

Abstract
Affinity for DNA and cross-reactivity with renal antigens are associated with enhanced renal pathogenicity of lupus autoantibodies. In addition, certain IgG subclasses are enriched in nephritic kidneys, suggesting that isotype may determine the outcome of antibody binding to renal antigens. To investigate if the isotype of DNA antibodies affects renal pathogenicity by influencing antigen binding, we derived IgM, IgG1, IgG2b and IgG2a forms of the PL9–11 antibody (IgG3 anti-DNA) by in vitro class switching or PCR cloning. The affinity and specificity of PL9–11 antibodies for nuclear and renal antigens were analyzed using ELISA, Western blotting, surface plasmon resonance (SPR), binding to mesangial cells, and glomerular proteome arrays. Renal deposition and pathogenicity were assayed in mice injected with PL9–11 hybridomas. We found that PL9–11 and its isotype-switched variants had differential binding to DNA and chromatin (IgG3 > IgG2a > IgG1 > IgG2b > IgM) by direct and competition ELISA, and SPR. In contrast, in binding to laminin and collagen IV the IgG2a isotype actually had the highest affinity. Differences in affinity of PL9–11 antibodies for renal antigens were mirrored in analysis of specificity for glomeruli, and were associated with significant differences in renal pathogenicity in vivo and survival. Our novel findings indicate that the constant region plays an important role in the nephritogenicity of antibodies to DNA by affecting immunoglobulin affinity and specificity. Increased binding to multiple glomerular and/or nuclear antigens may contribute to the renal pathogenicity of anti-DNA antibodies of the IgG2a and IgG3 isotype. Finally, class switch recombination may be another mechanism by which B cell autoreactivity is generated.
Keywords: Systemic lupus erythematosus (SLE), Lupus nephritis, Anti-DNA antibodies, Isotype switching
1. Introduction
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease driven by B cells, and characterized by the presence of autoantibodies. Among the most distinctive autoantibodies in SLE are antibodies directed against double stranded (ds) DNA; antibodies to dsDNA are believed to play an important role in the pathogenesis of lupus nephritis (LN), one of the most severe disease manifestations. Anti-dsDNA antibodies have multiple possible contributions to renal injury, including indirect and direct cross-reactivity with glomerular antigens [1], penetration into living cells [2], modulation of gene expression and cell metabolism [3], and enhancement of kidney cell proliferation [4].
However, not all antibodies to dsDNA share the same pathogenic potential. Antibody characteristics that have been experimentally linked to renal damage include affinity for DNA, charge of the antigen binding region, cross-reactivity with particular glomerular antigens, and antibody class [5]. Moreover, certain subclasses of anti-dsDNA antibodies have also been closely associated with pathogenic potential. In human lupus, the titers of IgG1 anti-dsDNA antibodies frequently rise prior to a renal relapse in LN patients [6]. In lupus-prone mice, IgG2a, IgG2b, and IgG3 are enriched in immunoglobulin eluates of kidneys with active nephritis, or otherwise shown to be highly pathogenic [7,8]. Clearly, the association between particular antibody subclasses and lupus nephritis may be a function of significant differences in Fc mediated effects, including complement activation. However, another possibility which has yet to be explored is that the link of pathogenic antibodies with particular subclasses is the result of isotype related effects on antigen affinity and specificity.
Isotype switching ordinarily progresses through an IgM→IgG3→IgG1→IgG2b→IgG2a switch recombination sequence, generating antibodies of different isotypes which share the identical variable region (variable-joining-diversity, or VDJ) of the original antibody. While traditional immunological dogma has been that antigenic specificity is conferred solely by the antibody variable region whereas the constant (C) region confers effector functions, based on kinetic and thermodynamic studies it was recently demonstrated that specificity and affinity of Abs are properties not exclusively determined by the V region [9]. If such a phenomenon would hold true for autoantibodies as well this would suggest a novel mechanism for generating autoreactivity and cross-reactivity, thereby significantly impacting current views on the pathobiology of anti-dsDNA antibodies. Support for the potential of this mechanism comes from the observation that isotype switching of mAbs to fungal polysaccharide was associated with emergent reactivity for self-antigens [9]. The purpose of this study is to investigate the effects of anti-DNA antibody isotype on binding to nuclear and glomerular antigens. To address this question, we generated a panel of antibodies that all share their variable region but differ in the constant regions, and compared their affinityand specificity for nuclear and glomerular antigens in vitro and in vivo.
2. Materials and methods
2.1. Isolation of IgG1, IgG2b and IgG2a variants of PL9–11
The hybridoma clone PL9–11 (IgG3) was isolated previously from a MRL/+ mouse, and exhibits significant binding to dsDNA [10]. To obtain IgG1, IgG2b and IgG2a isotypes that would share identical variable regions, the PL9–11 clone was stimulated with 100 ng/ml of IL-4, 100 μg/ml of LPS, and 10 ng/ml of TGF-β for 7 days. Class-switched hybridoma cells were identified by an ELISpot assay, followed by sib selection and agarose cloning to isolate positive clones as previously described in detail [11–13]. Murine monoclonal antibody isotype controls were obtained from Southern Biotech (Birmingham, AL): IgM, 11E10 (specificity for LPS); IgG3, B10 (unkown); IgG1, 15H6 (T-2 mycotoxin); IgG2b, A-1 (chicken IgA); IgG2a, HOPC-1 (unknown). All of the isotype controls were verified not to bind to dsDNA. As an IgG3 hybridoma control we used 3E5, an irrelevant IgG3 antibody that also does not bind dsDNA [9].
2.2. Generating an IgM PL9–11
The IgM variant of the PL9–11 clone was constructed by cloning the PL9–11 VDJ into the pHC-msCu IgM expression vector. PL9–11 VH cDNA was first generated, and nested PCR of RNA ligase-mediated rapid amplification of cDNA ends (Applied Biosystems, Carlsbad, CA) was performed using TACGTTGCAGATGACAGTCTGGCT (5') and AGTCACCAAGCTGCTGAGGGAATA (3') as gene specific primers. After gel analysis, the VDJ fragment was purified with the QIAquick PCR purification kit (Qiagen, Gaithersburg, MD). HindIII and NheI restriction sites were introduced into the VDJ fragment by a second PCR, using TATAAAGCTTTCGATTCCCAGTTCCTCACGTTCA and ATATAGCTAGCTGAGGAGACGGTGACTGAGGTT as forward and reverse primers, respectively. PCR products were confirmed by their expected size on SDS–PAGE, and subsequently by sequencing of the cloned VDJ from the reconstituted PL9–11 IgM hybridoma.
A mutant, heavy chain non-producing, light chain only hybridoma clone was obtained by growing PL9–11 cells in poor DMEM media (2% FBS) for two weeks, followed by screening for heavy chain loss by agarose cloning. Loss of the heavy chain in the selected clone was confirmed by ELISA and PCR.
After digestion with HindIII and NheI, the VDJ PCR product was ligated into the IgM vector, followed by transfection into the light chain only clone. G418 selection was done at a concentration of 0.5 mg/ml. A reconstituted PL9–11 IgM clone was then isolated, expressing the original PL9–11 light chain and an exogenous IgM heavy chain inserted with the PL9–11 VDJ.
2.3. SCID mice
Female SCID mice at 8 weeks of age (Jackson Laboratory, Bar Harbor, ME) were primed intraperitoneally with 0.5 ml of pristane (Sigma Aldrich, St. Louis, MO) per mouse. Two weeks later, they were randomly divided into groups of 5 mice, each receiving 107 cells by intraperitoneal injection from the PL9–11 IgM, IgG3, IgG1, IgG2b, and IgG2a hybridoma clones. Two additional groups of mice were injected with 3E5 (IgG3), or were not injected with cells after pristane priming. Urine and blood were collected at the end point when mice were sacrificed (based on moribund appearance, as judged by an external individual not involved in the study). Urine albumin (Bethyl Laboratories, Montgomery, TX) was determined by ELISA according to the manufacturer's instructions. Animal studies were approved by the appropriate institutional review committee at the Albert Einstein College of Medicine.
2.4. Antibody purification
Ascites from hybridoma injected mice was harvested and precipitated in saturated ammonium sulfate solution. Antibodies of the IgG isotype were purified using the Melon gel kit (Pierce Biotech, Rockford, IL). The PL9–11 IgM antibody was purified using the HiTrap purification kit (GE Healthcare, Port Washington, NY). Antibody purity was confirmed by Coomassie blue staining, and was above 90% for all antibodies used in the subsequent assays.
2.5. ELISAs
Anti-single stranded/dsDNA and chromatin ELISAs were performed as previously described [14]. For the other ELISAs, Matrigel (Becton-Dickinson, Franklin Lakes, NJ), laminin (Southern Biotech), collagen IV (Becton-Dickinson), total histone, histone H2A, and histone H2B (Roche, Pleasanton, CA) were coated, respectively, onto 96-well microtiter plates (Dynatech Laboratories, Chantilly, VA) at a concentration of 5 μg/ml. Plates were blocked with 2% BSA for 1 hour at 37 °C and incubated with mAb for 2 h at room temperature (RT). Plates were then washed five times with PBS-0.05% Tween, and alkaline-phosphatase-conjugated goat anti-mouse kappa chain (Southern Biotech) diluted 1:1000 in 1% BSA was added for 1 h at 37 °C, followed by phosphatase substrate (Sigma Aldrich). The OD values were read at 405 nm.
To test antibody binding to MC, a cell surface ELISA was performed as previously described [15]. To use DNase-treated MC in this assay, cells growing in 96-well plates were fixed with 4% paraformaldehyde. After three washes, cells were incubated with DNase buffer for 5 min at RT, followed by a solution of DNase I at 0.1 mg/ml for 30 min at 37 °C. The plates were then washed for 5 min with 1 mM EDTA/PBS before blocking with 2% BSA, and the ELISA was then continued as above.
Competition ELISAs were done as described previously [16]. Briefly, 96-well plates were coated with a salmon sperm dsDNA solution at a concentration of 100 μg/ml. After blocking with 2% BSA, the competing antibodies were added at a beginning concentration of 0.5 μg/ml and then serially diluted. This was followed immediately by the addition of the test antibody (0.5 μg/ml for all wells). Plates were incubated for 2 h at 37 °C, followed by heavy chain isotype-specific alkaline-phosphatase-conjugated goat anti-mouse antibodies and substrate.
2.6. Glomerular proteome array
Purified anti-dsDNA antibodies were reacted with a glomerular proteome array [17], which includes 76 lupus related autoantigens and 4 control proteins. Antibody samples were diluted to 5 μg/ml and added to the arrays in duplicate. Detection was performed using Cy5 labeled anti-mouse kappa chain antibodies (Jackson ImmunoResearch, West Grove, PA). A Genepix 4000B scanner (Sunnyvale, CA) at a laser wavelength of 635 nm was used to generate images for analysis. Images were analyzed using Genepix Pro 6.0 software to generate a GenePix Results file. Net fluorescence intensities were normalized using anti-mouse IgG or IgM spotted onto each array. Values obtained from duplicate spots were averaged, and normalized by subtracting background signals. Fluorescence intensities that were higher than the row means were designated with red color, those below the mean were colored green, and those with signals close to the mean were left black.
2.7. Surface plasmon resonance (SPR) analysis
dsDNA affinity of purified antibodies from the PL9–11 mAb panel at a concentration of 5 nM in running buffer (20 mM KPO4, 130 mM KCl, 3.4 mM EDTA and 0.005% Tween 20) was determined by SPR analysis using the Biacore 3000 instrument (Biacore, Piscataway, NJ). The antibodies were immobilized on a CM sensor chip (GE Healthcare) according to the manufacturer's instructions. Each antibody had a response signal of 21,000 response units (RU) at immobilization. Plasmid DNA was digested with XmnI, generating 8 kb linear dsDNA with blunt ends. DNA was injected over the chip at concentrations of 0–200 nM at a flow rate of 30 μl/min, with 10 min for contact time and 5 min for dissociation. The maximum value obtained for binding to linear dsDNA was 2200 RU. The chips were regenerated with 50 mM NaOH/1 M NaCl. In separate experiments, SPR analysis was also performed by immobilization of dsDNA to a streptavidin coated sensor chip, as described [18]. Briefly, the sensor chip was incubated overnight with 5 nM of streptavidin, a concentration empirically determined to provide an optimal signal/noise ratio. Linearized plasmid dsDNA was biotinylated using a 3′ end labeling kit (Thermo Scientific, Rockford, IL), and reacted with the streptavidin coated chip at a concentration of 60 nM. Purified antibodies at a concentration of 0–200 nm were then incubated with the chip, and the assay continued as above. All responses were normalized and expressed relative to the baseline defined by the running buffer. Non-specific and bulk responses were subtracted from an in-line blank reference surface. The simple Langmuir model was used for calculating the binding kinetics of the antibodyedsDNA complex. Calculations were performed using the Biacore evaluation software 1.1 [18].
2.8. Flow cytometry
MC were detached from tissue culture plates using a 0.25% trypsin-EDTA solution, blocked with 2% BSA/PBS for 1 hour at 4 °C, and then washed twice in cold PBS. The purified mAbs at a final concentration of 0.5 μg/ml were added for 2 h at 4 °C. The cells were washed, incubated with 0.5 μg/ml goat anti-mouse kappa-PE (Southern Biotech) for 1 h at 4 °C, and analyzed by flow cytometry using an LSRII instrument (BD Biosciences, San Jose, CA). For flow cytometry with DNase-treated cells, MC cells were first incubated with DNase buffer (0.01 M Tris, 0.01 M MgCl2, 0.01 M CaCl2, pH 7.5) for 10 min at RT, followed by the application of 0.1 mg/ml of DNase I for 30 min at 37 °C. After incubation with 1 mM EDTA/PBS for 5 min cells were washed again, and the assay continued as above.
2.9. Western blotting
Histone H2A or purified mAb (the latter for assessing antibody concentration) were heated for 10 min at 95 °C in Laemmli buffer supplemented with ß-mercaptoethanol, then run in a 4–15% Mini-proTEAN TGX precast gel (Bio-Rad, San Ramon, CA) in Tris/Glycine SDS buffer. After protein transfer, the membrane was cut into strips that were then incubated with each of the different purified antibodies. Following incubation of the strips with biotinylated goat anti-mouse κ chain (Southern Biotech) (1:2000) for 1 h, HRP-streptavidin (Thermo Scientific) was added followed by ECL reagents for color development. Bands were quantitated by ImageJ.
2.10. Glomerular binding assay
Glass slides containing fixed mouse glomeruli were prepared as described previously [15,19], thawed at RT and washed for 2 min using PBS-0.05% Tween 20. After blocking with a 2.5% goat serum solution and rat anti-mouse CD16/32 antibody (PharMingen, San Diego, CA), the slides were incubated with purified PL9–11 mAbs at a concentration of 0.5 μg/ml. After a 30 minute incubation at RT slides were washed again, and incubated with FITC-labeled IgG goat anti-mouse kappa chain for 45 min at RT. Slides were washed twice, and DAPI solution (1 μg/ml) was added. Slides were sealed, air dried, and viewed with an Axioskop II fluorescence microscope (Carl Zeiss, Jena, Germany).
For the DNase treatment, the slides and/or mAb were incubated for 5 min in DNase buffer, followed by the application of 0.1 mg/ml of DNase I for 30 min at 37 °C. Slides and/or mAb were then washed for 5 min in 1 mM EDTA/PBS to inactivate the DNase, and the assay continued as described. Effective DNAsing was verified for each slide by the lack of DAPI staining.
2.11. Immunohistochemistry
Immunohistochemical staining was performed to detect glomerular antibody deposition in SCID mice injected with the PL9–11 hybridomas [20]. In brief, kidneys were fixed in 10% formalin, embedded in paraffin, and sectioned at 4 μm. After deparaffinization and rehydration, sections were blocked with 2% BSA in PBS in a moist chamber for 1 h at RT. After a wash with PBS-0.05% Tween 20, biotinylated goat anti-mouse IgG (Vector Laboratories, Burlingame, CA) was added at a 1:200 dilution for 45 min at RT. Sections were washed, incubated with alkaline-phosphatase-ABC reagent (Vector Laboratories) for 5 min, followed by an NBTBCIP solution (Roche). The sections were mounted as above and viewed with an Axioskop II microscope.
For immunofluorescent staining, sections of kidneys frozen in O.C.T (Sakura Finetek, Torrance, CA) were cut at a thickness of 3 μm, and then fixed in precooled acetone. The slides were air dried for 30 min before blocking with 10% BSA in PBS, followed by incubation with goat anti-mouse IgG-FITC (Southern Biotech) 1:500 for 1 h at RT. The slides were rinsed twice with PBS-0.5% Tween 20, and covered with glass. Fluorescence was detected at room temperature using the Axioskop II microscope.
2.12. Transmission electron microscopy
Kidney tissue (1 mm × 1 mm) from PL9–11 hybridoma injected SCID mice was fixed in 2% glutaraldehyde and 4% formaldehyde in a 0.1 M phosphate buffered saline (pH 7.2) overnight at 4 °C, and postfixed in 1% osmium tetroxide in s-Collidine buffer. Thin sections (0.1 μm) were cut, stained with lead citrate and uranyl acetate, and examined at 80 kV with a JEOL 1200EX electron microscope (JEOL, Peabody, MA).
2.13. Immunogold staining
Sections were incubated in saturated sodium metaperiodate solution for 30 min, and then washed in 0.15 M glycine buffer for 10 min to inactivate residual aldehyde. The sections were blocked with 1% BSA in PBS for 10 min. Donkey anti-mouse IgG labeled with 6 nm gold (Electron Microscopy Sciences, Hatfield, PA) at a 1:20 dilution was used at 4 °C overnight to detect deposited mouse IgG. Control sections were stained with gold-labeled donkey anti-rabbit IgG. After five washes, sections were postfixed in 2% glutaraldehyde in PBS, and washed again before examination with electron microscopy.
2.14. Sequencing of heavy and light chain variable regions
For heavy and light chain sequencing, total RNA was extracted (Invitrogen, Grand Island, NY) from the PL9–11 cell lines. cDNA was generated and nested PCR of RNA ligase-mediated rapid amplification of cDNA ends (Applied Biosystems) was performed using the following primers:
VDJ (heavy chain): CTGTGCAGCCTCTGGATTCACTTT (forward) and TACTATAGGCCCGTCTTGCACAGT (reverse).
VJ (light chain): TCCCTGAGTGTGTCAACAGGAGAT (forward) and AATTGCCAGGTCTTCAGTCTGCAC (reverse).
After gel analysis, the PCR products of the expected size were purified, and sent for sequencing at the Albert Einstein College of Medicine sequencing facility.
2.15. Normalization of antibody concentrations
To normalize antibody concentrations for the binding assays, the concentrations of purified antibodies were determined by ELISA and Western blotting. In brief, 96-well microtiter plates were coated with goat anti-mouse κ chain antibody at a concentration of 10 μg/ml overnight at 4 °C. After blocking, PL9–11 antibodies (diluted to a presumptive concentration of 1 μg/ml according to the absorbance at 280 nm) were separately added, and incubated for 2 h at room temperature. Alkaline-phosphatase-labeled goat anti-mouse κ chain was used as the secondary antibody, and the OD values were read at 405 nm. For quantitation by Western blotting, the same concentrations of PL9–11 antibodies as were used in the ELISA were run. Proteins were transferred to the PVDF membrane as described above, and incubated with biotinylated anti-mouse κ chain for 30 min. The ABC solution kit and NBT/BCIP substrate were applied to develop the dark-brown color. Band intensity was measured by ImageJ.
3. Results
3.1. Generation of the PL9–11 mAb panel
IgG1, IgG2b and IgG2a isotype switch variants of the PL9–11 (IgG3) mAb were isolated by sib selection and agarose cloning [11]. The spontaneous rate of isotype switching of the PL9–11 mAb in vitro was <1/106; with cytokine stimulation, the frequency increased to between 1/104 and 1/105.
To generate a PL9–11 antibody of the IgM isotype, a spontaneously arising mutant PL9–11 hybridoma cell line expressing only the original light chain was first isolated. Heavy chain loss was confirmed by ELISPOT [21] and PCR. The variable region of the original PL9–11 IgG3 antibody was cloned by PCR, and ligated into an IgM expression vector which was then transfected into the light chain only line. Following G418 selection, reconstitution of the heavy and light chain of the PL9–11 IgM variant was confirmed by ELISA and Western blotting.
In this manner, we generated a PL9–11 antibody panel (IgM, IgG3, IgG1, IgG2b, and IgG2a) that all shared the original PL9–11 VDJ, but that differed in the identity of the constant region. Expression of a single heavy chain of the desired isotype was confirmed for each cell line. We did not find significant differences in the size of purified antibodies or in cryocrit formation, indicating no meaningful self-association in the members of the PL9–11 antibody panel (data not shown). Antibody concentrations were carefully and repeatedly normalized by Coomassie Blue, ELISA, and Western blotting, based on detection of the κ light chain which was shared by all the PL9–11 antibodies (data not shown).
3.2. The constant region significantly modulates binding to nuclear and glomerular antigens
We analyzed the effect of class switch recombination (CSR) on the antigen specificity of the PL9–11 antibody by ELISA. Fig. 1A demonstrates that isotype switching significantly decreased the dsDNA affinity of the original PL9–11 mAb; the relative affinity of PL9–11 for dsDNA was IgG3>IgG2a >IgG1 > IgG2b=IgM. Very similar results were found when binding of the PL9–11 panel to single stranded DNA (not shown) and chromatin were determined using the same technique (Fig. 1A). In contrast, in terms of binding to total histone (Fig. 1A), histone H2A (Fig. 1B) or histone H2B (not shown), isotype switching generated an IgG isotype with increased affinity relative to the parent antibody [10], with IgG2a> IgG3. Enhanced binding of the PL9–11 IgG2a mAb to histone H2A was confirmed by Western blotting (Fig. 1B).
Fig. 1.

Members of the PL9–11 antibody panel display variable affinity to nuclear antigens. (A) Serial dilutions of normalized mAbs were reacted with dsDNA, chromatin, and total histone bound to ELISA plates, and detected using an anti-kappa secondary antibody as described in Section 2. Data are representative of five experiments performed in duplicate. (B) mAb binding to purified histone H2A was assayed by ELISA and Western blot. For the Western blot, purified histone H2A was run by SDS–PAGE, and probed with isotype-specific anti-heavy chain antibodies and with an anti-kappa secondary antibody. The density ratios of the histone and kappa chain bands (as measured by ImageJ) for each isotype were calculated, and are provided at the bottom of the panel. ELISA data are representative of five experiments done in duplicate, and the Western blotting data is representative of two experiments.
To confirm that the differential binding to nuclear antigens by PL9–11 mAbs is indeed a function of their switched isotypes alone rather than arising from VDJ mutations occurring in vitro, we directly compared the variable region of the original PL9–11 IgG3 antibody to that of the IgG2b mAb which demonstrated greatly decreased affinity for nuclear antigens (Fig.1). The VDJ sequences of the PL9–11 IgG3 and IgG2b antibodies were identical, as were the sequences of the other members of the PL9–11 panel. Similarly, the light chain sequences of the PL9–11 IgG3 and IgG2b were identical as well (data not shown).
Affinity for dsDNA is closely linked to the renal pathogenicity of lupus autoantibodies, and is associated with cross-reactivity with kidney structural antigens [22]. To determine whether the altered affinity for DNA that arose with isotype switching is associated with changes in the binding to renal antigens, we assayed for binding of the PL9–11 antibody panel to several representative kidney antigens implicated in anti-dsDNA binding to renal tissue. With regards to renal antigens, PL9–11 IgG2a showed similar binding to IgG3 for Matrigel, and even higher binding than IgG3 for laminin and collagen IV (Fig. 2). This increased binding of the IgG2a PL9–11 to the latter two antigens is significant, since it confirms that the decreased binding to dsDNA and chromatin of the isotypes distal to IgG3 is not an artifact related to isotype switching in vitro. Indeed, class switching in the case of laminin and collagen IV increased affinity relative to the parent IgG3 antibody. PL9–11 IgG1, IgG2b, and IgM displayed weak or no binding to these glomerular antigens (Fig. 2).
Fig. 2.

Members of the PL9–11 antibody panel display variable affinity to glomerular antigens. Binding of PL9e11 and its class switch variants to representative glomerular antigens relevant to anti-DNA antibody binding to renal tissue, including Matrigel, laminin, and collagen IV, was assayed by serial dilutions on antigen precoated ELISA plates. Data are representative of five experiments performed in duplicate.
3.3. Competitive inhibition of antibodies in binding to dsDNA
To further elucidate potential specificity differences between various isotypes, we performed competition ELISA experiments. Each member of the PL9–11 antibody panel was tested for dsDNA binding under the same conditions, and analyzed for their ability to compete out binding of the other isotypes. These assays demonstrated similar differences among the antibodies in how effective they were in competing for dsDNA binding (Fig. 3). As a rule, the IgG3 isotype was most potent (i.e. a given concentration of antibody had the greatest effect in displacing the binding of the test antibody), followed by IgG2a, IgG1, IgG2b and IgM (Fig. 3). The finding that these switch variants manifest variable efficacy in competition studies further supports isotype related differences in specificity.
Fig. 3.

Competitive binding to dsDNA among the different PL9–11 mAbs. Serial dilutions of PL9–11 mAbs (“competing antibodies”) were bound to dsDNA coated ELISA plates, followed by each individual isotype (“test antibody”) at a fixed concentration of 0.5 μg/ml to assess the relative affinities of the test and competing antibodies to dsDNA. The title of each panel denotes the test antibody against which each of the other isotypes is competing for binding in that particular experiment. The amount of the test antibody that remained bound to dsDNA was detected with an isotype-specific secondary antibody, as shown in the illustration. Binding of the test antibody to dsDNA without the presence of a competitor was defined as 100%. Each dilution of the competing antibody was done in triplicate, and the mean and SD values are presented on the graph. Data are representative of three experiments performed in duplicate.
3.4. Binding kinetics of anti-dsDNA variants
To confirm and further quantitate the altered binding induced by CSR, we interrogated the affinity for dsDNA of the PL9–11 antibody panel by surface plasmon resonance (SPR) (Supplementary data, Fig. S1). Similar to the ELISA results, SPR analysis showed that IgG3 and IgG2a had relatively high affinity to dsDNA (Supplementary data, Fig. S1A, B). As compared to IgM, the relative affinities for dsDNA of the PL9–11 IgG isotypes were 2.30 (IgG1; calculated by KD IgM/KD IgG1, or 12.6/5.47), 1.58 (IgG2b), 3.57 (IgG2a), and 9.84 (IgG3), respectively. To assure that the differences in binding constants were not a function of variability in antibody adherence to the chip, the SPR analyses were repeated using streptavidin bound to the chip, followed by biotinylated dsDNA and antibody (Supplementary data, Fig. S1C). Once again, the IgG3 and IgG2a PL9–11 isotypes had the highest affinity for dsDNA (Supplementary data, Fig. S1C, D).
3.5. Isotype switching alters antigenic specificity
Multiplexed protein arrays can facilitate the simultaneous assay of antibody binding to multiple antigens. Using the glomerular proteome array we had described previously [17], we assayed for the binding of the PL9–11 panel to 76 lupus related nuclear, cytoplasmic, and extracellular antigens (Supplementary data, Fig. S2). PL9–11 IgG3 bound strongly to Ro/SS-A, La/SS-B, and U1-snRNP-A, while the IgG2a variant displayed greater binding to cytoplasmic proteins including cytochrome C and myosin. In terms of extracellular antigens, IgG3 had the highest affinity to glomerular extract, heparin and proteoglycan, while IgG2a showed stronger binding to fibrinogen and elastin (Supplementary data, Fig. S2). The IgM and IgG2b isotypes reacted weakly with the aforementioned antigens. These results of the slide-based array in Supplementary data, Fig. S2, showing IgG3 as the isotype with the highest affinity to dsDNA and chromatin and IgG2a for collagen IV, laminin and histone, were identical with the results of the ELISA assays presented above (Figs. 1, 2).
3.6. Members of the PL9–11 antibody panel show differential binding to mesangial cells
Cell surface binding to mesangial cells (MC) has been suggested as an important determinant of the renal pathogenicity of anti-DNA antibodies [23,24]. We examined the binding of the PL9–11 antibody panel to MC by cell surface ELISA and flow cytometry. In the cell surface ELISA (where non-DNase-treated MC contain both nuclear and non-nuclear antigens), MC were most strongly bound by IgG3. However, following DNAse treatment, MCs were bound by IgG3 and IgG2a with equal affinity (Fig. 4A).
Fig. 4.

Binding of the PL9–11 antibody panel to MC. Primary MRL/lpr MC were grown in culture, and used to assess binding of the PL9–11 isotype variants, with and without DNAse treatment. (A) Binding to MC bound to ELISA plates, with or without cell DNAse pre-treatment. Data are representative of four experiments performed in duplicate. (B) Binding to MC by members of the PL9–11 antibody panel as assessed by flow cytometry, with or without cell DNAse pre-treatment. Data are representative of three experiments performed in triplicate.
Similarly, we analyzed the binding of the PL9–11 antibody panel to MC by flow cytometry (Fig. 4B, left). IgG3-treated cells had the highest percentage of bound (positive) cells, followed by IgG2a. In contrast, IgM, IgG1 and IgG2b had significantly lower percentages of positive MC when compared to IgG3 or IgG2a, with no major difference between these former isotypes. Similar to the results in the cell surface ELISA, DNAse treatment of MC prior to flow cytometry significantly diminished the binding differences between IgG3 and IgG2a (Fig. 4B, right).
3.7. Differential binding affinity to isolated glomeruli
Quantifying binding to isolated glomeruli has been proposed as a model for the binding of anti-DNA antibodies in vivo, and may in fact better reflect their nephritogenic potential than affinity for DNA [19,25]. Accordingly, we studied the PL9–11 mAb panel in the glomerular binding assay (Fig. 5). IgG3 and IgG2a showed the highest affinity for isolated glomeruli, followed by much weaker binding by the other isotypes. In contrast, glomerular sections incubated with isotype controls showed no apparent staining. Finally, relative binding patterns were materially unchanged following DNAse pre-treatment of either the glomerular preparation or of the antibody (Fig. 5, and data not shown).
Fig. 5.

Antibody binding to glomeruli ex vivo. Glass slides containing fixed mouse glomeruli were blocked with 2.5% goat serum and rat anti-mouse CD16/32 antibody, and incubated with purified Abs at a concentration of 0.5 μg/ml. Antibody binding was detected with a FITC-labeled IgG goat anti-mouse kappa chain. Slides were then washed, stained with DAPI solution, and viewed at room temperature with a Zeiss fluorescence microscope. The mean fluorescent intensities for the binding of each PL9–11 antibody and its corresponding isotype control were measured using ImageJ software, and are provided in white font in the lower left corner of each panel. For the experiment depicted in the bottom part of the figure, glomeruli were first DNAsed as described in the Materials and Methods, and the assay then continued as described. Data are representative of three experiments performed in duplicate (200× magnification, numerical aperture = 0.5, bar = 50 μm).
3.8. Pathogenicity of PL9–11 antibodies in SCID mice
In experiments reported above we established that isotype switching led to significant changes in antibody affinity to nuclear antigens. Furthermore, we determined that considerable variations exist among members of the PL9–11 panel in binding to glomerular antigens in vitro, intact MC, and glomeruli ex vivo. To investigate whether altered affinity for glomerular antigens translated into differences in actual pathogenicity in vivo, we injected pristane-primed SCID mice with each of the cell lines corresponding to the members of the PL9–11 mAb panel. We found that PL9–11 injected mice, but not mice receiving pristane alone, developed considerable increases in proteinuria, significant versus baseline for PL9–11 IgG3 (p = 0.015), IgG1 (p = 0.004), and IgG2a (p = 0.019), and borderline for IgM (p = 0.09) (Fig. 6A). Proteinuria in PL9–11 IgG3 injected mice was also significantly higher than in mice injected with 3E5, an IgG3 non-DNA or glomerular binding hybridoma (p = 0.009). When comparing between the different IgG isotypes, PL9–11 IgG3 injected mice had significantly increased proteinuria as compared to IgG2b (p = 0.02) (Fig. 6A). Furthermore, survival analysis showed that PL9–11 IgM injected mice lived longer than mice receiving any of the other isotypes (p < 0.01), among whom there were no significant differences (Fig. 6B).
Fig. 6.

Renal disease and survival in SCID mice injected with PL9–11 hybridomas. Eight week old SCID mice received pristane 0.5 ml intraperitoneally, followed two weeks later by intraperitoneal injection of 107 hybridoma cells of each of the PL9–11 clones. Control groups of mice received pristane alone (“Blank”), or an IgG3-secreting hybridoma that does not bind DNA or glomerular antigens (3E5). (A) Ten days after the hybridoma cells were injected, urine albumin concentrations were determined. (B) Mice were sacrificed when they appeared moribund. Although there are no significant differences in the survival of mice injected with IgG3, IgG1, IgG2b, and IgG2a, the difference in survival between IgM and the IgG isotype injected mice groups is significant (p < 0.01). Note the overlap between the survival curves of IgG3 (in blue) and IgG1 (in green). (C) The concentration of the particular isotype each mouse group was injected with, in serum obtained at sacrifice, was measured by ELISA. For panels (A) and (C), mean and SEM are depicted.
3.9. Glomerular immunoglobulin deposition in PL9–11 panel injected mice
By both immunofluorescence staining (on frozen sections) and immunohistochemistry (on paraffin sections), we found that mice injected with the PL9–11 IgG3 and IgG2a cell lines had the most intense glomerular immunoglobulin deposition in vivo (Fig. 7). This finding is in line with the higher affinity for glomerular antigens demonstrated for these isotypes in the in vitro studies. Specific glomerular staining, albeit much weaker, could be seen in some PL9–11 IgG1 clone injected mice as well. There was little difference between mice injected with PL9–11 IgM or IgG2b, 3E5 clone injected mice, or pristane-primed mice not receiving hybridoma injections, all of which did not display significant immunoglobulin deposits (Fig. 7).
Fig. 7.
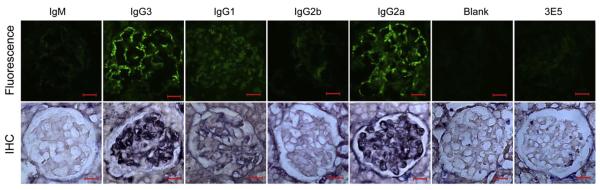
Immunoglobulin deposition in SCID mice injected with PL9–11 hybridoma clones. Frozen and paraffin sections of kidneys from mice injected with PL9–11 hybridoma cell lines were obtained, and stained for isotype-specific deposition by immunofluorescence and immunohistochemistry, respectively. By immunofluorescence, all mice injected with the PL9–11 clones of the IgM and IgG2b isotypes were completely negative. PL9–11 IgG3 and IgG2a injected mice all showed segmental to global granular glomerular basement membrane and mesangial staining. Three out of the five PL9–11 IgG1 injected mice showed faint staining. Staining for immunoglobulin deposition by immunohistochemistry showed similar differences between the groups. Kidneys of mice injected with pristane alone (not followed by a PL9–11 panel hybridoma) or with the non-DNA binding 3E5 clone (IgG3) did not demonstrate glomerular immunoglobulin deposition by either method. A representative staining pattern is shown for each mouse group. Immunofluorescence and immunohistochemistry images are representative of two experiments performed in duplicate (200× magnification, numerical aperture = 0.5, bar = 20 μm).
3.10. PL9–11 injected mice display ultrastructural evidence of renal damage
To evaluate for induction of renal damage at the ultrastructural level, we studied kidneys of PL9–11 injected mice by transmission electron microscopy. Pristane-primed mice not receiving hybridoma injections, or 3E5 injected mice, showed no ultrastructural abnormalities. In contrast, mice injected with members of the PL9–11 antibody panel showed an increase in podocyte foot process effacement (Fig. 8A). Furthermore, we observed subepithelial and intra-membranous electron dense deposits in PL9–11 IgG3 and IgG2a clone injected mice (Fig. 8A), correlating with the immunofluorescence and immunohistochemical positivity observed in these mice. In contrast, no immune type electron dense deposits were seen in the IgM, IgG1 or IgG2b injected mice. Consistent with the identification of the electron dense deposits as immune complexes, PL9–11 IgG3 but not 3E5 hybridoma injected mice demonstrated glomerular immunoglobulin deposition by immunogold staining (Fig. 8B–D), as was observed also in the immunofluorescence and immunohistochemistry studies (Fig. 7).
Fig. 8.

Transmission electron microscopy and immunogold staining of glomeruli from SCID mice injected with PL9–11 hybridoma clones. (A) Kidneys of mice injected with pristane alone (not followed by a PL9–11 panel hybridoma; “blank control”) or with the non-DNA binding 3E5 clone (IgG3) demonstrated normal kidney morphology, with preserved podocyte foot processes and no deposits along the glomerular basement membrane (bottom right). In contrast, patchy effacement of podocyte foot processes was present in glomeruli of SCID mice injected with PL9–11 hybridomas (arrows). In addition, electron dense deposits (arrowheads) were seen in IgG3 and IgG2a injected mice. In PL9–11 IgG3 injected mice, deposits were most often seen in a subepithelial location (between the glomerular basement membrane and the podocytes). In glomeruli of IgG2a injected mice deposits were more common, and found in subepithelial, intra-membranous, and subendothelial locations. A representative image from 5 mice injected with each cell line is shown (10K magnification, except where indicated on the image). (B)–(D) Mice injected with the PL9–11 IgG3 hybridoma display kidney immunoglobulin deposition, as demonstrated by immunogold staining. Sections were stained with gold-labeled donkey anti-mouse IgG labeled at 4 °C overnight and postfixed in 2% glutaraldehyde in PBS, as described in Section 2. (B), (C) Immunogold staining shows scattered intra-membranous clusters of gold particles (black dots; arrows) within glomeruli of the PL9–11 IgG3 injected mice, consistent with early immune complex deposition (40K magnification). (D) Significant immunogold particle staining is not observed in a 3E5 (IgG3) injected mouse (40K magnification, bar = 500 nm). A representative image from five mice injected with each cell line is shown.
3.11. Circulating antibody levels do not explain the renal immunoglobulin deposition and histopathology findings
We considered whether variability in circulating antibody levels in the hybridoma injected mice may underlie the differences found between members of the PL9–11 panel in their glomerular deposition and induction of renal injury. Several findings mitigate this theoretical concern. There were only minor, insignificant discrepancies between titers of the PL9–11 IgG3 and 3E5 (IgG3 as well) injected mice (Fig. 6C; p = 0.46), yet the differences detected by immunofluorescence, immunohistochemistry, and electron microscopy were highly significant. Similarly, titers of circulating PL9–11 IgG2a in the mice injected with the corresponding hybridoma were not different than that of IgM (p = 0.17) and IgG1 (p = 0.75) injected mice (Fig. 6C), yet distinct variations were seen in their pathogenicity. While the lowest antibody titers were indeed present in PL9–11 IgG2b injected mice which also had minimal kidney injury, nevertheless electron dense deposits along the GBM were only seen in mice injected with cell lines secreting the IgG3 and IgG2a mAb which have the highest affinity for glomeruli in vitro. Furthermore, no correlation was found between antibody titers and the degree of proteinuria, or the number of days until sacrifice (data not shown). Therefore, we believe that circulating antibody concentrations did not play a significant role in the observed differences in renal immunoglobulin deposition and damage.
4. Discussion
In this study we demonstrated that both the affinity and specificity of an anti-dsDNA Ab were altered when the antibody class was switched to either proximal (IgM) or distal (IgG1, IgG2b and IgG2a) isotypes. The PL9–11 variants all shared the same light chain and heavy chain variable region as the parent IgG3 clone, yet bound to dsDNA with substantially different affinity in both solid and fluid phases. Binding to other nuclear antigens showed a similar pattern. Given the close association between anti-dsDNA antibodies and lupus nephritis that may be mediated by cross-reactivity with renal antigens, it was important to determine whether modified affinity for DNA induced by isotype switching would translate as well into alterations in binding specificity for glomerular antigens and in pathogenicity in vivo. Indeed, we found significant variability in the binding of PL9–11 panel members to cross-reactive kidney antigens in vitro. Furthermore, PL9–11 IgG2a and IgG3 showed greatly enhanced affinity for MC and glomeruli, features which strongly predict pathogenicity in vivo [15,19,23–26]. Moreover, when injected into SCID mice, the PL9–11 IgG3 mAb induced significantly more proteinuria than IgG2b, and shortened survival relative to the IgM clone. While it may have theoretically been possible to show differences in proteinuria and survival between additional isotypes, the variability in circulating antibody levels between, and indeed within, groups of injected mice would have likely required a prohibitive number of animals for such a demonstration.
Although there were no significant renal histopathologic differences at the light microscopy level between PL9–11 switch variants administered in vivo (data not shown), at this resolution only rarely do glomerular binding anti-DNA hybridomas injected intraperitoneally induce renal injury over the short term [20,27,28]. The likely explanation is that additional elements lacking or modulated in the SCID mouse, such as B cells, T cells, and cytokines, are necessary for the full blown histological picture of nephritis to develop. We did find however glomerular immune deposits and podocyte injury by electron microscopy in PL9–11 IgG3 and IgG2a injected mice. The relatively small number of mice and the difficulty in controlling antibody levels are likely explanations for the lack of clear divergence in survival curves between nephrophilic and non-nephrophilic antibodies. Nevertheless, there was an exact correspondence between binding to renal antigens, MC, and glomeruli in vitro and glomerular immunoglobulin deposition and ultrastructural damage in vivo, convincingly demonstrating a meaningful difference in actual renal pathogenicity between PL9–11 switch variants.
The prevailing understanding of the pathogenesis of lupus nephritis is that it involves glomerular deposition of immune complexes, followed by recruitment of inflammatory cells via Fc receptors and complement activation [29–31]. However, there are several possible mechanisms by which these immune complexes can form and target renal tissue. Passive trapping of circulating immune complexes is not currently believed to play an important role, since they are not detectable, and in fact are normally cleared in lupus [22]. Careful studies by Rekvig et al. have implicated nucleosomal binding as being crucial for kidney immunoglobulin deposition, and in fact have determined that autoantibodies in lupus localize to chromatin-containing electron dense deposits in the kidney [32–35]. Alternatively, antibody binding to DNA may be irrelevant to the nephrotoxic potential. Rather, it is direct (non-DNA mediated) binding to renal antigens (e.g. α-actinin, laminin) which creates an immune complex in situ and initiates glomerular inflammation [5,29,36,37]. Whatever the case may be, there remains significant disagreement regarding the relative importance of binding to DNA/chromatin/nucleosomes versus cross-reactive renal antigens in mediating the pathogenicity of anti-DNA antibodies. Certainly, we need to consider whether the variability we observed in binding of the PL9–11 switch variants to MC and glomeruli is really not separate of the changes induced with isotype switching in affinity to DNA, but simply reflects binding to DNA which is present in the cell/glomerular preparations. However, while following DNAse treatment there was some attenuation of antibody binding, the IgG3 and IgG2a isotypes in particular still demonstrated significant binding to glomeruli and MC, and the noteworthy differences between these and the other isotypes in the panel were maintained. Therefore, it appears that isotype switching can independently modify the affinity for glomerular antigens, a conclusion further supported by the different subcellular localization of antibody deposition of IgG3 and IgG2a seen by electron microscopy.
While convincing experimental evidence demonstrates the nephritogenicity of anti-dsDNA antibodies, not all antibodies are created equal. Avidity for DNA, specificity for particular cross-reactive kidney antigens, identity of the idiotype, the nature and position of particular amino acids in complementarity determining regions (CDR), and cationicity are among antibody characteristics that have been linked to pathogenic potential [5,8,22,38,39]. Moreover, considering the centrality of complement and Fc receptor activation in lupus nephritis, antibody subclass which determines complement fixation and Fc binding is also considered to play a crucial role. Isotypes that fix complement well, including IgG1 and IgG3 in human and IgG2a and IgG2b in mouse, are enriched in glomeruli during active lupus nephritis [8]. Although we did not formally demonstrate that changes in antibody binding to renal tissue, rather than complement activation conferred by the constant region, are responsible for the differences in pathogenicity demonstrated in the SCID mice, the observed correlation with the changes in specificity observed in vitro indicate that the former explanation is the most likely. Interestingly, IgG subclass is also believed to play a role in other lupus manifestations as well. IgG2a and IgG2b which display the strongest complement activation and binding to the activating Fc receptors III and IV are the most potent at inducing autoantibody-mediated hemolytic anemia in vivo [7]. In SLE patients, particular IgG isotypes of serum anti-phospholipid antibodies (associated with thrombosis) have been linked to specific clinical manifestations [40].
Although IgG2a is believed to be the most pathogenic isotype in murine lupus, the ability of IgG3 antibodies to self-associate in antigen dependent and independent contexts and display cryoglobulin activity, and the high IgG3 titers in MRL/lpr mice, have focused attention also on this particular isotype. Several monoclonal IgG3 cryoglobulins with rheumatoid factor activity derived from MRL/lpr mice, including the 6–19 mAb, can induce glomerular wire loop lesions and cutaneous vasculitis when injected into normal mice [41–43]. Self-association of the 6–19 IgG3 mAb may be essential for its renal pathogenicity, as a spontaneous IgG1 switch variant (with an identical light chain) which does not display cryoglobulin activity is no longer pathogenic [44]. These studies supplied convincing evidence for the renal pathogenicity of IgG3 autoantibodies, and provided the first hint that the heavy chain constant region may play a role in determination of nephritogenic potential among lupus associated autoantibodies. However, we did not find evidence of self-association or cryoglobulin activity in the parent PL9–11 IgG3 antibody. Our results greatly expand on these previous studies, and prove that CSR can lead to increased binding to glomerular antigens and enhanced pathogenicity. While we acknowledge that our findings here relate to a single monoclonal antibody, together with the results of Fulpius et al. it would appear that this is not a restricted phenomenon.
Activation-induced deaminase (AID) is an enzyme that through cytosine deamination promotes isotype switching in vivo [45]. Considering the importance of class-switched antibodies in tissue injury in SLE, investigators explored the role of dysregulated AID in lupus-prone mice. Zan et al. reported that more than 20% of splenic B cells in MRL/lpr lupus mice are switched to IgG2a and more than 25% to IgA [46]. This increased proportion of class-switched B cells was due to an increased percentage of B cells undergoing switching in germinal centers rather than an increase in the actual numbers of germinal centers, and was mediated by increased AID expression. Upregulated CSR was also found in lymph nodes and Peyer's patches in this lupus strain. One of the factors controlling transcriptional regulation of AID is the HoxC4 transcription factor, which directly activates the AID gene promoter. Indeed, in both MRL/lpr mice and human SLE patients, HoxC4 expression is significantly increased [47].
To directly address the importance of CSR in induction of tissue injury in murine lupus, Jiang et al. generated AID deficient MRL/lpr mice [48], and compared the development and severity of disease to MRL/lpr AID wild type mice. AID deficient mice had a significant decrease in IgG autoantibodies, accompanied by a dramatic rise in autoantibodies of the IgM isotype. Furthermore, glomerulonephritis and mortality were significantly decreased. Interestingly, in AID heterozygous MRL/lpr mice immunoglobulin switching to IgG3 was maintained (apparently more distal switch regions are more sensitive to decreases in AID levels), as were total serum IgG levels, yet nephritis was still less severe [49]. Nevertheless, the lower apparent affinity of serum IgG for dsDNA in AID heterozygous MRL/lpr mice suggested that defective generation of higher affinity antibodies through the effects of AID on somatic hypermutation may have been primarily responsible for the attenuated phenotype in AID deficient mice [49]. Lastly, we recently were able to show that a selective genetic knockout of γ3 decreased the absolute amount of kidney-deposited immunoglobulins, diminished glomerulosclerosis, and significantly improved survival of MRL/lpr mice [50]. This latter study provides additional evidence for the role of IgG3 anti-DNA in the pathogenesis of glomerulonephritis in lupus mice.
What are the immunological implications of the observations in this study? Generation of altered (or novel) specifities through CSR can be an additional method by which the immune system creates diversity. Increasing the number of antigens recognized by a given variable region is clearly advantageous in host defense against foreign antigens [9,51], serving as a means of improving response efficiency without needing to further increase the number of variable regions genes. However, amplifying the antibody immune response by CSR would at the same time also create the risk of generating recognition of self-antigens by particular variable region-heavy chain combinations. Furthermore, given that isotype can affect specificity, antigen binding to the B cell during a memory response may favor the proliferation of isotype-restricted, antigen specific cells. This model can explain the favoring of isotype-restricted protective responses (i.e. IgG3 predominance in the anti-polysaccharide responses in mice), as well as the relative restriction in dominant isotypes in the murine and human lupus autoantibody responses. Finally, biologic response modifiers in the form of chimeric or fully human monoclonal antibodies are being increasingly utilized in the treatment of autoimmune disease, and have already revolutionized the treatment of several diseases in this category. In developing potential therapeutic antibodies, it will be however important to weigh not only the desired effector functions of the particular isotype being considered, but also its functional affinity and binding specificity [51–53].
The strength of association between antibody and antigen is characterized by its affinity, a measure of the binding strength between the antibody binding site and a monovalent epitope. Avidity, or functional affinity, measures the binding intensity between the two (or more) antigen binding sites and a multivalent antigen presenting multiple epitopes. Increased avidity due to the multivalent, pentameric structure of the IgM antibody cannot explain our findings, since this isotype exhibited low binding to the different classes of antigens tested in this study. Similarly, while IgG3 antibodies which can exhibit increased functional affinity through Fc–Fc interactions displayed the strongest binding to nuclear antigens, IgG2a was the dominant isotype in binding to glomerular antigens.
Several possible mechanisms can be advanced to explain the contributions of the heavy chain isotype to antigenic specificity. Indeed, for each isotype there may be a different explanation, reflecting changed Fab structure, different immunoglobulin surface properties, and altered Fab flexibility [52,53]. In fact, several studies suggest that local differences in the CH microenvironment can have electrostatic and hydrophobic effects which affect the flexibility of the antigen binding site even without marked conformational change. In other words, the constant region can impose structural constraints on variable region binding and directly influence the paratope–epitope interaction. Studies by Torres et al. have carefully documented significant effects of antibody subclass on the kinetics of antibody-antigen binding, and on thermodynamic parameters [9]. Moreover, several regions of structural differences within the CH1 domain of a particular antibody panel were observed by molecular modeling, in areas that could affect the structure of the antigen binding site [9].
Another option to consider in explaining the differences in antibody binding we observed lies in glycosylation, a characteristic of all IgG antibodies which occurs at a specific location in the CH2 domain [54–56]. Glycosylation is required for stability of the antibody molecule, and is important in several crucial effector functions including complement activation and binding to Fc receptors [54,56]. Since glycosylation may influence antibody conformation, the observation that IgG subclasses display distinct glycosylation differences may be pertinent to the finding of altered affinity for antigen between isotypes [55]. Nevertheless, several studies have shown that deglycosylated antibodies are no different from the parent antibody in antigen binding [54,57], although effector functions can be prominently affected [57]. Be that as it may, any possible role for glycosylation differences in altering antibody structure and function in the PL9–11 panel will need to be addressed experimentally in future studies.
Finally, besides the varied possible contributions of antibody subclass to interaction with antigen, it is important to mention another possible immunologic consequence. Formation of antibodies against the idiotype (antigenic determinants of the immunoglobulin structure itself) may have a key regulatory role in keeping immune responses, once initiated, in check. The fact that anti-idiotypic antibodies may recognize only certain isotypes [53] implies that the constant region also determines the formation of anti-idiotypic epitopes and hence may affect immunoregulatory processes.
5. Conclusions
Our data suggest that the constant region plays an important role in the affinity and specificity of anti-dsDNA antibodies, and that increased potential for binding to multiple glomerular and nuclear antigens may contribute to the nephritogenicity of anti-dsDNA antibodies of the IgG2a and IgG3 isotype. Studying the crystal structure of members of this mAb panel can further elucidate the mechanism by which CSR is actually modifying the antigen binding site. We hope to be able in such future studies to shed light on the cross-reactive tendencies displayed by pathogenic antibodies, and improve our understanding of the mechanistic basis for the renal pathogenicity of anti-DNA antibodies.
Supplementary Material
Acknowledgements
We thank Dr. Michael D. Brenowitz and Dr. Huiyong Cheng (Department of Biochemistry, Albert Einstein College of Medicine) for help with the Biacore analysis, Dr. Xingyi Guo (Department of Genetics, Albert Einstein College of Medicine) and the UT Southwestern Microarray Core for assistance in performance and analysis of the glomerular proteome array.
Funding This work was supported by grants from the NIH (AR048692 and DK090319 to C. Putterman, and HL059842, AI033774, AI033142, AI052733 to A. Casadevall), a Post-doctoral Fellowship Award from the Arthritis Foundation (to R. Pawar), and the Center for AIDS Research at the Albert Einstein College of Medicine (A. Casadevall).
Footnotes
Conflict of interest The authors have no conflicting financial interests.
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jaut.2012.06.005.
References
- [1].Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Sem Arthritis Rheum. 2004;34:501–37. doi: 10.1016/j.semarthrit.2004.07.002. [DOI] [PubMed] [Google Scholar]
- [2].Jang EJ, Nahm DH, Jang YJ. Mouse monoclonal autoantibodies penetrate mouse macrophage cells and stimulate NF-kappaB activation and TNF-alpha release. Immunology Lett. 2009;124:70–6. doi: 10.1016/j.imlet.2009.04.005. [DOI] [PubMed] [Google Scholar]
- [3].Qing X, Pitashny M, Thomas DB, Barrat FJ, Hogarth MP, Putterman C. Pathogenic anti-DNA antibodies modulate gene expression in mesangial cells: involvement of HMGB1 in anti-DNA antibody-induced renal injury. Immunology Lett. 2008;121:61–73. doi: 10.1016/j.imlet.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yung S, Tsang RC, Sun Y, Leung JK, Chan TM. Effect of human anti-DNA antibodies on proximal renal tubular epithelial cell cytokine expression: implications on tubulointerstitial inflammation in lupus nephritis. J Amer Soc Nephrol. 2005;16:3281–94. doi: 10.1681/ASN.2004110917. [DOI] [PubMed] [Google Scholar]
- [5].Hahn BH. Antibodies to DNA. N Engl J Med. 1998;338:1359–68. doi: 10.1056/NEJM199805073381906. [DOI] [PubMed] [Google Scholar]
- [6].Bijl M, Dijstelbloem HM, Oost WW, Bootsma H, Derksen RH, Aten J, et al. IgG subclass distribution of autoantibodies differs between renal and extra-renal relapses in patients with systemic lupus erythematosus. Rheumatology. 2002;41:62–7. doi: 10.1093/rheumatology/41.1.62. [DOI] [PubMed] [Google Scholar]
- [7].Baudino L, Azeredo da Silveira S, Nakata M, Izui S. Molecular and cellular basis for pathogenicity of autoantibodies: lessons from murine monoclonal autoantibodies. Semin Immunopathol. 2006;28:175–84. doi: 10.1007/s00281-006-0037-0. [DOI] [PubMed] [Google Scholar]
- [8].Ebling FM, Hahn BH. Pathogenic subsets of antibodies to DNA. Int Rev Immunol. 1989;5:79–95. doi: 10.3109/08830188909086990. [DOI] [PubMed] [Google Scholar]
- [9].Torres M, Fernandez-Fuentes N, Fiser A, Casadevall A. The immunoglobulin heavy chain constant region affects kinetic and thermodynamic parameters of antibody variable region interactions with antigen. J Biol Chem. 2007;282(13):917–27. doi: 10.1074/jbc.M700661200. [DOI] [PubMed] [Google Scholar]
- [10].Losman MJ, Fasy TM, Novick KE, Monestier M. Relationships among antinuclear antibodies from autoimmune MRL mice reacting with histone H2A–H2B dimers and DNA. Int Immunol. 1993;5:513–23. doi: 10.1093/intimm/5.5.513. [DOI] [PubMed] [Google Scholar]
- [11].Spira G, Bargellesi A, Teillaud JL, Scharff MD. The identification of monoclonal class switch variants by sib selection and an ELISA assay. J Immunol Methods. 1984;74:307–15. doi: 10.1016/0022-1759(84)90298-9. [DOI] [PubMed] [Google Scholar]
- [12].Iglesias-Ussel MD, Fan M, Li Z, Martin A, Scharff MD. Forced expression of AID facilitates the isolation of class switch variants from hybridoma cells. J Immunol Methods. 2006;316:59–66. doi: 10.1016/j.jim.2006.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Iglesias-Ussel MD, Zavadil J, Scharff MD. Molecular characterization of hybridoma subclones spontaneously switching at high frequencies in vitro. J Immunol Methods. 2009;350:71–8. doi: 10.1016/j.jim.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Deocharan B, Zhou Z, Antar K, Siconolfi-Baez L, Angeletti RH, Hardin J, et al. Alpha-actinin immunization elicits anti-chromatin autoimmunity in nonautoimmune mice. J Immunol. 2007;179:1313–21. doi: 10.4049/jimmunol.179.2.1313. [DOI] [PubMed] [Google Scholar]
- [15].Zhao Z, Weinstein E, Tuzova M, Davidson A, Mundel P, Marambio P, et al. Cross-reactivity of human lupus anti-DNA antibodies with alpha-actinin and nephritogenic potential. Arthritis Rheum. 2005;52:522–30. doi: 10.1002/art.20862. [DOI] [PubMed] [Google Scholar]
- [16].Winterroth L, Rivera J, Nakouzi AS, Dadachova E, Casadevall A. Neutralizing monoclonal antibody to edema toxin and its effect on murine anthrax. Infec Immun. 2010;78:2890–8. doi: 10.1128/IAI.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li QZ, Xie C, Wu T, Mackay M, Aranow C, Putterman C, et al. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005;115:3428–39. doi: 10.1172/JCI23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hedberg A, Fismen S, Fenton KA, Fenton C, Osterud B, Mortensen ES, et al. Heparin exerts a dual effect on murine lupus nephritis by enhancing enzymatic chromatin degradation and preventing chromatin binding in glomerular membranes. Arthritis Rheum. 2011;63:1065–75. doi: 10.1002/art.30211. [DOI] [PubMed] [Google Scholar]
- [19].Budhai L, Oh K, Davidson A. An in vitro assay for detection of glomerular binding IgG autoantibodies in patients with systemic lupus erythematosus. J Clin Invest. 1996;98:1585–93. doi: 10.1172/JCI118952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Putterman C, Limpanasithikul W, Edelman M, Diamond B. The double edged sword of the immune response: mutational analysis of a murine anti-pneumococcal, anti-DNA antibody. J Clin Invest. 1996;97:2251–9. doi: 10.1172/JCI118666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Spira G, Scharff MD. Identification of rare immunoglobulin switch variants using the ELISA spot assay. J Immunol Methods. 1992;148:121–9. doi: 10.1016/0022-1759(92)90165-p. [DOI] [PubMed] [Google Scholar]
- [22].Putterman C. New approaches to the renal pathogenicity of anti-DNA antibodies in systemic lupus erythematosus. Autoimmunity Rev. 2004;3:7–11. doi: 10.1016/S1568-9972(03)00082-X. [DOI] [PubMed] [Google Scholar]
- [23].Seret G, Le Meur Y, Renaudineau Y, Youinou P. Mesangial cell-specific antibodies are central to the pathogenesis of lupus nephritis. Clin Dev Immunol. 2012;2012:579670. doi: 10.1155/2012/579670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yung S, Cheung KF, Zhang Q, Chan TM. Anti-dsDNA antibodies bind to mesangial annexin II in lupus nephritis. J Am Soc Nephrol. 2010;21:1912–27. doi: 10.1681/ASN.2009080805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bernstein K, Bolshoun D, Gilkeson G, Munns T, Lefkowith JB. Detection of glomerular-binding immune elements in murine lupus using a tissue-based ELISA. Clin Exp Immunol. 1993;91:449–55. doi: 10.1111/j.1365-2249.1993.tb05923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Qing X, Zavadil J, Crosby MB, Hogarth MP, Hahn BH, Mohan C, et al. Nephritogenic anti-DNA antibodies regulate gene expression in MRL/lpr mouse glomerular mesangial cells. Arthritis Rheum. 2006;54:2198–210. doi: 10.1002/art.21934. [DOI] [PubMed] [Google Scholar]
- [27].Katz JB, Limpanasithikul W, Diamond B. Mutational analysis of an autoantibody: differential binding and pathogenicity. J Exp Med. 1994;180:925–32. doi: 10.1084/jem.180.3.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ehrenstein MR, Katz DR, Griffiths MH, Papadaki L, Winkler TH, Kalden JR, et al. Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int. 1995;48:705–11. doi: 10.1038/ki.1995.341. [DOI] [PubMed] [Google Scholar]
- [29].Isenberg DA, Manson JJ, Ehrenstein MR, Rahman A. Fifty years of anti-ds DNA antibodies: are we approaching journey's end? Rheumatology. 2007;46:1052–6. doi: 10.1093/rheumatology/kem112. [DOI] [PubMed] [Google Scholar]
- [30].Davidson A, Aranow C. Lupus nephritis: lessons from murine models. Nat Rev Rheumatol. 2010;6:13–20. doi: 10.1038/nrrheum.2009.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tsokos GC. Systemic lupus erythematosus. New Engl J Med. 2011;365:2110–21. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- [32].Mortensen ES, Rekvig OP. Nephritogenic potential of anti-DNA antibodies against necrotic nucleosomes. J Am Soc Nephrol. 2009;20:696–704. doi: 10.1681/ASN.2008010112. [DOI] [PubMed] [Google Scholar]
- [33].Fenton KA, Tommeras B, Marion TN, Rekvig OP. Pure anti-dsDNA mAbs need chromatin structures to promote glomerular mesangial deposits in BALB/c mice. Autoimmunity. 2010;43:179–88. doi: 10.3109/08916930903305633. [DOI] [PubMed] [Google Scholar]
- [34].Mjelle JE, Rekvig OP, Van Der Vlag J, Fenton KA. Nephritogenic antibodies bind in glomeruli through interaction with exposed chromatin fragments and not with renal cross-reactive antigens. Autoimmunity. 2011;44:373–83. doi: 10.3109/08916934.2010.541170. [DOI] [PubMed] [Google Scholar]
- [35].Hedberg A, Mortensen ES, Rekvig OP. Chromatin as a target antigen in human and murine lupus nephritis. Arthritis Res Ther. 2011;13:214. doi: 10.1186/ar3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mostoslavsky G, Fischel R, Yachimovich N, Yarkoni Y, Rosenmann E, Monestier M, et al. Lupus anti-DNA autoantibodies cross-react with a glomerular structural protein: a case for tissue injury by molecular mimicry. Eur J Immunol. 2001;31:1221–7. doi: 10.1002/1521-4141(200104)31:4<1221::aid-immu1221>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- [37].Deocharan B, Qing X, Lichauco J, Putterman C. Alpha-actinin is a cross-reactive renal target for pathogenic anti-DNA antibodies. J Immunol. 2002;168:3072–8. doi: 10.4049/jimmunol.168.6.3072. [DOI] [PubMed] [Google Scholar]
- [38].Ebling F, Hahn BH. Restricted subpopulations of DNA antibodies in kidneys of mice with systemic lupus. Comparison of antibodies in serum and renal eluates. Arthritis Rheum. 1980;23:392–403. doi: 10.1002/art.1780230402. [DOI] [PubMed] [Google Scholar]
- [39].Dang H, Harbeck RJ. The in vivo and in vitro glomerular deposition of isolated anti-double-stranded-DNA antibodies in NZB/W mice. Clin Immunol Immunopathol. 1984;30:265–78. doi: 10.1016/0090-1229(84)90061-8. [DOI] [PubMed] [Google Scholar]
- [40].Samarkos M, Davies KA, Gordon C, Walport MJ, Loizou S. IgG subclass distribution of antibodies against beta(2)-GP1 and cardiolipin in patients with systemic lupus erythematosus and primary antiphospholipid syndrome, and their clinical associations. Rheumatology. 2001;40:1026–32. doi: 10.1093/rheumatology/40.9.1026. [DOI] [PubMed] [Google Scholar]
- [41].Fulpius T, Berney T, Lemoine R, Pastore Y, Reininger L, Brighouse G, et al. Glomerulopathy induced by IgG3 anti-trinitrophenyl monoclonal cryoglobulins derived from non-autoimmune mice. Kidney Int. 1994;45:962–71. doi: 10.1038/ki.1994.130. [DOI] [PubMed] [Google Scholar]
- [42].Lemoine R, Berney T, Shibata T, Fulpius T, Gyotoku Y, Shimada H, et al. Induction of “wire-loop” lesions by murine monoclonal IgG3 cryoglobulins. Kidney Int. 1992;41:65–72. doi: 10.1038/ki.1992.9. [DOI] [PubMed] [Google Scholar]
- [43].Berney T, Fulpius T, Shibata T, Reininger L, Van Snick J, Shan H, et al. Selective pathogenicity of murine rheumatoid factors of the cryoprecipitable IgG3 subclass. Int Immunol. 1992;4:93–9. doi: 10.1093/intimm/4.1.93. [DOI] [PubMed] [Google Scholar]
- [44].Fulpius T, Spertini F, Reininger L, Izui S. Immunoglobulin heavy chain constant region determines the pathogenicity and the antigen-binding activity of rheumatoid factor. Proc Natl Acad Sci USA. 1993;90:2345–9. doi: 10.1073/pnas.90.6.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Durandy A. Activation-induced cytidine deaminase: a dual role in class-switch recombination and somatic hypermutation. Eur J Immunol. 2003;33:2069–73. doi: 10.1002/eji.200324133. [DOI] [PubMed] [Google Scholar]
- [46].Zan H, Zhang J, Ardeshna S, Xu Z, Park SR, Casali P. Lupus-prone MRL/faslpr/lpr mice display increased AID expression and extensive DNA lesions, comprising deletions and insertions, in the immunoglobulin locus: concurrent upregulation of somatic hypermutation and class switch DNA recombination. Autoimmunity. 2009;42:89–103. doi: 10.1080/08916930802629554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].White CA, Seth Hawkins J, Pone EJ, Yu ES, Al-Qahtani A, Mai T, et al. AID dysregulation in lupus-prone MRL/Fas(lpr/lpr) mice increases class switch DNA recombination and promotes interchromosomal c-Myc/IgH loci trans-locations: modulation by HoxC4. Autoimmunity. 2011;44:585–98. doi: 10.3109/08916934.2011.577128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jiang C, Foley J, Clayton N, Kissling G, Jokinen M, Herbert R, et al. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J Immunol. 2007;178:7422–31. doi: 10.4049/jimmunol.178.11.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jiang C, Zhao ML, Diaz M. Activation-induced deaminase heterozygous MRL/lpr mice are delayed in the production of high-affinity pathogenic antibodies and in the development of lupus nephritis. Immunology. 2009;126:102–13. doi: 10.1111/j.1365-2567.2008.02882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Greenspan NS, Lu MA, Shipley JW, Ding X, Li Q, Sultana D, et al. IgG3 deficiency extends lifespan and attenuates progression of glomerulonephritis in MRL/lpr mice. Biology Direct. 2012;7:3. doi: 10.1186/1745-6150-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Torres M, Casadevall A. The immunoglobulin constant region contributes to affinity and specificity. Trends Immunol. 2008;29:91–7. doi: 10.1016/j.it.2007.11.004. [DOI] [PubMed] [Google Scholar]
- [52].McLean GR, Torres M, Elguezabal N, Nakouzi A, Casadevall A. Isotype can affect the fine specificity of an antibody for a polysaccharide antigen. J Immunol. 2002;169:1379–86. doi: 10.4049/jimmunol.169.3.1379. [DOI] [PubMed] [Google Scholar]
- [53].Torres M, May R, Scharff MD, Casadevall A. Variable-region-identical antibodies differing in isotype demonstrate differences in fine specificity and idiotype. J Immunol. 2005;174:2132–42. doi: 10.4049/jimmunol.174.4.2132. [DOI] [PubMed] [Google Scholar]
- [54].Torres M, Fernandez-Fuentes N, Fiser A, Casadevall A. Exchanging murine and human immunoglobulin constant chains affects the kinetics and thermodynamics of antigen binding and chimeric antibody autoreactivity. PloS One. 2007;2:e1310. doi: 10.1371/journal.pone.0001310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wuhrer M, Stam JC, van de Geijn FE, Koeleman CA, Verrips CT, Dolhain RJ, et al. Glycosylation profiling of immunoglobulin G (IgG) subclasses from human serum. Proteomics. 2007;7:4070–81. doi: 10.1002/pmic.200700289. [DOI] [PubMed] [Google Scholar]
- [56].Wright A, Morrison SL. Effect of glycosylation on antibody function: implications for genetic engineering. Trends Biotechnol. 1997;15:26–32. doi: 10.1016/S0167-7799(96)10062-7. [DOI] [PubMed] [Google Scholar]
- [57].Nose M, Wigzell H. Biological significance of carbohydrate chains on monoclonal antibodies. Proc Natl Acad Sci U S A. 1983;80:6632–6. doi: 10.1073/pnas.80.21.6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.