

RSV reinfection increases IL-4Rα expression on T helper cells, responsible for the enhanced immunopathologies in mice initially infected as neonates.
Keywords: viral infection, infants, T helper cells
Abstract
RSV is the major cause of severe bronchiolitis in infants, and severe bronchiolitis as a result of RSV is associated with subsequent asthma development. A biased Th2 immune response is thought to be responsible for neonatal RSV pathogenesis; however, molecular mechanisms remain elusive. Our data demonstrate, for the first time, that IL-4Rα is up-regulated in vitro on human CD4+ T cells from cord blood following RSV stimulation and in vivo on mouse pulmonary CD4+ T cells upon reinfection of mice, initially infected as neonates. Th cell-specific deletion of Il4ra attenuated Th2 responses and abolished the immunopathophysiology upon reinfection, including airway hyper-reactivity, eosinophilia, and mucus hyperproduction in mice infected initially as neonates. These findings support a pathogenic role for IL-4Rα on Th cells following RSV reinfection of mice initially infected as neonates; more importantly, our data from human cells suggest that the same mechanism occurs in humans.
Introduction
RSV is the leading cause of acute respiratory tract infections in infants younger than 1 year of age [1, 2]. Typically children are infected with RSV in their first RSV season, and 50% of them are reinfected by 2 years of age [3, 4]. Long-term resistance to RSV infection does not develop, and reinfection is common, particularly in those with chronic lung or heart diseases, where severity of the illness is much greater [5]. Although there is considerable worldwide, clinical impact from RSV infection, we have neither vaccines nor specific therapeutics against RSV infection. Development of vaccines or therapeutics has been slow as a result of limited understanding of the virus, the host immune system at the time of initial infection, and the interaction between the virus and the host immune system.
Studies have demonstrated the importance of age at the time of initial RSV infection in determining the pathophysiological responses [6–11]. In humans, severe RSV infection in infants leads to respiratory dysfunction that can persist into early adulthood [12–15]. In mice, we have demonstrated that age of initial infection with RSV determines respiratory function in later life [8, 9]. Mice infected with RSV as neonates (<7 days of age) develop long-term airway dysfunction [8], whereas mice infected initially as weanlings fail to do so [9]. Studies from other laboratories have demonstrated further that mice infected as neonates respond to RSV reinfection with a skewed Th2 immune response [10, 11]. Increased susceptibility to RSV and disease severity in infants are thought to be, in part, a result of the developmental state of their immune system [16, 17], although other factors, such as airway size and maturation status of the lung, may also contribute to the pathology observed.
In humans, Th2 cytokines IL-4 and IL-13 and their receptors have been associated with severe RSV disease [18–23]. In mouse models, IL-4/13 and their receptors have been shown to play a key role in the multiple pathophysiologies associated with infant RSV infection, including airway hyper-reactivity, eosinophilic inflammation, and mucus hyperproduction [18–23]. Interestingly, IL-4 and IL-13 share a common receptor, the type II IL-4R, which is composed of IL-4Rα and IL-13Rα1 and is thought to be responsible for the pathological changes observed in asthma and RSV-associated disease [20, 24, 25]. IL-4 also binds to a second receptor, the type I receptor, which is composed of IL-4Rα and a γ chain and is believed to initiate Th2 differentiation. All of these data point to a potential role of IL-4Rα in RSV pathogenesis in infants.
Here, we confirm that upon reinfection, mice infected initially with RSV as neonates responded with a skewed Th2 immune response associated with airway hyper-reactivity, eosinophilia, and mucus hyperproduction. This is not the case when the initial infection occurs in adults. The immunophysiological changes following reinfection of neonatally infected mice correlated with increased expression of IL-4Rα on pulmonary CD4+ Th cells and elevated proliferation of these cells, especially the Th2 subset. Specific deletion of Il4ra from Th cells attenuated pulmonary Th2 responses and abolished the immunopathophysiology associated with RSV reinfection. Validating these data in the human system, we observed elevated IL-4Rα expression on CD4+ Th cells from cord blood following stimulation with RSV in vitro. Collectively, our data reveal a mechanism, whereby IL-4Rα signaling on Th cells in neonates results in an exaggerated Th2 immune response upon reinfection with RSV and adds significantly to our understanding of RSV pathogenesis, which may be applicable to humans.
MATERIALS AND METHODS
Mice
BALB/c breeder pairs and female adult mice (6–8 weeks old) were purchased from Harlan Laboratories (Indianapolis, IN, USA) and were maintained under a specific, pathogen-free condition in the Division of Animal Care at LSUHSC (New Orleans, LA, USA). Breeders were time-mated, and pups born on the same date were used for experiments. The CD4+ T cell-specific, IL-4Rα-deficient mice (LckcreIL4Rα−/Lox) were generated by crossing IL4RαLox/Lox BALB/c mice and LckcreIL4Rα−/− BALB/c mice (gifts from Drs. Debroski Herbert and Frank Brombacher, University of California, San Francisco, USA, and University of Cape Town, South Africa). In these mice, Cre recombinase is under the control of the T cell-specific promoter Lck; however, LckcreIL4Rα−/Lox mice showed effective deletion of Il4ra in CD4+ T cells but incomplete and variable deletion in other T cells, including CD8+ T cells, NKT cells, and γδT cells [26].
All animal protocols were prepared in accordance with the Guide for the Care and Use of Laboratory Animals [27] and were approved by the Institutional Animal Care and Use Committee at LSUHSC.
Human cord or peripheral blood samples
Cord and peripheral blood samples were obtained with informed, written consent from term neonatal subjects (between 38 and 41 weeks of gestation), delivered at the Children's & Women's Health Centre of British Columbia (Vancouver, BC, Canada), and from healthy adult volunteers at the Child & Family Research Institute (Vancouver, BC, Canada), respectively. Consent was obtained for children by their parent or legal guardian. Protocols were approved by the Clinical Research Ethics Board (H07-02681).
RSV infection
The original human RSV strain A-2 was purchased from Advanced Biotechnologies (Columbia, MD, USA). For all mouse studies, the virus was propagated in Vero cells (American Type Culture Collection, Manassas, VA, USA) using serum-free medium (SFM4MegaVir; HyClone, Logan, UT, USA) and stored at −80°C. Five-day-old pups or adults were anesthetized with 5% isofluorane and infected intranasally with 2 × 105 50% tissue culture-infective dose/g body weight of RSV in 10 μl (for pups) or 50 μl (for adults) serum-free medium. In some experiments, mice were sham-infected with serum-free medium. For stimulation of human PBMCs and CBMCs, the virus was propagated in HEp-2 cells (Hybrid Diagnostics, Athens, OH, USA) using minimal essential medium with Earle's salts, containing 2% FBS and 1 mM sodium pyruvate (HyClone), and purified further by ultracentrifugation on a discontinuous sucrose gradient [28].
Pulmonary function test
Airway resistance upon MeCh (Sigma-Aldrich, St. Louis, MO, USA) challenge was measured using the flexiVent system (Scireq, Montreal, QC, Canada). Mice were anesthetized with ketamine/xylazine (180/10 mg/kg body weight), tracheotomized, and ventilated with a computer-controlled piston pump. The pressure and volume of the airway were recorded and fitted into a single compartment model to calculate airway resistance. Raw data were normalized by subtracting individual baseline values (resistance at 0 mg/ml MeCh) and plotted as normalized resistance. Baseline values were not statistically different among the groups.
Lung histopathology
Mice were killed and retrograde perfusion performed to remove blood from the lungs, which were then gently inflated and fixed with zinc formalin (Thermo Fisher Scientific, Waltham, MA, USA). After fixation, lungs were embedded in paraffin, sectioned, and mounted on slides; then, the slides were stained with H&E or PAS to identify cellular infiltrates or mucus production in the airway epithelial cells, respectively. Lung sections were stained by H&E and visualized under high magnification (60×). Eosinophils were quantified in the peribronchial tissues of lung specimens in a blinded manner and expressed as number of eosinophils/surface area of the lung parenchyma. Mucus expression in the airway epithelial cells was detected by PAS staining and images acquired. Images were analyzed with ImageJ [29]. Mucus expression was quantified and expressed as area of mucus-positive cells/surface area of airway epithelial cells.
T cell activation assay
To measure the proliferative ability of lung Th cells in vitro, T cell activation assay was performed [30]. Briefly, pulmonary CD11c+ cells were isolated from naïve, adult mice with a mouse CD11c-positive selection kit from Stemcell Technologies (Vancouver, BC, Canada) and infected with RSV (MOI=50) for 18 h. Lung CD4+ T cells were selected with a mouse CD4-positive selection kit (Stemcell Technologies) and stained with 0.5 μM CFSE (Life Technologies, Carlsbad, CA, USA). CD4+ cells (5×104) were then cocultured with 5 × 103 CD11c+ cells in the presence of 1 μg/ml CD3 antibody (clone #17A2; eBioscience, San Diego, CA, USA) for 72 h. The cells were stimulated, stained, and subjected to flow cytometry to determine the Th populations and CFSE signals in each population. The feasibility of this T cell activation assay was tested using increasing infectious doses of RSV, with or without CD3 antibody (Supplemental Fig. 1).
BALF cellularity
BALFs were isolated by flushing lungs with 1 ml PBS containing 2% heat-inactivated FBS. Total cell numbers were counted with a hemocytometer. BALF cells were centrifuged onto slides and stained with a Hema-3 staining kit (Thermo Fisher Scientific). Differential cell counts were determined by two unbiased observers using standard morphological criteria.
Cytokine assay
Cytokine levels in the BALF were measured using a Milliplex mouse cytokine/chemokine assay kit (Millipore, Billerica, MA, USA) on the Bioplex system (Bio-Rad Laboratories, Hercules, CA, USA), as per the manufacturer's instruction. Each sample was assayed in duplicate for type I cytokines, IFN-γ and IL-12 (p40), and type II cytokines, IL-4 and IL-13. Data presented herein excluded any number outside of the sensitivity level of each cytokine.
Cell staining and flow cytometry
To determine the Th cell population in the lung, lung single-cell suspension was isolated using the gentleMACS Octo Dissociator and lung dissociation kit (Miltenyi Biotec, Auburn, CA, USA). The cells were then stimulated for 5 h at 37°C in stimulation media containing RPMI 1640 (HyClone), 10% heat inactivated FBS (Life Technologies), 100 U/ml penicillin (HyClone), 100 mg/ml streptomycin (HyClone), 5 ng/ml PMA (Sigma-Aldrich), and 500 ng/ml ionomycin (Sigma-Aldrich) in the presence of a protein transport inhibitor (1 μl/106 cells; GolgiPlug; BD Biosciences, Franklin Lakes, NJ, USA). After stimulation, the cells were stained with fixable viability dye (eBioscience); fixed and permeabilized with intracellular fixation and permeabilization buffer (eBioscience); and stained with antibodies to CD3 (clone #17A2; eBioscience), CD4 (clone #RM4-4; BioLegend, San Diego, CA, USA), IFN-γ (clone #XMG1.2; eBioscience), and IL-4 (clone #BVD6-24G2; eBioscience). The flow data were acquired with a Canto II flow cytometer (BD Biosciences), analyzed, and plotted with FlowJo software (version 7.6.5 for Windows; Tree Star, Ashland, OR, USA).
To determine the proliferation of Th cell populations in the T cell activation assay, the cells collected from the CD11c+ and CD4+ cell coculture were stimulated and stained as above and subjected to flow cytometry to measure the CFSE signal in each Th population.
To measure IL-4Rα expression level on pulmonary Th cells, lung cells were isolated as above. The single-cell suspension was then stained with fixable viability dye and antibodies to CD3 (clone #17A2; eBioscience), CD4 (clone #RM4-4; BioLegend), IL-4Rα (clone #mIL-4R-M1; BD Biosciences), and PE-labeled streptavidin (eBioscience) and subjected to flow cytometry.
To correlate our mouse data to humans, we also measured the expression level of IL-4Rα on Th cells from CBMCs and adult PBMCs, with or without RSV stimulation in vitro. For this purpose, PBMCs from seven healthy adult volunteers and CBMCs from seven term neonatal subjects were isolated by Ficoll extraction. PBMCs or CBMCs were infected with RSV or media at a MOI of 2 in RPMI 1640 (HyClone), containing 10% heat-inactivated FBS (HyClone) and 1 mM sodium pyruvate. Cells were harvested after 48 h, resuspended in FACS lysing solution (BD Biosciences), and stored at −80°C until staining. For staining, cells were thawed; permeabilized in FACS permeabilization buffer (eBioscience); and stained by antibodies to CD3 (clone #SP34-2 from BD Biosciences or clone #OKT3 from eBioscience), CD4 (clone #RPA-T4; eBioscience), T-bet (clone #4B10; eBioscience), and IL-4Rα (clone #hIL4R-M57; BD Biosciences). The staining profile was acquired on an LSR II flow cytometer (BD Biosciences). Given the heterogeneity in human immune responses, we calculated the iMFI of IL-4Rα-positive cells by multiplying the relative frequency (percent positive) of cells expressing IL-4Rα with the MFI of that population. iMFI had been shown to correlate better with vaccine-induced protection from Leishmania major challenge than either the percentage or the MFI of the cytokine-positive population [31].
Statistics
All data were plotted as means ± sem using Prism5 (GraphPad Software, La Jolla, CA, USA). The statistics were analyzed with Prism5 or InStat 3 (GraphPad Software). Two-way ANOVA and Bonferroni post hoc tests were used to compare between the groups for the pulmonary function tests. Student's t-test was used for comparisons between ARR versus NRR mice for Th populations, BALF cellularity, cytokines, and IL-4Rα expression. Mann-Whitney test was used to compare the expression level of IL-4Rα between infant and adult Th cells. One-way ANOVA and Bonferroni post hoc tests were used to test for differences among IL4Rα−/Lox, LckcreIL4Rα−/Lox, IL4Rα−/LoxRR, and LckcreIL4Rα−/LoxRR mice. Differences were considered significant if P < 0.05.
Online Supplemental material
Supplemental Fig. 1 demonstrated the feasibility of using CD3 antibody in the in vitro Th activation assay. Supplemental Fig. 2 showed that pulmonary viral load and Th responses during primary infection were similar, between 5- and 12-week-old mice.
RESULTS
Neonatal RSV infection induced airway hyper-reactivity and lung histopathology upon reinfection
To model the unique pathophysiological effects that RSV infection has on infants, we infected 5-day-old pups or adult mice (6–8 weeks) with RSV and then reinfected these mice at 4 weeks postprimary infection. At 6 days postreinfection, we assessed pulmonary function and evaluated pulmonary pathology. Pulmonary viral load data from other groups have shown that RSV infects and replicates in the lung during secondary infection [11]. As shown in Fig. 1A, NRR mice exhibited significantly greater airway resistance following MeCh challenge than ARR mice (6.16±0.99 vs. 1.88±0.26 cm H2O·s/ml at 50 mg/ml MeCh), indicating that neonatal primary infection but not adult primary infection results in airways hyper-reactivity upon reinfection. Histological analysis of lung sections from NRR and ARR mice revealed similar levels of perivascular and peribronchiolar inflammation between NRR and ARR mice (Fig. 1B). However, significantly more eosinophils were observed in the lungs of NRR versus ARR mice (932±151 vs. 46±15 cells/mm2 surface area of epithelium). Additionally, we observed a substantial amount of mucus cell hyperplasia and mucus hyperproduction in mice infected as neonates, whereas ARR mice exhibited little-to-moderate levels of mucus-positive staining cells in the airways (Fig. 1C). In fact, the area of mucus-positive cells in the airways of NRR mice was significantly greater than in ARR mice (0.47±0.04 vs. 0.08±0.02 μm2/μm2 epithelium). Occasionally, we observed sloughing epithelial cells in the airway of NRR mice (data not shown), and this was not observed in any of the lung sections from ARR mice.
Figure 1. Neonatal RSV infection induced pulmonary immunopathophysiology upon reinfection.
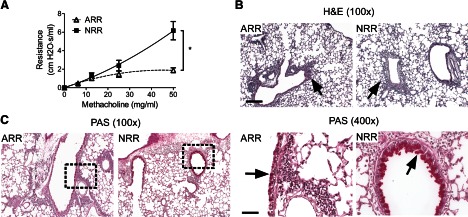
Neonates (5 days old; NRR) or adults (6–8 weeks old; ARR) were infected and reinfected with RSV at 4 weeks postprimary infection. Airway resistance and lung histopathology were determined at 6 days postsecondary infection. (A) Airway resistance was measured at baseline (0 mg/ml MeCh) and increasing doses of intratracheally delivered MeCh quantify airway hyper-reactivity. At baseline, airway resistance was not different between the groups; therefore, individual responses were normalized to their prospective baselines (i.e., subtracting resistance at 0 mg/ml MeCh from resistance value at dose) and plotted as normalized resistance; n = 5; *P < 0.05. (B) H&E staining showing peribronchial inflammation. The arrows point to cellular infiltrates. Original scale bar = 50 μm. (C) PAS staining showing airway mucus. Arrows point to mucus staining. The right panels are an enlargement of the rectangles in the left panels. Original scale bar = 200 μm.
Based on our previous studies in neonates and weanlings [9], we thought it was highly unlikely that age at reinfection (i.e., 5 or 10–12 weeks) had a substantial effect on Th cell responses following reinfection. However, to address this, we monitored pulmonary viral loads at 1, 2, and 4 dpi and Th cell responses at 6 days postprimary infection from mice infected at 5 or 12 weeks old. As expected, there was no difference in pulmonary viral load or primary T cell response (Supplemental Fig. 2) between these two groups. These data sustain that the differences we are observing are associated with the age difference at primary but not secondary infection.
Neonatal RSV infection induced an exaggerated Th2 response upon reinfection
We further examined the immune responses to RSV reinfection in the lungs of mice infected as neonates (NRR) or adults (ARR). Six days postreinfection, lung cells were isolated from NRR and ARR mice and Th subsets analyzed by flow cytometry (Fig. 2A and B). RSV reinfection induced substantial amounts of Th1 (IFN-γ-expressing CD4+ T) cells in both NRR and ARR mice (21.03±2.65 and 16.19±2.21%). However, NRR mice recruited six-fold more Th2 (IL-4-expressing CD4+ T) cells to the lung compared with ARR mice (3.8±0.98 vs. 0.46±0.12%). More interestingly, NRR mice had a subset of “multifunctional” Th cells that coexpressed the prototypical Th1 cytokine IFN-γ and Th2 cytokine IL-4. These data demonstrate that mice infected as neonates elicited a Th2-biased immune response in the lung upon secondary RSV infection.
Figure 2. Neonatal RSV infection induced an exaggerated Th2 response upon reinfection.

Neonates (5 days old; NRR) or adults (6–8 weeks old; ARR) were infected and reinfected with RSV at 4 weeks postprimary infection. Pulmonary Th cell subsets and BALF cellularity and cytokine levels were measured at 6 days postsecondary infection. (A) Pulmonary Th profile during secondary infection. Lung cells were isolated from NRR and ARR mice, stimulated in vitro with PMA and ionomycin, and stained with dead cell, excluding dye and antibodies to CD3, CD4, IFN-γ, and IL-4. The cells were then analyzed by flow cytometry. Lines indicate the mean of each group; n = 10. (B) Representative dot plots showing the IL-4- and IFN-γ-expressing Th cells in ARR and NRR mice. (C) BALF cellularity; n = 5. (D) Cytokine levels in the BALF. Th1 cytokines: IFN-γ and IL-12; Th2 cytokines: IL-4 and IL-13; n = 4–8. N.D., Not detected. (E) Pulmonary Th profile during primary infection. Lung cells were isolated from NR or AR mice and stimulated, stained, and analyzed as in B. Lines indicate the mean of each group; n = 4; *P < 0.05.
There was no significant difference in the total number of leukocytes recovered from the BALF between NRR and ARR mice (Fig. 2C). Significantly more macrophages were observed in the BALF of NRR compared with ARR mice (79,987±12,516 vs. 38,456±5367). In concordance with the exaggerated Th2 response, NRR mice also possessed a substantially greater number of eosinophils in the BALF compared with ARR mice (33,217±14,988 vs. 0; Fig. 2C). Finally, Th2 cytokines, such as IL-4 and IL-13, were readily detectable in the BALF of NRR mice and were secreted at significantly elevated levels compared with ARR mice (9.40±2.09 pg/ml vs. nondetectable and 68.42±10.56 vs. 23.46±1.85 pg/ml, respectively; Fig. 2D). Consistent with increased Th2 cytokine secretion, NRR mice secreted significantly less IL-12(p40) (15.81±1.6 vs. 47.72±6.7 pg/ml) compared with ARR mice. No significant difference was observed in IFN-γ levels between NRR and ARR mice.
It is possible that the exaggerated Th2 response observed in mice infected initially as neonates was a result of memory responses resulting from a skewed Th2 primary response. To test this, we measured the primary CD4+ T cell response in the lungs of neonatal or adult mice at 6 dpi. As shown in Fig. 2E, there were no difference in IL-4+ or IL-4+IFN-γ+ expressing CD4+ T cells between NR and AR. Interestingly, RSV infection of neonates resulted in significantly lower levels of pulmonary Th1 (IFN-γ-expressing CD4+) cells compared with adults (2.99±0.35 vs.0.60±0.08%), suggesting that the exaggerated Th2 response upon reinfection in neonatally infected mice was not a result of a biased Th2 primary response.
IL-4Rα was up-regulated on Th cells upon reinfection of mice infected initially as neonates
As IL-4 signaling is a determinant for Th cell development, we measured the expression of the common component of the IL-4R and IL-13R—IL-4Rα—on pulmonary Th cells of RSV-infected mice. Five-day-old pups or adults were infected with RSV and reinfected 4 weeks postprimary infection. At 2 days postreinfection, lung cells were isolated and the expression of IL-4Rα analyzed by flow cytometry. IL-4Rα expression was higher on Th cells from NRR versus ARR mice (MFI: 735.60±29.58 vs. 618.20±8.18; Fig. 3A and B). Furthermore, Th1 and Th2 cells from NRR mice expressed more IL-4Rα than those cells from the lungs of ARR mice (MFI: 891.00±15.55 vs. 810.60±15.33 and 1208.60±44.07 vs. 767.80±20.33, respectively; Fig. 3C). Interestingly, the expression of IL-4Rα on Th2 cells from NRR mice was significantly higher than its expression on Th1 cells from the same mice.
Figure 3. IL-4Rα was up-regulated upon reinfection on Th cells from mice infected originally as neonates.

Neonates (5 days old; NRR) or adults (6–8 weeks old; ARR) were infected and reinfected with RSV at 4 weeks postprimary infection. The expression of IL-4Rα on Th cells and Th subsets was determined by flow cytometry at 2 days postsecondary infection. Lung cells were isolated from NRR and ARR mice, stimulated in vitro, and stained with dead cell, excluding dye and antibodies to CD3, CD4, IFN-γ, IL-4, and IL-4Rα. (A) Relative expression level of IL-4Rα on CD4+ Th cells indicated by MFI of IL-4Rα; n = 5. (B) Representative histogram showing the staining profile of IL-4Rα on Th cells. IL-4Rα fluorescence minus one (FMO) serves as a negative control for IL-4Rα staining. (C) Relative expression level of IL-4Rα on CD4+ Th1 or Th2 cells, indicated by MFI of IL-4Rα; n = 5. (D) Relative expression level of IL-4Rα on circulating Th cells from cord blood of healthy term infants or from peripheral blood of healthy adults at baseline or upon in vitro stimulation with RSV. iMFI was calculated by multiplying the relative frequency (percent positive) of cells expressing IL-4Rα with MFI of that population; n = 7. (E) IL-4Rα expression on neonatal or adult CD4+ T cells during primary RSV infection in mice. Neonates (5 days) or adults (6–8 weeks) were infected with RSV (NR and AR, respectively) or media (NS and AS, respectively). IL-4Rα expression on CD4+ T cells was measured at 2 dpi; n = 5. NS, pups infected with media; AS, adults infected with media. (F) IL-4Rα expression on neonatal or adult Th1 cells during primary RSV infection. Mice were treated and IL-4Rα measured as in E; n = 5. (G) IL-4Rα expression on neonatal or adult Th1 cells during primary RSV infection. Mice were treated and IL-4Rα measured as in E; n = 5; *P < 0.05.
It is possible that IL-4Rα expression actually increased on neonatal Th cells during primary infection. To test this, we measured IL-4Rα expression on neonatal and adult CD4+ T cells at 2 days after primary infection. Surprisingly, IL-4Rα expression was decreased on naïve neonatal versus adult CD4+ T cells. However, with RSV infection, we observed a decrease in IL-4Rα expression on adult but not neonatal CD4+ T cells, and as a result, IL-4Rα expression was similar on CD4+ T cells between RSV-infected neonates and adults. More importantly and relevantly, neonatal Th1 and Th2 cells expressed higher levels of IL-4Rα compared with their adult counterparts at primary infection. Without RSV infection, neonatal Th2 cells showed significantly greater expression of IL-4Rα compared with adult Th2 cells.
IL-4Rα expression was up-regulated on Th cells purified from cord blood and stimulated with RSV in vitro
To begin to explore the translational potential of our findings, we compared the expression level of IL-4Rα on CD4+ T cells purified from cord blood or adult peripheral blood. As shown in Fig. 3D, Th cells from CBMCs or PBMCs expressed similar levels of IL-4Rα without stimulation, as indicated by the comparable iMFI values (3462±1132 vs. 4766±1186). However, when stimulated with RSV in vitro, IL-4Rα levels on Th cells from CBMCs increased substantially and significantly compared with IL-4Rα levels on Th cells from PBMCs (iMFI: 13,931±3095 vs. 3462±1132). These data demonstrate that IL-4Rα expression on infant CD4+ T cells was up-regulated, responding to RSV infection in humans.
Elevated IL-4Rα on CD4+ T cells upon reinfection was associated with hyperproliferation in vitro
To determine the mechanism by which Th2 bias occurs to RSV reinfection in neonatally infected mice, we examined the proliferative ability of the Th cells from mice infected as neonates (NRR) or adults (ARR) in vitro. Five-day-old pups or adults were infected with RSV and reinfected at 4 weeks postprimary infection. At 6 days after reinfection, lung CD4+ cells were isolated and cocultured with RSV-infected lung CD11c+ cells as APCs. Three days after coculture, CD4+ T cell subsets and their proliferation were measured. As shown in Fig. 4A and B, CD4+ T cells from NRR mice, when stimulated in vitro with RSV, possessed the ability to differentiate into Th1 (20.68±0.31%) and Th2 (7.57±0.48%) cells. In contrast, CD4+ T cells from ARR mice developed predominantly into Th1 cells (50.19±0.85%), with only 0.09% developing into Th2 cells. With the use of CFSE dilution, we analyzed the proliferative capacity of Th cells from NRR and ARR mice. We observed that 86.9 ± 0.44% of the Th2 cells from NRR mice had undergone cell division, whereas only 39.04 ± 7.59% of the Th2 cells from ARR mice divided into further generations (Fig. 4C and D). Th1 cells also proliferated with 65.9 ± 0.76% from NRR mice and 17.17 ± 1.33% from ARR mice undergoing cell division. In agreement with the IL-4Rα expression data, these data indicated that CD4+ T cells from NRR mice were more proliferative, that there was increased proliferation of Th2 cells from NRR mice, and that Th2 cells predominated in NRR mice.
Figure 4. CD4+ T cells from mice, infected initially as neonates versus adults, were more proliferative and were more prone to develop into Th2 cells.

Neonates (5 days old; NRR) or adults (6–8 weeks old; ARR) were infected and reinfected with RSV at 4 weeks postprimary infection. At 6 days postsecondary infection, pulmonary CD4+ T cells from NRR and ARR mice were isolated, stained with CFSE, and cocultured with RSV-infected lung CD11c+ cells (APCs) for 3 days. Cells were then collected and stained with dead cell, excluding dye and antibodies to CD3, CD4, IFN-γ, and IL-4. The cells were analyzed by flow cytometry to access the Th subsets and proliferation. Cytokines secreted into the medium were measured with multiplex cytokine assays. (A) Th cell profile. Lines indicate the mean of each group; n = 5. (B) Representative dot plots showing Th cells expressing IFN-γ or IL-4. (C) Proliferation of Th1 and Th2 cells, as indicated by CFSE dilution; n = 5. (D) Representative histogram showing the division of Th1 and Th2 cells; *P < 0.05.
Th cell-specific deletion of Il4ra abolished airway hyper-reactivity and lung histopathology upon reinfection in mice infected initially as neonates
As greater levels of IL-4Rα expression on pulmonary CD4+ T cells were associated with neonatal RSV infection-induced pathophysiological consequences, we hypothesized that IL-4Rα signaling was responsible for the observed phenotype. To investigate the role of IL-4Rα on CD4+ T cells, we infected mice specifically lacking IL-4Rα on CD4+ T cells (LckcreIL4Rα−/Lox) and their littermate controls (IL-4Rα−/lox) at 5 days of age and subsequently reinfected those mice with RSV, 4 weeks later (LckcreIL4Rα−/Lox-RR and IL4Rα−/Lox-RR, respectively). Control mice, including IL-4Rα-deficient and -competent mice, were mock-infected with viral production media (LckcreIL4Rα−/Lox-Sham and IL4Rα−/Lox-Sham, respectively). Pulmonary function and lung histopathology were determined at 6 days after reinfection.
As shown in Fig. 5A, no statistical difference in airways resistance was observed between the two sham-infected mouse genotypes (i.e., LckcreIL4Rα−/Lox-Sham and IL4Rα−/Lox-Sham), indicating that specific deletion of Il4ra in CD4+ T cells did not induce a baseline change in pulmonary function. As expected, RSV reinfection of the IL-4Rα-competent mice (IL4Rα−/Lox-RR) induced airway hyper-reactivity, as evidenced by the increase in airways resistance compared with the sham-infected control mice (IL4Rα−/Lox-Sham) in response to MeCh challenge. While in the absence of CD+ T cell-specific IL-4Rα, RSV reinfection failed to induce airway hyper-reactivity (LckcreIL4Rα−/Lox-RR vs. IL4Rα−/Lox-RR; 2.17±0.44 vs. 3.01±0.34 cm H2O·s/ml). Furthermore, there was no difference between CD4+ T cell-specific, IL-4Rα-deficient mice that were RSV- or SHAM-reinfected (LckcreIL4Rα−/Lox-RR vs. LckcreIL4Rα−/Lox-Sham; 2.17±0.44 vs. 2.43±0.18 cm H2O·s/ml), indicating a complete abrogation of airway hyper-reactivity in the CD4+ T cell-specific, IL-4Rα-deficient mice.
Figure 5. CD4+ T cell-specific deletion of IL4ra abolished neonatal RSV infection-induced immunopathophysiology upon reinfection.
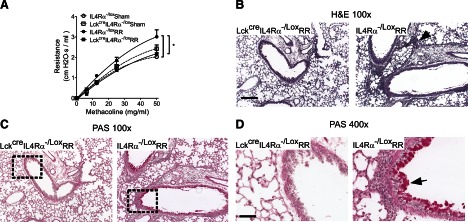
Th-specific, IL-4Rα-deficient (LckcreIL4Rα−/Lox-RR) and -competent mice (IL4Rα−/Lox-RR) were infected with RSV at 5 days old and reinfected at 4 weeks postprimary infection. Control mice of each genotype were treated with serum-free medium (LckcreIL4Rα−/Lox-sham and IL4Rα−/Lox-sham, respectively). Pulmonary function and lung histopathology were determined at 6 days postreinfection. (A) Airway resistance was measured at baseline (0 mg/ml MeCh) and increasing doses of intratracheally delivered MeCh to quantify airway hyper-reactivity. At baseline, airway resistance was not different between the groups; therefore, individual responses were normalized to their prospective baselines and normalized resistance plotted; n = 5–7; *P < 0.05. (B) H&E staining, demonstrating pulmonary inflammation. The arrow points to cellular infiltrates. Original scale bar = 50 μm. (C) PAS staining showing airway mucus. The arrow points to mucus staining. The right panels are enlargements of the rectangles in the left panels. Original scale bar = 200 μm.
Supporting the pulmonary function data, we observed a substantial decrease in perivascular and peribronchiolar inflammation in CD4+ T cell-specific, IL-4Rα-deficient mice (LckcreIL4Rα−/Lox-RR) compared with the IL-4Rα-competent mice (IL4Rα−/Lox-RR; Fig. 5B). In particular, substantially less eosinophils were observed in LckcreIL4Rα−/Lox-RR mice compared with IL4Rα−/Lox-RR mice (10±6 vs. 559±128 cells/mm2 surface area). Additionally, little-to-no mucus was observed in the airways of the CD4+ T cell-specific, IL-4Rα-deficient mice, whereas we observed a significant amount of mucus production and mucus cell hyperplasia in the IL-4Rα-competent mice (Fig. 5C; 0.006±0.005 vs. 0.25±0.04 μm2 area of mucus+cells/μm2 area of epithelium). No pulmonary inflammation or airway mucus was observed in IL-4Rα-deficient or -competent, sham-infected mice (data now shown). These data demonstrated a requirement for CD4+ T cell-specific IL-4Rα in pulmonary dysfunction and histopathology associated with RSV reinfection in neonatally infected mice.
Th-specific deletion of Il4ra attenuated Th2 responses to reinfection in mice infected initially as neonates
To understand how the absence of IL-4Rα on CD4+ T cells affected the pulmonary immune response to RSV reinfection, we analyzed the pulmonary immune response in the above mice. Pulmonary immunophenotyping, including CD4+ T cell profile and BALF cellularity and cytokine levels, were measured 6 days after reinfection. As shown in Fig. 6A and B, sham-infected, IL-4Rα-deficient and -competent mice failed to induce effector CD4+ T cells (i.e., few CD4+ T cells expressed effector cytokines: IFN-γ or IL-4. Reinfection with RSV induced substantial and similar amounts of the IFN-γ-expressing Th1 cells in IL-4Rα-deficient and -competent mice (21.80±1.41 vs. 20.48±3.52%) but failed to induce significant amounts of IL-4-expressing Th2 cells or multifunctional Th cells coexpressing IFN-γ and IL-4 (1.94±0.46 vs. 4.48±0.81% and 1.86±0.33 vs. 5.3±1.07%, respectively).
Figure 6. CD4+ T cell-specific deletion of IL4ra attenuated neonatal RSV infection-induced Th2 responses upon reinfection.

Th-specific, IL-4Rα-deficient (LckcreIL4Rα−/Lox-RR) and -competent mice (IL4Rα−/Lox-RR) were infected at 5 days of age and reinfected with RSV at 4 weeks postprimary infection. Control mice of each genotype were treated with serum-free medium (LckcreIL4Rα−/Lox-sham and IL4Rα−/Lox-sham, respectively). Pulmonary Th profile and BALF cellularity and cytokine levels were measured at 6 days postsecondary infection. (A) Pulmonary Th profile. Lung cells were isolated, stimulated, and stained with dead cell, excluding dye and antibodies to CD3, CD4, IFN-γ, and IL-4. The cells were then analyzed by flow cytometry; n = 3–7. (B) Representative dot plots showing the IL-4- and IFN-γ-expressing Th cells. (C) BALF cellularity; n = 5. (D) Cytokine levels in BALF; n = 4–12; *P < 0.05.
Consistent with the reduction in pulmonary Th2 cells, we observed a significant reduction in eosinophils in the BALF of RSV-reinfected, IL-4Rα-deficient versus -competent mice (1527±541 vs. 19,200±12,089; Fig. 6C). No differences in total BALF cell numbers, alveolar macrophages, lymphocytes, or neutrophils were observed between the two groups of mice. In addition, we observed a significant decrease in Th2 cytokine IL-13 and an increase in Th1 cytokine IL-12(p40) in the BALF of IL-4Rα-deficient compared with -competent mice (59.43±11.35 vs. 32.34±5.36 pg/ml and 21.58±2.47 vs. 12.18±2.28 pg/ml, respectively; Fig. 6D). There was no difference in IFN-γ between IL-4Rα-deficient and -competent mice upon RSV reinfection. These data demonstrated that CD4+ T cell-specific ablation of Il4ra attenuated the pathogenic, Th2-biased immune responses associated with RSV reinfection in the neonatally infected mice.
DISCUSSION
It has been proposed that a biased Th2 immune response is responsible for RSV-induced pulmonary pathophysiology in infants. In this study, we first confirmed that an exaggerated Th2 response was induced by RSV reinfection in mice originally infected as neonates but not as adults, and this Th2 immune response correlated with airway hyper-reactivity, mucus hyperproduction, and eosinophilia. We further observed increased expression of IL-4Rα on CD4+ T cells from mice infected initially with RSV as neonates and reinfected with RSV as adults. Increased expression of IL-4Rα on Th cells was associated with an increased ability of these cells to proliferate and develop into Th2 cells. Specific deletion of Il4ra on Th cells abolished airway hyper-reactivity and mucus hyperproduction and attenuated airway eosinophilia and pulmonary Th2 responses. We demonstrated further that IL-4Rα expression was increased on human CD4+ Th cells from cord blood but not peripheral blood from adults upon RSV stimulation in vitro, suggesting that the mechanisms that we have identified in mice are translatable to humans.
Whereas human epidemiological studies suggest a gain-of-function association of IL4RA polymorphisms with severe RSV disease in infants [22], few studies have investigated the function of IL-4Rα in a neonatal model. We clearly showed that the expression of IL-4Rα on CD4+ T cells, including Th1 and Th2 cells, was increased upon reinfection when the mice were infected initially as neonates. This observation is in line with the study of Li et al. [32], which showed increased expression of IL-13Rα1, the other component of the type II IL-4R complex, on neonatal Th1 cells upon antigen recall. Whereas Li and coworkers ' data demonstrated increased expression of IL-13Rα1, correlated with enhanced Th1 apoptosis, and suggested this as a mechanism skewing the neonatal immune response toward a Th2-dominant response, we were unable to observe any changes in Th1 apoptosis between RSV-infected and noninfected mice after secondary RSV infection (data not shown). What we did observe is that Th1 and Th2 cells from mice infected initially as neonates possess better proliferative ability than cells from mice infected as adults. Moreover, Th2 cells from mice infected initially as neonates showed the greatest ability to proliferate. This is consistent with the observation that RSV reinfection resulted in increased numbers of pulmonary Th2 cells without an increase in the numbers of Th1 cells in mice infected initially as neonates compared with mice infected initially as adults. Cumulatively, these data suggest exaggerated Th2 cell proliferation rather than Th1 apoptosis as a mechanism inducing biased Th2 responses during reinfection in neonatally infected mice.
It was unexpected that in mice, primary RSV infection did not increase the expression of IL-4Rα on neonatal CD4+ T cells but rather decreased IL-4Rα expression on adult CD4+ T cells. This is in contrast to the ex vivo human CD4+ T cell data, which demonstrated an increase in IL-4Rα expression on CD4+ T cells from CBMCs versus adult PBMCs exposed to RSV. Although not significant but similar to the mouse in vivo data, IL-4Rα expression appeared to decrease on PBMC CD4+ T cells. This discrepancy between mouse and human data could be a result of a variety of factors. First, humans are reinfected by RSV throughout life [4], and therefore, a portion of the PBMC CD4+ T cells is likely effector or memory cells masking the inhibitory effect of RSV infection on IL-4Rα expression on adult CD4+ T cells. Second, mouse data were derived from in vivo experiments, whereas human data were derived from in vitro stimulations and studies. Finally, it is difficult to achieve a MOI of two in vivo as a result of the fact that there are much more cells in actual airways than cultured cells. Therefore, no change in IL-4Rα expression on neonatal CD4+ T cells may reflect the fact that not enough cells were infected. Nevertheless, Th1 and Th2 cells from RSV-infected neonates showed higher expression of IL-4Rα than cells from RSV-infected adult mice, and this is consistent with the human data, in that CBMC CD4+ T cells expressed more IL-4Rα than PBMC CD4+ T cells when stimulated with RSV. Intriguingly, Th2 cells also expressed higher levels of IL-4Rα than Th1 cells from RSV-infected neonates. Although beyond the scope of these studies, this suggests that the elevated expression of IL-4Rα may increase the survival of memory Th2 cells preferentially and that those cells may undergo favored proliferation during reinfection.
The absence of IL-4Rα on CD4+ T cells abolished the immunopathophysiology associated with RSV reinfection in mice originally infected as neonates, including airway hyper-reactivity, eosinophilia, and mucus hyperproduction. Surprisingly, unlike the drastic effects on pathophysiology, the deletion of Il4ra from CD4+ T cells did not inhibit completely the development of Th2 or multifunctional Th cells coexpressing IFN-γ and IL-4. One possible explanation is the often-reported, partial efficiency of Cre recombinase [33]; however, the original report on this mouse line demonstrated 95% efficiency for disruption of CD4+ T cell-specific IL-4Rα [26]. It seems unlikely that the residual 5% expression of full-length IL-4Rα could explain the percentage of Th2 and multifunctional Th cells that were induced upon reinfection of these mice. Instead, we believe that the Th2 and multifunctional Th cells observed following RSV reinfection indicate an IL-4Rα-independent Th2 commitment/maintenance mechanism, as has been suggested previously in the context of different pathogens or allergens [34–38]. In addition, the abrogation of the Th2-associated pathophysiology, despite the presence of residual Th2 cells in the lungs, suggests that either Th2 effector functions require a minimal number of Th2 cells (or a minimal release of Th2 effector cytokines) to induce functional responses or current measurements of Th2-induced pathophysiology are not sensitive enough to detect these responses. It is also possible that undefined mediators released by IL-4Rα-expressing CD4+ T cells are responsible for RSV-induced pathophysiology and that upon ablation of these cells, those mediators are lost, resulting in a reduction in pathophysiology.
As mentioned above, reinfection of neonatally infected mice induced not only Th2 cells expressing IL-4 but also a multifunctional Th subset expressing the prototypical Th2 and Th1 cytokines, IL-4, and IFN-γ. Intriguingly, this multifunctional subset was only observed in neonatally infected mice, where IL-4Rα was up-regulated upon reinfection and not in mice infected as adults. Furthermore, the number of multifunctional Th cells was reduced drastically in Th-specific, IL-4Rα-deficient mice. These observations indicated that IL-4Rα is partially required for the development of these cells. The exact role of these multifunctional cells is uncertain, and a few studies suggest that they are simply an immature cell type (i.e., Th0 cells) in the process of committing to a Th1 or Th2 cell [25, 39, 40]. From our data, it is unclear if and how these cells might function in the pathogenesis of neonatal RSV infection.
In summary, we confirmed that skewed Th2 immune responses with airway hyper-reactivity, mucus hyperproduction, and eosinophilia are induced upon RSV reinfection, specifically in neonatally infected mice. This skewed Th2 response correlated with increased IL-4Rα expression on CD4+ T cells. Furthermore, deletion of Il4ra from Th cells attenuated Th2 responses and abolished Th2-associated immunopathophysiology. The increase in IL-4Rα expression was also observed in CD4+ T cells from CBMCs, suggesting that IL-4Rα may also have a role in RSV pathogenesis in human infants. These findings elucidate a pathogenic role for Th-specific IL-4Rα during RSV reinfection in mice infected as neonates and provide important insight for infant RSV vaccine development.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health grants (R01AI090059, R01ES015050, and P42ES013648) to S.A.C.
We thank Dr. Barney Graham (Vaccine Research Center, Bethesda, MD, USA) and Dr. Teresa Johnson (GenVec, Gaithersburg, MD, USA) for thoughtful discussions and insightful comments.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- AR
- respiratory syncytial virus-infected adults
- ARR
- mice initially infected as adults and reinfected as adults
- BALF
- BAL fluid
- CBMC
- cord blood mononuclear cell
- dpi
- days postinfection
- iMFI
- integrated mean fluorescent intensity
- LSUHSC
- Louisiana State University Health Sciences Center
- MeCh
- methacholine
- MOI
- multiplicity of infection
- NR
- respiratory syncytial virus-infected neonates
- NRR
- mice initially infected as neonates and reinfected as adults
- PAS
- periodic acid-Schiff
- RSV
- respiratory syncytial virus
AUTHORSHIP
S.A.C. was the principal investigator in this study, designed the research, analyzed the data, and wrote the manuscript. D.Y. designed and performed most of the experiments, analyzed the data, and wrote the manuscript draft. B.S. and G.L. assisted in some of these experiments. N.M. and S.E.T. collected and analyzed human Th cell data and assisted in preparing the manuscript. J.S.S. studied the Th profile of the Th-specific, IL-4Ra-deficient mice. F.B. and D.H. provided the Th-specific, IL-4Ra-deficient mice.
DISCLOSURES
A U.S. patent application has been filed by LSUHSC as a result of this research, with S.A.C. as the inventor. The other authors have no financial conflicts of interest. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the U.S. National Institutes of Health.
REFERENCES
- 1. Kristjansson S., Bjarnarson S. P., Wennergren G., Palsdottir A. H., Arnadottir T., Haraldsson A., Jonsdottir I. (2005) Respiratory syncytial virus and other respiratory viruses during the first 3 months of life promote a local TH2-like response. J. Allergy Clin. Immunol. 116, 805–811 [DOI] [PubMed] [Google Scholar]
- 2. Simoes E. A. (1999) Respiratory syncytial virus infection. Lancet 354, 847–852 [DOI] [PubMed] [Google Scholar]
- 3. Boeck K. D. (1996) Respiratory syncytial virus bronchiolitis: clinical aspects and epidemiology. Monaldi Arch. Chest Dis. 51, 210–213 [PubMed] [Google Scholar]
- 4. Glezen W. P., Taber L. H., Frank A. L., Kasel J. A. (1986) Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child 140, 543–546 [DOI] [PubMed] [Google Scholar]
- 5. Bont L., Versteegh J., Swelsen W. T., Heijnen C. J., Kavelaars A., Brus F., Draaisma J. M., Pekelharing-Berghuis M., van Diemen-Steenvoorde R. A., Kimpen J. L. (2002) Natural reinfection with respiratory syncytial virus does not boost virus-specific T-cell immunity. Pediatr. Res. 52, 363–367 [DOI] [PubMed] [Google Scholar]
- 6. Wu P., Dupont W. D., Griffin M. R., Carroll K. N., Mitchel E. F., Gebretsadik T., Hartert T. V. (2008) Evidence of a causal role of winter virus infection during infancy in early childhood asthma. Am. J. Respir. Crit. Care Med. 178, 1123–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gern J. E., Rosenthal L. A., Sorkness R. L., Lemanske R. F., Jr., (2005) Effects of viral respiratory infections on lung development and childhood asthma. J. Allergy Clin. Immunol. 115, 668–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. You D., Becnel D., Wang K., Ripple M., Daly M., Cormier S. A. (2006) Exposure of neonates to respiratory syncytial virus is critical in determining subsequent airway response in adults. Respir. Res. 7, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Becnel D., You D., Erskin J., Dimina D. M., Cormier S. A. (2005) A role for airway remodeling during respiratory syncytial virus infection. Respir. Res. 6, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Culley F. J., Pollott J., Openshaw P. J. (2002) Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J. Exp. Med. 196, 1381–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dakhama A., Park J. W., Taube C., Joetham A., Balhorn A., Miyahara N., Takeda K., Gelfand E. W. (2005) The enhancement or prevention of airway hyperresponsiveness during reinfection with respiratory syncytial virus is critically dependent on the age at first infection and IL-13 production. J. Immunol. 175, 1876–1883 [DOI] [PubMed] [Google Scholar]
- 12. Sigurs N., Gustafsson P. M., Bjarnason R., Lundberg F., Schmidt S., Sigurbergsson F., Kjellman B. (2005) Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am. J. Respir. Crit. Care Med. 171, 137–141 [DOI] [PubMed] [Google Scholar]
- 13. Sigurs N., Aljassim F., Kjellman B., Robinson P. D., Sigurbergsson F., Bjarnason R., Gustafsson P. M. (2010) Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 65, 1045–1052 [DOI] [PubMed] [Google Scholar]
- 14. Korppi M., Piippo-Savolainen E., Korhonen K., Remes S. (2004) Respiratory morbidity 20 years after RSV infection in infancy. Pediatr. Pulmonol. 38, 155–160 [DOI] [PubMed] [Google Scholar]
- 15. Ruotsalainen M., Piippo-Savolainen E., Hyvarinen M. K., Korppi M. (2010) Respiratory morbidity in adulthood after respiratory syncytial virus hospitalization in infancy. Pediatr. Infect. Dis. J. 29, 872–874 [DOI] [PubMed] [Google Scholar]
- 16. Forster J., Streckert H. J., Werchau H. (1995) The humoral immune response of children and infants to an RSV infection: its maturation and association with illness. Klin. Padiatr. 207, 313–316 [DOI] [PubMed] [Google Scholar]
- 17. Cormier S. A., You D., Honnegowda S. (2010) The use of a neonatal mouse model to study respiratory syncytial virus infections. Expert Rev. Anti. Infect. Ther. 8, 1371–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dakhama A., Lee Y. M., Ohnishi H., Jing X., Balhorn A., Takeda K., Gelfand E. W. (2009) Virus-specific IgE enhances airway responsiveness on reinfection with respiratory syncytial virus in newborn mice. J. Allergy Clin. Immunol. 123, 138.e5–145.e5 [DOI] [PubMed] [Google Scholar]
- 19. Choi E. H., Lee H. J., Yoo T., Chanock S. J. (2002) A common haplotype of interleukin-4 gene IL4 is associated with severe respiratory syncytial virus disease in Korean children. J. Infect. Dis. 186, 1207–1211 [DOI] [PubMed] [Google Scholar]
- 20. Tekkanat K. K., Maassab H. F., Cho D. S., Lai J. J., John A., Berlin A., Kaplan M. H., Lukacs N. W. (2001) IL-13-induced airway hyperreactivity during respiratory syncytial virus infection is STAT6 dependent. J. Immunol. 166, 3542–3548 [DOI] [PubMed] [Google Scholar]
- 21. Puthothu B., Krueger M., Forster J., Heinzmann A. (2006) Association between severe respiratory syncytial virus infection and IL13/IL4 haplotypes. J. Infect. Dis. 193, 438–441 [DOI] [PubMed] [Google Scholar]
- 22. Hoebee B., Rietveld E., Bont L., Oosten M., Hodemaekers H. M., Nagelkerke N. J., Neijens H. J., Kimpen J. L., Kimman T. G. (2003) Association of severe respiratory syncytial virus bronchiolitis with interleukin-4 and interleukin-4 receptor α polymorphisms. J. Infect. Dis. 187, 2–11 [DOI] [PubMed] [Google Scholar]
- 23. Ripple M. J., You D., Honnegowda S., Giaimo J. D., Sewell A. B., Becnel D. M., Cormier S. A. (2010) Immunomodulation with IL-4R α antisense oligonucleotide prevents respiratory syncytial virus-mediated pulmonary disease. J. Immunol. 185, 4804–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zurawski S. M., Chomarat P., Djossou O., Bidaud C., McKenzie A. N., Miossec P., Banchereau J., Zurawski G. (1995) The primary binding subunit of the human interleukin-4 receptor is also a component of the interleukin-13 receptor. J. Biol. Chem. 270, 13869–13878 [DOI] [PubMed] [Google Scholar]
- 25. Bucy R. P., Karr L., Huang G. Q., Li J., Carter D., Honjo K., Lemons J. A., Murphy K. M., Weaver C. T. (1995) Single cell analysis of cytokine gene coexpression during CD4+ T-cell phenotype development. Proc. Natl. Acad. Sci. USA 92, 7565–7569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Radwanska M., Cutler A. J., Hoving J. C., Magez S., Holscher C., Bohms A., Arendse B., Kirsch R., Hunig T., Alexander J., Kaye P., Brombacher F. (2007) Deletion of IL-4Rα on CD4 T cells renders BALB/c mice resistant to Leishmania major infection. PLoS Pathog. 3, e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Council N. R. (1996) Guide for the Care and Use of Laboratory Animals. Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council. National Academy, Washington, DC, USA [Google Scholar]
- 28. Ueba O. (1978) Respiratory syncytial virus. I. Concentration and purification of the infectious virus. Acta Med. Okayama 32, 265–272 [PubMed] [Google Scholar]
- 29. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Csillag A., Boldogh I., Pazmandi K., Magyarics Z., Gogolak P., Sur S., Rajnavolgyi E., Bacsi A. (2010) Pollen-induced oxidative stress influences both innate and adaptive immune responses via altering dendritic cell functions. J. Immunol. 184, 2377–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Darrah P. A., Patel D. T., De Luca P. M., Lindsay R. W., Davey D. F., Flynn B. J., Hoff S. T., Andersen P., Reed S. G., Morris S. L., Roederer M., Seder R. A. (2007) Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat. Med. 13, 843–850 [DOI] [PubMed] [Google Scholar]
- 32. Li L., Lee H. H., Bell J. J., Gregg R. K., Ellis J. S., Gessner A., Zaghouani H. (2004) IL-4 utilizes an alternative receptor to drive apoptosis of Th1 cells and skews neonatal immunity toward Th2. Immunity 20, 429–440 [DOI] [PubMed] [Google Scholar]
- 33. Kyronlahti A., Vetter M., Euler R., Bielinska M., Jay P. Y., Anttonen M., Heikinheimo M., Wilson D. B. (2011) GATA4 deficiency impairs ovarian function in adult mice. Biol. Reprod. 84, 1033–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jankovic D., Kullberg M. C., Noben-Trauth N., Caspar P., Paul W. E., Sher A. (2000) Single cell analysis reveals that IL-4 receptor/Stat6 signaling is not required for the in vivo or in vitro development of CD4+ lymphocytes with a Th2 cytokine profile. J. Immunol. 164, 3047–3055 [DOI] [PubMed] [Google Scholar]
- 35. Jankovic D., Kullberg M. C., Noben-Trauth N., Caspar P., Ward J. M., Cheever A. W., Paul W. E., Sher A. (1999) Schistosome-infected IL-4 receptor knockout (KO) mice, in contrast to IL-4 KO mice, fail to develop granulomatous pathology while maintaining the same lymphokine expression profile. J. Immunol. 163, 337–342 [PubMed] [Google Scholar]
- 36. Mohrs M., Holscher C., Brombacher F. (2000) Interleukin-4 receptor α-deficient BALB/c mice show an unimpaired T helper 2 polarization in response to Leishmania major infection. Infect. Immun. 68, 1773–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Noben-Trauth N., Shultz L. D., Brombacher F., Urban J. F., Gu H., Paul W. E. (1997) An interleukin 4 (IL-4)-independent pathway for CD4+ T cell IL-4 production is revealed in IL-4 receptor-deficient mice. Proc. Natl. Acad. Sci. USA 94, 10838–10843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Muller U., Piehler D., Stenzel W., Kohler G., Frey O., Held J., Grahnert A., Richter T., Eschke M., Kamradt T., Brombacher F., Alber G. (2012) Lack of IL-4 receptor expression on T helper cells reduces T helper 2 cell polyfunctionality and confers resistance in allergic bronchopulmonary mycosis. Mucosal Immunol. 5, 299–310 [DOI] [PubMed] [Google Scholar]
- 39. Firestein G. S., Roeder W. D., Laxer J. A., Townsend K. S., Weaver C. T., Hom J. T., Linton J., Torbett B. E., Glasebrook A. L. (1989) A new murine CD4+ T cell subset with an unrestricted cytokine profile. J. Immunol. 143, 518–525 [PubMed] [Google Scholar]
- 40. Kamogawa Y., Minasi L. A., Carding S. R., Bottomly K., Flavell R. A. (1993) The relationship of IL-4- and IFN γ-producing T cells studied by lineage ablation of IL-4-producing cells. Cell 75, 985–995 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.