

Abstract
Photochemistry has the potential to significantly impact multiple aspects of chemical synthesis, in part because photoinduced reactions can be used to construct molecular architectures that would otherwise be difficult to produce. Nevertheless, organic chemists have been slow to embrace photochemical synthesis because of technical complications associated with the use of ultraviolet light. Our laboratory has been part of an effort to design synthetically useful reactions that utilize visible light. This strategy enables the synthesis of a diverse range of organic structures by generation of a variety of reactive intermediates under exceptionally mild conditions. This Perspective article describes the reasoning that led to the conception of our first experiments in this area, the features of our reaction design that have been most powerful in the discovery of new processes, and a few of the possible future areas in which visible light photocatalysis might have a large impact.
Keywords: Photochemistry, Cycloadditions, Radical ions, Photocatalysis, Natural Products
1. Introduction
Photochemical cycloaddition reactions were first discovered in the early 20th century1 and have been recognized as useful reactions in synthetic organic chemistry since at least the 1960s. The synthetic community’s interest in these transformations has been motivated by a number of factors. Several recent reviews of synthetic organic photochemistry have focused on the fact that light is a traceless, non-polluting reagent and have thus argued that many photochemical reactions can potentially be considered “green” chemical reactions, compared to their thermal counterparts.2 This is a historically important consideration that has been a central motivation for photochemical research for over a century.3
An equally valid — and arguably more fundamental — rationale for the development of organic photochemical reactions, however, is their distinctive synthetic utility. The structures produced by photochemical reactions can be quite different from those resulting from analogous thermal processes. The input of light energy that initiates a photochemical reaction can produce high-energy reactive intermediates, which in turn are uniquely suited for the production of strained and unusual molecular architectures that cannot be accessed using standard thermal reaction pathways. For example, many of the most straightforward synthetic routes to cyclobutanes, oxetanes, and other four-membered ring systems are photochemically initiated cycloaddition reactions. In addition, photochemically generated intermediates often react with stereochemical and regiochemical outcomes that differ from their ground-state counterparts. Thus, photochemical methods are an important complement to the conventional thermal reactions because they enable the synthesis of molecules with structures that would be inaccessible using non-photochemical synthetic methods alone.
There have been a number of elegant and now classic total syntheses of natural products that highlight the synthetic value of photocycloaddition reactions (Figure 1). These include, perhaps most obviously, the syntheses of cyclobutane-containing natural products such as grandisol (1)4 and caryophyllene (2).5 However, the utility of photocycloaddition reactions has also been demonstrated in syntheses of complex natural products such as isocomene (3)6 and ingenol (4)7 where strain-releasing reactions of photochemically assembled cyclobutanes are key to the construction of complex polycyclic carbon skeletons. The use of photochemical reactions in organic synthesis has been highlighted in several excellent recent reviews.8
Figure 1.

Representative structures of natural products prepared by photochemical synthesis.
Why, then, after a century of sustained interest, has the development of photochemical cycloaddition reactions lagged so far behind corresponding advances in thermal cycloaddition chemistry? Given the synthetic community’s increasing interest in “green” chemical strategies, and given the unique ability of photochemical reactions to assemble strained and novel molecular scaffolds, it seems that photochemistry could be serving a central role in transforming modern synthetic chemistry. Nevertheless, photochemical reactions continue not to be considered standard techniques in the repertoire of most synthetic organic chemists.
Many reasons have been cited for the reluctance of organic chemists to use photochemical reactions in synthesis. Among the most significant of these are the various technical complications associated with the use of traditional UV light sources.9 Conventional photochemical reactors are energy intensive, use lamps with limited lifetimes, and require considerable efforts to dissipate the heat of the light source. In addition, the energy of a UV photon is of the same order as the energy of a carbon–carbon sigma bond. One of the most problematic complications of traditional photochemical synthesis is the occurrence of undesired photoinduced radical decomposition processes, which can negatively impact the overall yield of a photochemical transformation and also result in the deposition of optically opaque polymeric material on the reactor walls. However, many of these technical disadvantages have been mitigated by modern innovations in reactor design, particularly the use of microflow reactors10 and energy-efficient LED light sources.11 Moreover, the technical obstacles that have historically been associated with UV photolysis become prohibitive only on large, industrially relevant scales; thus, these concerns do not fully explain why academic labs have also been slow to adopt photochemical methods.
A second possible explanation is the common assumption is that the structures accessible using photochemical synthesis are somewhat esoteric and unlikely to possess interesting biological activity. The evidence, however, contradicts this notion. Over 1600 cyclobutane-containing natural products have been reported to date;12 these have been isolated from organisms ranging from archaea to invertebrate animals and from both marine and terrestrial environments. The bioactivity profiles of these compounds include potent antibiotic, cytotoxic, anti-inflammatory, pheromonal, antiproliferative, and antineurodegenerative activity. Thus, this is an area of chemical diversity space that is likely to be rich in potential drug candidates but that has remained relatively unexplored by medicinal chemists, largely because the photochemical methods that are arguably the most efficient routes to these structures are not well developed and, consequently, are not widely utilized.
A final, though admittedly superficial, obstacle for academic researchers may simply be the requirement for specialized photochemical equipment, which effectively creates an operational barrier to the use of these techniques by non-specialists. Photoreactors are not standard instrumentation in most synthetic labs, and they can be costly to purchase. The quartz glassware required for short-wavelength UV photoreactions are also much more expensive than normal borosilicate glass vessels. As a consequence, new photochemical methods have been developed in a small number of groups with a specialized expertise in the area. For labs that do not generally conduct photochemical experiments, the acquisition of the necessary supplies coupled with a lack of familiarity with the general reactivity of photochemically generated intermediates may constitute a significant impediment to the use of these techniques.
When our laboratory began to study photocycloaddition reactions, our primary goal was to develop a strategy to utilize visible light in photochemical applications that would facilitate the widespread adoption of photochemistry by synthetic chemists. We felt this would be enabling for several reasons. Household light bulbs are cheap and easy to obtain, and every synthetic lab almost certainly has artificial lighting sources in abundance. These commercial visible light sources are also neither as bright nor as harmful as the mercury lamps used in traditional UV photoreactors, so the use of a protective housing would not be required for their use. The clear borosilicate reaction flasks that are ubiquitous in synthetic research laboratories are transparent to visible light, which would obviate the need for specialized quartz glassware. Finally, irradiation with long-wavelength visible light sources is less likely to suffer from issues associated with radical decomposition and poor heat dissipation.
Our approach was inspired by the remarkable developments made by researchers investigating the photochemical properties of transition metal complexes. In particular, Ru(bpy)32+ (5) and related transition metal polypyridyl complexes exhibit broad, strong absorbances in the visible range, their photoexcited states are redox-active, and they possess significantly longer excited state lifetimes and chemical stabilities than most common organic photosensitizers (Figure 2).13 Thus, they are ideally suited for initiating photoredox processes using visible light. The most important applications involving Ru(bpy)32+ and related transition metal complexes have exploited the visible light photoredox properties of these chromophores to initiate a variety of one-electron transfer processes. In the realm of solar energy conversion, photoactive transition metal complexes have been integral components of systems for reductive reactions such as H+ to H2 and CO2 to formate, or for oxidation of water to O2. In biological systems, these light-activated transition metal photocatalysts have been extensively utilized to study the dynamics of electron transfer through biomolecules such as proteins and DNA. However, prior to 2008, the use of Ru(bpy)32+ photochemistry in organic synthesis had received limited attention.14
Figure 2.
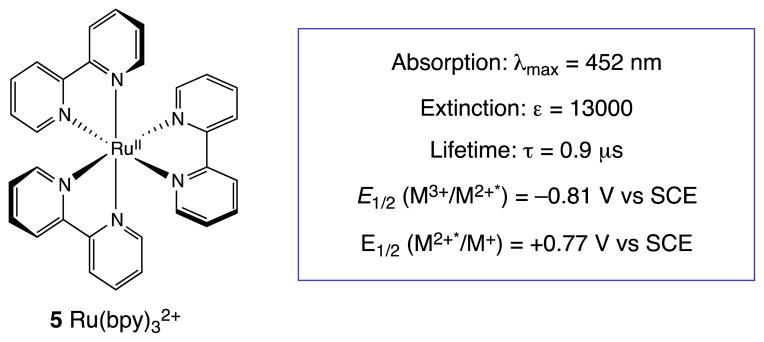
Photochemical properties of Ru(bpy)32+ in MeCN.
We hypothesized that the well studied photoredox behavior of Ru(bpy)32+ and its derivatives could also be exploited to initiate synthetically useful reactions of photogenerated organic radical ions. These charged, open-shell reactive intermediates have commonly been generated via photoinduced electron transfer (PET) processes initiated by organic photosensitizers.15 The study of their reactivity has resulted in a vast array of mechanistically intriguing, synthetically powerful, complexity-building transformations that are not accessible to their closed-shell counterparts.16 The most frequently utilized photosensitizers, however, are transparent to visible light, and photoinduced radical ion reactions have thus typically required UV irradiation. As a consequence, the chemistry of photogenerated radical ions has largely remained the province of specialists, and their synthetic potential has not yet been fully exploited by the broader synthetic community.
The past several years have witnessed a rapid growth in interest in transition metal mediated photochemical reactions from the labs of MacMillan17 and Stephenson,18 among many others,19,20 in addition to our own. This Perspective summarizes the logic leading to our first experiments in this area, outlines the features that make the resulting system adaptable to a wide variety of photocatalytic processes, and describes the outlook for what we believe is a rapidly emerging area with the potential to impact numerous aspects of the chemical synthesis enterprise.
2. Photocatalysis of Redox-Neutral Cycloadditions
Our starting point for the development of photocatalytic cycloaddition reactions was the intramolecular [2+2] cycloaddition of bis(enone) 6 (Scheme 1). Krische had speculated that cobalt-catalyzed [2+2] cycloaddition of 6 involved a radical anion intermediate (9) resulting from one-electron reduction of the easily reducible aryl enone.21 To corroborate this finding, Krische also showed that the same cycloaddition could be promoted by cathodic reduction22 or by reaction with a substoichiometric one-electron reductant,23 although the yields of 7 were modest due to the formation of a number of side products. We became intrigued by the observation that [2+2] cycloaddition reactions could be catalyzed by one-electron reduction. If the necessary reductant could be generated by photoexcitation of Ru(bpy)32+, this would constitute a general strategy to couple visible light irradiation to formal [2+2] cycloaddition reactions.
Scheme 1.

Mechanism of intramolecular radical anion [2+2] cycloaddition reaction.
Our initial publication in this area reported that bis(enone) 6 undergoes efficient [2+2] cycloaddition upon irradiation with visible light in the presence of Ru(bpy)32+, LiBF4, and i-Pr2NEt.24 The mechanism we have proposed for this cycloaddition is summarized in Scheme 1. Photoexcitation of the Ru(bpy)32+ chromophore with visible light generates a long-lived, redox-active excited state. Reductive quenching of Ru*(bpy)32+ with i-Pr2NEt results in the formation of Ru(bpy)3+, which we propose is the catalytically relevant reductant in this process (Ered = −1.2 V vs SCE). We measured the reduction potential of enone 6 to be −1.4 V vs SCE, which is outside of a reasonable range for reduction by Ru(bpy)3+. However, coordination of the enone to the Lewis acidic lithium cation activates the enone towards one-electron reduction25 and stabilizes the resulting radical anion, which can undergo the same [2+2] cycloaddition proposed by Krische and Bauld. Finally, the immediate product of the cycloaddition is ketyl radical 11, and formation of the neutral cycloadduct 7 requires loss of an electron, either to a second equivalent of 8 in a chain-propagation step, or to the photogenerated amine radical cation in a chain-termination step.
Importantly, we have found that a number of visible light sources are suitable for this and other photocatalytic reactions developed in our laboratory. Although we generally conduct these [2+2] cycloadditions using household 20 W compact fluorescent lightbulbs, we have also shown that the reaction proceeds smoothly in ambient sunlight. From the perspective of reproducibility and practicality, of course, sunlight is not an ideal reagent for controlled laboratory investigations, as reactions will proceed at different rates depending on the time of day and on current meteorological conditions. Nevertheless, we hope the demonstration that photocycloaddition reactions can be performed without specialized photochemical instrumentation makes these strategies more tractable for a broader synthetic audience than conventional photochemical reactions with ultraviolet light.
One obvious question about this reaction arises from the striking difference between the results of the radical anion cycloaddition under photocatalytic conditions and under electrochemical conditions. While we were able to obtain high yields and excellent diastereoselectivities by using the photocatalytic protocol, Krische and Bauld had reported modest yields and poor diastereoselectivity in reactions of the same substrates initiated by cathodic reduction. This difference is even more striking considering the fact that both experiments were conducted in acetonitrile in the presence of a Lewis acidic lithium additive. As the structure of the putative radical anion intermediate would be expected to be very similar in both cases, we speculated that the difference in the results of these experiments might arise from a secondary process following the cycloaddition event. Indeed, extended exposure of authentic, diastereomerically pure cycloadduct 7 to the conditions of photocatalysis resulted in a slow erosion of the diastereomeric purity of the cyclobutane (Scheme 2). This epimerization was not observed in the presence of i-Pr2NEt and LiBF4 alone, and thus, we concluded that the cis cycloadduct is the kinetic product of cycloaddition, and that the trans isomer arises from re-reduction of the aryl ketone to a ketyl radical, which can undergo fragmentation and subsequent re-cyclization to approach the thermodynamic ratio of diastereomers. In addition, in this experiment, we also observed the formation of various by-products including 14 and 15, which were also reported by Krische and Bauld, that account for the low yield of the electrochemical cyclization protocol.
Scheme 2.

Slow photocatalytic erosion of the diastereomeric purity of [2+2] cycloadducts.
We speculate that the superior yields and selectivity of the photocatalytic radical anion cycloaddition can be attributed to the in situ generation of an amine radical cation, which is a strong oxidant that can shorten the lifetime of the reactive cyclobutane ketyl radical by producing both the neutral cycloadduct and regenerating i-Pr2NEt, both in closed-shell form. The formation of the amine radical cation is a necessary consequence of the mechanism proposed in Scheme 1; for every equivalent of Ru(bpy)3+ generated that can initiate a radical anion chain, there is formed an equivalent of i-Pr2NEt•+ that can terminate it. In other words, the photocatalytic protocol is particularly suited for a redox-neutral processes such as cycloaddition reactions because the net redox balance of the reagents is also neutral, unlike the conditions at the surface of a cathode, where the local environment is net reducing and the concentration of potential oxidizers is quite low. Empirically, we have found that redox-neutral cycloaddition reactions seem particularly well-suited to the net redox-neutral photocatalytic conditions that our lab has been investigating; we have described a variety of [2+2], [3+2], and [4+2] radical ion cycloadditions that are high-yielding under photocatalytic conditions. These are described in the following section.
3. Modular Multicomponent Photocatalytic Systems
An analysis of the mechanistic hypothesis depicted in Scheme 1 reveals that the photocatalytic system is composed of three important components, each of which serves a different role in the overall transformation:
Ru(bpy)32+ is a sensitizer for photoinduced electron transfer; it absorbs the energy from an incident photon and converts it to electrochemical potential.
i-Pr2NEt is the reductive quencher that reacts with the excited photocatalyst to provide the ultimate source of the reducing equivalent required to generate the key radical anion intermediate.
LiBF4 serves as a Lewis acid that activates the enone towards reduction and stabilizes the resulting radical anion.
In our investigations of radical ion chemistry, we have found that one of the most attractive features of this reaction system is the ability to independently tune each of these three reaction components to optimize conditions for a particular transformation. Some of these changes are relatively modest perturbations that do not alter the overall properties of the photocatalytic reaction, while others lead to profound changes in mechanism.
Scheme 3 depicts two examples of the former category. First, we have reported an intramolecular [4+2] hetero-Diels–cycloaddition between two enone moieties (eq 1).26 An empirical screen of Lewis acid additives revealed that the highest-yielding reactions were obtained using MgClO4, which appears to balance the rate of productive cycloaddition with the rate of a non-photochemical Lewis-acid catalyzed product decomposition pathway. We also reported a formal [3+2] cycloaddition between aryl cyclopropyl ketones and enones (eq 2).27 In this case, both the identity of the La(OTf)3 Lewis acid and TMEDA, which presumably serves as both a reductive quencher and a ligand for La3+, were found to be critical for optimal reactivity. The development of both processes was facilitated by the ability to alter these reaction conditions without impacting the photophysical properties of the Ru(bpy)32+ chromophore.
Scheme 3.

Representative photocatalytic radical anion cycloadditions.
On the other hand, we observed a more profound change in the course of the reaction when the Lewis acid component of the catalyst system was replaced with a Brønsted acid (Scheme 4). Rather than generating [2+2] cycloadducts, reactions conducted in the presence of protic acids instead of LiBF4 afforded reductive coupling products;28 no cyclobutane side products were observed in the reaction mixtures. We attribute the divergence in reactivity in these two systems to the different reactive intermediates involved in each case. One-electron reduction of a Lewis acid-activated enone affords a radical anionic intermediate; the analogous reduction of a protonated oxocarbenium ion produces a neutral radical, which exhibits reactivity that is fundamentally different from that of radical anions. Thus the [2+2] cycloaddition and the reductive coupling differ in several important ways. First, the redox balance of the two reactions differ; cycloadditions are redox-neutral reactions, but the reductive coupling is a net two-electron reductive process. Second, the stereochemical outcome in each process is quite distinct; the β,β bond in the reductive coupling product is formed with high trans selectivity, while the analogous bond at the ring junction of [2+2] cycloadducts is necessarily cis. Finally, the scope of each reaction is also different. While neither aliphatic enones nor styrenes participate in the radical anion [2+2] cycloaddition, the two undergo facile reductive coupling under photocatalytic conditions in the presence of a Brønsted acid. Thus a reasonably simple perturbation of the acid additive can lead to a dramatic change in the reactivity observed.
Scheme 4.

Photocatalytic reductive coupling of enones.
The tertiary amine quencher can also be varied, and we have found that the identity and loading of the amine can affect the efficiency of production of the Ru(I) complex as well as the activity of the Lewis acid co-catalyst, and this variable can be tuned for optimal rates and yields. However, variation of the amine structure does not affect the reduction potential of Ru(bpy)3+, which is the species that delivers the reducing electron to the organic substrate. A more interesting question, therefore, is whether variation of the quencher could produce a different ruthenium species with distinct electrochemical properties.
As a demonstration of this possibility, we investigated the radical cation mediated [2+2] cycloaddition summarized in Scheme 5. The [2+2] cycloaddition of a variety of electron-rich olefins is known to be initiated by one-electron oxidation, and Bauld has studied the aminium radical promoted [2+2] cycloaddition of styrenes such as 22 in great detail.29 Many electron-deficient species have been shown to react efficiently with Ru*(bpy)32+ to afford Ru(bpy)33+, which is a strong oxidant (Eox = +1.3 V vs SCE), which should be thermodynamically capable of oxidizing the electron-rich styryl moiety of 22 and initiating the [2+2] cycloaddition. We screened a variety of compounds known to be efficient oxidative quenchers of Ru*(bpy)32+, and upon irradiation of 22 in the presence of Ru(bpy)32+ and methyl viologen (25) as an oxidative quencher, the desired [2+2] cycloadduct 24 is obtained in good yield with excellent diastereoselectivity.30 Thus, both oxidative and reductive quenching of the photoexcited state can be incorporated into the design of photocatalytic reactions.
Scheme 5.

Photocatalytic radical cation [2+2] cycloadditions of electron-rich alkenes.
Finally, many derivatives of Ru(bpy)32+ with known photophysical and electrochemical properties are known.31 The syntheses of these complexes are straightforward, and the effects of ligand modifications on the photoredox properties of the catalyst are well understood. The ability to rationally tune catalyst structure in this fashion has been essential in the success of a variety of processes developed in our lab. A recent example is highlighted in Scheme 6. We had been studying the [4+2] cycloaddition of photogenerated styrene radical cations (eq 3) and found that some attempted cycloadditions failed despite the fact that the oxidation potential of the starting styrene lay well within the range accessible by Ru(bpy)33+, a result that we attributed to competitive deactivation of the radical cation by back electron transfer from reduced methyl viologen. For successful cycloaddition of these challenging substrates, we found that the use of a more strongly oxidizing photocatalyst (Ru(bpz)32+) whose excited state could directly oxidize the styrene enabled efficient cycloaddition upon irradiation under ambient air.32 The identity of the photocatalyst is critical; reactions using Ru(bpy)32+ under otherwise identical conditions failed to produce any cycloadduct.
Scheme 6.

Representative photocatalytic radical cation cycloadditions.
The ability to tune the oxidation potential of the photocatalyst also proved to be essential to the development of intermolecular [2+2] radical cation cycloadditions (eq 4).33 Our preliminary investigations involved the application of the Ru(bpz)2+-catalyzed conditions we had developed for the radical cation Diels–Alder reaction to the cycloaddition of 26 and 29. Surprisingly, these early experiments consistently failed to provide high yields of the expected cyclobutane; the reactions inevitably stalled at partial conversion even when the irradiation time was increased, the catalyst loading was varied, or catalyst was introduced in multiple batches.
The key insight arose from control experiments in which we investigated the stability of the cycloadduct to the reaction conditions. When 30 was isolated and irradiated in the presence of Ru(bpz)32+, we observed that the monomeric styrene 26 slowly begins to appear in the reaction mixture. We wondered if the appearance of the monomer was due to over-oxidation of subsequent cycloreversion of 30. Consistent with this interpretation, we measured the peak oxidation potential of the cycloadduct to be +1.27 V vs SCE. Thus, the cyclobutane product could indeed be oxidized by photoexcited Ru*(bpz)32+, whose oxidation potential has been estimated to be +1.45 V vs SCE.34
We speculated, therefore, that high yields could be obtained by using a photocatalyst whose photoexcited state was oxidizing enough to generate the radical cation of the methoxystyrene (+1.1 V) but not powerful enough to trigger the oxidative fragmentation of the cycloadduct. We selected the known tris(bipyrimidine) complex, Ru(bpm)32+, whose photoexcited oxidation potential (+1.2 V) is intermediate between that of the substrate and the cycloadduct. Upon irradiation of a mixture of 26 and 29 in the presence of this catalyst, the crossed cyclobutane product 30 can be isolated in 79% yield.
The ability to tune multiple parameters of the photocatalytic system, and in particular the electrochemical potential of the photocatalyst, has been an important factor in our ability apply of this strategy beyond the [2+2] enone cycloadditions we originally reported. Because of the interest in the use of transition metal polypyridyl complexes as chromophores in solar energy conversion, hundreds of analogues of Ru(bpy)32+ with known electrochemical and photophysical properties have been prepared and fully characterized in the literature. Importantly, this flexibility enables us to perturb a variety of reaction conditions without significantly impacting the attractive photophysical properties of the photocatalyst itself.
4. Radical Ion Cycloadditions in Natural Product Synthesis
Our initial forays into natural product synthesis using photocatalytic radical ion cycloaddition reactions have focused on relatively small, simple targets to showcase methods developed in our group, but our few examples have been instructive. In the context of our studies on radical ion Diels–Alder cycloadditions, we became intrigued by the structure of heitziamide A, a bioactive compound isolated from the African medicinal plant Fagara heitzii (Scheme 7).35 The compound clearly arises from [4+2] cycloaddition between another amide natural product co-isolated from the same plant (fagaramide, 34) and the ubiquitous terpene myrcene. The possibility that heitziamide A arises in nature via this Diels–Alder cycloaddition was introduced in the isolation paper describing this natural product.
Scheme 7.

Heitziamide A does not arise from a thermal Diels–Alder cycloaddition.
Nevertheless, the polarity of the dienophile in this putative Diels–Alder cycloaddition predicts that the cycloaddition with myrcene would produce a [4+2] cycloadduct with the opposite regiochemical outcome from that necessary to produce heitziamide A. Indeed, a control experiment in which 34 and 35 were heated together for 72 h provided modest yields of the expected “thermal” regioisomer without any trace of heitziamide A. On the other hand, the presence of the electron-rich aryl ring on the dienophile suggested to us that heitziamide A may arise in nature from a radical cation Diels–Alder reaction. Indeed, a Diels–Alder cycloadduct with the appropriate stereochemistry and regiochemistry for heitziamide A is produced in 80% yield when styrene 37 and myrcene are irradiated in the presence of Ru(bpz)32+ (Scheme 8). Cleavage of the silyloxy group, oxidation to the corresponding carboxylic acid, and coupling with isobutylamine affords material that is spectroscopically identical to the natural product.
Scheme 8.

Synthesis of heitziamide A enabled by a radical cation Diels–Alder cycloaddition.
The synthesis of this simple natural product is intriguing from several points of view. First, it highlights the fact that photoinduced radical ion cycloadditions can be used to prepare compounds that cannot be directly accessed by standard, closed-shell cycloaddition reactions. In the synthesis of heitziamide A, conventional Diels–Alder reactions were found to be inefficient and to produce the regiochemistry opposite that required for the synthesis. Thus, it appears that the reactions accessible using photoredox catalysis do indeed complement existing methods for the synthesis of bioactive small molecules, as we had hoped when first developing this project area.
A second, and arguably more important, question that is raised by this synthesis is whether radical cation processes are more widespread in natural product biosynthesis than generally appreciated. Many biological oxidation reactions involve species capable of inducing one-electron oxidation events; perhaps the structures of natural products that cannot easily be understood using standard retrosynthetic logic arise in nature from straightforward reactions of radical cations generated by biological one-electron oxidants. Heitziamide A, for example, is co-isolated along with a second compound, heitziamide B, which is an oxidized analogue of fagaramide (Scheme 9). We speculate that both heitziamide A and B might arise in nature from interception of the radical cation of fagaramide by either water or myrcene. We have begun to wonder what other classes of natural products might be related to one another by radical cation processes, and whether photoredox catalysis presents a strategy to replicate their biosynthesis in the laboratory.
Scheme 9.

Plausible biosynthetic origin of heitziamide A and B via one-electron oxidation of fagaramide.
Outlook
One of the underlying goals of our laboratory’s research in photocatalysis over the past several years has been to design strategies that make photochemical reactions more easily accessible to the synthetic chemistry community. While it is too early to assess the impact of any of the particular methods developed by us or any of the active research groups in this area, it has been gratifying to note that many synthetic research groups have begun to investigate photocatalytic reactions in their own labs. We feel that the rapid adoption of this strategy in the synthetic community is an affirmation of our belief that visible light photocatalysis represents an exceptionally convenient method to access interesting new reactivity with great potential utility in organic synthesis.
We feel there is significant promise for the continued growth of this field in many directions. First, investigations of visible light photoredox catalysts in the context of organic synthesis are still at an early stage. Over the next several years, it will be important to continue elucidating general strategies to control the stereoselectivity of photocatalytic reactions and to demonstrate the amenability of these reactions to applications in complex target synthesis. The most fundamental goal of research in this area, however, will continue to be the discovery of new synthetic reactions in which photocatalytic activation affords a significant advantage over other, nonphotochemical methods to achieve the same transformations.36 The interaction between photocatalysts and other organic17ab,37 or transition metal38 catalysts holds particular promise for the development of novel methods.
References
- 1.Ciamician G, Silber P. Chem Ber. 1908;41:1928–1935. [Google Scholar]
- 2.(a) Esser P, Pohlmann B, Scharf HD. Angew Chem Int Ed Engl. 1994;33:2009–2023. [Google Scholar]; (b) Albini A, Fagnoni M. Green Chem. 2004;6:1–6. [Google Scholar]; (c) Fagnoni M, Dondi D, Ravelli D, Albini A. Chem Rev. 2007;107:2725–2756. doi: 10.1021/cr068352x. [DOI] [PubMed] [Google Scholar]; (d) Oelgemöller M, Jung C, Mattay J. Pure Appl Chem. 2007;79:1939–1947. [Google Scholar]; (e) Albini A, Fagnoni M. ChemSusChem. 2008;1:63–66. doi: 10.1002/cssc.200700015. [DOI] [PubMed] [Google Scholar]; (f) Protti S, Fagnoni M. Photochem Photobiol Sci. 2009;8:1499–1516. doi: 10.1039/b909128a. [DOI] [PubMed] [Google Scholar]; (g) Yoon TP, Ischay MA, Du J. Nature Chem. 2010;2:527–532. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]
- 3.Ciamician G. Science. 1912;36:385–394. doi: 10.1126/science.36.926.385. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zurflüh R, Dunham LL, Spain VL, Siddall JB. J Am Chem Soc. 1970;92:425–427. doi: 10.1021/ja00705a636. [DOI] [PubMed] [Google Scholar]; (b) Meyers A1, Fleming SA. J Am Chem Soc. 1986;108:306–307. [Google Scholar]
- 5.(a) Corey EJ, Mitra RB, Uda H. J Am Chem Soc. 1963;85:362–363. [Google Scholar]; (b) Corey EJ, Mitra RB, Uda H. J Am Chem Soc. 1964;86:485–492. [Google Scholar]
- 6.(a) Pirrung MC. J Am Chem Soc. 1979;101:7130–7131. [Google Scholar]; (b) Pirrung MC. J Am Chem Soc. 1981;103:82–87. [Google Scholar]
- 7.(a) Winkler JD, Henegar KE, Williard PG. J Am Chem Soc. 1987;109:2850–2851. [Google Scholar]; (b) Winkler JD, Kim S, Harrison S, Lewin NE, Blumberg PM. J Am Chem Soc. 1999;121:296–300. [Google Scholar]; (c) Winkler JD, Rouse MG, Greaney MF, Harrison SJ, Jeon YT. J Am Chem Soc. 2002;124:9726–9728. doi: 10.1021/ja026600a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Bach T. Synthesis. 1998:683–703. [Google Scholar]; (b) Iriondo-Alberdi J, Greaney MF. Eur J Org Chem. 2007:4801–4815. [Google Scholar]; (c) Hoffmann N. Chem Rev. 2008;108:1052–1103. doi: 10.1021/cr0680336. [DOI] [PubMed] [Google Scholar]; (d) Bach T, Hehn JP. Angew Chem Int Ed. 2011;50:1000–1045. doi: 10.1002/anie.201002845. [DOI] [PubMed] [Google Scholar]
- 9.(a) Fisher M. Angew Chem Int Ed Engl. 1978;17:16–26. [Google Scholar]; (b) Pfoertner KH. J Photochem Photobiol A: Chemistry. 1990;51:81–86. [Google Scholar]; (c) Braun AM, Maurette M, Oliveros E. Photochemical Technology. Wiley; New York: 1991. [Google Scholar]
- 10.Kreisel G, Meyer S, Tietze D, Fidler T, Gorges R, Kirsch A, Schäfer B, Rau S. Chem Ing Tech. 2007;79:153–159. [Google Scholar]
- 11.Oelgemöller M. Chem Eng Technol. 2012;35:1144–1152. [Google Scholar]
- 12.A query of the Reaxys database returned 1671 known cyclobutane-containing natural products (accessed Jan 28, 2013).
- 13.Kalyanasundaram K. Coord Chem Rev. 1982;46:159–244. [Google Scholar]
- 14.(a) Hedstrand DM, Kruizinga WH, Kellogg RM. Tetrahedron Lett. 1978;19:1255–1258. [Google Scholar]; (b) van Bergen TJ, Hedstrand DM, Kruizinga WH, Kellogg RM. J Org Chem. 1979;44:4953–4962. [Google Scholar]; (c) Ishitani O, Pac C, Sakurai H. J Org Chem. 1983;48:2941–2942. [Google Scholar]; (d) Goren Z, Willner I. J Am Chem Soc. 1983;105:7764–7765. [Google Scholar]; (e) Pac C, Ihama M, Yasuda M, Miyauchi Y, Sakurai H. J Org Chem. 1984;49:26–34. [Google Scholar]; (f) Hironaka K, Fukuzumi S, Tanaka T. J Chem Soc, Perkin Trans 2. 1984:1705–1709. [Google Scholar]; (g) Cano-Yelo H, Deronzier A. J Chem Soc, Perkin Trans II. 1984:1093–1098. [Google Scholar]; (h) Cano-Yelo H, Deronzier A. Tetrahedron Lett. 1984;25:5517–5520. [Google Scholar]; (i) Tomioka H, Ueda K, Ohi H, Izawa Y. Chem Lett. 1986;15:1359–1362. [Google Scholar]; (j) Ishitani O, Yanagida S, Takamuku S, Pac C. J Org Chem. 1987;52:2790–2796. [Google Scholar]; (k) Fukuzumi S, Mochizuki S, Tanaka T. J Phys Chem. 1990;94:722–726. [Google Scholar]; (l) Willner I, Tsfania T, Eichen Y. J Org Chem. 1990;55:2656–2662. [Google Scholar]; (m) Okada K, Okamoto K, Morita N, Okubo K, Oda M. J Am Chem Soc. 1991;113:9401–9402. [Google Scholar]; (n) Barton DHR, Csiba MA, Jaszberenyi JC. Tetrahedron Lett. 1994;35:2869–2872. [Google Scholar]; (o) Zen JM, Liou SL, Kumar AS, Hsia MS. Angew Chem Int Ed. 2003;42:577–579. doi: 10.1002/anie.200390166. [DOI] [PubMed] [Google Scholar]
- 15.(a) Chanon M, Fox M-A. Photoinduced Electron Transfer: Organic Substrates, Part C. 1988 [Google Scholar]; (b) Kavarnos GJ. Fundamentals of Photoinduced Electron Transfer. Wiley-VCH; New York: 1993. [Google Scholar]
- 16.For reviews of radical ion chemistry in synthesis, see: Shono T. Tetrahedron. 1984;40:811–850.Bauld NL. Tetrahedron. 1989;45:5307–5363.Dalko PI. Tetrahedron. 1995;51:7579–7653.Schmittel M, Burghart A. Angew Chem Int Ed Engl. 1997;36:2550–2589.Staettel NJ, Oxgaard J, Wiest O. Eur J Org Chem. 2001:1429–1439.Sperry JB, Wright DL. Chem Soc Rev. 2006;35:605–621. doi: 10.1039/b512308a.
- 17.For representative publications, see: Nicewicz DA, MacMillan DWC. Science. 2008;322:77–80. doi: 10.1126/science.1161976.Nagib DA, Scott ME, MacMillan DWC. J Am Chem Soc. 2009;131:10875–10877. doi: 10.1021/ja9053338.Nagib DA, MacMillan DWC. Nature. 2011;480:224–228. doi: 10.1038/nature10647.
- 18.For representative publications, see: Narayanam JMR, Tucker JW, Stephenson CRJ. J Am Chem Soc. 2009;131:8756–8757. doi: 10.1021/ja9033582.Condie AG, González-Gómez JC, Stephenson CRJ. J Am Chem Soc. 2010;132:1464–1465. doi: 10.1021/ja909145y.Nguyen JD, Tucker JW, Konieczynska MD, Stephenson CRJ. J Am Chem. 2011;133:4160–4163. doi: 10.1021/ja108560e.Furst L, Narayanam JMR, Stephenson CRJ. Angew Chem Int Ed. 2011;50:9655–9659. doi: 10.1002/anie.201103145.
- 19.For a selection of key results from other laboratories, see: Andrews RS, Becker JJ, Gagné MR. Angew Chem Int Ed. 2010;49:7274–7276. doi: 10.1002/anie.201004311.Chen Y, Kamlet AS, Steinman JB, Liu DR. Nat Chem. 2011;3:146–153. doi: 10.1038/nchem.932.Larraufie MH, Pellet R, Fensterbank L, Goddard JP, Lacôte E, Malacria M, Ollivier C. Angew Chem Int Ed. 2011;50:4463–4466. doi: 10.1002/anie.201007571.Rueping M, Vila C, Koenigs RM, Poscharny K, Fabry DC. Chem Comm. 2011;47:2360–2362. doi: 10.1039/c0cc04539j.Zou YQ, Lu LQ, Fu L, Chang NJ, Rong J, Chen JR, Xiao WJ. Angew Chem Int Ed. 2011;50:7171–7175. doi: 10.1002/anie.201102306.Hailton D, Nicewicz DA. J Am Chem Soc. 2012;134:18577–18580. doi: 10.1021/ja309635w.Fors BP, Hawker CJ. Angew Chem Int Ed. 2012;35:8850–8853. doi: 10.1002/anie.201203639.Leung JCT, Chatalova-Sazepin C, West JG, Rueda-Becerril M, Paquin JF, Sammis GM. Angew Chem Int Ed. 2012;51:10804–10807. doi: 10.1002/anie.201206352.Kohls P, Jadhav D, Pandey G, Reiser O. Org Lett. 2012;14:672–675. doi: 10.1021/ol202857t.Zhu S, Das A, Bui L, Zhou H, Curran DP, Rueping M. J Am Chem Soc. 2013;135:1823–1829. doi: 10.1021/ja309580a.
- 20.For reviews, see: Zeitler K. Angew Chem Int Ed. 2009;48:9785. doi: 10.1002/anie.200904056.Yoon TP, Ischay MA, Du J. Nature Chem. 2010;2:527. doi: 10.1038/nchem.687.Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n.Teply F. Collect Czech Chem Commun. 2011;76:859.Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617. doi: 10.1021/jo202538x.Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013 doi: 10.1021/cr300503r. in press.
- 21.Baik TG, Luis AL, Wang LC, Krische MJ. J Am Chem Soc. 2001;123:6716–6717. doi: 10.1021/ja010800p. [DOI] [PubMed] [Google Scholar]
- 22.Roh Y, Jang HY, Lynch V, Bauld NL, Krische MJ. Org Lett. 2002;4:611–613. doi: 10.1021/ol0172065. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Felton GAN, Bauld NL, Krische MJ. J Am Chem Soc. 2004;126:1634–1635. doi: 10.1021/ja030543j. [DOI] [PubMed] [Google Scholar]
- 24.Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- 25.Fournier F, Fournier M. Can J Chem. 1986;64:881–890. [Google Scholar]
- 26.Hurtley AE, Cismesia MA, Ischay MA, Yoon TP. Tetrahedron. 2011;67:4442–4448. doi: 10.1016/j.tet.2011.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu Z, Shen M, Yoon TP. J Am Chem Soc. 2011;133:1162–1164. doi: 10.1021/ja107849y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du J, Ruiz Espelt L, Guzei IA, Yoon TP. Chem Sci. 2011;2:2115–2119. doi: 10.1039/C1SC00357G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauld NL. Tetrahedron. 1989;45:5307–5363. [Google Scholar]
- 30.Ischay MA, Lu Z, Yoon TP. J Am Chem Soc. 2010;132:8572–8574. doi: 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Juris A, Balzani V, Barigelletii F, Campagna S, Belser P, von Zelewsky A. Coord Chem Rev. 1988;84:85–277. [Google Scholar]
- 32.Lin S, Ischay MA, Fry CG, Yoon TP. J Am Chem Soc. 2011;133:19350–19353. doi: 10.1021/ja2093579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ischay MA, Ament MS, Yoon TP. Chem Sci. 2012;3:2807–2811. doi: 10.1039/C2SC20658G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crutchley RJ, Lever ABP. J Am Chem Soc. 1980;102:7128–7129. [Google Scholar]
- 35.Mbaze LM, Lado JA, Wansi JD, Shiao TC, Chiozem DD, Mesaik MA, Choudhary MI, Lacaille-Dubois MA, Wandji J, Roy R, Sewald N. Phytochemistry. 2009;70:1442–1447. doi: 10.1016/j.phytochem.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 36.For an intriguing recent report of a photooxidative [3+2] cycloaddition reaciton, see: Maity S, Zhu M, Shinabery RS, Zheng N. Angew Chem Int Ed. 2012;51:222–226. doi: 10.1002/anie.201106162.
- 37.DiRocco DA, Rovis T. J Am Chem Soc. 2012;134:8094–8097. doi: 10.1021/ja3030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.(a) Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J Am Chem Soc. 2011;133:18566–18569. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ye Y, Sanford MS. J Am Chem Soc. 2012;134:9034–9037. doi: 10.1021/ja301553c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.For a recent example, see: Wever WJ, Cinelli MA, Bowers AA. Org Lett. 2013:30–33. doi: 10.1021/ol302941q.
- 40.For a recent example, see: Fors BP, Hawker CJ. Angew Chem Int Ed. 2012;51:8850–8853. doi: 10.1002/anie.201203639.