

Abstract
Pasteurella multocida toxin (PMT) is a mitogenic protein that hijacks cellular signal transduction pathways via deamidation of heterotrimeric G proteins. We previously showed that rPMT activates mTOR signalling via a Gαq/11/PLCβ/PKC mediated pathway, leading in part to cell proliferation and migration. Herein, we show that mTOR and MAPK, but not membrane-associated tyrosine kinases, are activated in serum-starved 3T3 cells by an autocrine/paracrine substance(s) secreted into the conditioned medium following rPMT treatment. Surprisingly, this diffusible factor(s) is capable of activating mTOR and MAPK pathways even in MEF Gαq/11 double knockout cells. Microarray analysis identified connective tissue growth factor (CTGF) mRNA as the most upregulated gene in rPMT-treated serum-starved 3T3 cells relative to untreated cells. These results were further confirmed using RT-PCR and Western blot analysis. In accord with rPMT-induced mTOR activation, upregulation of CTGF protein was observed in WT MEF, but not in Gαq/11 double knockout MEF cells. Although CTGF expression is regulated by TGFβ, rPMT did not activate TGFβ pathway. In addition, MEK inhibitors U0126 or PD98059, but not mTOR specific inhibitors, rapamycin and Torin 1, inhibited rPMT-induced upregulation of CTGF. Importantly, CTGF overexpression in serum-starved 3T3 cells using adenovirus led to phosphorylation of ribosomal protein S6, a downstream target of mTOR. However, despite the ability of CTGF to activate the mTOR pathway, upregulation of CTGF alone could not induce morphological changes as those observed in rPMT-treated cells. Our findings reveal that CTGF plays an important role, but there are additional factors involved in the mitogenic action of PMT.
Keywords: mTOR, PMT, CTGF, TGFβ, Gαq/11
1. Introduction
Pasteurella multocida toxin (PMT) is an intracellular acting bacterial protein known for its mitogenic properties and its ability to induce anchorage-independent growth for certain type of cells like fibroblasts and osteoblasts. It is a 146 kDa single-chain protein that belongs to the A-B family of bacterial protein toxins. The C-terminal region of rPMT comprised of three distinct domains designated as C1, C2, and C3 [1]. While the C2 domain has no known function, the C1 domain was found to be the intracellular membrane-targeting domain of rPMT [2] and C3 is the minimal functional catalytic domain [3]. The amino-terminus of rPMT is the binding domain [4] and contains a putative membrane insertion motif assumed to be involved in membrane translocation into the cytosol [5].
PMT has been shown to exert its biological effects, in part, via the deamidation of a conserved glutamine residue in the α-subunit of heterotrimeric G proteins catalyzed by its C3 domain [6, 7]. The deamidation causes an inhibition of the inherent GTPase activity and leads to a constitutively active phenotype of the G proteins. The fact that rPMT is known to activate various families of heterotrimeric G proteins including Gαq, Gαi, Gα12 and Gα13, and subsequently mediate the activation of various signal transduction pathways, including MAPK, STAT, PLCβ, PKC, FAK and calcium mobilization [8-14]. As a consequence rPMT leads to cell growth and proliferation in vitro and in vivo. Recently, we showed that rPMT activates mTOR signalling pathway through a Gαq/PLCβ/PKC pathway [15]. In addition, we found that rPMT-induced mTOR activation is at least in part responsible for the increased protein synthesis, cell proliferation and migration observed when cells were exposed to rPMT.
An increasing body of evidence supports the notion that proteins of the extracellular matrix (ECM) are major players in the global control of intercellular communication and integration of environmental signals. However it is not known whether rPMT-treated cells are able to express and secret into the conditioned medium substance(s) capable of activating autocrine or paracrine signalling pathways.
Connective tissue growth factor (CTGF) was initially cloned in 1991 as growth factor inducible immediate-early gene [16] in cultured human vascular endothelial cells. CTGF has been shown to regulate a variety of cellular functions including proliferation, migration, adhesion, survival, differentiation and induction of ECM proteins in various cell types [17, 18]. CTGF has also been implicated in more complex biological and pathological processes, including angiogenesis, osteogenesis, wound healing, tumorigenesis, and fibrosis [18-21]. The diverse biological activities of CTGF are consistent with its modular structure, where each module can regulate different cellular functions and some modules can function interdependently. It is a secreted, cysteine-rich and heparin-binding protein containing 349 amino acid residues with an apparent molecular weight of 36–38 kDa. Initial sequence analysis revealed that CTGF was related to a v-src-induced peptide expressed in chick embryo fibroblasts (cef- 10) [22]. Subsequently, work by several groups demonstrated that CTGF and Cef-10 belong to the same family of structurally related peptides that included the serum-induced immediate early gene products cysteine rich 61 (Cyr61) [23], Fisp12, a CTGF ortholog in mouse [24], and nephroblastoma overexpressed (Nov) [25]. The first letter from the three founding members, which include Cyr 61, CTGF and Nov, was used to create the acronym CCN. The CCN family members are characterized by an absolute conservation of 38 cysteine residues that corresponding to more than 10% of the total amino acid residues.
With these considerations, we investigated whether rPMT treated cells could secrete autocrine or paracrine substance(s) which has/have the potential to activate mTOR in serum-starved 3T3 cells. Using microarray, RT-PCR, and Western blot analysis we found that CTGF was the most significantly upregulated gene induced by rPMT in serum-starved 3T3 cells. Interestingly, rPMT-mediated CTGF gene expression was observed in MEF wild type but not in MEF deficient in Gαq/11. Furthermore, CTGF expression in serum-starved 3T3 cells using an adenoviral expression system led to S6 phosphorylation, a downstream target of mTOR, but not ERK activation. The significance of these findings will be discussed below.
2. Experimental Procedures
2.1 Materials
Polyclonal antibody directed against CTGF protein and polyclonal antibody against ERK1/2 were from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Phosphor-ERK1/2 (Thr202/Tyr 204) polyclonal antibody, phosphor-MEK (Ser 217/221) monoclonal antibody, MEK monoclonal antibody, MEK inhibitors, U0126 and PD58059 were from Cell Signaling Technology (Beverly, MA). IRDye 800CW-conjugated affinity-purified anti-rabbit IgG and IRDye 700 CW conjugated affinity-purified anti-mouse IgG secondary antibodies were from LI-COR Biosciences (Lincoln, NE). RC DC protein assay kit was from Bio Rad Laboratories (Hercules, CA). Thermoscript cDNA synthesis and DNA polymerase were purchased from Invitrogen (Carlsbad, CA). Torin 1 was purchased from Tocris Bioscience (Minneapolis, MN). Monoclonal antibody, 3G3, against deamidated form of Gαq and mouse embryonic fibroblasts (MEF) double deficient in Gαq/11-/- were generous gifts from Dr. Shigeki Kamitani, (Research Institute for Microbial Diseases, Osaka University, Japan) and from Professor Stefan Offermanns (Pharmakologisches Institute, Universitat Heidelberg, Germany), respectively. Mouse receptor tyrosine kinases array was from RD system (Minneapolis, MN). Swiss 3T3 cells and calf fetal serum were obtained from American Type Culture Collection (Manassas, VA). Culture media and antibiotics were from Invitrogen (Carlsbad, CA).
2.2 Cell culture and PMT treatment
Swiss 3T3 and MEF cells were cultured in DMEM containing 10% fetal bovine serum (ATCC), L-glutamine, and antibiotics (50 U/ml penicillin, 50 mg/ml streptomycin) and incubated at 37°C in a humidified atmosphere of 5% CO2 in air. Swiss 3T3 cells were grown in DMEM medium containing 10% calf fetal serum (CFS). The same number of cells were seeded in 10 cm Petri dishes and grown to confluency. Confluent cells were then incubated in serum-free medium for 24h and then treated with rPMT in the presence or absence of the indicated inhibitor. Treated cells were rinsed with ice-cold phosphate-buffered saline and scraped into loading buffer. Protein concentrations were determined by the Lowry method using the RC DC protein assay kit according to the manufacturer’s instructions. Equal amounts of proteins were subjected to SDS-PAGE. Separated proteins were then transferred onto a nitrocellulose membrane, probed with an appropriate antibody, and detected by using the LI-COR Odyssey (Lincoln, NE, USA). To detect CTGF protein in the medium, 100 μl of heparin/agarose were added to 6 ml of conditioned medium from control untreated or rPMT-treated cells in the presence or absence of indicated inhibitor. The mixtures were then left at 4°C under agitation for 60 min. Heparin agarose was pelted by centrifugation and washed three times with phosphate buffer saline. The protein bound to heparin agarose was eluted by using 100 μl of loading buffer containing mercaptoethanol. Equal volumes were loaded and separated by SDS PAGE under reducing condition. Alternatively, 200 μl of condition medium from rPMT-treated or untreated cells were dried using speed-Vac. The results presented in the figures represent a typical observation from two to four independent experiments.
2.3 Reverse-transcriptase Polymerase Chain-reaction (RT-PCR) Analysis
Swiss 3T3 cells were grown in 100-mm tissue culture plates in DMEM media containing 10% FCS. When the cells were confluent, they were maintained in serum-free medium for 24 hrs, followed by treatment with rPMT at a final concentration of 0, 0.1, 1, or 10 μg/mL for 12, 24, and 48 hrs. Total RNA was isolated, and the concentration and purity of RNA were determined by spectrophotometric absorption at wavelengths of 260/280 nm. Double-stranded cDNA was synthesized using ThermoScript II (Invitrogen). The expression of CTGF mRNA transcripts was semi-quantitatively analyzed by RT-PCR (95°C for 5 min followed by 59°C for 40s, 72°C for 1 min, 95°C for 40s), and was normalized to the expression level of PP2A transcripts. PCR products were separated on a 1.4% agarose gel.
2.4 Mouse receptor tyrosine kinases array
To study the effect of rPMT treatment on membrane-associated receptor tyrosine kinases in serum-starved Swiss 3T3 cells, we used the mouse phosphor-receptor tyrosine kinase (phosphor-RTK) array. Each membrane array contains 39 different anti-RTK antibodies, three positive controls and one negative control printed in duplicate. Cell lysates from control non-treated and rPMT treated cells for 24h were diluted and incubated with the membranes according to the manufacturer recommendation. The membranes were washed to remove any unbound material and a pan anti-phospho-tyrosine antibody conjugated to horseradish peroxidase was used to detect phosphorylated tyrosines on activated receptors by chemiluminescence. Serum-starved 3T3 cells were treated for the indicated time with 6 ml of conditioned medium taken either from control non-treated or from cells treated with rPMT for 24h. After treatment, cell lysates were prepared as above, applied to mouse RTK microarray membranes and activated RTK revealed as described above.
2.5 Affimetrix Microarray Analysis
Total RNA was prepared from control non-treated and rPMT treated cells for 24h using the RNasy Kit (QIAGEN) including the optional DNase treatment before elution from purification columns. RNA was processed according to NHLBI core facility and hybridized to oligonucleotide microarrays (Mouse Genome 430 2.0 array, Affymetrix, Santa Clara, CA). Experiments were performed in quadruplicates, and average induction values were obtained. Scanned image files were quantified with GENECHIP 3.2 (Affymetrix). Genes that increased or decreased at least 2-fold (p < 0.008) in rPMT treated samples relative to the control non treated samples were used to generate the heatmaps illustrated in Figure 4A.
Figure 4. rPMT induces CTGF mRNA in quiescent 3T3 cells.
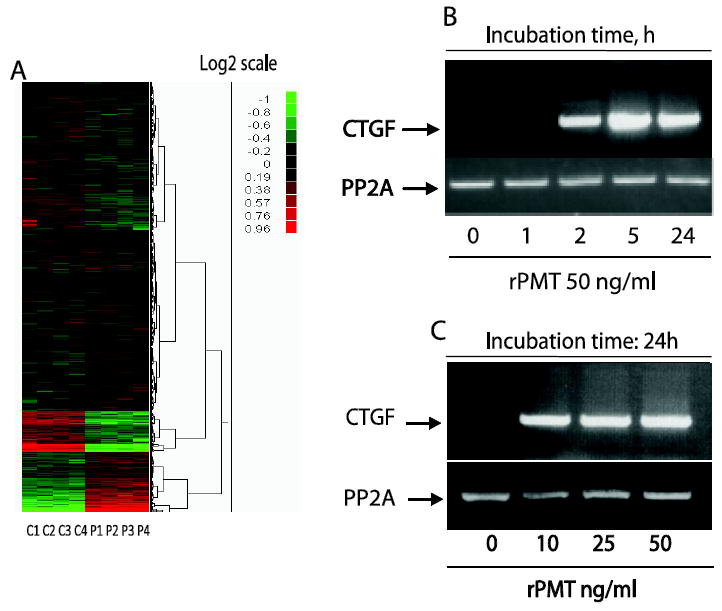
Serum-starved 3T3 cells were treated with 50 ng/ml of rPMT for 24h. Total RNA was prepared from control and rPMT treated cells, reverse transcribed, and applied to Affymetrix arrays. (A) shows cluster of genes that are altered due to rPMT treatment. Experiments were performed in quadruplicates, and average induction values were obtained. Data analysis showed that 1774 probe sets representing 1185 genes were up-regulated and 1599 probe set representing 1110 genes were decreased by at least 2-fold (treated vs. non-treated). CTGF, a CCN family member, is the most upregulated gene following rPMT treatment (Table 1). RNA was isolated from both control and cells exposed to either a fixed concentration of rPMT for indicated period of times (B) or to different concentrations of rPMT for 24h (C) and analyzed by RT-PCR using CTGF primers.
2.6 Cell transfection
3T3 cells were grown in DMEM containing 10% of serum till confluency then switched to serum free media and immediately infected with empty adenovirus or adenovirus expressing CTGF at a multiplicity of 5 PFU/cell for 24h. Cells were lysed in loading buffer as above and equal amount of protein were separated on SDS PAGE electrophoresis
3.Results and discussion
3.1 Conditioned medium from rPMT-treated 3T3 cells activates mTOR in 3T3 and MEF cells
rPMT is known to exert some of its cellular effects via heterotrimeric G protein-coupled signalling pathways, especially those involving Gαq/11. Some of the cellular effects of rPMT are abrogated in both Xenopus oocytes and HEK293 cells using either antibodies directed against the α subunit (αq and α11) of Gq family and Gαq antisense RNA or by overexpressing of the C-terminal peptide inhibitor of the α subunit (αq and α11) of Gq, respectively [10, 26]. Furthermore, studies in fibroblasts deficient in Gαq and/or Gα11 suggest that rPMT’s action is mediated through Gαq, but not its homolog Gα11 [11, 27]. Using mass spectrometry, it has been shown that rPMT deamidates Gαq and Gαi leading to their constitutively active forms [6]. Recently, an antibody against deamidated form of Gαq showed that rPMT can deamidate several heterotrimeric G proteins including Gαq, Gα11, Gα12, and Gα13 in vitro and in transfected cells [7]. The fact that rPMT is able to activate a variety of G proteins may lead to a simultaneous activation of several signalling pathways.
We have shown recently that rPMT persistently activates the mTOR signalling pathway in a manner dependent on the Gαq/11/PLCβ/PKC pathway [15]. We next set out to determine whether the observed mTOR pathway activation is due to a direct activation of Gαq/11/PLCβ/PKC signalling pathway as a result of Gαq/11 deamidation or alternatively to the production and secretion of one or more autocrine/paracrine substance(s) into the medium in response to rPMT treatment. Figure 1A shows that rPMT treatment for 24h led to a drastic increase in the phosphorylation of rpS6, a read out of mTOR activation, in comparison to the control untreated cells. Concomitantly, an increase in Gαq/11 deamidation was also observed, over that of control non-treated cells. Interestingly, S6 phosphorylation was also observed, without any increase in the deamidated Gαq/11 levels, when serum-starved Swiss 3T3 cells were treated for 30 minutes with conditioned medium taken from cells pretreated with rPMT for 24h. In contrast, conditioned medium taken from control non-treated cells had no effect on S6 phosphorylation or Gαq/11 deamidation when added to quiescent 3T3 cells for the same period of time. As a further control, exposure of quiescent cells to rPMT for 30 minutes did not induce any increase in S6 phosphorylation or Gαq/11 deamidation. This result rules out the involvement of a possible remaining rPMT in the condition medium to induce the observed S6 phosphorylation. The lag phase in the action of recombinant P. multocida toxin reflects its binding to the plasma membrane [4], followed by cellular entry and possible processing and activation. To determine whether conditioned medium-induced S6 phosphorylation is mTOR dependent, we treated serum-starved cells with conditioned medium in the presence or absence of rapamycin or Torin1, two specific inhibitors of mTOR, for the indicated time. As shown in figure 1B, S6 phosphorylation in response to conditioned medium from rPMT-treated cells is rapamycin and Torin1 sensitive ruling out the involvement of other serine threonine kinases other than mTOR in the phosphorylation of its downstream target, S6.
Figure 1. Conditioned medium from rPMT treated cells phosphorylates S6 in serum-starved 3T3 cells.
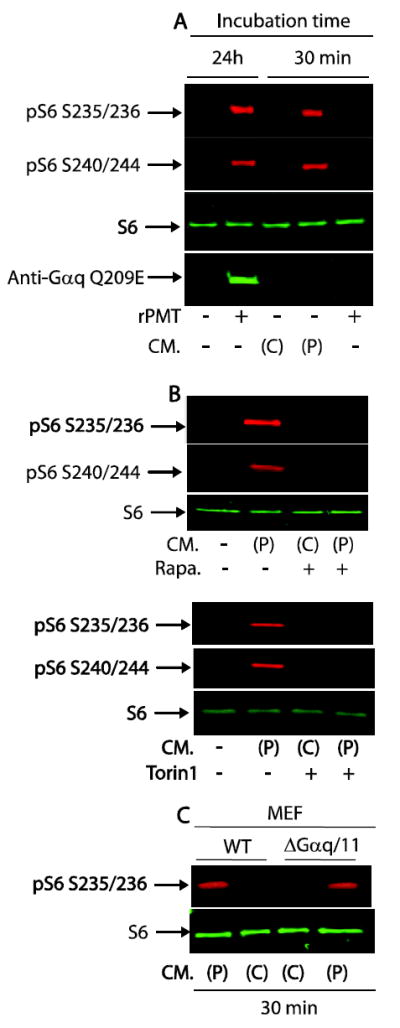
Confluent 3T3 cells were serum-starved for 24h then either left untreated or treated with 50 ng/ml of rPMT for another 24h. Conditioned medium from control non-treated or rPMT-treated cells were collected and added to confluent and serum-starved 3T3 cells for 30 min (A). Quiescent 3T3 cells were incubated with conditioned medium from control non-treated or rPMT-treated cells for 30 min in the presence or absence of rapamycin (50 nM) or Torin1 (100 nM), two specific inhibitors of mTOR (B). Quiescent Gαq/11 deficient and WT MEF cells treated with conditioned medium from control non-treated or rPMT-treated cells for 30 min (C). The cells were then lysed in SDS PAGE loading buffer and equal amounts of proteins were separated on SDS polyacrylamid gel elepctrophoresis, and transferred onto nitrocellulose membranes. Transferred proteins were detected using an antibody against S6, phosphor-rpS6 and deamidated form of Gαq. When treated with CM, conditioned medium, (P) and (C) represent medium from rPMT treated and control non-treated cells.
The observed protracted mTOR phosphorylation following rPMT treatment could be attributed at least to two different, yet related, mechanisms: (i) rPMT catalyzed deamidation of glutamine 209 of Gαq/11 to glutamic acid [7], which subsequently leads to its persistent activation [6]. The GTP bound deamidated Gαq/11 in turn binds and activates PLCβ leading to the production of inositol phosphates and diacylglycerol (DAG) [8, 26, 28]. The rPMT-induced Gαq/11/PLCβ activation has been shown to be responsible for the observed increase in S6 phosphorylation in a manner sensitive to rapamycin or Torin1 [15]. The role of Gαq/11 in rPMT-induced mTOR activation was further supported by the fact that activation of the mTOR pathway was observed in MEF wild type cells but not in MEF Gαq/11 deficient cells following rPMT treatment [15]. (ii) A diffusible factor(s) is/are present in the conditioned medium taken from rPMT-treated cells that is, at least in part, responsible for mTOR activation in serum-starved cells independence of Gαq/11 deamidation and activation. However this observation does not exclude the possibility that the conditioned medium contains a factor capable of activating G protein coupled receptor (GPCR) independent of Gαq/11 deamidation. To explore this possibility, serum-starved 3T3 cells were either left untreated or treated with rPMT for 24h, then the conditioned medium was collected from control and rPMT-treated cells, and used to treat serum-deprived wild type and Gαq/11 double knockout MEF cells for 30 min. The data in Figure 1C show that conditioned medium from untreated 3T3 cells did not induce S6 phosphorylation in either wild type or in Gαq/11 knockout MEF. In contrast, conditioned medium from 3T3 cells treated with rPMT induced an increase in S6 phosphorylation in both WT and Gαq/11 double knock out MEF cells, despite the fact that rPMT failed to induce S6 phosphorylation in the double knocked cells. Together, these results clearly support the existence of a diffusible factor(s) in the rPMT-treated conditioned medium that is capable of inducing the S6 phosphorylation in a manner independent of Gαq/11 mediated signalling pathway. This observation is in line with a previous report showing that while rPMT treatment leads to an increase in inositol phosphates in serum-starved 3T3 cells [7] probably caused by the rPMT-mediated Gαq deamidation and activation [6], while cells exposed to the conditioned medium from cells treated for 4 h with 20 ng/ml rPMT failed to provoke an increase in InsP [8]. Thus, it is possible that mTOR activation observed after cell treatment with conditioned medium from rPMT treated cells is independent of inositol phosphates signalling. Contrary to quiescent 3T3 and MEF WT cells, it is likely that rPMT treatment did not induce the synthesis and/or the secretion of mTOR activating paracrin/autocrin substance(s) in Gαq/11 deficient cells, reflecting the requirement of Gαq/11. However, we could not exclude a direct involvement of Gαq/11/PLCβ/PKC pathway in rPMT-mediated S6 phosphorylation that might be a result of several concomitant mechanisms deployed by rPMT. These results reveal for the first time that rPMT-treated cells produce and secrete into the culture medium a mediator(s) capable of activating mTOR.
3.2 Conditioned medium from rPMT-treated 3T3 cells activates MEK signalling pathway in 3T3 and MEF cells
Since the conditioned medium from rPMT treated cells is able to activate mTOR signalling in serum-starved 3T3, MEF WT as well as Gαq/11-deficient MEF cells, we investigated whether conditioned medium from rPMT-treated cells can activate other signalling pathways such as MAPK and EGFR. In fact, rPMT is known to activate the MAPK signalling pathway [10, 14, 29]. However, the mechanisms behind rPMT-induced MAPK phosphorylation are still largely unknown. Thus we analyzed the effect of rPMT on the phosphorylation of ERK signalling and its upstream kinase MEK, by Western blot analysis using serum-starved 3T3 cells. We found that rPMT induced a strong and sustained phosphorylation of MEK1 (Fig. 2A) and ERK1/2 (Fig. 2B) when quiescent cells were treated with concentrations of rPMT ranging from 0 to 100 ng/ml for 24h. Furthermore, this observed rPMT-induced ERK1/2 phosphorylation is also confirmed by the results showing rPMT mediated ERK1/2 phosphorylation was totally inhibited by the presence of 10 to 20 μM MEK inhibitors shown in Fig.8A.
Figure 2. rPMT induced MEK signalling pathway activation in serum-starved 3T3 and WT MEF cell.
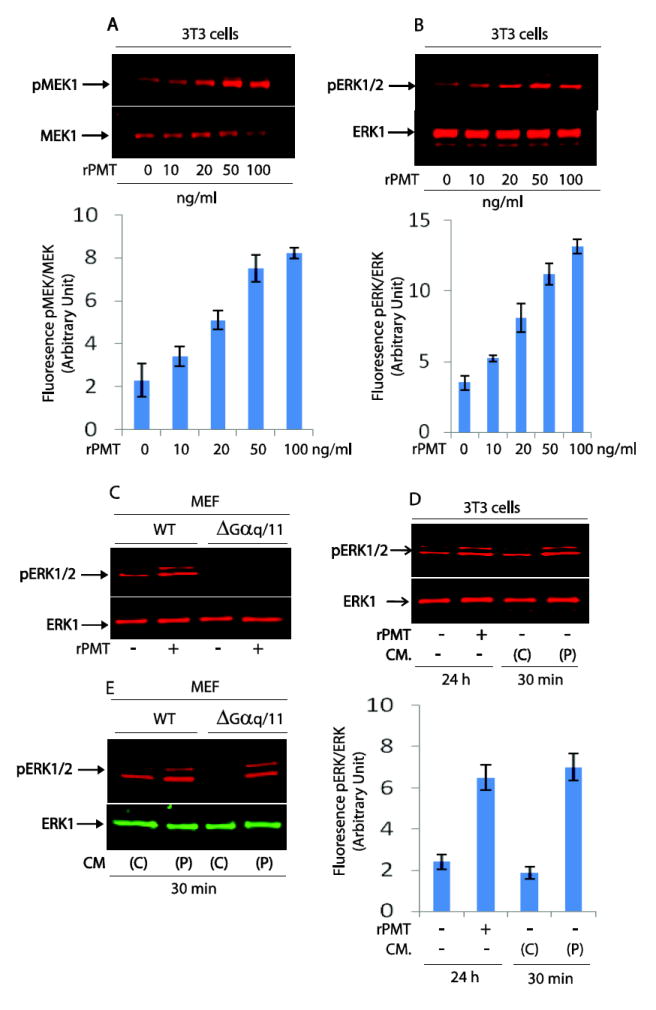
Confluent cells were serum-starved over night and thereafter treated with indicated concentrations of rPMT for 24h. rPMT treated and non-treated control cells were harvested and lysed directly in loading buffer. The same amount of proteins were separated on SDS PAGE and transferred onto nitrocellulose membranes. Shown are representative immunoblots developed with antibodies that recognize pMEK1 and MEK1 protein, (A); pERK1/2 and ERK1, (B). MEF wild type and MEF Gαq/11 knockout cells were serum-deprived, treated with 100 ng/ml of rPMT, and lysed as described above. Equal amount of proteins were subjected to immunoblotting with antibodies against pERK1/2 and ERK1 (C). Conditioned medium (CM) from untreated (C) and rPMT (50 ng/ml) treated (P) 3T3 cells were collected and used to treat serum-starved 3T3 cells (D), WT MEF and MEF Gαq/11 knockout cells (E) for 30 min. Cells were lysed as described above and immunoblots were developed using antibodies that recognize ERK1 and pERK1/2. The bar charts shown in 2A, 2B and 2D represent the mean +/− SEM from three independent experiments.
Figure 8. rPMT-induced CTGF expression is inhibited by MEK inhibitors, but not EGFR inhibitor.
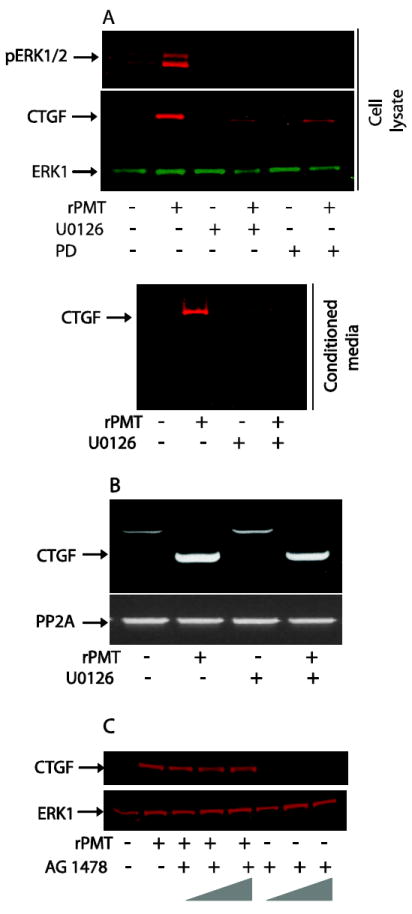
Confluent and serum-starved 3T3 cells were treated with 50 ng/ml of rPMT for 24h in the absence or presence of MEK inhibitor U0126 (10 μM) or PD 98059 (20 μM). Cells were then lysed in loading buffer. The same amount of protein (A upper panel) or the same volume of conditioned medium (A lower panel) were separated and transferred to nitrocellulose membranes. The immunoblots were developed using antibodies against CTGF, ERK and pERK. Quiescent and serum-starved cells were treated with 50 ng/ml of rPMT for 24h in the absence or presence of 10 μM of MEK inhibitors, U0126. Total RNA was prepared as described above, reverse transcribed, and analyzed by RT-PCR using CTGF primers (B). Quiescent Swiss 3T3 cells were treated with 50 ng/ml of rPMT for 24h in the absence or presence of 1.0, 5.0 and 10.0 μM of AG1476. Thereafter, cells were lysed and equal amount of proteins were separated on SDS PAGE and transferred to the nitrocellulose membrane. The immunoblots were developed using antibodies against CTGF and ERK1 (C).
Importantly, rPMT treatment (50 ng/ml) did not activate ERK signalling pathway in Gαq/11-deficient MEF cells compared to MEF WT cells (Fig. 2C). This result is in agreement with the notion that rPMT-induced ERK phosphorylation is Gαq/11 dependent [10]. We next carried out experiments to determine whether conditioned medium from rPMT treated cells can activate ERK in serum-starved 3T3 cells. The data in Fig. 2D show that ERK signalling pathway was activated when serum-starved Swiss 3T3 cells were treated for 30 minutes with conditioned medium taken from cells treated with rPMT for 24h. However, conditioned medium from control non-treated cells had no effect on ERK phosphorylation when added to quiescent 3T3 cells. Intriguingly, conditioned medium from PMT-treated 3T3 cells did induce ERK activation in Gαq/11 deficient MEF cells, despite the fact that rPMT itself failed to induce ERK phosphorylation in these cells (Fig. 2E).
Using a mouse RTK array consisting of anti-bodies against 39 RTKs, we previously observed that rPMT induces tyrosine phosphorylation of EGFR 1 and 2 [15]. We therefore investigated the effect of conditioned medium on RTK phosphorylation. Figure 3 shows that while rPMT treatment for 24h led to EGFR phosphorylation, conditioned medium collected from untreated 3T3 cells or cells treated with rPMT (50 ng/ml) for 24h exerted no effect on RTK phosphorylation after the indicated incubation time. This observation is in agreement with previous result showing that rPMT treatment leads to the EGFR transactivation [10].
Figure 3. Receptor tyrosine kinase activation by rPMT.
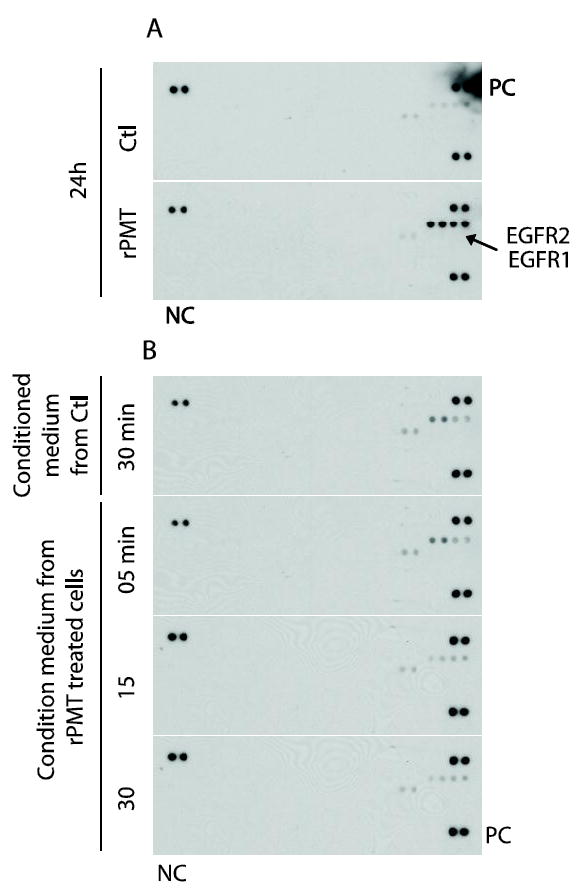
(A) Serum-starved Swiss 3T3 cells were either left untreated or treated with 50 ng/ml of rPMT for 24h. (B) serum-starved 3T3 cell were treated with conditioned medium from control untreated cells or from rPMT treated cells for indicated time. Cells were lysed and 100 μg of protein were applied to the receptor tyrosine kinase array that contained 39 different anti-RTK antibodies, three positive controls (PC) and one negative control (NC) printed in duplicate. The membranes were then washed and revealed using an anti-phosphor-tyrosine antibody conjugated to horseradish peroxidase.
Taken together, these results reveal that rPMT-treated cells produce and secrete into the culture medium a mediator(s) capable of activating mTOR and MAPK signalling but not the RTK pathway in serum-starved cells in a manner independent of Gαq/11. It would be interesting to see whether other biological effects of rPMT including STAT, Rho, and PDK activation [30], observed in rPMT treated cells can also be mediated by factors secreted into the conditioned medium.
3.3 rPMT induces CTGF mRNA in quiescent 3T3 cells
To identify the putative factor(s) secreted by rPMT-treated cells that is (are) responsible for mTOR activation, total RNA was prepared from control and rPMT treated cells for 24h, reverse transcribed, and applied to Affymetrix arrays. Data analysis revealed that 1774 probe sets representing 1185 genes were up-regulated, and 1599 probe sets representing 1110 genes were decreased by 2-fold or more (in rPMT-treated vs. untreated cells). Gene cluster analysis revealed that transcripts involved in cell proliferation and cancer biology were significantly affected by rPMT treatment (Fig 4A). The mRNA with the greatest mean elevation in rPMT treated cells was that of the connective tissue growth factor (CTGF), which was elevated 128-fold relative to that of the control non-treated cells. CTGF is a 36-38 kDa secreted, cysteine-rich and haperin-binding protein, known to play essential roles in the development of many tissues. It is barely expressed in normal adult tissue. However, it is strongly up-regulated in fibrotic tissue independently of its etiology, during development, in wound healing, and in certain types of cancer [18-21]. Our microarray results showed that in addition to CTGF mRNA, rPMT treatment induced an increase in Cyr61 and Wisp2 mRNA albeit at lower magnitude than that of CTGF. However, contrary to CTGF, Cyr61, and Wisp2, Nov mRNA levels were downregulated by rPMT treatment (see Table 1). This observation is in agreement with previous studies showing that CTGF and Nov can mutually regulate their expression in an Yin/Yang manner and that either the addition or over-expression of one produces a marked down-regulation of the other [31, 32]. However, the mechanism for this regulation of CTGF and Nov is still not known.
Table I.
Expression of CCN mRNA induced by rPMT in 3T3 cells
| Gene | Symbole | Fold increase |
|---|---|---|
| Connective tissue Growth Factor | CTGF | 140 |
| Cysteine Rich Protein | Cyr61 | 8 |
| Wnt1 Inducible Signaling Pathway Protein 2 | Wisp2 | 6 |
| Nephroblastoma Overexpressed Gene | Nov | -6 |
To confirm the microarray results, RNA was isolated at the indicated time following rPMT stimulation of quiescent Swiss 3T3 cells and analyzed by RT-PCR. Figure 4B shows that CTGF mRNA levels were very low in quiescent cells, but rapidly increased in response to rPMT treatment, reaching a maximal expression level of approximately 120-fold over the untreated control by 2h. CTGF mRNA levels remained significantly high at 24 h (Fig. 4B). To confirm its identity, the observed 600-bp band was sequenced and found to correspond to CTGF cDNA. It is worth noting that in addition to the CTGF replicon whose seize is around 600 bp, we also observed an additional band with a molecular size of 1000 bp in the control untreated samples. This band was not observed in rPMT or serum treated cells. This slow migrating band may correspond to the immature unprocessed CTGF mRNA or to a CTGF splice variant. Attempts to amplify this band by PCR after gel purification yield a truncated 600 bp fragment (data not shown). This observation suggests that the CTGF transcript may form a secondary structure that is hard to PCR as shown for chicken Cyr61 [33]. In addition, induction of CTGF mRNA by rPMT was also concentration dependent (Fig. 4C). Indeed, exposure of quiescent Swiss 3T3 cells to as low as 10 ng/ml of rPMT for 24h was sufficient to cause CTGF mRNA induction. Increasing rPMT concentration leads to a slight but reproducible decrease in the CTGF mRNA.
Unlike most genes that have been shown to be controlled by growth factors, rPMT-induced CTGF mRNA expression is not transient and lasted at least 24h. CTGF is coded by an early response gene and as such levels of CTGF mRNA are induced in a manner independent of de novo protein synthesis, when quiescent human skin fibroblast cells are exposed to TGFβ or serum [34]. Furthermore, treatment of quiescent cells with TGFβ or serum in the presence of protein synthesis inhibitor leads to long lasting up-regulation of CTGF mRNA typical of immediate early response genes suggesting that CTGF mRNA is somehow an unstable molecule and its half life is determined to be between 10 and 15 min [24].To investigate whether rPMT-induced CTGF mRNA expression is altered by de novo protein synthesis, experiments were carried out in the present of the protein synthesis inhibitor, cycloheximide. The results showed that the expression of CTGF mRNA is independent of de novo protein synthesis, and that there was no super-induction in the presence of cycloheximide (data not shown). In addition, cycloheximide alone at a concentration of 20 μg/ml increases significantly CTGF mRNA level in comparison to the control non-treated cells. Taken together, these results indicate that CTGF gene expression is regulated by rPMT at the transcriptional level. To determine the half-life of CTGF mRNA, quiescent Swiss 3T3 cells were treated with rPMT for 5h and subsequently treated with actinomycin D for different period of time. The levels of CTGF mRNA did not decrease significantly after transcriptional inhibition by actinomycin D for 4h suggesting that rPMT-induced CTGF mRNA is a relatively stable transcript (data not shown).
3.4 Release of CTGF Protein by rPMT-treated cells
To determine whether the effects of rPMT on CTGF mRNA levels were correlated with its protein levels, CTGF protein expression was assessed using Western blot analysis. Serum-starved Swiss 3T3 cells were incubated with concentrations of rPMT ranging from 10 to 50 ng/ml for 24h as shown in figure 5A. Samples of culture medium and cell lysates from control non-treated and rPMT treated cells were subjected to Western blot analysis using an anti-CTGF antibody. An immunoreactive protein of 36-38 kDa was observed in both cell lysate and conditioned medium of the cells exposed to rPMT. As low as 10 ng/ml of rPMT, was sufficient to induce a substantial accumulation of CTGF protein in whole cell lysate and culture medium of quiescent Swiss 3T3 cells. Above this concentration of toxin, we observed a slight decrease in the amount of CTGF protein expression. However, nearly undetectable amount of CTGF were observed in both whole cell lysates and conditioned medium when the cells were cultured in the absence of rPMT.
Figure 5. rPMT induces CTGF protein expression and secretion in quiescent 3T3 cells.
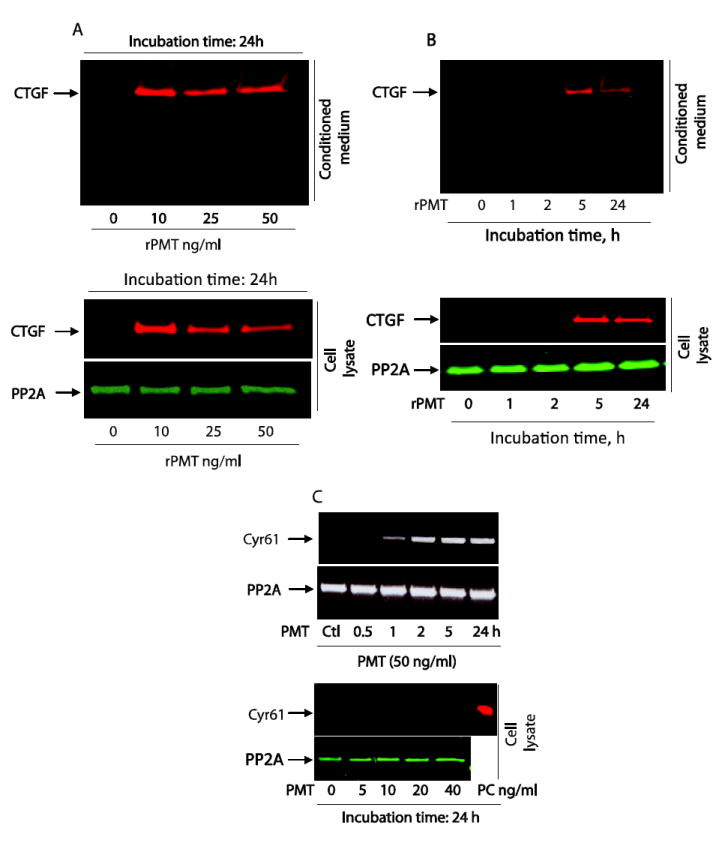
Quiescent 3T3 cells were treated with indicated concentrations of rPMT for 24 h (A) or with 50 ng/ml of rPMT and incubated for indicated h. (B). The cells were then lysed and equal amounts of proteins were separated on SDS polyacrylamid gel elepctrophoresis. Proteins in conditioned medium were also separated on SDS polyacrylamid gel elepctrophoresis. CTGF protein levels were monitored by Western blot in both whole cell extract and conditioned medium. An antibody that recognizes protein phosphatase 2A was used for loading control. Quiescent 3T3 cells were treated with indicated concentration of rPMT for different period of times. Total RNA was prepared from control and rPMT treated cells and analyzed by RT-PCR using Cyr61 primers. Cell extracts were prepared from control untreated and PMT treated cells as above. Equal amount of protein was separated on SDS PAGE electrophoresis. Cyr61 protein is detected in control untreated and PMT treated samples, along with a positive control sample (PC), by immunobloting using an antibody the specifically recognize Cyr61 protein (C).
To study the kinetics of CTGF expression, quiescent cells were treated with 50 ng/ml of rPMT for different periods of time up to 24h and CTGF protein levels were monitored by Western blot in both whole cell extract and conditioned medium. A huge accumulation of CTGF protein was observed in both whole cell lysates and conditioned medium by 5h after exposure of quiescent cells to rPMT as depicted in figure 5B. Increasing the exposure time of rPMT leads to a small decrease in CTGF levels in cell lysates and culture medium, but still much higher than that of the control non-treated cells. The half-life of rPMT-induced CTGF protein was determined by incubating quiescent Swiss 3T3 cells with rPMT for 5h followed by treatment with15 μg/ml of cycloheximide, for various time ranging from 30 min to 4h. After cycloheimide treatment for 4h, the CTGF protein levels decreased roughly by 30%, indicating a half-life of about 2h (data not shown). However, it should be pointed out that an earlier study estimated the half-life of CTGF to be 60 to 90 min [24].
Taken together, these results indicate that CTGF, a relatively stable protein, was strongly induced by rPMT treatment, and secreted into the medium where it may function as an autocrine/paracrine mediator in rPMT treated fibroblast cells. It is worth noting that kinetics of both rPMT-induced CTGF mRNA and protein expression is quite different. While CTGF mRNA accumulation in the cells was observed 2h after rPMT treatment (Fig. 4B and 5B), CTGF protein accumulation was actually observed in both cells and conditioned medium after 2h, and significantly accumulated to maximum levels 5h after exposure of the cells to rPMT, reflecting a delay in the translation of CTGF protein. Despite the fact that CTGF is known to be upregulated in fibrosis diseases independently of their etiology, to the best of our knowledge, there is no report linking PMT to fibrosis in vivo.
It is worth noting that rPMT also upregulates Cyr61 mRNA in time and concentration dependent manner in quiescent 3T3 cells (Table 1 and Figure 5C). Using Western blot analysis, however, we did not observe any increase in Cyr61 protein in rPMT treated cells in comparison to the control non-treated cells (Figure 5C). In fact, Cyr61 protein level was very low and was nearly undetectable in control cells and did not increase upon rPMT treatment, suggesting that rPMT treatment regulates the Cyr61 gene at the transcriptional. Thus, in this study, we decided to concentrate on the effects of CTGF induced by rPMT.
3.5 rPMT-induced CTGF protein synthesis is Gαq/11 dependent
On one hand, rPMT is known to exert at least some of its cellular effects by modulating the activity of Gαq/11 heterotrimeric G proteins [14, 26] through the deamidation of a specific glutamine residue in the α subunit of Gq and Gi of the heterotrimeric G proteins and leads to their persistent activation [6]. On the other hand, it has been recently shown in human endometrium that Gαq participate in the regulation of CTGF protein by prokineticin receptor, a Gαq-coupled receptor that mediates extracellular signal-regulated kinase phosphorylation [35, 36]. We next carried out experiments to study the role of Gαq/11 in rPMT-induced CTGF production. MEF WT and Gαq/11 deficient cells were treated with 50 ng/ml of rPMT for 24h and whole cell lysates and culture medium were subjected to Western blot analysis using an anti-CTGF antibody. As depicted in figure 6A, MEF WT cells treated with rPMT led to a significant increase in CTGF protein expression and release into conditioned medium in comparison to control non-treated cells. However, under the same conditions, rPMT treatment did not induce any noticeable increase in CTGF protein expression in Gαq/11 deficient cells. This result suggests that rPMT-induced activation of Gαq/11 is required for rPMT-mediated CTGF generation.
Figure 6. rPMT-induced CTGF protein is Gαq/11 dependent.
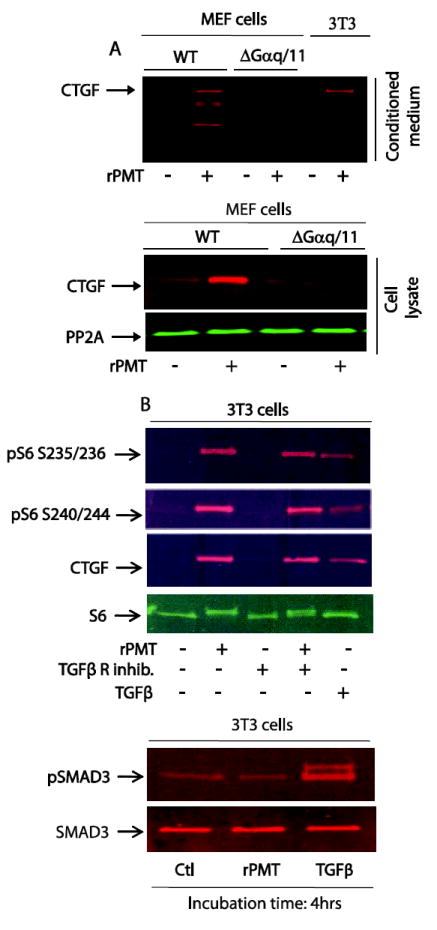
MEF wild type and MEF Gαq/11 deficient cells were grown in DMEM containing 10% of FBS till confluency. Confluent cells were switched to serum-free DMEM media for 48h and then treated with 100 ng/ml of rPMT for 24h. Control and rPMT treated cells were lysed in SDS PAGE loading buffer and equal amounts of proteins were separated on SDS polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. Proteins in conditioned medium were also separated and transferred as described. CTGF protein levels were monitored by Western blot in both whole cell extract and conditioned medium. An antibody that recognizes protein phosphatase 2A was used for loading control (A). (B) 3T3 cells were serum-starved as above and treated with 50 ng/ml of rPMT or 20 ng/ml of TGFβ for 4 hours in the presence or absence of TGFβ receptor inhibitor. Cells were lysed and proteins were separated by SDS PAGE. Immunoblots were developed with antibodies that recognized unphosphorylated S6 (S6), phosphorylated S6 (pS6), CTGF, SMAD3 and phoaphorylated SMAD3 (pSMAD3).
In addition to the full length CTGF protein in MEF WT cells, we observed CTGF immunoreactive fast migrating bands that may correspond to byproducts of CTGF in the conditioned medium. In fact CTGF is known for its multimodular structure [37], which is prone to proteolysis by several proteases. The linkers that separate the modules may be the target for proteases as documented by the cleavage of CTGF by ADAM28 [38] and MMP2 metalloproteinase [39] at the junction between the VWC and TSP1 modules. Thus the low molecular weight bands observed in MEF cells treated with rPMT may correspond to truncated fragments or individual modules of the full length CTGF protein [40] that may display biological properties distinct from that of the full length protein or may be evidence of regulation of the biological activity of CTGF protein [41]. These low molecular weight bands, however, are rarely seen in Swiss 3T3 cells treated with rPMT (see Fig. 6A, upper panel). This may reflect the difference between MEF and Swiss 3T3 cells in term of proteases expression, basal or induced by rPMT, that are able to process CTGF.
3.6 rPMT did not activate the TGFβ signalling pathway
It was originally thought that CTGF is exclusively regulated by TGFβ[42-44]. Figure 6B (upper panel) shows that, like rPMT treatment, exposure of serum-starved 3T3 cells to 20 ng/ml of TGFβ for 4 hours leads to a substantial increase in the levels of S6 phosphorylation in contrast to the control untreated cells. Concomitantly, TGFβ treatment increased CTGF protein expression in serum-starved 3T3 cells as the case observed with rPMT treatment. However, CTGF level in untreated control cells was undetectable. This result raises the issue whether rPMT somehow activates TGFβ signalling, which in turn leads to CTGF expression. To explore this possibility, we treated serum-starved cells with rPMT or TGFβ for 4 hours and monitored the phosphorylation status of SMAD3, one of known downstream targets of TGFβ signalling pathway. As shown in Figure 6B (lower panel) TGFβ treatment caused a substantial increase in pSMAD3 levels in comparison to the untreated cells. However, in rPMT-treated cells, SMAD3 phosphorylation remained low and was comparable to the control untreated cells. This result was further confirmed using a TGFβ receptor inhibitor. Results depicted in Fig. 6B (upper panel) show that TGFβ receptor specific inhibitor (20 μg/ml) exerted no effect on the levels of CTGF generation or of S6 phosphorylation induced by rPMT. Together, these results indicate that rPMT does not activate TGFβ signalling pathway mediated by Smad phosphorylation in serum-starved 3T3 cells. In other words, rPMT-mediated CTGF protein expression and secretion is dependent on Gαq/11 but independent of TGFβ signalling.
3.7 mTOR inhibitor exerts no effect on CTGF expression induced by rPMT or TGFβ
Since rPMT induced mTOR activation and concomitantly upregulated CTGF expression and excretion into the conditioned medium, we investigated whether inhibition of mTOR signalling might affect the expression of CTGF protein. Figure 7A shows that rPMT treatment upregulates CTGF protein level in Swiss 3T3 cells relative to that observed with the control non-treated cells. However, rapamycin, at concentrations that abrogate rPMT-mediated mTOR activation, exerted no effect on the rPMT-induced CTGF protein expression. This result together with those shown in Fig. 6A, suggest that while rPMT-induced CTGF protein expression is Gαq/11 dependent, yet is independent of mTOR signalling pathway. Furthermore, these observations imply that rPMT-mediated CTGF expression may not be strictly linked to cell growth and proliferation induced by rPMT because mTOR inhibitor, rapamycin, exerts a partial inhibitory effect on cell growth and proliferation [15] without altering CTGF expression. It should be pointed out that rapamycin has been reported to induce CTGF expression mediated by TGFβ1 in renal mesangial cells [45], a phenomenon we did not observed with serum-starved Swiss 3T3 cells.
Figure 7. mTOR inhibitors exert no effect on rPMT- or TGFβ-mediated CTGF expression.
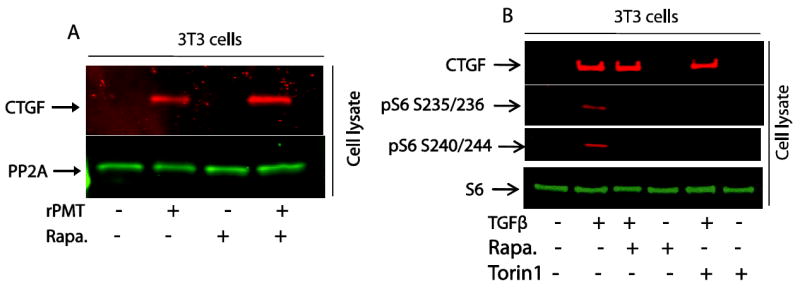
Confluent 3T3 were serum-starved over night then treated with 50 ng/ml of rPMT for 24 hours in the presence or absence or 50 nM of rapamycin (A) or treated with 20 ng/ml of TGFβ for 4 hours in the presence or absence of 50 nM rapamycin or 100 nM Torin1 (B). After separation, proteins were probed with antibodies that recognized CTGF, PP2A, rpS6 and phosphrylated rpS6.
Similarly, when serum-starved 3T3 cells were treated with 20 ng/ml of TGFβ for 4 hours, it induced CTGF expression, and this induction was independent of rapamycin (50 nM) and Torin1 (100 nM) as shown in Fig. 7B. Interestingly, this observation reveals a correlation between CTGF production and S6 phosphorylation in a manner sensitive to rapamycin and Torin1. In view of the fact that TGFβ induces the upregulation of CTGF, this observation may suggest the existence of a CTGF-mediated cross-talk between TGFβ and mTOR signalling pathways.
3.8 MEK/ERK pathway is involved in rPMT-induced CTGF expression in 3T3 cells
It has been reported that the TGFβ-mediated CTGF expression is dependent on protein kinase C, Ras/MEK/ERK and src kinase signalling pathways in human foreskin fibroblasts, primary osteoblasts, and human epithelial cells [46-49]. However, it is important to note that the role of MAPKs in the TGFβ-induced CTGF in fibroblasts may differ according to their cellular origin [50]. Inhibition of any two MAP kinases, ERK, p38, or JNK, completely block the TGFβ-mediated induction of CTGF in lung fibroblasts, while only JNK is involved in CTGF regulation in gingival fibroblasts [50]. To probe the role of ERK pathway in rPMT-induced CTGF mRNA and protein expression, we incubated serum-starved Swiss 3T3 cells with or without rPMT in the presence or absence of MEK inhibitor, U0126 or PD 098059, for 24 h. As shown in figure 8A, immunoblot analysis revealed that quiescent 3T3 cells stimulated with 50 ng/ml of rPMT for 24h induced a huge increase in the phosphorylation of ERK as well as the production of CTGF. However, rPMT-induced ERK phosphorylation was abolished and the level of CTGF formed was also greatly reduced when cells were treated with rPMT in the presence of PD98059 (20 μM) or U0126 (10 μM). In accord with these observations, Fig. 5A and 7A, show that rPMT alone induced a huge increase in intracellular CTGF levels as well as in CTGF secreted into the conditioned medium in comparison to control non treated cells. Interestingly, while U0126 and PD 098059 were capable of inhibiting the CTGF protein expression induced by rPMT, neither inhibitor had any effect on rPMT-induced CTGF mRNA expression (Fig. 8B). This suggests that ERK regulates CTGF expression at the translational level in rPMT treated cells. Nevertheless, using airway smooth muscle cells, inhibition of ERK or JNK but not p38 MAPK or PI3K, reportedly blocks the effect of TGFβ1 on CTGF mRNA and protein expression [51].
We have previously shown, using a receptor tyrosine kinase array, that PDGF receptor phosphorylation remains unchanged in both control and rPMT-treated cells. However, rPMT treatment induced a significant increase in the phosphorylation of both EGFR and Erb2 [15]. Despite the ability of rPMT to activate EGF receptor, Fig. 8C shows that when serum-starved 3T3 cells were treated with 50 ng/ml of rPMT for 24 h in the presence of 1.0, 5.0 and 10.0 μM of AG1476, a known EGFR specific inhibitor, we observed no decrease in the levels of CTGF expression. This result is in agreement with the previously published observation showing that while TGFβ stimulates CTGF expression, other growth factors including PDGF, EGF, and basic fibroblast growth factor have no effect on CTGF expression at least in human foreskin fibroblasts [34]. This difference is probably attributable to the fact that transduction pathways produced by activation of the TGFβ receptor are different from that of PDGF, FGF or EGF and to the TGFβ response element identified in the CTGF promoter [42].
Together, these results demonstrates that rPMT-stimulated CTGF expression and secretion into the conditioned medium of quiescent Swiss 3T3 cells is mediated by mechanism(s) involving ERK, but not EGFR or mTOR pathways. These are in agreement with the observations showing that rPMT induces ERK activation and CTGF production in MEF WT but not in Gαq/11 deficient cells. In addition they suggest that MEK inhibitors block rPMT-induced CTGF protein expression at the translational level and not at the transcriptional level.
3.9 CTGF expression in serum-starved cells leads to mTOR activation
The TGFβ signalling pathway controls numerous cellular responses such as proliferation, differentiation, migration and apoptosis [52]. Aberrant TGFβ has been implicated in tumor formation and progression, fibrotic diseases and atherosclerosis [53, 54]. The results in Figs. 6B and 7B reveal that TGFβ is capable of inducing not just CTGF upregulation, but also mTORC1 activation monitored by S6 phosphorylation. However, treatment with an mTOR specific inhibitor, rapamycin or Torin1 abolished the TGFβ-induced S6 phosphorylation while exerting no effect on CTGF protein expression. These observations are consistent with the notion that upregulation of CTGF expression is independent of mTORC1 activation mediated by either rPMT or TGFβ.
We investigated whether the secreted CTGF following rPMT or TGFβ treatment could activate mTOR and ERK signalling pathways in culture cells. To this end, we infected serum-starved 3T3 cells with either adenovirus control (Ad-Ctl) or adenovirus expressing human CTGF (Ad-CTGF) for 24h. The expression of CTGF was monitored in whole cell lysates using Western blot analysis. As shown in figure 9, CTGF was expressed in Ad-CTGF-transfected cells, but not in Ad-Ctl transfected cells. In addition, we observed a significant increase in S6 phosphorylation but no change in ERK phosphorylation in Ad-CTGF transfected cells relative to that found in control cells. This observation together with those shown in Fig. 6B and 7B provide the first demonstration of mTOR activation by CTGF, a downstream mediator of TGFβ-induced cellular signalling. However the mechanism by which CTGF mediates S6 phosphorylation remains to be elucidated.
Figure 9. CTGF overexpression induces S6 phosphorylation.
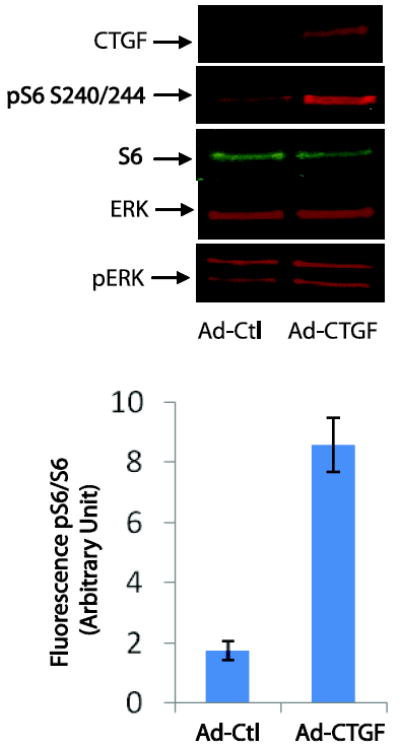
Swiss 3T3 cells were grown in DMEM containing 10% of FBS till confluency. Confluent cells were switched to serum-free DMEM media and immediately transfected with Adenovirus ctl (Ad-Ctl) or adenovirus-CTGF (Ad-CTGF) for 24h. Cells were then harvested and lysed in SDS PAGE loading buffer. The same amount of protein were separated on SDS PAGE and transferred to nitrocellulose membranes. Proteins were probed using antibodies that recognize CTGF, ERK, pERK, S6 and pS6. The bar graph represents the mean +/- SEM from three independent experiments.
Interestingly, we did not observed any morphological change in Ad-CTGF transfected as well as TGFβ treated cells despite the fact that in each case S6 phosphorylation was elevated. Recently, it has been reported that combinatorial signalling pathways determine whether fibroblasts respond to CTGF as a mitogen or differentiation-inducing factor [55]. Indeed, the presence or of other growth factors, such as epidermal growth factor (EGF) or insulin-like growth factor-2 (IGF-2), was essential to determine the biological response of the target cell [55]. Together, these findings indicate that upregulation of CTGF alone is insufficient to recapitulate all of the cellular effects induced by rPMT.
4. Concluding Remarks
Pasteurella multocida toxin, an intracellular acting bacterial protein, has been shown to exert its biological effects, in part, via the deamidation of a conserved glutamine residue in the α-subunit of heterotrimeric G proteins leading to their persistent activation. Its biological effects on culture cells were observed after a lag phase, indicating that PMT does not act through an extracellular receptor. In this study, we reveal for the first time that rPMT treated Swiss 3T3 cells generate and secrete into the conditioned medium an autocrine/paracrine substance(s) capable of inducing mTORC1 and MAPK in a manner independent on Gαq/11 in 30 min or less. Microarray and RT-PCR analysis of rPMT-treated serum-starved 3T3 cells showed that CTGF mRNA levels upregulated followed by two other members of the CCN family, Cry61 and Wisp2. However, Nov mRNA levels, another CCN family member was suppressed, in agreement with the reciprocal expression reported for these two CCN proteins [31]. Western blot analysis showed that CTGF protein was strongly induced by rPMT treatment and successfully secreted into the medium. CTGF is an ECM protein known to regulate a diverse array of cellular functions including cell proliferation, adhesion, migration and differentiation [17, 18]. It has also been implicated in more complex biological processes such as angiogenesis, chondrogenesis, osteogenesis, wound healing, fibrotis diseases, and tumorigenesis [20, 21].
Transcriptional activation of the CTGF gene following rPMT treatment is Gαq/11 and ERK dependent (Fig. 6A and Fig. 8). In a stark contrast with MEF WT, exposure of MEF Gαq/11 deficient cells to rPMT did not lead to the ERK activation or CTGF expression. In addition, MAPK inhibitors abrogate rPMT-induced CTGF expression and secretion in serum starved 3T3 cells (Fig. 8). However, rPMT-induced CTGF expression is independent of mTOR signaling pathway. Addition of rPMT to serum starved 3T3 cells in the presence of well established mTOR inhibitors have no effect on rPMT-induced CTGF protein expression. Furthermore, despite the fact that CTGF is known to be upregulated by TGFβ1 [43], several line of evidence suggest that PMT-induced CTGF protein expression is independent of TGFβ signaling. We found that contrary the TGFβ, rPMT did not induced the phosphorylation of SMAD3, a downstream target of TGFβ and PMT-induced CTGF expression was not affected by TGFβ receptor inhibitor (Fig. 6B).
CTGF overexpression in serum-starved 3T3 cells led to mTOR activation as judged by S6 phosphorylation, a downstream target of mTOR. However the mechanism by which CTGF activates the mTOR pathway remains to be elucidated. In view of the fact that PKC inhibitor totally blocked the rPMT-induced mTOR activation [15] and that PKC has been linked to CTGF-mediated signalling [56] suggests that CTGF may activate mTOR via a PKC-mediated pathway. Since CTGF expression led to mTOR but not ERK activation while conditioned medium taken from rPMT treated cells activates both mTOR and ERK suggest that there are other factors in the conditioned medium that activate other signaling pathways including mTOR.
Based on our study and those reported in the literatures, we propose a signalling scheme (Fig. 10) to depict a possible mechanism by which PMT mediates CTGF protein expression and mTORC1 activation. In this scheme, upon entry into the cell, PMT catalyzes the deamidation of the conserved Q209 in the Gαq/11 subunit of a heterotrimeric G protein. The active and GTP-bound Gαq/11 then binds to PLCβ to form an active complex which catalyzes the hydrolysis of phosphatidyl inositol diphosphate (PIP2) into two well known second messengers, DAG and IP3. Both DAG and IP3 can activate PKC leading to rpS6 phosphorylation through the activation of mTOR. Accordingly, we observed the PKC and mTOR inhibitors, Gö 6976 and rapamycin or Torin1, abrogate PMT-mediated rpS6 phosphorylation. It is worth noting that the sustained rpS6 phosphorylation induced by PMT is consistent with the notion that PMT inhibits the hydrolysis of Gαq/11(E209)- bound GTP and leads to its persistent activation. Furthermore, the dependence of PMT on Gαq/11 to activate mTOR and MEK signaling pathways is supported by the observation that PMT activates mTOR and MEK in MEF WT but not in MEF Gαq/11 deficient cells. Surprisingly, the condition medium from PMT-treated 3T3 cells is able to activate both mTOR and ERK in MEF Gαq/11 deficient cells. This observation led to the discovery that PMT upregulates CTGF expression in a manner dependent on Gαq/11 and ERK but not mTOR. Interestingly, CTGF overexpression induces rpS6 but not ERK phosphorylation. Together, these observations suggest that additional unidentified and diffusible factors, X and Y, may be responsible for enhancing the activity of ERK and mTOR.
Figure 10. A proposed signalling scheme depicting PMT mediated CTGF expression and mTORC1 activation.
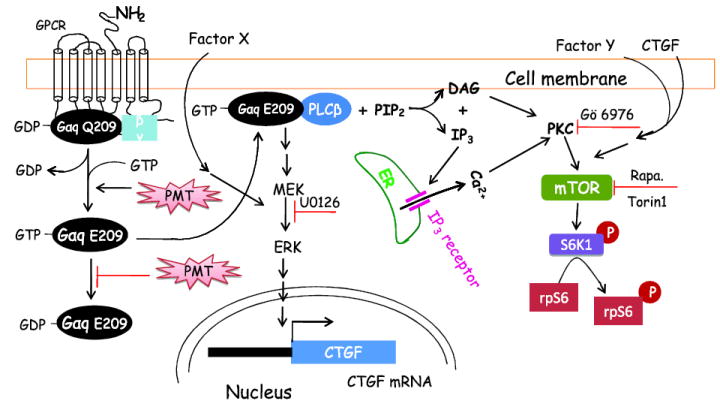
Here PMT is shown to enter the cell to activate the heterotrimeric G protein persistently by deamidating Gαq(Q-209) and inhibit the hydrolysis of Gαq(E-209)-bound GTP. The active GTP-bound Gαq(E-209) forms a complex with PLCβ and activates its phospholipase C activity to catalyze the hydrolysis of PIP2 to form DAG and IP3. IP3 can bind to IP3 receptor to trigger the release of Ca2+ from the ER. Both DAG and Ca2+ can activate PKC and lead to the activation of mTORC1, which can also activated by CTGF. The active Gαq(E-209)-PLCβ complex also leads to TFGβ- independent and MEK-mediated CTGF expression and secretion. Additional secreted unidentified factors, X and Y, may participate in activating the ERK and mTOR pathways.
Overall, we revealed for the first time that CTGF protein was expressed and successfully secreted into conditioned medium to serve as an autocrine/paracrine molecule in rPMT treated cell. rPMT-induced CTGF protein expression is dependent on Gαq/11 and MAPK but not mTOR, RTK or TGFβ, a well known inducer of CTGF. Importantly, CTGF expression using adenovirus led to mTOR activation. Despite the fact that CTGF is known to be upregulated in fibrosis diseases, to the best of our knowledge, there is no report linking PMT to fibrosis in vivo.
Highlights.
CTGF is secreted to serve as an autocrine/paracrine mediator in rPMT treated cells.
rPMT-induced CTGF expression is Gαq/11 dependent and TGFβ independent.
CTGF activates mTORC1 signalling.
Acknowledgments
We thank Dr. Shigeki Kamitani and Prof. Stefan Offermanns for providing us with the 3G3 monoclonal antibody that recognizes deamidated form of Gαq and MEF WT and Gαq/11 deficient cells, respectively.
Funding Sources---This research was supported by the Intramural Research Program of the NIH, NHLBI.(BAW is supported by NIH/NIAID grant #A1038396).
The abbreviations used are
- CM
conditioned medium
- CCN
a family of cysteine rich matricellular proteins including CTGF, Cyr61 and Nov
- GPCR
G-protein coupled receptor
- mTORC1
mammalian target of rapamycin complex 1
- PMT
Pasteurella multocida toxin
- S6K1
ribosomal S6 kinase
- DAG
diacylglycerol
- IP3
inositol trisphosphate
- RTK
receptor tyrosine kinase
- RSK
ribosomal S6 kinase
- EGFR
epidermal growth factor receptor
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PKC
protein kinase C
- rpS6
ribosomal protein S6
- PLCβ
phospholipase C β
- MAPK
mitogen-activated protein kinase
- ERK
extracellular-signal-regulated kinase
- 4E-BP1
eIF4E binding protein 1
- PI3K
phosphatidylinositol 3-kinase
- PMA
phorbol-12-myristate-13-acetate
- rpS6
ribosomal S6 protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kitadokoro K, Kamitani S, Miyazawa M, Hanajima-Ozawa M, Fukui A, Miyake M, Horiguchi Y. Proc Natl Acad Sci USA. 2007;104:5139–5144. doi: 10.1073/pnas.0608197104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kamitani S, Kitadokoro K, Miyazawa M, Toshima H, Fukui A, Abe H, Miyake M, Horiguchi Y. J Biol Chem. 2010;285:25467–25475. doi: 10.1074/jbc.M110.102285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aminova LR, Luo S, Bannai Y, Ho M, Wilson BA. Protein Sci. 2008;17:945–949. doi: 10.1110/ps.083445408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brothers MC, Ho M, Maharjan R, Clemons NC, Bannai Y, Waites MA, Faulkner MJ, Kuhlenschmidt TB, Kuhlenschmidt MS, Blanke SR, Rienstra CM, Wilson BA. FEBS J. 2011;278:4633–4648. doi: 10.1111/j.1742-4658.2011.08365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baldwin MR, Lakey JH, Lax AJ. Mol Microbiol. 2004;54:239–250. doi: 10.1111/j.1365-2958.2004.04264.x. [DOI] [PubMed] [Google Scholar]
- 6.Orth JH, Preuss I, Fester I, Schlosser A, Wilson BA, Aktories K. Proc Natl Acad Sci USA. 2009;106:7179–7184. doi: 10.1073/pnas.0900160106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamitani S, AO S, Toshima H, Tachibana T, Hashimoto M, Kitadokoro K, Fukui-Miyazaki A, Abe H, Horiguchi Y. FEBS J. 2011;278:2702–2712. doi: 10.1111/j.1742-4658.2011.08197.x. [DOI] [PubMed] [Google Scholar]
- 8.Staddon JM, Barker CJ, Murphy AC, Chanter N, Lax AJ, Michell RH, Rozengurt E. J Biol Chem. 1991;266:4840–4847. [PubMed] [Google Scholar]
- 9.Murphy AC, Rozengurt E. J Biol Chem. 1992;267:25296–25303. [PubMed] [Google Scholar]
- 10.Seo B, Choy EW, Maudsley S, Miller WE, Wilson BA, Luttrell LM. J Biol Chem. 2000;275:2239–2245. doi: 10.1074/jbc.275.3.2239. [DOI] [PubMed] [Google Scholar]
- 11.Zywietz A, Gohla A, Schmelz M, Schultz G, Offermanns S. J Biol Chem. 2001;276:3840–3845. doi: 10.1074/jbc.M007819200. [DOI] [PubMed] [Google Scholar]
- 12.Wilson BA, Aminova LR, Ponferrada VG, Ho M. Infect Immun. 2000;68:4531–4538. doi: 10.1128/iai.68.8.4531-4538.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas W, Pullinger GD, Lax AJ, Rozengurt E. Infect Immun. 2001;69:5931–5935. doi: 10.1128/IAI.69.9.5931-5935.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orth JH, Lang S, Taniguchi M, Aktories K. J Biol Chem. 2005;280:36701–36707. doi: 10.1074/jbc.M507203200. [DOI] [PubMed] [Google Scholar]
- 15.Oubrahim H, Wong A, Wilson BA, Chock PB. J Biol Chem. 2013;288:2805–2815. doi: 10.1074/jbc.M112.427351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradham DM, Igarashi A, Potter RL, Grotendorst GR. J Cell Biol. 1991;114:1285–1294. doi: 10.1083/jcb.114.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brigstock DR. J Endocrinol. 2003;178:169–175. doi: 10.1677/joe.0.1780169. [DOI] [PubMed] [Google Scholar]
- 18.de Winter P, Leoni P, Abraham D. Growth Factors. 2008;26:80–91. doi: 10.1080/08977190802025602. [DOI] [PubMed] [Google Scholar]
- 19.Chen CC, Lau LF. Int J Biochem Cell Biol. 2009;41:771–783. doi: 10.1016/j.biocel.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubota S, Takigawa M. Angiogenesis. 2007;10:1–11. doi: 10.1007/s10456-006-9058-5. [DOI] [PubMed] [Google Scholar]
- 21.Cicha I, Goppelt-Struebe M. Biofactors. 2009;35:200–208. doi: 10.1002/biof.30. [DOI] [PubMed] [Google Scholar]
- 22.Simmons DL, Levy DB, Yannoni Y, Erikson RL. Proc Natl Acad Sci USA. 1989;86:1178–1182. doi: 10.1073/pnas.86.4.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Brien TP, Yang GP, Sanders L, Lau LF. Mol Cell Biol. 1990;10:3569–3577. doi: 10.1128/mcb.10.7.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryseck RP, Macdonald-Bravo H, Mattei MG, Bravo R. Cell Growth Differ. 1991;2:225–233. [PubMed] [Google Scholar]
- 25.Joliot V, Martinerie C, Dambrine G, Plassiart G, Brisac M, Crochet J, Perbal B. Mol Cell Biol. 1992;12:10–21. doi: 10.1128/mcb.12.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson BA, Zhu X, Ho M, Lu L. J Biol Chem. 1997;272:1268–1275. doi: 10.1074/jbc.272.2.1268. [DOI] [PubMed] [Google Scholar]
- 27.Orth JH, Lang S, Aktories K. J Biol Chem. 2004;279:34150–34155. doi: 10.1074/jbc.M405353200. [DOI] [PubMed] [Google Scholar]
- 28.Staddon JM, Chanter N, Lax AJ, Higgins TE, Rozengurt E. J Biol Chem. 1990;265:11841–11848. [PubMed] [Google Scholar]
- 29.Sabri A, Wilson BA, Steinberg SF. Circ Res. 2002;90:850–857. doi: 10.1161/01.RES.0000016165.23795.1F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waldron RT, Innamorati G, Torres-Marquez ME, Sinnett-Smith J, Rozengurt E. Cell Signal. 2012;24:914–921. doi: 10.1016/j.cellsig.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riser BL, Najmabadi F, Perbal B, Peterson DR, Rambow JA, Riser ML, Sukowski E, Yeger H, Riser SC. Am J Pathol. 2009;174:1725–1734. doi: 10.2353/ajpath.2009.080241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bohr W, Kupper M, Hoffmann K, Weiskirchen R. PLoS One. 2010;5:e16000. doi: 10.1371/journal.pone.0016000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukudai Y, Kubota S, Eguchi T, Sumiyoshi K, Janune D, Kondo S, Shintani S, Takigawa M. J Cell Biochem. 2010;111:1607–1618. doi: 10.1002/jcb.22894. [DOI] [PubMed] [Google Scholar]
- 34.Igarashi A, Okochi H, Bradham DM, Grotendorst GR. Mol Biol Cell. 1993;4:637–645. doi: 10.1091/mbc.4.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Negri L, Lattanzi R, Giannini E, Colucci MA, Mignogna G, Barra D, Grohovaz F, Codazzi F, Kaiser A, Kreil G, Melchiorri P. Br J Pharmacol. 2005;146:625–632. doi: 10.1038/sj.bjp.0706376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waddell JM, Evans J, Jabbour HN, Denison FC. Hum Reprod. 2011;26:67–75. doi: 10.1093/humrep/deq294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holbourn KP, Acharya KR, Perbal B. Trends Biochem Sci. 2008;33:461–473. doi: 10.1016/j.tibs.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mochizuki S, Tanaka R, Shimoda M, Onuma J, Fujii Y, Jinno H, Okada Y. Biochem Biophys Res Commun. 2010;402:651–657. doi: 10.1016/j.bbrc.2010.10.077. [DOI] [PubMed] [Google Scholar]
- 39.Dean RA, Butler GS, Hamma-Kourbali Y, Delbe J, Brigstock DR, Courty J, Overall CM. Mol Cell Biol. 2007;27:8454–8465. doi: 10.1128/MCB.00821-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brigstock DR. Endocr Rev. 1999;20:189–206. doi: 10.1210/edrv.20.2.0360. [DOI] [PubMed] [Google Scholar]
- 41.Perbal B. Lancet. 2004;363:62–64. doi: 10.1016/S0140-6736(03)15172-0. [DOI] [PubMed] [Google Scholar]
- 42.Grotendorst GR, Okochi H, Hayashi N. Cell Growth Differ. 1996;7:469–480. [PubMed] [Google Scholar]
- 43.Grotendorst GR. Cytokine Growth Factor Rev. 1997;8:171–179. doi: 10.1016/s1359-6101(97)00010-5. [DOI] [PubMed] [Google Scholar]
- 44.Chen Y, Blom IE, Sa S, Goldschmeding R, Abraham DJ, Leask A. Kidney Int. 2002;62:1149–1159. doi: 10.1111/j.1523-1755.2002.kid567.x. [DOI] [PubMed] [Google Scholar]
- 45.Osman B, Doller A, Akool el S, Holdener M, Hintermann E, Pfeilschifter J, Eberhardt W. Cell Signal. 2009;21:1806–1817. doi: 10.1016/j.cellsig.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 46.Secker GA, Shortt AJ, Sampson E, Schwarz QP, Schultz GS, Daniels JT. Exp Cell Res. 2008;314:131–142. doi: 10.1016/j.yexcr.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 47.Leask A, Holmes A, Black CM, Abraham DJ. J Biol Chem. 2003;278:13008–13015. doi: 10.1074/jbc.M210366200. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X, Arnott JA, Rehman S, Delong WG, Jr, Sanjay A, Safadi FF, Popoff SN. J Cell Physiol. 2010;224:691–701. doi: 10.1002/jcp.22173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arnott JA, Zhang X, Sanjay A, Owen TA, Smock SL, Rehman S, DeLong WG, Safadi FF, Popoff SN. Bone. 2008;42:871–885. doi: 10.1016/j.bone.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Black SA, Jr, Palamakumbura AH, Stan M, Trackman PC. J Biol Chem. 2007;282:15416–15429. doi: 10.1074/jbc.M610432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie S, Sukkar MB, Issa R, Oltmanns U, Nicholson AG, Chung KF. Am J Physiol Lung Cell Mol Physiol. 2005;288:L68–76. doi: 10.1152/ajplung.00156.2004. [DOI] [PubMed] [Google Scholar]
- 52.Derynck R, Zhang YE. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 53.Massague J. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobaczewski M, Chen W, Frangogiannis NG. J Mol Cell Cardiol. 2011;51:600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grotendorst GR, Rahmanie H, Duncan MR. FASEB J. 2004;18:469–479. doi: 10.1096/fj.03-0699com. [DOI] [PubMed] [Google Scholar]
- 56.Arnott JA, Lambi AG, Mundy C, Hendesi H, Pixley RA, Owen TA, Safadi FF, Popoff SN. Crit Rev Eukaryot Gene Expr. 2011;21:43–69. doi: 10.1615/critreveukargeneexpr.v21.i1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]