

Abstract
C3-arylated indazole and pyrazoles are privileged structural motifs in agrochemicals and pharmaceuticals. C-3 C-H arylation of (1H) indazole and pyrazole has been a significant challenge due to the poor reactivity of the C-3 position. Herein, we report a practical Pd(II)/Phen catalyst and conditions for direct C-3 arylation of indazole and pyrazole with ArI or ArBr without using Ag additives as halide scavengers. The use of toluene, chlorobenzene, trifluoromethylbenzene and mesitylene as the solvent was found to be crucial for the selectivity and reactivity. We further demonstrate the robustness of this protocol through the first total synthesis of Nigellidine hydrobromide as well as expedient preparation of heterocycles structurally related to pesticides and drug molecules.
Introduction
Transition metal-catalyzed arylation of heteroarene C-H bonds has regained considerable attention over recent years.1 These reactions can potentially provide concise routes for constructing bi(hetero)aryls, as well as late-stage diversification of bioactive target molecular scaffolds.2 As an important class of heteroarenes, indazole and pyrazole motifs are embedded in numerous pharmaceuticals and agrochemicals with broad range of biological activities,3 including anti-inflammatory (Celebrex, Fig. 1),3a antiemetic (Granisol),3b antipsychotic (CDPPB),3c and anticancer (Pyrazolanthrone)3d drugs, as well as herbicide (Fluazolate),3e and insecticide (Chlorantraniliprole).3f
Fig. 1.

Biologically active indazoles and pyrazoles.
As a result, extensive efforts have been directed towards the development of practical and efficient catalytic systems for direct arylation of indazole and pyrazole in recent years. These protocols have been applied to the rapid syntheses of indazole or pyrazole bi(hetero)aryls, or late-stage modification of complex indazole or pyrazole scaffolds.4–9 Despite the development of several efficient protocols for C-4,4,5 C-5 arylation of pyrazole,6 and C-3 arylation of (2H) indazole,7,8 C-3 arylations of pyrazole and (1H) indazole remain formidable challenges due to lack of reactivity (Fig. 2).9 Indeed, installation of an aryl group at C-3 of pyrazole via C-H activation can only be achieved by using a elegantly devised protecting-group-switch strategy.6a This strategy essentially converted the C-3 position into the more reactive C-5 position. It has also been shown that an efficient arylation protocol for C-3 arylation of (2H) indazole is not applicable to (1H) indazole substrates.8
Fig. 2.

Reactivity of indazole and pyrazole towards C-H arylation.
Building on our previous success of C-3 selective arylation and olefination of pyridines using Pd(II)/Phen(phenanthroline) catalysts,10 we embarked on the development of related protocols for C-3 arylation of (1H) indazoles and pyrazoles. While our manuscript was in preparation, the Itami and Kazzouli groups independently reported Pd/Phen (phenanthroline) catalysts for C-3 arylation of (1H) indazole.11 In Itami’s protocol, the reaction was run with 10 mol% PdCl2, 10 mol% Phen, 2.0 equiv K3PO4, and 1.5 equiv Ag2CO3 in DMA.11a Kazzouli’s conditions required 20 mol% Pd(OAc)2 and 40 mol% Phen in polar DMA.11b While these reactions represent exciting development for C-3 arylation of (1H) indazole, both yields and substrate scope remain to be substantially improved. The use of stoichiometric amount of Ag additive as halide scavenger, or high catalyst loading (20 mol% Pd) is also a drawback. Herein, we report a highly efficient Pd/Phen catalytic system for the C-3 arylation of (1H) indazole and pyrazole without using Ag additive (eq 1). We demonstrate the utility of this protocol by achieving the first total synthesis of Nigellidine.
 |
(1) |
Results and Discussion
Initial Studies and Reaction Optimization
There exist two major challenges in the development of selective C-H functionalization methods for heterocycles, namely, controlling the regioselectivity among C-H bonds with subtle electronic and steric differences, and preventing the non-productive coordination of the heteroatom of the substrates with the metal catalysts. In search for solutions to these two problems, we found phenanthroline ligand coordinate strongly with Pd(II) center and weakens coordination of the N-atom of pyridine substrates, most likely through sterics.10 The disscociation of the pyridine enhances the local concentration of substrate nearby the Pd(II) catalysts for productive π-binding which triggers the C-3 C-H functionalizations. Computational studies suggest that the C-3 selectivity can be attributed to the recognition of the highest electron density at this carbon by the catalyst.12 Encouraged by these preliminary results, we began to test if Pd(II)/Phen system could be adapted for C-3 arylation of (1H) indazole and pyrazole. We were aware of several potential problems that we could encounter in applying this Pd/Phen system to C-3 arylation of pyrazole and indazole. Firstly, N-N bond is known to be unstable under previously reported conditions (eq 2).13 Secondly, the sp2 nitrogen at the 2-position could direct further arylation of the newly installed aryl groups (eq 3).
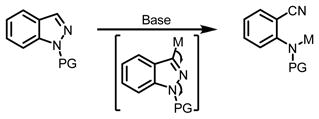 |
(2) |
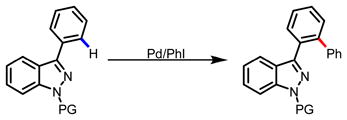 |
(3) |
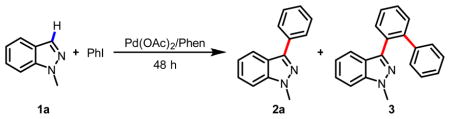
Based on our previous protocol for C-3 arylation of pyridines, we began to screen conditions for arylation of 1-methyl indazole 1a. We found that the reaction of 1 equiv 1a with 1 equiv iodobenzene in DMF in the presence of 10 mol% Pd(OAc)2, 30 mol% Phen and 3.0 equiv Cs2CO3 under air at 140 °C gave the desired arylated product 2a in 24% yield after 48 h (entry 1, Table 1). The undesired byproduct 3 from further directed arylation of product 2a was also isolated in 3% yield. Increasing the amount of iodobenzene to 2 equiv improved the yield of 2a to 51%. Unfortunately, the yield of side product 3 also increased to 14% (entry 2, Table 1). We reasoned that bulkier or tridentate ligand would prevent the nitrogen at the 2-position from directing further arylation of product 2a. Indeed, bulkier ligand (Phen-PhMe, entry 3) and tridentate (Dpya, entry 4) inhibited the formation of 3, with ligand Dpya affording 2a in 42% yield (entry 4). Although the result obtained with Dpya was promising, we further optimized conditions using phenanthroline due to its ready availability, and found that the use of K3PO4 as the base in DMF diminished the formation of 3 (entry 5) while affording 2a in 47% yield. Disappointingly, the material balance under all these conditions was only around 60%.
Table 1.
Discovery of C-3 C-H arylation of indazolea
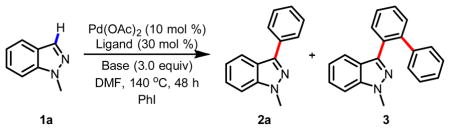
| |||||
|---|---|---|---|---|---|
| entry | PhI (equiv) | ligand | base | 2a (%)b | 3 (%)b |
| 1 | 1.0 | Phen | Cs2CO3 | 24 | 3 |
| 2 | 2.0 | Phen | Cs2CO3 | 51 | 14 |
| 3 | 2.0 | Phen-PhMe | Cs2CO3 | 21 | 0 |
| 4 | 2.0 | Dpya | Cs2CO3 | 42 | 0 |
| 5 | 2.0 | Phen | K3PO4 | 47 | 6 |
| 6 | 2.0 | Phen | K2HPO4 | 3 | 0 |
 Phen |
 Phen-PhMe |
 Dpya |
|||
Reaction conditions: Indazole (0.25 mmol) in solvent (1 mL) at 140 °C for 48 h.
Yield was determined by 1H NMR using CH2Br2 as an internal standard.
To improve the material balance, we examined the solvent effect using K3PO4 as a base (entries 1–4, Table 2). Strikingly, the use of PhCl as the solvent completely suppressed the formation of the side product 3 (entry 4). Most importantly, the material balance in PhCl reached above 90%. The stability of indazole substrate in this solvent prompted us to raise the reaction temperature to 160 °C which improved the yield to 60% (entry 5). This dramatic solvent effect urged us to revisit the use Cs2CO3 as the base. To our delight, Cs2CO3 in PhCl improved the yield to 89% with the formation of only 3% of byproduct 3 (entry 6). Either reducing the loading of ligand (entry 7) or the amount of base resulted in a lower yield (entry 8). We also observed that PhCl reacted with indazole in the absence of PhI (entry 9) which could be a potential problem in synthetic applications. In light of these two drawbacks, we further screened arene-based solvents and found that toluene, PhCF3 and mesitylene also gave comparable results (entries 10–12). Importantly, the loading of phen ligand and base can be reduced to 10 mol% and 1 equiv, respectively only when toluene is used as solvent (entries 13, 14). Control experiments showed that the Pd(OAc)2, Phen and base are essential for the observed reactivity (entries 15, 16).
Table 2.
Optimization for C-3 arylation of indazolea
| ||||||
|---|---|---|---|---|---|---|
| entry | Pd(OAc)2 (mol %) | Phen (mol %) | base (equiv) | solvent (1 mL) | temp. (C) | yield (%)b 2a (3) |
| 1 | 10 | 30 | K3PO4 (3.0) | Dioxane | 140 | 0 (0) |
| 2 | 10 | 30 | K3PO4 (3.0) | DMSO | 140 | 45 (0) |
| 3 | 10 | 30 | K3PO4 (3.0) | o-xylene | 140 | 19 (0) |
| 4 | 10 | 30 | K3PO4 (3.0) | PhCl | 140 | 45 (0) |
| 5 | 10 | 30 | K3PO4 (3.0) | PhCl | 160 | 60 (0) |
| 6 | 10 | 30 | Cs2CO3 (3.0) | PhCl | 160 | 89 (3) |
| 7 | 10 | 10 | Cs2CO3 (3.0) | PhCl | 160 | 72 (1) |
| 8 | 10 | 30 | Cs2CO3 (1.0) | PhCl | 160 | 64 (0) |
| 9 | 10 | 30 | Cs2CO3 (3.0) | PhCl | 160 | 29 (0)c |
| 10 | 10 | 30 | Cs2CO3 (3.0) | PhCF3 | 160 | 87 (0) |
| 11 | 10 | 30 | Cs2CO3 (3.0) | mesitylene | 160 | 77 (4) |
| 12 | 10 | 30 | Cs2CO3 (3.0) | toluene | 160 | 85 (3) |
| 13 | 10 | 10 | Cs2CO3 (1.0) | toluene | 160 | 93 (0) |
| 14 | 5 | 5 | Cs2CO3 (1.0) | toluene | 160 | 23 (0) |
| 15 | 10 | 0 | Cs2CO3 (1.0) | toluene | 160 | 0 |
| 16 | 0 | 10 | Cs2CO3 (1.0) | toluene | 160 | 0 |
Reaction conditions: Indazole (0.25 mmol) and PhI (0.5 mmol) in solvent (1 mL) for 48 h.
Yield was determined by 1H NMR using CH2Br2 as an internal standard.
No PhI was added.
Substrate Scope
With this significantly improved protocol in hand, we examined the scope of arylhalide coupling partners for C-3 arylation of indazoles. Both aryl iodides and bromides worked well to give the corresponding 3-arylated indazoles in high to excellent yield (2a-2p, Table 3). Aryl chlorides also reacted under these conditions to give the arylated product in 29% yield, albeit requiring use of excess phenyl chloride (entry 9, Table 2). The possibility of using aryl chlorides as coupling partners upon further development is appealing from the practical perspective. A wide range of functional groups on the aryliodides were tolerated including electron-donating and -withdrawing groups, OMe, MeS, F, Cl, CF3, CN and CO2Et (2g-2m). The tolerance of a strongly chelating MeS group is a valuable feature for making sulfur containing compounds. Most importantly, arylation with heteroaryl bromides also proceeded smoothly to afford biologically important biheteroaryls in good to excellent yields (2n-2p), which speaks to the robustness of the Pd/Phen catalysts and conditions.
Table 3.
Various aryl halides in C-3 arylationa
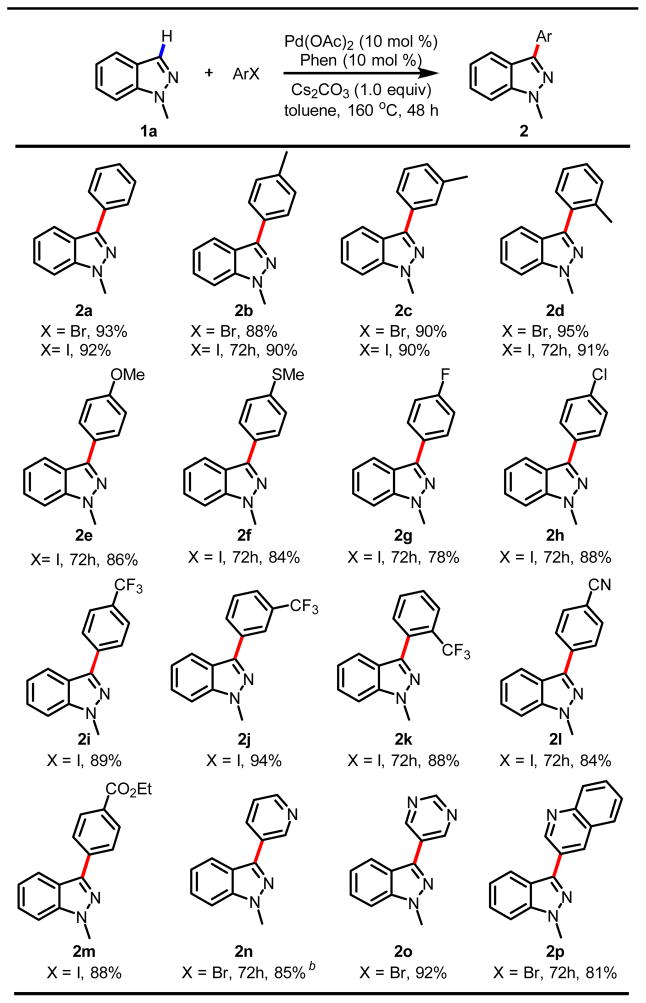
|
Reaction conditions: Pd(OAc)2 (10 mol %), Phen (10 mol %), Cs2CO3 (1.0 equiv), indazole (0.25 mmol), ArX (0.5 mmol) in toluene (1 mL) for 48 h and then isolated yield was obtained.
3-Bromopyridine (1.0 mmol) was used.
Next, we focused our attention on the scope of the indazoles and pyrazoles. Unprotected indazole and pyrazole gave no reaction under the optimal conditions. To improve the versatility of this protocol, we tested various synthetically useful N-protecting groups. Unfortunately, electron-withdrawing protecting groups such as Ac, Boc, and Ts inhibit the reaction severely. To our delight, the practically useful alkyl protecting group, such as Bn (2q, 2r), SEM (2s), and THP (2t) were compatible with this reaction (Table 4).14 It is worth noting that the undesired directed ortho-arylation of the benzyl groups was not observed. The synthetic utility C-3 arylation of Bn-protected indazole was demonstrated by a one-step synthesis of biologically active agent YD-3 (2r) in 89% yield using para-methoxylcarbonylaryliodide coupling partner.15 Various substituents including fluoro, chloro and ester on indazole phenyl rings were tolerated (2u–2y), although 4-Cl substituted indazole reacted very slowly due to the sterics, giving 44% yield in 72 h (2u). We were pleased that azaindazole, a biologically important structural motif, was also successfully arylated to give the desired product in 81% yield (2y), without any reaction of the highly activated chloro group at the 2-position of the pyridyl ring, thus allowing subsequent further cross couplings at this position. Finally, C-3 arylations of substituted pyrazoles including a hindered triphenyl pyrazole were also successful (4a–4c).16 Prior to our work, a single example of tristrifluoromethylphenylation of N-methylpyrazole in a moderate yield (52%) using [Pd(Phen)2](PF6)2 catalyst was reported by Murai.17
Table 4.
Various substituted indazoles and pyrazolesa
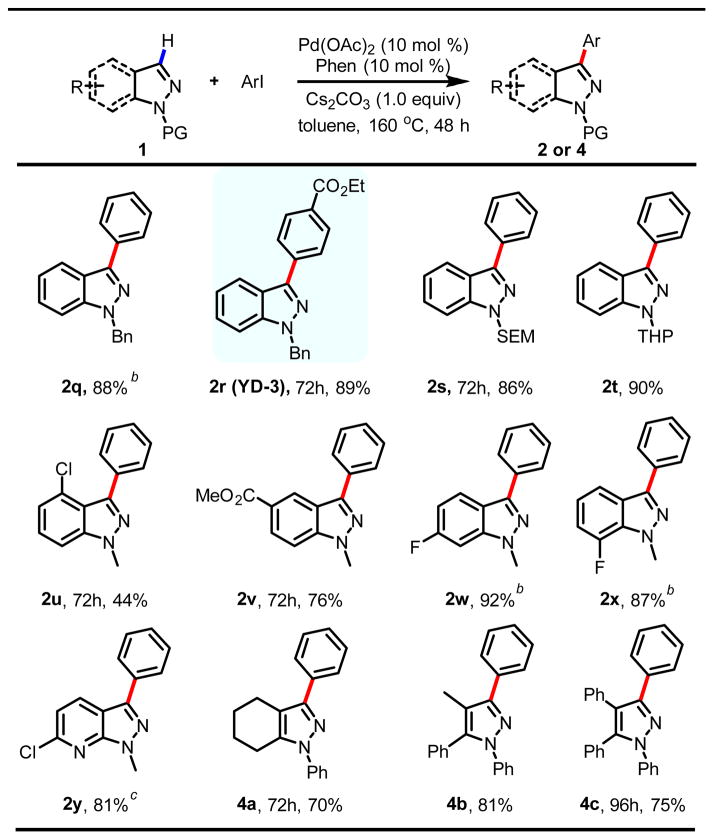
|
Reaction conditions: Pd(OAc)2 (10 mol %), Phen (10 mol %), Cs2CO3 (1.0 equiv), indazole (0.25 mmol), ArI (0.5 mmol) in toluene (1 mL) for 48 h and then isolated yield was obtained.
30 mol % of Phen was used.
Reaction was run for 12 h to give the same yield when chlorobenzene was used.
As expected, arylation of (2H) indazole under these reaction conditions gave the desired products in 91% yield (Fig. 3). Competition experiments using (1H) indazole and (2H) indazole in the same reaction afforded 75% C3-arylated (2H) indazole and 8% C-3 arylated (1H) indazole, confirming that the reactivity of (2H) indazole is superior to that of (1H) indazole.
Fig. 3.
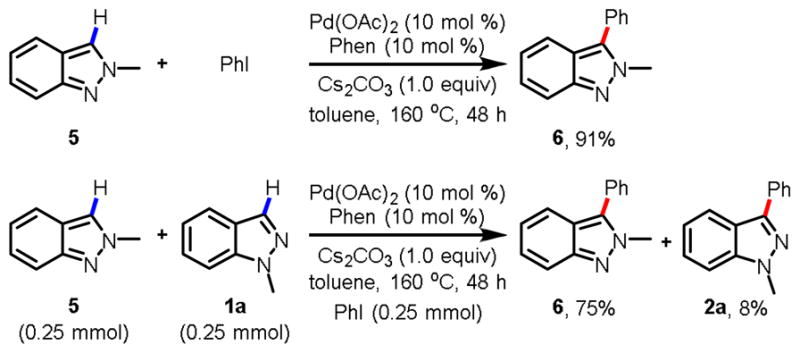
Competition reaction between (1H) indazole and (2H) indazole.
Synthetic Application
Developing C-H activation reactions as enabling synthetic disconnections to allow for highly convergent synthesis is one major impetus for our efforts in this field.18,19 We therefore sought to demonstrate the utility of our C-3 indazole arylation in a total synthesis of the natural alkaloid nigellidine (12). Nigellidine was isolated in 1995 from the seeds of Nigella sativa, which are used to treat various diseases.20 While related indazole natural products have been synthesized by Kelly,21 no total synthesis of nigellidine has yet been reported. We envisioned a convergent synthesis of nigellidine could be rapidly accomplished using this C-3 arylation reaction as a key step. Thus, commercially available 4-methoxy-6-methyl-(1H)-indazole was first protected using the readily removable THP group to give 7. C-3 arylation of 7 under the optimized conditions furnished 8 in 54% isolated yield on gram scale (Fig. 4). Deprotection of the THP group and stepwise N-alkylations with 1,4-dibromobutane afforded precursor 11. Subsequent demethylation by treating 11 with BBr3 afforded the natural product nigellidine hydrobromide (12), which was confirmed by X-ray crystallographic analysis. Thus, the first total synthesis of nigellidine hydrobromide was completed in six steps with an overall yield of 18%.
Fig. 4.

Total synthesis of nigellidine hydrobromide.
Conclusions
In summary, we have developed a practical Pd/Phen catalyst system for C-3 arylation of indazole and pyrazole. This protocol in general gives high yields and accommodates broad range of substrates and coupling partners. The robustness of this catalytic reaction was demonstrated in the first total synthesis of natural alkaloid Nigellidine hydrobromide as well as one-step synthesis of drug molecule YD-3.
Supplementary Material
Acknowledgments
We gratefully acknowledge The Scripps Research Institute, Syngenta and the National Institutes of Health (NIGMS 1 R01 GM102265-01) for financial support.
Footnotes
Electronic Supplementary Information (ESI) available: Data for new compounds and experimental procedures. See DOI: 10.1039/b000000x/
General procedure for C-3 Arylation of Indazoles and Pyrazoles: To a 35 mL sealed tube, were added Pd(OAc)2 (5.6 mg, 0.025 mmol), 1,10-phenanthroline (4.5 mg, 0.025 mmol), Cs2CO3 (82 mg, 0.25 mmol), aryl halide (0.25 mmol), indazole derivative (0.25 mmol), and toluene (1 mL). The tube was capped and stirred at 160 °C for 48–72 h. The reaction mixture was cooled to room temperature and diluted with EtOAc, filtered through a short pad of Celite, washed with EtOAc, and concentrated in vacuo. The resulting residue was purified by PTLC using hexanes: EtOAc (10:1 to 6:1, depending on different substrates) as the eluent.
Notes and references
- 1.For recent reviews on arylation of heteroarenes, see: Roger J, Gottumukkala AL, Doucet H. ChemCatChem. 2010;2:20–40.Ackermann L, Vicente R, Kapdi AR. Angew Chem, Int Ed. 2009;48:9792–9826. doi: 10.1002/anie.200902996.Bellina F, Rossi R. Tetrahedron. 2009;65:10269–10310.McGlacken GP, Bateman LM. Chem Soc Rev. 2009;38:2447–2464. doi: 10.1039/b805701j.Lewis JC, Bergman RG, Ellman JA. Acc Chem Res. 2008;41:1013–1025. doi: 10.1021/ar800042p.Seregin IV, Gevorgyan V. Chem Soc Rev. 2007;36:1173–1193. doi: 10.1039/b606984n.Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174–238. doi: 10.1021/cr0509760.Satoh T, Miura M. Chem Lett. 2007;36:200–205.
- 2.(a) Meyer C, Neue B, Schepmann D, Yanagisawa S, Yamaguchi J, Wuerthwein EU, Itami K, Wuensch B. Org Biomol Chem. 2011;9:8016–8029. doi: 10.1039/c1ob06149f. [DOI] [PubMed] [Google Scholar]; (b) Dai HX, Stepan AF, Plummer MS, Zhang YH, Yu JQ. J Am Chem Soc. 2011;133:7222–7228. doi: 10.1021/ja201708f. [DOI] [PubMed] [Google Scholar]
- 3.(a) Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, Graneto MJ, Lee LF, Malecha JW, Miyashiro JM, Rogers RS, Rogier DJ, Yu SS, Anderson GD, Burton EG, Cogburn JN, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Veenhuizen AW, Zhang YY, Isakson PC. J Med Chem. 1997;40:1347–1365. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]; (b) Plosker GL, Goa KL. Drugs. 1991;42:805–824. doi: 10.2165/00003495-199142050-00007. [DOI] [PubMed] [Google Scholar]; (c) de Paulis T, Hemstapat K, Chen Y, Zhang Y, Saleh S, Alagille D, Baldwin RM, Tamagnan GD, Conn PJ. J Med Chem. 2006;49:3332–3344. doi: 10.1021/jm051252j. [DOI] [PubMed] [Google Scholar]; (d) Okuno S, Saito A, Hayashi T, Chan PH. J Neurosci. 2004;24:7879–7887. doi: 10.1523/JNEUROSCI.1745-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Maxwell BD. J Labelled Compd Radiopharm. 2000;43:645–654. [Google Scholar]; (f) Lahm GP, Cordova D, Barry JD. Bioorg Med Chem. 2009;17:4127–4133. doi: 10.1016/j.bmc.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 4.For intramoleular C-4 direct arylation of pyrazole, see: Kumar S, Ila H, Junjappa H. J Org Chem. 2009;74:7046–7051. doi: 10.1021/jo901309t.
- 5.For intermolecular C-4 direct arylation of pyrazole, see: Fall Y, Doucet H, Santelli M. Synthesis. 2010:127–135.
- 6.For intermolecular C-5 direct arylation of pyrazole, see: Gikhman R, Jacques TL, Sames D. J Am Chem Soc. 2009;131:3042–3048. doi: 10.1021/ja8096114.Mateos C, Mendiola J, Carpintero M, Minguez JM. Org Lett. 2010;12:4924–4927. doi: 10.1021/ol1020898.Beladhria A, Beydoun K, Ben Ammar H, Ben Salem R, Doucet H. Synthesis. 2011:2553–2560.
- 7.For intramolecular direct arylation of 2H indazole, see: Laleu B, Lautens M. J Org Chem. 2008;73:9164–9167. doi: 10.1021/jo8017236.
- 8.For intermolecular direct arylation of 2H indazole, see: Ohnmacht SA, Culshaw AJ, Greaney MF. Org Lett. 2010;12:224–226. doi: 10.1021/ol902537d.
- 9.For intramolecular C-3 arylation of pyrazole, see: Brnardic EJ, Garbaccio RM, Fraley ME, Tasber ES, Steen JT, Arrington KL, Dudkin VY, Hartman GD, Stirdivant SM, Drakas BA, Rickert K, Walsh ES, Hamilton K, Buser CA, Hardwick J, Tao W, Beck SC, Mao X, Lobell RB, Sepp-Lorenzino L, Yan Y, Ikuta M, Munshi SK, Kuo LC, Kreatsoulas C. Bioorg Med Chem Lett. 2007;17:5989–5994. doi: 10.1016/j.bmcl.2007.07.051.
- 10.(a) Ye M, Gao GL, Yu JQ. J Am Chem Soc. 2011;133:6964–6967. doi: 10.1021/ja2021075. [DOI] [PubMed] [Google Scholar]; (b) Ye M, Gao GL, Edmunds AJF, Worthington PA, Morris JA, Yu JQ. J Am Chem Soc. 2011;133:19090–19093. doi: 10.1021/ja209510q. [DOI] [PubMed] [Google Scholar]
- 11.For most recent intermolecular C-3 arylation of 1H indazole, see Hattori K, Yamaguchi K, Yamaguchi J, Itami K. Tetrahedron. 2012;68:7605–7612.therein, a protocol using CuI/Phen/LiOtBu for this arylation has also been reported; Ben-Yahia A, Nass M, El Kazzouli S, Essassi EK, Guillaumet G. Eur J Org Chem. 2012;36:7075–7081.
- 12.Petit A, Flygare J, Miller AT, Winkel G, Ess DH. Org Lett. 2012;14:3680–3683. doi: 10.1021/ol301521n. [DOI] [PubMed] [Google Scholar]
- 13.Unsinn A, Knochel P. Chem Commun. 2012;48:2680–2682. doi: 10.1039/c2cc17804d.Also see ref 11 (a).
- 14.Arylation of phenyl protected indazole also proceeded to give the desired product in 50% isolated yield.
- 15.For previous synthesis of YD-3, see: Chen HS, Kuo SC, Teng CM, Lee FY, Wang JP, Lee YC, Kuo CW, Huang CC, Wu CC, Huang LJ. Bioorg Med Chem. 2008;16:1262–1278. doi: 10.1016/j.bmc.2007.10.070.Lee FY, Lien JC, Huang LJ, Huang TM, Tsai SC, Teng CM, Wu CC, Cheng FC, Kuo SC. J Med Chem. 2001;44:3746–3749. doi: 10.1021/jm010001h. also see ref 11a.
- 16.Arylation of THP-protected unsubstituted pyrazole under these conditions occurs at C-5 predominantly (C5:C4:C3 = 12/1.5/1), consistent with previous reports.
- 17.Shibahara F, Yamaguchi E, Murai T. J Org Chem. 2011;76:2680–2693. doi: 10.1021/jo200067y. [DOI] [PubMed] [Google Scholar]
- 18.(a) Wang DH, Yu JQ. J Am Chem Soc. 2011;133:5767–5769. doi: 10.1021/ja2010225. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem Int Ed. 2012;51:8960–9009. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]
- 19.Total synthesis using Pd-catalyzed C-H functionalizations of heterocycles: Trost BM, Godleski SA, Genêt JP. J Am Chem Soc. 1978;100:3930–3931.Cushing TD, Sanz-Cervera JF, Williams RM. J Am Chem Soc. 1993;115:9323–9324.Baran PS, Corey EJ. J Am Chem Soc. 2002;124:7904–7905. doi: 10.1021/ja026663t.Garg NK, Caspi DD, Stoltz BM. J Am Chem Soc. 2004;126:9552–9553. doi: 10.1021/ja046695b.Lu J, Tan X, Chen C. J Am Chem Soc. 2007;129:7768–7769. doi: 10.1021/ja072844p.Beck EM, Hatley R, Gaunt MJ. Angew Chem, Int Ed. 2008;47:3004–3007. doi: 10.1002/anie.200705005.Bowie AL, Jr, Trauner D. J Org Chem. 2009;74:1581–1586. doi: 10.1021/jo801791j.Mandal D, Yamaguchi AD, Yamaguchi J, Itami K. J Am Chem Soc. 2011;133:19660–19663. doi: 10.1021/ja209945x.
- 20.Atta-ur-Rahman, Malik S, Hasan SS, Choudhary MI, Ni CZ, Clardy J. Tetrahedron Lett. 1995;36:1993–1996. [Google Scholar]
- 21.For relevant synthesis of Nigellidine family, see: Elliott EL, Bushell SM, Cavero M, Tolan B, Kelly TR. Org Lett. 2005;7:2449–2451. doi: 10.1021/ol050769m.Schmidt A, Habeck T, Kindermann MK, Nieger M. J Org Chem. 2003;68:5977–5982. doi: 10.1021/jo0344337.Inamoto K, Katsuno M, Yoshino T, Arai Y, Hiroya K, Sakamoto T. Tetrahedron. 2007;63:2695–2711.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.