

Key Points
Tat acts as an inflammatory cytokine and as an antiviral factor via interaction with MAP2K6, MAP2K3, and IRF7 promoters in APCs.
These interactions are the molecular events that link HIV to p38 MAPK activation, which is a mediator of HIV-associated pathology and immune activation.
Abstract
As a result of its interaction with transcription factors, HIV type 1 (HIV-1) Tat can modulate the expression of both HIV and cellular genes. In antigen-presenting cells Tat induces the expression of a subset of interferon (IFN)-stimulated genes (ISGs) in the absence of IFNs. We investigated the genome-wide Tat association with promoters in immature dendritic cells and in monocyte-derived macrophages. Among others, Tat associated with the MAP2K6, MAP2K3, and IRF7 promoters that are functionally part of IL-1 and p38 mitogen-activated protein kinase (MAPK) signaling pathways. The association correlated with their increased gene expression, increased activation of p38 MAPK and of phosphorylated signal transducer and activator of transcription 1 (STAT1), and consequent induction of ISGs. Probing these pathways with RNA interference, pharmacological p38 MAPK inhibition, and in cell lines lacking STAT1s or the type I IFN receptor chain confirmed the role of MAPKKs and IRF7 in Tat-mediated modulation of ISGs and excluded the involvement of IFNs in this modulation. Tat interaction with the 2 MAPKK and IRF7 promoters in HIV-1–infected cells and the resulting persistent activation of ISGs, which include inflammatory cytokines and chemokines, can contribute to the increased immune activation that characterizes HIV infection.
Introduction
HIV–infected cells respond to the infection with different outcomes depending on their cell type; this is due to the interplay of cellular and viral proteins.1 Of particular interest are the interactions of the HIV Tat protein, which is the viral transcriptional activator that interacts with many cellular factors.2,3 Tat is essential for efficient transcription of HIV type 1 (HIV-1) genes, which is accomplished by binding to the transactivation-responsive region (TAR), recruitment of P-TEFb to TAR, and interaction with enzymes such as histone acetyl transferases (HATs).4-6 Tat can also modulate cellular gene expression and affect cytokine production, induction of apoptosis, and immunosuppression in infected and noninfected cells.3,7 We have shown that Tat mediates HIV-induced apoptosis in CD4+ T cells by activating the Phosphatase and Tensin homolog-Forkhead box O3 (PTEN-FOXO3a) pathway via association with PTEN and protein phosphatase 2A (PP2A) promoters.8 We also found that HIV-1 infection and Tat expression in primary antigen-presenting cells (APCs) can induce expression of a subset of interferon (IFN)-stimulated genes (ISGs) that encode transcription factors as the IFN regulatory factors 7 (IRF7), signal transducer and activator of transcription 1 (STAT1), and chemokines that recruit activated T cells and monocyte-derived macrophages (MDM). (N.K., S.K., M.D.P.M.-V., M.M., and A.A., Journal of Virology, December 6, 2012).9 This reprogramming facilitates expansion of the infection. A species-specific increase of some ISGs was observed in human and Rhesus macaque immature dendritic cells (iDC) and MDM but not in the same cells from chimpanzee, sooty mangabeys, and African green monkeys, in which simian immunodeficiency virus infection is asymptomatic or less severe.10 Our results established a correlation between the Tat-mediated differential induction of ISGs and species-specific differences in disease susceptibility.
The mechanism of transcriptional regulation of ISGs involves different cellular pathways including Janus kinase–STAT (JAK–STAT) and the mitogen-activated protein (MAP) kinase signaling cascades. Although virus infections result in expression of type I IFNs, which induces expression of ISGs, some ISGs are also directly induced independently of IFN signaling, via Toll-like receptor (TLR) activation.11-13
To investigate how Tat stimulates the transcription of ISGs, we evaluated the genome-wide promoter association of Tat in iDC and MDM. We report that in these APCs, Tat modulates expression of ISGs in the absence of type I IFNs by triggering activation of the p38 MAPK-STAT1 pathway via association with the promoters of 2 MAPKKs, MAP2K6 and MAP2K3, and of IRF7.
Materials and methods
Cell lines, primary cells
The human myelomonocytic cell line KG-1,14 the monocytic cell line THP-1,15 and CD14+ monocytes derived from human peripheral blood mononuclear cell were differentiated into iDC (supplemental Table 1) or MDM as described in supplemental Methods. Dr George Stark (Cleveland Clinic) provided the 2fTGH, U5A, U5A-R, and U6A cell lines.16 Infections were carried out with either the dual tropic HIVSF2 or the CCR5 tropic HIVBal as described in supplemental Methods.
Adenoviruses, plasmids, transfections, and luciferase assay
Flagged, recombinant adenoviruses Ad-tTA, Ad-TatSF2, and Ad- TatSF2G48-R57A were constructed according to published procedures.17 The tat coding DNA was cloned into the vector pAd-TRE-MCS1 under a tetracycline-inducible promoter. The corresponding adenovirus requires coinfection with Ad-tTA, expressing the tetracycline-responsive transactivator. For luciferase assays, cells were transfected with MAP2K6- (−991 to −1 nucleotides from MAP2K6 start site), MAP2K3- (−1013 to −1), and IRF7- (−1018 to −1) luciferase vectors and then infected with Ad-tTA or Ad-TatSF2/Ad-tTA. Cell lysates were assayed for firefly and Renilla luciferase activities (Promega, Madison, WI).
RNA isolation and quantitative reverse transcription-polymerase chain reaction
RNA (100 ng) was reverse transcribed and amplified using iScript and SYBR Green SuperMix with the ROX cDNA kit (Bio-Rad). Supplemental Table 2 lists the primers used in the amplification.
Genome-wide promoter association analysis
Chromatin immunoprecipitation (ChIP) and promoter association analysis (ChIP-on-Chip) was performed as described.18 Kim et al8 and the supplemental Methods describe how the analysis was carried out and how significant values were selected (GEO accession number: GSE42191).
RNAi and chemical inhibition experiments
Primary cells and cell lines were transfected with 100 nM siRNAs (Dharmacon) and then infected with adenoviruses at a multiplicity of infection of 107. For chemical inhibition studies, cells were treated with 10 μM SB203580 (EMD Millipore), 50 μM PD98059 (EMD Millipore), or 0.5 μM AS601245 (Enzo Life Sciences) and infected with adenoviruses (see also supplemental Methods).
Immunofluorescence staining and flow cytometry
Cells were stained with the antibodies described previously,7 except for the human anti-IRF7, anti-IP10, anti-HuMIG, anti-STAT1, anti-phospho-STAT1 (pY701), and anti-phospho-STAT1 (pS727) antibodies (BD Biosciences) and the anti-MAP2K3, anti-MAP2K6, anti-phospho-p38 (pThr180/Thr182), and anti-phospho-p44/42 MAPK (ERK1/2) (pThr202/pThr204) antibodies (Cell Signaling or Millipore). MCP2, MAP2K3, and MAP2K6 antibodies were labeled using the Tandem conjugation kit (Innova Biosciences).
Results
Tat association with the IRF7, MAP2K6, and MAP2K3 promoters in KG1-derived iDC, THP1-derived macrophages, and primary iDC and macrophages
Our previous findings indicate that HIV Tat modulates the expression of the ISGs in iDC and that this modulation of gene expression could be different in different species.9,10 To establish a reproducible system relevant to primary cells and amenable to study the mechanisms by which Tat affects gene expression in APCs, we evaluated whether HIV-1 Tat modifies gene expression of KG-1–derived iDC (KG-iDC) and THP-1–derived macrophages (THP-Mac) in a way that is similar to that observed with Tat or HIVBAL in primary human iDC and MDM.9,19,20 KG-iDC were infected with adenoviral vectors expressing a 101 aa FLAG-tagged Tat protein from HIVSF2 (Ad-TatSF2) or TatSF2G48-R57A (Ad-TatSF2G48-R57A), a mutant in which the nuclear localization signal residues were substituted by alanines,21 in combination with Ad-tTA or with Ad-tTA alone as controls. Tat expression was comparable in all cultures examined (Figure 1A), and Tat-intracellular staining ranged from 30% to 40% (data not shown). Expression levels of selected ISGs were evaluated in Tat-expressing KG-iDC by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and were found to be similarly modulated as in primary iDC infected with Ad-Tat or HIVBAL (Figure 1B).9 These data support the use of KG-iDC as an in vitro model for the study of molecular mechanisms critical to Tat-mediated gene modulation in iDC.
Figure 1.

Tat-mediated gene modulation in KG-iDC and THP-Mac and its association with cellular promoters. (A) mRNA levels of Tat in KG-1 cells infected with Ad-TatSF2 24 hours after infection at multiplicities of infection (MOI) of 0.1, 1, and 10, analyzed by qRT-PCR. Results are normalized to actin and reported as fold induction relative to Ad-Tat samples infected at MOI of 0.1. The means ± standard error of the mean (SEM) derived from 3 independent experiments are reported. (B) mRNA levels of selected ISGs 24 hours after infection with adenoviral vectors Ad-TatSF2 and negative control Ad-TatSF2G48R57A. Results are reported as fold induction relative to Ad-TatSF2G48R57A samples. (C) Tat association with the MAP2K6 promoter in KG-iDC, detected by ChIP and qPCR. DNA from input (90, 30, and 10 ng of DNA) and immunoprecipitated samples (10 ng of DNA) was amplified by standard PCR using the P1, P2, and P3 set of primers. DNA from cells infected with Ad-TatSF2G48-R57A and Ad-tTA was used as a negative control. One representative experiment is shown. (D) Average fold enrichments of the indicated promoters in the immunoprecipitated DNA relative to input DNA ± SEM from 3 independent experiments are reported. Cycle threshold (Ct) values obtained with 10 ng of immunoprecipitated DNA were compared with the Ct values obtained with 10 ng of the corresponding input DNA. (E) MAP2K6 and Tat mRNA levels in KG-1 cells infected with Ad-TatSF2 and Ad-TatSF2G48-R57A, analyzed by RT-qPCR. Results are normalized to actin and reported as fold induction relative to samples infected with Ad-tTA alone. The means ± SEM of 3 experiments are shown. (F) Luciferase activity in lysates from cells expressing wild-type TatSF2 or TatSF2 G48R57A and transfected with an IRF7- or a MAP2K6-luciferase reporter vector. Firefly luciferase activity was normalized to Renilla luciferase activity. Results are reported as fold induction relative to those in the cells infected with Ad- tTA. (G) mRNA levels of selected ISGs 48 hours after infection with Ad-TatSF2 and Ad-TatSF2G48R57A. Results are reported as fold induction relative to Ad-TatSF2G48R57A samples. (H) Tat association with the IRF7, MAP2K3, and MAP2K6 promoters in THP-Mac expressing TatSF2. DNA from input (90, 30, and 10 ng of DNA) and immunoprecipitated samples (10 ng of DNA) was amplified by qPCR using the P2 and P3 set of primers. DNA from cells infected with Ad-TatSF2G48-R57A and Ad-tTA was used as a negative control. One representative experiment is shown. (I) Average fold enrichments of IRF7, MAP2K3, and MAP2K6 promoters using ChIP-qPCR in THP-Mac expressing TatSF2 relative to input DNA + SEM from 3 independent experiments. (J) Luciferase activity of lysates from THP-Mac expressing the wild-type TatSF2 or TatSF2 G48R57A and transfected with IRF7-, MAP2K6-, or MAP2K3-luciferase reporter vectors.
To gain insight into the effect of Tat on cellular gene expression during HIV-1 infection, we used ChIP of Tat coupled with promoter DNA microarray analysis (ChIP-Chip) to identify what promoters are associated with Tat. KG-iDC were infected with Ad-TatSF2+Ad-tTA or with the negative controls Ad-TatSF2G48-R57A+Ad-tTA and Ad-tTA. Two independent experiments were performed. In cells expressing TatSF2, we identified 342 promoters that were occupied by Tat (P < .001) but not enriched in controls.
Using the ingenuity pathway analysis (IPA) application, we identified the cellular pathways affected by the genes driven by Tat-associated promoters. These genes are part of a few significant pathways, including IL-1 and p38 MAPK signaling, dendritic cell maturation, and TLR signaling (Table 1). Among these genes were MAP2K4 and MAP2K6, which encode dual-specificity protein kinases that, together with MAP2K3, function as mitogen-activated protein (MAP) kinase kinases (MAPKKs) and phosphorylate c-Jun NH2-terminal kinase (JNK) and p38 MAP kinase (MAPK), which play an important role in ISG transcription regulation via the activation of STATs or downstream effectors that cooperate with STATs to induce optimal transcription of ISG.11,22-27 Associations with additional pathways are listed in supplemental Table 3.
Table 1.
Analysis of promoters found associated with HIV-Tat in KG-iDC and THP-Mac
| Cell type | Ingenuity canonical pathway | P value | Molecules |
|---|---|---|---|
| KG-iDC | IL-1 signaling | 1.86E-04 | MAP2K4,PRKACB,MAP2K6,IL1A |
| p38 MAPK signaling | 3.62E-03 | MAP2K4,MAP2K6,IL1A,TGFB3 | |
| cAMP-mediated signaling | 4.80E-03 | PRKACB,PKIA,AKAP7,RAP1A | |
| Dendritic cell maturation | 8.40E-03 | MAP2K4,IL1A,PIK3R1 | |
| TLR signaling | 1.01E-02 | MAP2K4,MAP2K6 | |
| THP-Mac | p38 MAPK signaling | 1.78E-02 | MAP2K6,H3F3A/H3F3B,IL1RAPL1 |
| IL-10 signaling | 6.20E-02 | MAP2K6,IL1RAPL1 | |
| IL-1 signaling | 1.67E-01 | IL1RAPL1 |
Tat association with the MAP2K6 promoter was further validated by conventional ChIP, using site-specific primers on the same chromatin samples used for ChIP-Chip. MAP2K6 promoter sequences were consistently enriched in IP-captured chromatin from cells expressing TatSF2 compared with controls (Figure 1C-D). We did not observe any Tat association with MA2PK3 and IRF7 promoters in KG-iDC. These associations were significant in primary MDM and IRF7 association in iDC (see below). Despite evidence of Tat association with the MAP2K4 promoter in ChIP-Chip analysis, this association was not consistently reproduced in conventional ChIP experiments, possibly due to variation in infection rate from one experiment to another, with detection being significant in 3 of 7 independent experiments carried out in KG-iDC and in primary iDC. As Tat association with the MAP2K4 promoter did not affect MAP2K4 expression and siRNA inhibition did not significantly affect ISG modulation (data not shown), further investigation of this gene was abandoned. To determine whether the association of Tat with the MAP2K6 promoter affected MAP2K6 gene expression, we evaluated MAP2K6 mRNA levels in TatSF2-expressing cells and found that they were increased (Figure 1E). In addition, TatSF2 expression significantly increased the activity of the MAP2K6 promoter when the activity of a luciferase gene regulated by this promoter was evaluated (Figure 1F). These experiments support the association of Tat with the MAP2K6 promoter as the key event leading to the activation of this kinase.
HIV infection and Tat expression also induce a subset of ISGs in MDM, and the profile of Tat-mediated gene modulation in iDC and MDM is similar.9,19,20 The major gene expression pathways found to be affected in iDC and in MDM are listed in supplemental Table 4.
As a system to use for further studies, THP-1 cells were differentiated into macrophages (THP-Mac) and used to evaluate cellular gene expression modulation induced by Tat. We found that genes upregulated in primary iDC and MDM were similarly modulated in THP-Mac infected with Ad-TatSF2. Tat expression increased mRNA levels of selected ISGs, including STAT1, IRF7, IP10, MCP2, MIG, and TRAIL (Figure 1G).
We carried out ChIP-Chip analysis to identify cellular promoters associated with Tat in THP-Mac infected with Ad-TatSF2/Ad-tTA and with Ad-TatSF2G48-R57A/Ad-tTA or Ad-tTA alone as negative controls. We identified 308 promoters that were occupied by Tat. IPA of the corresponding genes revealed that they are linked to 3 major pathways (Table 1), and 1 is the p38 MAPK signaling pathway (P = .0178). Associations with other pathways are listed in supplemental Table 3. Tat was found associated with the MAP2K6, MAP2K3, and IRF7 promoters, and the association was confirmed by ChIP-qPCR (Figure 1H-I). This association resulted in increased expression of the corresponding genes (Figure 1G). To evaluate whether the MAP2K6, MAP2K3, and IRF7 promoter sequences conferred Tat-dependent stimulation of transcription of the luciferase gene, approximately 1000 nucleotides of the promoter sequences located at the 5′ end of the transcriptional start site were cloned at the 5′ end of the luciferase gene and evaluated in the presence or absence of TatSF2. TatSF2 expression increased MAP2K6, MAP2K3, and IRF7 promoter activities approximately 3- to 4-fold compared with that observed in control cells (Figure 1J).
We investigated whether TatSF2 association with the promoters identified in KG-iDC and THP-MAC could be confirmed by ChIP-qPCR analysis in primary iDC infected with Ad-TatSF2 or Ad-TatSF2G48R57A. Tat did associate with MAP2K6 and IRF-7 promoters in primary iDC expressing TatSF2 (Figure 2A-B) or infected with HIVSF2 carrying a flagged tat gene (HIVSF2-FLAG Tat; Figure 2D-E) but not in the controls. No association with MAP2K3 promoter was observed in these cells. As observed in KG-iDC, TatSF2 expression in iDC increased MAP2K6- and IRF7-luciferase transcription by approximately 3-fold compared with that observed in controls (Figure 2C). Tat association with the IRF7, MAP2K3, and MAP2K6 promoters was detected in primary MDM infected with Ad-TatSF2 (Figure 2F-G) or infected with HIVSF2-FLAG Tat (Figure 2H-I). These results indicate that Tat associates with IRF7 and MAP2K6 promoters in primary iDC and MDM and with the MAP2K3 promoter in MDM and that this association increases their transcriptional activity.
Figure 2.

Tat association with the IRF7, MAP2K6, and MAP2K3 promoters in primary iDC and MDM. (A) Signals obtained by PCR in iDC expressing wild type TatSF2 or the mutant TatSF2G48R57A with two or three sets of primers carried out with input DNA (90, 30, 10 ng) or with 3 ng of DNA extracted from immunoprecipitated samples are shown. (B) The average fold enrichment of the IRF7, MAP2K3, and MAP2K6 promoter in primary iDC in the immunoprecipitated DNA relative to input DNA +/− SEM from three independent qPCR experiments is reported. All Ct values obtained with immunoprecipitated DNAs were compared with the Ct value obtained with the same amount of the corresponding input DNA. (C) Luciferase activity of lysates from primary iDC expressing no Tat (tTA), wild type TatSF2, TatSF2 G48R57A and transfected with an IRF7- or MAP2K6-luciferase reporter vector. (D,E) Enrichments of the IRF7, MAP2K3, and MAP2K6 promoters after Tat immunoprecipitation from chromatin lysates of primary iDC infected with a replication competent HIVSF2, expressing a flagged Tat. (F) Signals obtained by PCR in MDM expressing wild type TatSF2 or the mutant TatSF2G48R57A with two or three sets of primers carried out with input DNA (90, 30, 10 ng) or with 3 ng of DNA extracted from immunoprecipitated samples are shown. (G) The average fold enrichment of the IRF7, MAP2K3, and MAP2K6 promoter in primary MDM in the immunoprecipitated DNA relative to input DNA +/− SEM from three independent qPCR experiments is reported. (H,I) Enrichments of the IRF7, MAP2K3, and MAP2K6 promoters after Tat immunoprecipitation from chromatin lysates of MDM infected with a replication competent HIVSF2, expressing a flagged Tat.
siRNA-mediated inhibition of MAP2K6, MAP2K3, and IRF7 reduces Tat-mediated modulation of ISG gene expression in APCs
We investigated the contribution of MAP2K6 to Tat-mediated modulation of gene expression using siRNA-mediated knockdown of the MAP2K6. KG-iDC were transfected with siRNA targeting MAP2K6, IRF7, or nonspecific siRNA and then infected with Ad-tTA or Ad-TatSF2/Ad-tTA. MAP2K6 and IRF7 mRNA levels were reduced approximately 60% and 80%, respectively, by the corresponding siRNAs but not by the control siRNA (Figure 3A). This inhibition resulted in reduced mRNA expression of IP10, TRAIL, MIG, and MCP2, which are upregulated after TatSF2 expression (Figure 3A). These experiments support the role of both MAP2K6 and IRF7 in Tat-dependent modulation of ISGs in KG-iDC.
Figure 3.

MAP2K6 and IRF7 induce Tat-mediated cellular gene modulation via the p38-STAT1 pathway. mRNA levels of ISGs in KG-iDC (A) and type I IFNR2-deficient 5UA cells (B) transfected with MAP2K6 and IRF7 siRNAs and infected with Ad-TatSF2/Ad-tTA or Ad-tTA alone. Results are normalized to actin and reported as fold induction relative to Ad-tTA–infected cells treated with nonspecific (ns) siRNA. mRNA levels of selected ISGs in (C) STAT1-deficient U3A cells, (D) U3A-R cells (U3A reconstituted with STAT1) infected with Ad-Tat SF2/Ad-tTA or Ad-tTA alone and transfected with MAP2K6 siRNA, and (E) STAT2-deficient U6A cells treated with ns siRNA or siMAP2K6 RNA and infected with Ad-TatSF2/tTA or Ad-tTA alone.
A similar experiment was carried out in U5A cells, a human cell line that lacks the type I IFN receptor chain 2 (IFNAR2c),28 where type I IFN contribution to Tat modulation can be excluded. TatSF2 expression resulted in increased mRNA levels of IP10, TRAIL, HuMIG, and MCP2 and was reduced by treatment with MAP2K6 and IRF7 siRNAs (Figure 3B). These results and similar results obtained with the K562 cell line in which the type 1 IFN locus is deleted (N.K., S.K., M.D.P.M.-V., M.M., and A.A., Journal of Virology, December 6, 2012) indicate that Tat-mediated modulation of ISGs is independent of IFN signaling and further support the role of both MAP2K6 and IRF7 in the modulation of gene expression induced by Tat.
To investigate the role played by STAT1 and STAT2 in the activation of the MAP2K6-p38 pathway and in ISG modulation mediated by Tat, we used the human STAT1-deficient U3A cell line, the STAT-1-reconstituted U3A-R, and the STAT2-deficient U6A (Figure 3C-E).16 These cells were transfected with siRNAs and infected with Ad-TatSF2. MAP2K6 siRNA in U3A cells and U3A-R cells reduced mRNA levels of MAP2K6 by approximately 60% and 80% of its original level, respectively. Tat expression in U3A cells did not induce ISGs that were upregulated in primary iDC and KG-iDC cells, and MAP2K6 siRNA had no effect on their levels (Figure 3C). In contrast, TatSF2 expression increased the mRNA level of ISGs in STAT1-reconstituted U3A-R and in STAT2-deficient U6A cells, and inhibition of MAP2K6 expression resulted in decreased levels of the same genes (Figure 3D-E). Taken together, these results indicate that HIV-1 Tat modulation of ISGs occurs via increased transcription of MAP2K6 and IRF7 and modulation of p38 and STAT1 but not STAT2. The role played in modulation of ISG expression by other transcription factors such as NFκB, ATF, and CREB, whose activities are also affected by p38 kinase, was not explored.
To further confirm the role of MAP2K6, MAP2K3, and IRF7 in Tat-mediated gene modulation, we investigated the effects of targeting these genes with RNAi in primary iDC and MDM expressing Tat. The increased mRNA and protein levels of MAP2K6 and IRF7 in iDC (Figure 4A-B) and of MAP2K3, MAP2K6, and IRF7 in MDM (Figure 4C-E), observed after TatSF2 expression, were reduced by treatment with the corresponding siRNAs, as were the tested ISG mRNAs and TRAIL and HuMIG proteins. Inhibition of MAP2K3 and MAP2K6 resulted in p38 and STAT1 reduced phosphorylation, supporting their role in the activation (Figure 4B,D) and had limited impact on the expression of IRF7, which itself is an ISG, while a significant reduction of expression of a few tested ISGs was observed in these conditions (Figure 4A-D). This result was not surprising, as Tat can affect the transcription of IRF7 via association with its promoter. The inhibitory effect induced by the IRF7 siRNA on modulation of gene expression was similar to that of MAP2K3 or MAP2K6 siRNAs, indicating a similar contribution of 2 different pathways to ISG activation (Figure 4A-D). IRF7 siRNA treatment resulted in lower STAT1, TRAIL, HuMIG, IP10, and MCP2 mRNA levels and of the proteins we tested (TRAIL and HuMIG; Figure 4A-D). Similar results were observed when the same experiment was carried out in THP-Mac (supplemental Figure 1). These RNAi experiments in cell lines and primary cells confirm a role for IRF7, MAP2K6, and MAP2K3 in Tat-mediated modulation of ISGs.
Figure 4.

siRNA-mediated knockdown of IRF7, MAP2K6, and MAP2K3 reduces the expression of ISGs induced by Tat in primary iDC and primary MDM. (A) mRNA levels of different genes in iDC infected with Ad-tTA or Ad-TatSF2+Ad-tTA and treated with siRNA, reported as fold induction relative to those observed in Ad-tTA infected iDC, treated with nonspecific siRNA. The average of three independent experiments is reported. (B) Flow cytometric protein expression analysis in iDC infected with Ad-tTA or Ad-TatSF2+Ad-tTA and treated with siRNA, reported as median fluorescence intensity (MFI). (C) mRNA levels of different genes in MDM infected with Ad-tTA or Ad-TatSF2+Ad-tTA and treated with siRNA, reported as fold induction relative to those observed in Ad-tTA infected MDM, treated with nonspecific siRNA. The average of three independent experiments is reported. (D,E) Flow cytometric protein expression analysis in MDM infected with Ad-tTA or Ad-TatSF2+Ad-tTA and treated with siRNA, reported as MFI.
Pharmacological inhibition of Tat-mediated ISG modulation
To validate the role of the p38 pathway in the gene expression modulation induced by Tat and to explore the possibility of eventual therapeutic intervention to block Tat-modulating activity, primary iDC expressing Tat were treated with different pharmacological inhibitors of the p38 MAPK (SB203580), ERK, the extracellular signal-regulated kinases (PD98059), and JNK, the c-Jun NH2-terminal kinases (AS601245).11,22,24,29 Tat expression increased the levels of phosphorylated p38 and JNK via MAPK26 that can contribute to both p38 and JNK phosphorylation and possibly via the limited modulation of MAP2K4 that we observed at times in these experiments, but not of pERK (Figure 5A). When iDC expressing Tat were pretreated with SB203580, PD98059, or AS601245 and infected with Ad-TatSF2 or a control, a significant reduction of the ISG RNA levels was observed with SB203580 and to a lesser degree with AS601245 and PD98059, suggesting that JNK and ERK could also be involved in this event (Figure 5C). As ERK levels were not affected by Tat expression, we concluded that the modest contribution of PD98059 to ISG RNA reduction may depend on nonspecific activity of this compound or on the inhibition of baseline levels of this kinase and that Tat-modulation is not dependent on ERK modulation. Treatment with SB203580 significantly reduced the levels of phosphorylated p38, STAT1 tyrosine 701, and of the IRF7 protein, a target of STAT1 activity increased by Tat (Figure 5C). A similar experiment carried out in MDM (Figure 5D) and THP-Mac (data not shown) also resulted in the inhibition of Tat-mediated ISG induction.
Figure 5.

Pharmacological inhibition of the MAP2K6-p38 pathway reduces Tat-modulated gene expression in primary iDC and MDM. (A) Levels of phosphorylated p38 MAPK, JNK, and ERK in iDC expressing Tat. (B) mRNA expression of selected ISGs in iDC transfected with vectors expressing MAP2K6 or MAP2K6(glu) or infected with Ad-TatSF2+ Ad-tTA in the presence or absence of SB203580. (C) mRNA levels of selected ISGs in primary iDC infected with Ad-tTA alone or Ad-TatSF2/Ad-tTA and treated for 1 hour with the p38 inhibitor SB203580, the PD98059 inhibitor of ERK, or the JNK inhibitor AS601245. Levels of the indicated proteins (reported as median fluorescence intensity [MFI]) during p38 inhibitor treatment are also shown. (D) mRNAs levels and MFI of the indicated proteins in control and Tat-expressing MDM treated with or without SB203580. (E) mRNA levels of selected ISGs in primary MDM infected with HIVBAL or exposed to medium alone in the presence or absence of the p38 inhibitor SB203580. (F) Cytokine detection in primary MDM supernatants 7 days after HIVBAL infection+/− inhibitor (mean ± standard errors of 2 independent samples). (G) Flow cytometric analysis of CD38+/HLA-DR+ cells in CD4+ and CD8+ lymphocyte subsets, exposed for 3 days to the conditioned medium derived from HIVBAL-infected MDM +/− inhibitor. Peripheral blood lymphocytes were obtained from the same donor of MDM and analyzed by flow cytometry. The amount of CD4+ or CD8+ T cells coexpressing HLA-DR and CD38 is shown as a fold increase compared with the percentage of CD38+/HLA-DR+ cells stimulated with medium from uninfected macrophages. Cells stimulated with phytohemagglutinin (10 ng/mL) and inomycin (100 ng/mL) provide the positive control.
To establish that modulation of MAP2K6 is by itself sufficient to induce ISG, MAP2K6 was overexpressed in primary iDC using vectors expressing wild type MAP2K6 or MAP2K6(glu), a constitutively activated MAP2K6,30 in the presence or absence of SB203580. An increase of selected ISG mRNA levels similar to that found after Tat expression was observed and was reduced by SB203580 (Figure 5B). When we evaluated selected ISG RNA expression and IP10, MIG, and MCP2 in culture supernatant, we also found that HIVBal increases their amounts, as previously observed,9 and treatment with SB203580 reduced HIV-mediated gene modulation (Figure 5E-F). CD8+, but not CD4+, T-cell immune activation, was significantly higher after exposure to the conditioned medium from HIVBAL-infected MDM compared with that from the uninfected control. CD8+ T-cell immune activation did not increase when cells were exposed to medium from infected MDM also treated with the p38 inhibitor (Figure 5G). These results indicate that increased expression of MAP2K6, stimulated by Tat, can by itself activate ISGs via the p38 pathway and contribute to T-cell immune activation.
Taken together, these experiments provide the experimental evidence for how Tat triggers a signaling pathway that activates ISGs in APCs infected by HIV (Figure 6). In addition, they support the pharmacological inhibition of Tat or p38 MAPK to reduce ISG activation and the immunological effects it mediates.
Figure 6.
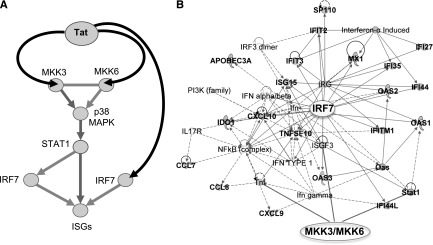
Schematic of the pathway triggered by Tat and leading to activation of ISGs by Tat in APCs infected by HIV. (A) Tat triggers signaling pathways that activate ISGs by associating with MAP2K3 and MAP2K6 kinases, which in turn activate p38 MAPK and STAT1, and with IRF7 in APCs infected by HIV. (B) Activation of MAP2K3, MAP3K6, and IRF7 leads to expression of many ISGs (shown here in connection with MAP2K3, MAP3K6, and IRF7), whose product can positively impact T-cell immunoactivation and negatively impact virus production. Bold labeling indicates genes found upregulated by HIV and Tat in APC (Figures 4,5).8,9
Discussion
We explored the effects of HIV-1 infection and Tat expression in APCs such as myeloid iDC and MDM, which are targets of HIV infection. We found that Tat association with the MAP2K6, MAP2K3, and IRF7 promoters leads to activation of the p38-STAT1 pathway and the subsequent transcriptional activation of a subset of ISGs. Inhibition of IRF7 and the 2 MAPKKs reduced Tat-induced expression of ISGs in iDC and MDM via a type I IFN-independent pathway.
Tat was found to be associated with approximately 300 cellular promoters in KG-iDC and THP-Mac. An IPA of both the gene expression analysis during Tat expression and of genome-wide Tat promoter association point to the modulation of ISGs as the most significant cluster of genes affected in these cell types, and our investigation focused on this specific observation (supplemental Tables 3 and 4). In an earlier study of Jurkat cells and CD4+ T cells, we found that Tat associated with the promoters of 2 phosphatases, PTEN and PP2A, and this association was key to the induction of apoptosis in these cells.8 We did not observe Tat association with these promoters in APCs. Accordingly, lack of Tat association with PTEN and PP2A promoters correlated with lack of significant modulation of FOXO3a target genes. It is unclear what determines the specificity of Tat association with different promoters in different cell types. An understanding of this issue is beyond the scope of this study, but it seems likely that Tat association with a specific promoter occurs in the context of defined transcription complexes that target different genes in different cells and not via direct DNA interaction. Different epigenetic status of chromatin in different cells may affect promoter accessibility by the same transcription complex. Future experiments aim at identifying the composition of different transcription complexes that include Tat in different cell types. The experiments that report of increased luciferase activity, when luciferase expression was driven by MAP2K6, MAP2K3, or IRF7 promoter sequences and Tat was expressed, suggest that Tat may not affect transcription of those genes via a TAR-like element. It is more likely that Tat increases RNAPII processivity by increasing the active pool of P-TEFb.31
Transcriptional regulation of ISGs relies on IFN-dependent and IFN-independent pathways. The IFN-dependent pathway involves activation of the IFN receptors by exogenous IFNs and triggering of JAK-STAT signal transduction.32 We did not detect secreted IFNs in the course of HIV infection or Tat expression in the cells investigated. We also found that Tat induced ISG expression in cells where the type I IFN response cannot occur because of deletion of the receptor gene or the type I IFN locus. These results exclude the fact that type I IFN in amounts below8,9 assay detection mediate the transcriptional activation of ISGs in an autocrine way and further support the role of Tat in modulating ISGs in an IFN-independent fashion. IFN-independent expression of some ISGs such as IP10 and TRAIL after TLR7 triggering is stimulated by STAT1, phosphorylated via the p38 MAPK pathway.33,34 This mechanism is consistent with our results. Induction of an IFN-like response can establish an antiviral state that may be key to the lower levels of virus production observed in these cells compared with T cells.35,36 Importantly, activation of cytokines that impact T-cell activation can also contribute to the chronic immunoactivation observed in HIV-infected patients.
Viral infection triggers a subset of ISGs as a consequence of activation of pattern recognition receptors, in particular, TLRs.37,38 TLR signaling can induce a subset of ISGs via activation of IRF3 and IRF7.39 In this study Tat-specific activation of IRF3 was not detected. IRF7 was the only Tat target among the different factors of the IRF family, as we detected association of Tat with the IRF7 but not with other IRFs by ChIP-Chip analysis.34,39,40 IRF7 transcription can be regulated by binding the virus-activated factor (VAF) to the IRF7 interferon-stimulated response element and IRF-binding element.41 The coactivators, CREB-binding protein and p300, together with IRF7 form the VAF complex and induce IRF7 transcription independently of IFN-triggered signaling. Tat may increase transcription of IRF7 by facilitating the recruitment of VAF, which is known to interact with CREB-binding protein and p300, to the IRF7 promoter. We found that modulation of IRF7 transcription was noticeably reduced when a Tat mutant that does not bind p300 or P-TEFb was expressed in iDC (data not shown).
p38 MAPK plays crucial roles in various pathological processes associated with HIV infection, including macrophage activation, neurotoxicity and impairment of neurogenesis, and lymphocyte apoptosis. p38 MAPK inhibition suppresses viral replication and cytopathic effects in T lymphocytes and MDM, possibly by preventing the activation of the HIV-1 Long Terminal Repeat, impairing reverse transcription and compromising proviral integration.42 An increased in active p38 MAPK has been reported in brains of simian immunodeficiency virus–infected macaques with encephalitis.43 Interestingly, p38 MAPK has also been implicated in the production and release of IP10 in astrocytes exposed to HIV-1 and Tat.44-46 In addition, HIV-1 and Tat were reported to activate p38 MAPK in infected or stimulated neurotoxic monocytes and MDM.47 Furthermore, p38 MAPK activation in iDC appears to be critical to the role played by these cells in the differentiation of Th17 helper T cells, which are preferentially targeted by HIV and ultimately are severely depleted, especially in the intestine.48 The relevance of an induced IFN-like phenotype in monocytes during HIV infection has also been suggested by others.49,50 Collectively, these in vivo and in vitro data indicate that p38 MAPK is an important mediator of HIV-associated pathology. Understanding the molecular events that link HIV to p38 MAPK activation may suggest Tat as a target for pharmacological treatment. Our results link the activation of p38 to Tat interaction with MAP2K6, MAP2K3, and IRF7 promoters, establishing how HIV mediates the modulation of the p38 MAPK pathway. Our results also provide a rationale for the development of Tat inhibitors, as they are likely to have fewer side effects than p38 MAPK inhibitors, which are already available, or MAP2K6 and MAP2K3 inhibitors.
In summary, Tat modulation of APCs appears to have 2 major consequences. First, Tat acts as an inflammatory cytokine, modulating chemokines and cytokines linked to increased immunoactivation. Second, Tat acts as an antiviral factor via the modulation proteins that reduce retroviral gene expression, facilitating immune evasion from the adaptive response but supporting sufficient virus production to spread to T cells where virus replication is abundant. This dual arrangement may be important to favor both the persistence and high-level replication of this pathogen in its infected host.
Supplementary Material
Acknowledgment
This work was supported by a grant from the National Institutes of Health, National Institute of Mental Health, to A.A. (MH095671).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: N.K. carried out the bulk of the experimental work with assistance in some experiments from M.D.P.M.-V., and wrote the first draft of the manuscript; S.K. constructed and produced the adenoviral vectors; S.G. analyzed all the ChIP-Chip files and derived the lists of selected genes reported in supplemental Tables 3 and 4; and A.A. designed the experiments, supervised their execution and interpretation, and edited the manuscript into its final form.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Anna Aldovini, Children’s Hospital Boston, 300 Longwood Ave, Boston, MA 02115; e-mail: anna.aldovini@childrens.harvard.edu.
References
- 1.Bieniasz PD. An overview of intracellular interactions between immunodeficiency viruses and their hosts. AIDS. 2012;26(10):1243–1254. doi: 10.1097/QAD.0b013e328353bd04. [DOI] [PubMed] [Google Scholar]
- 2.Van Duyne R, Kehn-Hall K, Carpio L, Kashanchi F. Cell-type-specific proteome and interactome: using HIV-1 Tat as a test case. Expert Rev Proteomics. 2009;6(5):515–526. doi: 10.1586/epr.09.73. [DOI] [PubMed] [Google Scholar]
- 3.Li L, Dahiya S, Kortagere S, et al. Impact of Tat Genetic Variation on HIV-1 Disease. Advances in virology. 2012;2012:123605. [DOI] [PMC free article] [PubMed]
- 4.Brès V, Yoh SM, Jones KA. The multi-tasking P-TEFb complex. Curr Opin Cell Biol. 2008;20(3):334–340. doi: 10.1016/j.ceb.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ott M, Geyer M, Zhou Q. The control of HIV transcription: keeping RNA polymerase II on track. Cell Host Microbe. 2011;10(5):426–435. doi: 10.1016/j.chom.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He N, Zhou Q. New insights into the control of HIV-1 transcription: when Tat meets the 7SK snRNP and super elongation complex (SEC). J Neuroimmune Pharmacol. 2011;6(2):260–268. doi: 10.1007/s11481-011-9267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dabrowska A, Kim N, Aldovini A. Tat-induced FOXO3a is a key mediator of apoptosis in HIV-1-infected human CD4+ T lymphocytes. J Immunol. 2008;181(12):8460–8477. doi: 10.4049/jimmunol.181.12.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim N, Kukkonen S, Gupta S, Aldovini A. Association of Tat with promoters of PTEN and PP2A subunits is key to transcriptional activation of apoptotic pathways in HIV-infected CD4+ T cells. PLoS Pathog. 2010;6(9):e1001103. doi: 10.1371/journal.ppat.1001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Izmailova E, Bertley FM, Huang Q, Makori N, Miller CJ, Young RA, Aldovini A. HIV-1 Tat reprograms immature dendritic cells to express chemoattractants for activated T cells and macrophages. Nat Med. 2003;9(2):191–197. doi: 10.1038/nm822. [DOI] [PubMed] [Google Scholar]
- 10.Kim N, Dabrowska A, Jenner RG, Aldovini A. Human and simian immunodeficiency virus-mediated upregulation of the apoptotic factor TRAIL occurs in antigen-presenting cells from AIDS-susceptible but not from AIDS-resistant species. J Virol. 2007;81(14):7584–7597. doi: 10.1128/JVI.02616-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5(5):375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 12.Rani MR, Ransohoff RM. Alternative and accessory pathways in the regulation of IFN-beta-mediated gene expression. J Interferon Cytokine Res. 2005;25(12):788–798. doi: 10.1089/jir.2005.25.788. [DOI] [PubMed] [Google Scholar]
- 13.Netea MG, Wijmenga C, O’Neill LA. Genetic variation in Toll-like receptors and disease susceptibility. Nat Immunol. 2012;13(6):535–542. doi: 10.1038/ni.2284. [DOI] [PubMed] [Google Scholar]
- 14.St Louis DC, Woodcock JB, Franzoso G, et al. Evidence for distinct intracellular signaling pathways in CD34+ progenitor to dendritic cell differentiation from a human cell line model. J Immunol. 1999;162(6):3237–3248. [PubMed] [Google Scholar]
- 15.Takashiba S, Van Dyke TE, Amar S, Murayama Y, Soskolne AW, Shapira L. Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor kappaB. Infect Immun. 1999;67(11):5573–5578. doi: 10.1128/iai.67.11.5573-5578.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung S, Qureshi SA, Kerr IM, Darnell JE, Jr, Stark GR. Role of STAT2 in the alpha interferon signaling pathway. Mol Cell Biol. 1995;15(3):1312–1317. doi: 10.1128/mcb.15.3.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chartier C, Degryse E, Gantzer M, Dieterle A, Pavirani A, Mehtali M. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J Virol. 1996;70(7):4805–4810. doi: 10.1128/jvi.70.7.4805-4810.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc. 2006;1(2):729–748. doi: 10.1038/nprot.2006.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woelk CH, Ottones F, Plotkin CR, et al. Interferon gene expression following HIV type 1 infection of monocyte-derived macrophages. AIDS Res Hum Retroviruses. 2004;20(11):1210–1222. doi: 10.1089/aid.2004.20.1210. [DOI] [PubMed] [Google Scholar]
- 20.Brown JN, Kohler JJ, Coberley CR, Sleasman JW, Goodenow MM. HIV-1 activates macrophages independent of Toll-like receptors. PLoS ONE. 2008;3(12):e3664. doi: 10.1371/journal.pone.0003664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hauber J, Malim MH, Cullen BR. Mutational analysis of the conserved basic domain of human immunodeficiency virus tat protein. J Virol. 1989;63(3):1181–1187. doi: 10.1128/jvi.63.3.1181-1187.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 23.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 24.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298(5600):1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Batra S, Sassano A, et al. Activation of mitogen-activated protein kinase kinase (MKK) 3 and MKK6 by type I interferons. J Biol Chem. 2005;280(11):10001–10010. doi: 10.1074/jbc.M410972200. [DOI] [PubMed] [Google Scholar]
- 26.Uddin S, Lekmine F, Sharma N, et al. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J Biol Chem. 2000;275(36):27634–27640. doi: 10.1074/jbc.M003170200. [DOI] [PubMed] [Google Scholar]
- 27.Goh KC, Haque SJ, Williams BR. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J. 1999;18(20):5601–5608. doi: 10.1093/emboj/18.20.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lutfalla G, Holland SJ, Cinato E, et al. Mutant U5A cells are complemented by an interferon-alpha beta receptor subunit generated by alternative processing of a new member of a cytokine receptor gene cluster. EMBO J. 1995;14(20):5100–5108. doi: 10.1002/j.1460-2075.1995.tb00192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15(1):11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- 30.Raingeaud J, Whitmarsh AJ, Barrett T, Dérijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16(3):1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barboric M, Yik JH, Czudnochowski N, et al. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007;35(6):2003–2012. doi: 10.1093/nar/gkm063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3(11):900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 33.Ramsauer K, Sadzak I, Porras A, Pilz A, Nebreda AR, Decker T, Kovarik P. p38 MAPK enhances STAT1-dependent transcription independently of Ser-727 phosphorylation. Proc Natl Acad Sci USA. 2002;99(20):12859–12864. doi: 10.1073/pnas.192264999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Domizio J, Blum A, Gallagher-Gambarelli M, Molens JP, Chaperot L, Plumas J. TLR7 stimulation in human plasmacytoid dendritic cells leads to the induction of early IFN-inducible genes in the absence of type I IFN. Blood. 2009;114(9):1794–1802. doi: 10.1182/blood-2009-04-216770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuniga EI, Hahm B, Oldstone MB. Type I interferon during viral infections: multiple triggers for a multifunctional mediator. Curr Top Microbiol Immunol. 2007;316:337–357. doi: 10.1007/978-3-540-71329-6_16. [DOI] [PubMed] [Google Scholar]
- 36.Schmid S, Mordstein M, Kochs G, García-Sastre A, Tenoever BR. Transcription factor redundancy ensures induction of the antiviral state. J Biol Chem. 2010;285(53):42013–42022. doi: 10.1074/jbc.M110.165936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brennan K, Bowie AG. Activation of host pattern recognition receptors by viruses. Curr Opin Microbiol. 2010;13(4):503–507. doi: 10.1016/j.mib.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 38.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1(2):135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 39.Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. 2011;12(6):399–414. doi: 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6(9):644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 41.Ning S, Huye LE, Pagano JS. Regulation of the transcriptional activity of the IRF7 promoter by a pathway independent of interferon signaling. J Biol Chem. 2005;280(13):12262–12270. doi: 10.1074/jbc.M404260200. [DOI] [PubMed] [Google Scholar]
- 42.Furler RL, Uittenbogaart CH. Signaling through the P38 and ERK pathways: a common link between HIV replication and the immune response. Immunol Res. 2010;48(1-3):99–109. doi: 10.1007/s12026-010-8170-1. [DOI] [PubMed] [Google Scholar]
- 43.Barber SA, Uhrlaub JL, DeWitt JB, Tarwater PM, Zink MC. Dysregulation of mitogen-activated protein kinase signaling pathways in simian immunodeficiency virus encephalitis. Am J Pathol. 2004;164(2):355–362. doi: 10.1016/S0002-9440(10)63125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kutsch O, Oh J, Nath A, Benveniste EN. Induction of the chemokines interleukin-8 and IP-10 by human immunodeficiency virus type 1 tat in astrocytes. J Virol. 2000;74(19):9214–9221. doi: 10.1128/jvi.74.19.9214-9221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams R, Yao H, Dhillon NK, Buch SJ. HIV-1 Tat co-operates with IFN-gamma and TNF-alpha to increase CXCL10 in human astrocytes. PLoS ONE. 2009;4(5):e5709. doi: 10.1371/journal.pone.0005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams R, Dhillon NK, Hegde ST, et al. Proinflammatory cytokines and HIV-1 synergistically enhance CXCL10 expression in human astrocytes. Glia. 2009;57(7):734–743. doi: 10.1002/glia.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sui Z, Fan S, Sniderhan L, et al. Inhibition of mixed lineage kinase 3 prevents HIV-1 Tat-mediated neurotoxicity and monocyte activation. J Immunol. 2006;177(1):702–711. doi: 10.4049/jimmunol.177.1.702. [DOI] [PubMed] [Google Scholar]
- 48.Huang G, Wang Y, Vogel P, Kanneganti TD, Otsu K, Chi H. Signaling via the kinase p38α programs dendritic cells to drive TH17 differentiation and autoimmune inflammation. Nat Immunol. 2012;13(2):152–161. doi: 10.1038/ni.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pulliam L, Rempel H, Sun B, Abadjian L, Calosing C, Meyerhoff DJ. A peripheral monocyte interferon phenotype in HIV infection correlates with a decrease in magnetic resonance spectroscopy metabolite concentrations. Aids. 2011;25(14):1721-1726. [DOI] [PMC free article] [PubMed]
- 50.Rempel H, Sun B, Calosing C, Pillai SK, Pulliam L. Interferon-alpha drives monocyte gene expression in chronic unsuppressed HIV-1 infection. Aids. 2010;24(10):1415-1423. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.