
- Amrani N, Sachs MS, Jacobson A 2006. Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol 7: 415–425 [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN 2006a. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444: 756–760 [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, Rich JN 2006b. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 66: 7843–7848 [DOI] [PubMed] [Google Scholar]
- Bradley RK, Merkin J, Lambert NJ, Burge CB 2012. Alternative splicing of RNA triplets is often regulated and accelerates proteome evolution. PLoS Biol 10: e1001229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CG, Ohi R, Mehta S, O'Toole ET, Winey M, Clark TA, Sugnet CW, Ares M Jr, Gould KL 2002. Removal of a single α-tubulin gene intron suppresses cell cycle arrest phenotypes of splicing factor mutations in Saccharomyces cerevisiae. Mol Cell Biol 22: 801–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, et al. 2013. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153: 139–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR 2010. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci 107: 1894–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrionero A, Minana B, Valcarcel J 2011. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev 25: 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Anczukow O, Akerman M, Krainer AR 2012. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep 1: 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Chen M, Assanah M, Canoll P, Manley JL 2010. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463: 364–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Hubert CG, Herman J, Corrin P, Toledo CM, Skutt-Kakaria K, Vazquez J, Basom R, Zhang B, Risler JK, et al. 2013. Cancer-specific requirement for BUB1B/BUBR1 in human brain tumor isolates and genetically transformed cells. Cancer Discov 3: 198–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato R, Miljan EA, Hines SJ, Aouabdi S, Pollock K, Patel S, Edwards FA, Sinden JD 2007. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci 8: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM 2011. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem Biol 6: 582–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Acheampong E, Dave R, Wang F, Mukhtar M, Pomerantz RJ 2005. The RNA helicase DDX1 is involved in restricted HIV-1 Rev function in human astrocytes. Virology 336: 299–307 [DOI] [PubMed] [Google Scholar]
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A 2004. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 64: 7011–7021 [DOI] [PubMed] [Google Scholar]
- Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, Ravetti GL, Zona GL, Daga A, Corte G 2009. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 27: 40–48 [DOI] [PubMed] [Google Scholar]
- Gooding C, Clark F, Wollerton MC, Grellscheid SN, Groom H, Smith CW 2006. A class of human exons with predicted distant branch points revealed by analysis of AG dinucleotide exclusion zones. Genome Biol 7: R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI 2003. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci 100: 15178–15183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyd F, Lynch KW 2011. Degrade, move, regroup: Signaling control of splicing proteins. Trends Biochem Sci 36: 397–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurica MS, Moore MJ 2003. Pre-mRNA splicing: Awash in a sea of proteins. Mol Cell 12: 5–14 [DOI] [PubMed] [Google Scholar]
- Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, et al. 2007. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol 3: 576–583 [DOI] [PubMed] [Google Scholar]
- Kendall SD, Linardic CM, Adam SJ, Counter CM 2005. A network of genetic events sufficient to convert normal human cells to a tumorigenic state. Cancer Res 65: 9824–9828 [DOI] [PubMed] [Google Scholar]
- Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y 2007. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol 3: 570–575 [DOI] [PubMed] [Google Scholar]
- Kramer A 1996. The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu Rev Biochem 65: 367–409 [DOI] [PubMed] [Google Scholar]
- Lagisetti C, Pourpak A, Goronga T, Jiang Q, Cui X, Hyle J, Lahti JM, Morris SW, Webb TR 2009. Synthetic mRNA splicing modulator compounds with in vivo antitumor activity. J Med Chem 52: 6979–6990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagisetti C, Pourpak A, Jiang Q, Cui X, Goronga T, Morris SW, Webb TR 2008. Antitumor compounds based on a natural product consensus pharmacophore. J Med Chem 51: 6220–6224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson MA, Kapoor TM 2005. The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nat Cell Biol 7: 93–98 [DOI] [PubMed] [Google Scholar]
- Latera J, Brem H 2002. Primary brain tumors in adults. In Diseases of the nervous system: Clinical neuroscience and theraputic principals (ed. A Asbury, et al.), pp. 1431–1466 Cambridge University Press, Cambridge, UK. [Google Scholar]
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. 2006. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9: 391–403 [DOI] [PubMed] [Google Scholar]
- Li B, Dewey CN 2011. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Nguyen DH, Dong Q, Shitaku P, Chung K, Liu OY, Tso JL, Liu JY, Konkankit V, Cloughesy TF, et al. 2009. Molecular properties of CD133+ glioblastoma stem cells derived from treatment-refractory recurrent brain tumors. J Neurooncol 94: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ 2009. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137: 835–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiola A, Lama G, Giannitelli C, De Bonis P, Anile C, Lauriola L, La Torre G, Sabatino G, Maira G, Jhanwar-Uniyal M, et al. 2007. Stem cell marker nestin and c-Jun NH2-terminal kinases in tumor and peritumor areas of glioblastoma multiforme: Possible prognostic implications. Clin Cancer Res 13: 6970–6977 [DOI] [PubMed] [Google Scholar]
- Mellor J 2006. It takes a PHD to read the histone code. Cell 126: 22–24 [DOI] [PubMed] [Google Scholar]
- Mizui Y, Sakai T, Iwata M, Uenaka T, Okamoto K, Shimizu H, Yamori T, Yoshimatsu K, Asada M 2004. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J Antibiot (Tokyo) 57: 188–196 [DOI] [PubMed] [Google Scholar]
- Musselman CA, Kutateladze TG 2009. PHD fingers: Epigenetic effectors and potential drug targets. Mol Interv 9: 314–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, Shimomura K 1996. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J Antibiot (Tokyo) 49: 1204–1211 [DOI] [PubMed] [Google Scholar]
- Oltra E, Pfeifer I, Werner R 2003. Ini, a small nuclear protein that enhances the response of the connexin43 gene to estrogen. Endocrinology 144: 3148–3158 [DOI] [PubMed] [Google Scholar]
- Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O'Shaughnessy A, Gnoj L, Scobie K, et al. 2004. A resource for large-scale RNA-interference-based screens in mammals. Nature 428: 427–431 [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. 2008. An integrated genomic analysis of human glioblastoma multiforme. Science 321: 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. 2009. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35: 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, et al. 2009. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4: 568–580 [DOI] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzymski T, Grzmil P, Meinhardt A, Wolf S, Burfeind P 2008. PHF5A represents a bridge protein between splicing proteins and ATP-dependent helicases and is differentially expressed during mouse spermatogenesis. Cytogenet Genome Res 121: 232–244 [DOI] [PubMed] [Google Scholar]
- Sawin KE, LeGuellec K, Philippe M, Mitchison TJ 1992. Mitotic spindle organization by a plus-end-directed microtubule motor. Nature 359: 540–543 [DOI] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR 2000. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 14: 2501–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz JC, Goehe RW, Wijesinghe DS, Murudkar C, Hawkins AJ, Shay JW, Minna JD, Chalfant CE 2010. Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res 70: 9185–9196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB 2003. Identification of a cancer stem cell in human brain tumors. Cancer Res 63: 5821–5828 [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB 2004. Identification of human brain tumour initiating cells. Nature 432: 396–401 [DOI] [PubMed] [Google Scholar]
- Son MJ, Woolard K, Nam DH, Lee J, Fine HA 2009. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 4: 440–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles CD, Rowitch DH 2008. Glioma stem cells: A midterm exam. Neuron 58: 832–846 [DOI] [PubMed] [Google Scholar]
- Trappe R, Ahmed M, Glaser B, Vogel C, Tascou S, Burfeind P, Engel W 2002. Identification and characterization of a novel murine multigene family containing a PHD-finger-like motif. Biochem Biophys Res Commun 293: 816–826 [DOI] [PubMed] [Google Scholar]
- Veiseh M, Gabikian P, Bahrami SB, Veiseh O, Zhang M, Hackman RC, Ravanpay AC, Stroud MR, Kusuma Y, Hansen SJ, et al. 2007. Tumor paint: A chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res 67: 6882–6888 [DOI] [PubMed] [Google Scholar]
- Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, Hjelmeland AB, Rich JN 2008. c-Myc is required for maintenance of glioma cancer stem cells. PLoS ONE 3: e3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weg-Remers S, Ponta H, Herrlich P, Konig H 2001. Regulation of alternative pre-mRNA splicing by the ERK MAP-kinase pathway. EMBO J 20: 4194–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Urlaub H, Achsel T, Gentzel M, Wilm M, Luhrmann R 2002. Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J 21: 4978–4988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, et al. 2008a. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455: 1129–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, et al. 2008b. Pten and p53 converge on c-Myc to control differentiation, self-renewal, and transformation of normal and neoplastic stem cells in glioblastoma. Cold Spring Harb Symp Quant Biol 73: 427–437 [DOI] [PubMed] [Google Scholar]