

Key Points
MAPK pathway activation and Bim loss may represent a fundamental mechanism of resistance to histone deacetylase inhibitors.
Combination of romidepsin with an MEK inhibitor may lead to greater responses in cancers in which the MAPK pathway is active.
Abstract
To identify molecular determinants of histone deacetylase inhibitor (HDI) resistance, we selected HuT78 cutaneous T-cell lymphoma (CTCL) cells with romidepsin in the presence of P-glycoprotein inhibitors to prevent transporter upregulation. Resistant sublines were 250- to 385-fold resistant to romidepsin and were resistant to apoptosis induced by apicidin, entinostat, panobinostat, belinostat, and vorinostat. A custom TaqMan array identified increased insulin receptor (INSR) gene expression; immunoblot analysis confirmed increased protein expression and a four- to eightfold increase in mitogen-activated protein kinase (MAPK) kinase (MEK) phosphorylation in resistant cells compared with parental cells. Resistant cells were exquisitely sensitive to MEK inhibitors, and apoptosis correlated with restoration of proapoptotic Bim. Romidepsin combined with MEK inhibitors yielded greater apoptosis in cells expressing mutant KRAS compared with romidepsin treatment alone. Gene expression analysis of samples obtained from patients with CTCL enrolled on the NCI1312 phase 2 study of romidepsin in T-cell lymphoma suggested perturbation of the MAPK pathway by romidepsin. Immunohistochemical analysis of Bim expression demonstrated decreased expression in some skin biopsies at disease progression. These findings implicate increased activation of MEK and decreased Bim expression as a resistance mechanism to HDIs, supporting combination of romidepsin with MEK inhibitors in clinical trials.
Introduction
Histone deacetylase inhibitors (HDIs) are potent antitumor agents that lead to increased histone acetylation and altered transcriptional activity.1 To date, most of the responses using HDIs as a single agent were observed in hematologic cancers, particularly T-cell lymphomas.2 Vorinostat and romidepsin have been approved by the US Food and Drug Administration for the treatment of cutaneous T-cell lymphoma (CTCL),3-5 and romidepsin is additionally approved for the treatment of peripheral T-cell lymphoma (PTCL).6
Romidepsin, a cyclic tetrapeptide isolated from the fermentation product of Chromobacterium violaceum, is a potent HDI that inhibits predominantly class I histone deacetylases (HDACs).2,7-10 Like other HDIs, romidepsin induces both cell cycle arrest and apoptosis in vitro.11 We observed the clinical efficacy of romidepsin in patients with PTCL or CTCL in phase 1 testing7 and in a follow-up multi-institutional phase 2 trial.5 However, both acquired and de novo resistance to romidepsin were observed.5 Thus, studies are needed to determine mechanisms of resistance and combination strategies that may lead to increased efficacy.
Early studies with the National Cancer Institute (NCI) Anticancer Drug Screen demonstrated that romidepsin was a substrate for the ATP-binding cassette transporter P-glycoprotein (Pgp).12 Although epigenetic effects of romidepsin, including upregulation of MDR-1/Pgp were observed in normal and malignant peripheral blood mononuclear cells (PBMCs) of patients treated with romidepsin, no evidence for Pgp-mediated resistance emerged in clinical samples obtained from patients with CTCL or PTCL at the time of disease progression.13,14 Other, non-Pgp mechanisms of resistance have been suggested, such as activation of the mitogen-activated protein kinase (MAPK) pathway. Lung cancer cells transfected with constitutively active MAPK kinase (MEK) acquired romidepsin resistance,15 and cell lines harboring activated MEK were more sensitive to combined treatment with romidepsin and an MEK inhibitor than to romidepsin alone.16 Given the frequent MAPK pathway activation in cancer, this could represent a significant mechanism of resistance.
To explore non-Pgp mechanisms of resistance to romidepsin, we generated three romidepsin-resistant sublines by selecting the CTCL cell line HuT78 with romidepsin in the presence of Pgp inhibitors verapamil or valspodar (PSC833) to prevent emergence of the transporter. Although the selected cell lines were highly resistant to romidepsin, they were also resistant to apoptosis mediated by treatment with apicidin, entinostat, panobinostat, belinostat, and vorinostat. Our results suggest that decreased expression of proapoptotic Bim via MAPK pathway activation is a mechanism of resistance to romidepsin as well as other HDIs and underscore the potential importance of Bim in romidepsin-mediated cell death.
Materials and methods
Drug source
Romidepsin (depsipeptide; NSC 630176) was obtained from the NCI Anticancer Drug Screen (Bethesda, MD). Belinostat, entinostat, panobinostat, ABT-737, OSI-906, AS703026, AZD6244, and PD0325901 were obtained from ChemieTek (Indianapolis, IN). AG1024 and LY294002 were obtained from Calbiochem (San Diego, CA). Vorinostat was purchased from Cayman Chemicals (Ann Arbor, MI). Valspodar (PSC833) was a kind gift from Novartis Pharmaceuticals (New York, NY). Puromycin and verapamil were purchased from Sigma Chemicals (St. Louis, MO).
Cell culture
The human CTCL cell line HuT78 and the myeloma cell line RPMI-8226 were obtained from American Type Culture Collection (Manassas, VA). The cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY) with antibiotics at 37°C in 5% CO2. The romidepsin-resistant sublines DpVp35 and DpVp50 were independently selected and were maintained in 35 and 50 ng/mL romidepsin, respectively, in the presence of 5 µM verapamil.17 The DpP75 subline was maintained in 75 ng/mL romidepsin and 3 µg/mL valspodar (PSC833). The Dp500 subline was cultured in 500 ng/mL romidepsin alone and was previously characterized as overexpressing Pgp.17
To develop cells with stable knockdown of Bim, HuT78 parental and DpP75 cells were transduced with MISSION short hairpin RNA (shRNA) lentiviral particles containing empty vector, nontargeting shRNA, or shRNA targeting BCL2L11 (Sigma-Aldrich, St. Louis, MO). Cells were incubated with 2 µg/mL puromycin, and the surviving cells were pooled to generate stably transduced cell lines.
Cytotoxicity assays
Cells were plated in 96-well plates (1000 cells/well), and 4-day cytotoxicity assays were performed by using the CellTiter 96 AQueous One kit (Promega, Madison, WI). Cells were incubated in quadruplicate in varying concentrations of drug for 96 hours.
Flow cytometry
Apoptosis was measured by using the Annexin V-FITC Apoptosis detection kit (BD Biosciences, San Diego, CA). Fluorescein isothiocyanate (FITC) and propidium iodide fluorescence were detected with a FACSort flow cytometer (BD Biosciences). An unpaired, two-tailed Student t test was used to determine significant differences in apoptosis induction, with P < .05 considered significant.
Immunoblot analysis
Whole cell lysates were prepared by using radioimmunoprecipitation assay buffer (50 mM Tris-HCl [pH 7.4],1% Triton X-100, 10% glycerol, 0.1% sodium dodecyl sulfate, 2 mM EDTA, and 0.5% deoxycholic acid, 50 mM NaCl, and 50 mM NaF) with protease inhibitor (Sigma-Aldrich) and phosphatase inhibitor cocktails (Roche). Extracted proteins were electrophoresed, transferred to nitrocellulose (Invitrogen, Carlsbad, CA), and incubated in 5% nonfat milk/Tris-buffered saline or, for phosphoproteins, in LI-COR blocking buffer (LI-COR, Lincoln, NE). Membranes were incubated overnight at 4°C with primary antibody, washed with 0.1% Tween-20 in Tris-buffered saline, and incubated with LI-COR secondary antibody conjugate; the signal was quantitated by using the Odyssey Infrared Imager (LI-COR). The primary antibodies were from Cell Signaling Technology (Beverly, MA), unless otherwise specified: IGF1R, IRS2, Mcl-1, Bcl-xL, Bid, Bax, Bak, and Bim; phosphorylated and total Akt, STAT3, mTOR, MEK, and ERK; total PARP, cleaved-PARP, insulin receptor beta (Santa Cruz Biotechnology, Santa Cruz, CA), and acetylated histone H3 (Millipore, Billerica, MA). GAPDH antibody (American Research Products, Belmont, MA) served as a loading control.
RNA isolation and polymerase chain reaction assays
Total RNA was isolated by using Trizol (Invitrogen), and 1 μg was reverse transcribed by using the High Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems, Foster City, CA) with reverse transcription conditions: 10 minutes at 25°C, 120 minutes at 37°C, 5 minutes at 85°C.
Expression levels of 381 MDR-associated genes were measured by using a custom-made TaqMan Low-Density Array (Applied Biosystems).18 The median expression of each sample was subtracted from all gene expression data for that sample. One of the genes (18S) was present as multiple probes. The expression data from the multiple probes for that gene were averaged together. Relative quantification of genes was performed by using the ΔΔ Ct method.19
Real-time polymerase chain reaction (RT-PCR) was performed by using the Univeral ProbeLibrary System. Complementary DNA (cDNA) was obtained by reverse transcription of 1 µg RNA using random primers, and amplification was done by using specific primers listed in supplemental Table 2. Amplification of RN28S1 served as an internal control. Quantitative RT-PCR was done by using TaqMan Master Mix (Light Cycler Taq Man Master #04535286001; Roche Applied Science) in a LightCycler 480 instrument. PCR amplification was carried out at 95°C for 10 minutes followed by 30 to 35 cycles of 95°C for 10 seconds and 60°C for 10 seconds. Fluorescent signal was acquired at the end of the elongation step of every PCR cycle (72°C for 1 second). PCR results were first normalized by RN28S1, and fold changes were determined by dividing expression values of the genes in the resistant cells by expression in the parental cells; in the patient samples, the treated samples were normalized by untreated controls.
Patient samples, array analysis, and immunohistochemistry
All patient samples were obtained from patients with CTCL enrolled on the NCI1312 phase 2 study of romidepsin administered as a 4-hour infusion at 14 mg/m2 on days 1, 8, and 15 of a 28-day schedule in T-cell lymphoma.5 PBMCs were obtained before infusion (pre), and at 4 hours or 24 hours after the start of the infusion of the first cycle of treatment. Levels of acetylated histone H3 and ABCB1 gene expression were previously reported.14 Samples were hybridized on Illumina WG-8v2 human whole-genome bead arrays by using a constant amount (400 ng) of total RNA. Illumina BeadStudio v.3.0 software was used to export expression levels and detection P values for each probe of each sample. Arrays were quantile normalized and filtered to remove noninformative probes. A probe was regarded as noninformative if it had a detection value of P > .05 in all samples or if the maximum ratio between all sample pairs was below 1.2. For heatmap generation, an annotated list of 17 genes shown to be under the control of the MAPK pathway was selected (supplemental Table 1). All genes in the data set were subject to log transformation and mean centering normalization. As a secondary filter, only genes with a twofold change in expression at 4 hours or 24 hours compared with pretreatment levels (pre) were considered. A heatmap was then generated with genes grouped by hierarchical clustering by using Pearson correlation for the similarity metric and centroid linkage. Transformations and clustering were done by using Cluster 3.0 software, and Java TreeView software was used for heatmap generation.
Immunohistochemical analyses were performed on formalin-fixed, paraffin-embedded skin biopsies obtained pretreatment and at the time the patients went off study (except where noted). After deparaffinization and rehydration, Bim immunohistochemistry was performed with citrate buffer antigen retrieval. Following an endogenous peroxidase block and normal goat serum block, Bim antibody (Cell Signaling Technology) was incubated overnight at 4°C and diluted 1:50. A biotinylated goat anti-rabbit secondary antibody and Vector’s Vectastain Elite ABC (Vector Laboratories) reagent were used in addition to diaminobenzidine for detection. Slides were counterstained in hematoxylin. Cell pellets of HuT78 were used as a positive control. Negative control slides were incubated with a normal rabbit immunoglobulin G monoclonal isotype control in place of Bim. Slides were subsequently scored by a pathologist.
Results
Characterization of romidepsin-resistant HuT78 sublines
We studied the resistant HuT78 sublines DpVp35, DpVp50, and DpP75, each an independent selection in romidepsin (Dp, depsipeptide) in the presence of the Pgp inhibitors verapamil (Vp) or valspodar (P; PSC833) to avoid selection for Pgp expression.13,17,20 Following a 48-hour exposure to romidepsin (1, 10, and 100 ng/mL), significant apoptosis was observed in parental cells at 1 ng/mL romidepsin, and the DpVp35, DpVp50, and DpP75 sublines exhibited minimal apoptosis, even when exposed to high levels of romidepsin (Figure 1A). Cells were then treated with apicidin, entinostat, vorinostat, belinostat, or panobinostat for 48 hours. Although HuT78 cells were sensitive to all HDIs tested, less apoptosis was observed with DpVp35, DpVp50, and DpP75 cells, as shown in Figure 1B. Notably, romidepsin, apicidin and entinostat target predominantly class I HDACs, and the others also target HDAC6.9 In 4-day cytotoxicity assays, the DpVp35, DpVp50, and DpP75 sublines were more than 150-fold resistant to romidepsin compared with parental cells (Table 1). The resistant sublines were not cross-resistant to panobinostat, vorinostat, or belinostat in the 96-hour assay (Table 1), suggesting that the longer exposure may lead primarily to growth arrest in the resistant lines.
Figure 1.
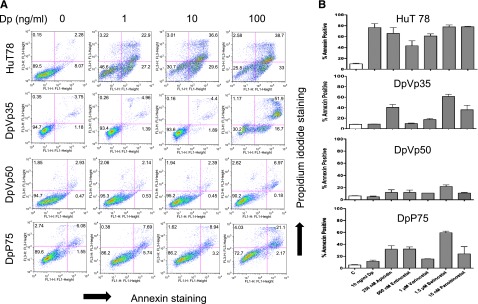
Romidepsin-resistant HuT78 cells are resistant to multiple HDIs. (A) HuT78, DpVp35, DpVp50, and DpP75 cells were incubated with 1, 10, or 100 ng/mL romidepsin (Dp) for 48 hours, after which cells were incubated with anti-annexin V antibody and propidium iodide (PI) as outlined in “Materials and methods.” The percentage of live cells is shown in the lower left quadrant. The percentage of annexin-positive cells was derived from the upper and lower right quadrants. (B) HuT78, DpVp35, DpVp50, and DpP75 cells were treated with the noted concentrations of romidepsin, apicidin, entinostat, vorinostat, belinostat, or panobinostat for 48 hours, and percent annexin-positive cells are presented. Results from at least 3 independent experiments are shown.
Table 1.
Cross-resistance profile of HuT78 parental and romidepsin-resistant sublines to HDIs
| HuT78 | DpVp35 | DpVp50 | DpP75 | |
|---|---|---|---|---|
| Romidepsin | 2.3 ± 0.28 nM | 250 ± 218 nM | 373 ± 162 nM | 387 ± 186 nM |
| Panobinostat | 23 ± 19 nM | 13 ± 1.6 nM | 8 ± 6 nM | 16 ± 2.3 nM |
| Vorinostat | 2 ± 0.5 µM | 1.7 ± 0.29 µM | 2.5 ± 0.51 µM | 1.6 ± 0.21 µM |
| Belinostat | 747 ± 274 nM | 550 ± 50 nM | 800 ± 141 nM | 583 ± 160 nM |
Concentrations at which growth is 50% inhibited (IC50) values were determined by using the 96 Aqueous One Solution Cell Proliferation Assay kit as outlined in “Materials and methods.”
We next examined global histone H3 acetylation as a marker of HDAC inhibition after treatment with various concentrations of romidepsin for 24 hours. Histone H3 acetylation was readily observed in DpP75 cells, although to a lesser extent than observed in the parental line (18-fold vs fourfold; Figure 2A). This was in contrast to Pgp-overexpressing Dp500, in which histone acetylation was not observed unless the Pgp inhibitor valspodar was added (Figure 2B). Flow cytometry for Pgp at the cell surface demonstrated slightly higher levels in DpVp50 and DpP75 cells than in Pgp-negative parental cells but much lower than in Dp500 cells (Figure 2C). The Pgp inhibitor tariquidar did not increase apoptosis in the DpVp50 or DpP75 cells when treated for 48 hours with romidepsin, in contrast to results in Dp500 cells (Figure 2D). Although the apparently reduced histone acetylation in the resistant cells could be due to altered uptake or adaptation to chronic exposure, these results show unequivocally that Pgp is not a significant mechanism of resistance in the DpVp and DpP sublines.
Figure 2.

Resistance to romidepsin in HuT78, DpVp50, and DpP75 cells does not involve P-glycoprotein but does involve the MAPK pathway. (A) Whole-cell lysates were prepared from HuT78 or DpP75 cells after a 24-hour incubation in the noted concentrations of romidepsin (Dp), subjected to polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose. Blots were probed with acetylated histone H3 (AcH3), histone H3 (H3), or GAPDH antibodies. Results from 1 of 3 independent experiments are shown; densitometry of AcH3, with normalization to histone H3 levels, is shown on the bottom row. (B) Pgp-expressing Dp500 cells were incubated with the noted concentrations of romidepsin in the presence or absence of 3 µg/mL of the Pgp inhibitor valspodar (PSC). Whole-cell lysates were prepared, subjected to PAGE, transferred to nitrocellulose, and probed with antibodies to AcH3 and GAPDH as in (A). Results from 1 of 3 independent experiments are shown. (C) HuT78 parental, DpVp50, and DpP75 cells were incubated with either an isotype control antibody (red histogram) or with the anti-Pgp antibody MRK-16 (Kamiya Biomedical, Seattle, WA) to detect cell surface expression (blue histogram) for 30 minutes, after which cells were washed and incubated with a phycoerythrin-labeled secondary antibody (Vector Laboratories, Burlingame, CA) and subsequently analyzed by flow cytometry. (D) HuT78, DpVp50, DpP75, and Dp500 cells were treated with 1 or 10 ng/mL of romidepsin (Dp) for 48 hours in the presence or absence of 200 nM of the Pgp inhibitor tariquidar (Tar.) or with tariquidar alone for 48 hours. Cells were harvested and incubated with anti-annexin V antibody and propidium iodide as outlined in Figure 1. Graphed results are from at least 3 independent experiments. Whole-cell lysates were prepared from HuT78 parental, DpVp35, DpVp50, and DpP75 cells, subjected to PAGE, and transferred to nitrocellulose membranes. The membranes were incubated with antibodies to (E) insulin receptor beta or GAPDH; (F) MEK, phosphorylated MEK (pMEK), ERK, or phosphorylated ERK (pERK); (G) Akt, phosphorylated Akt (pAKT), mTOR, phosphorylated mTOR (p-mTOR), IRS2, or GAPDH; and (H) STAT3, phosphorylated STAT3 (pSTAT3), or GAPDH. (I) HuT78 cells were treated with the noted concentrations of romidepsin for 24 hours. Expression of IR beta and pMEK was then examined by immunoblot analysis of whole-cell lysates. GAPDH served as a loading control. All experiments were performed at least 3 times. C, control.
Romidepsin-resistant HuT78 cells express increased levels of insulin receptor and phosphorylated MEK
Seeking mechanisms of resistance, we determined the expression of 381 genes associated with drug resistance by using a custom-made TaqMan low-density array.21 Gene expression data from untreated HuT78 parental cells was compared with DpVp35 and DpP75 cells (Gene Expression Omnibus [GEO] with accession number: GSE45018). Genes expressed at twofold or higher levels in the DpVp35 cells relative to parental cells are presented in supplemental Table 3; genes decreased by twofold or more are presented in supplemental Table 4. Increased expression of ABCB1, INSR, IGF1R, MMP9, and TGFA was found in DpVp35 cells compared with parental cells, although expression of several signaling pathway genes was downregulated. STAT3 and STAT5B, reported to correlate with resistance to vorinostat in a panel of lymphoma cell lines,22 were downregulated in DpVp35-resistant cells.
Several genes identified in the TaqMan array were examined by RT-PCR in the three sublines (Table 2). In agreement with the array data, expression of INSR was increased 390-fold or more over parental levels in the 3 drug-selected sublines. RT-PCR confirmed the decrease observed with the array in TNFSF10 and ABCC11 expression in the resistant sublines. However, increased expression of IGF1R was not observed by RT-PCR nor was the decrease in NTRK2 expression.
Table 2.
Gene expression results by RT-PCR
| INSR | IGF1R | TNFSF10 | NTRK2 | ABCC11 | ABCB1 | |
|---|---|---|---|---|---|---|
| HuT78 | 1 | 1 | 1 | 1 | 1 | 1 |
| DpVp35 | 390 ± 255 | 0.93 ± 0.30 | 0.52 ± 0.46 | 1.8 ± 0.63 | 0.71 ± 0.53 | 77 ± 31 |
| DpVp50 | 648 ± 262 | 0.69 ± 0.49 | 0.06 ± 0.06 | 1.6 ± 0.96 | 0.77 ± 0.06 | 4.1 ± 3.8 |
| DpP75 | 837 ± 202 | 0.65 ± 0.43 | 0.055 ± 0.057 | 0.48 ± 0.58 | 0.35 ± 0.02 | 14.4 ± 6.0 |
| INSR (fold increase) in HuT78 cells treated with romidepsin | ||||||
| Control | Dp 0.1 ng/mL | Dp 1 ng/mL | Dp 2 ng/mL | Dp 4 ng/mL | ||
| 1 | 0.99 ± 0.14 | 18.95 ± 6.01 | 34.25 ± 12.65 | 25.75 ± 5.86 | ||
Expression of various genes normalized to GAPDH levels as determined by RT-PCR. Gene expression values for resistant sublines were then divided by expression levels for parental cells to obtain the fold increase for each subline.
We explored the expression of insulin receptor (IR) and downstream signaling effectors by immunoblot analysis. Increased expression of IR was found in the resistant sublines, confirming array data (Figure 2E). The mitogenic signal of the IR is transmitted via the MAPK pathway, and the metabolic signal is transmitted via the phosphatidylinositol 3-kinase (PI3K) pathway,23 leading us to examine both the MAPK and the PI3K pathways in the resistant sublines. Phosphorylated MEK levels were five-, four- and eightfold higher in the DpVp35, DpVp50, and DpP75 cells, respectively, compared with parental cells (Figure 2F). In contrast, in the PI3K pathway, we observed low levels of IRS2, a decrease in the levels of pAkt, and no change in phosphorylated mTOR levels (Figure 2G). Additionally, STAT3 was dephosphorylated in the resistant cells (Figure 2H). No consistent changes in EIF4e or S6K phosphorylation were observed (data not shown).
Treatment of the parental cells for 24 hours with increasing concentrations of romidepsin caused enhanced expression of IR at both the transcriptional level (Table 2) and the protein level but no increase in MEK phosphorylation (Figure 2I).
Romidepsin-resistant HuT78 cells exhibit exquisite sensitivity to MEK inhibitors
Because MEK inhibitors induce growth arrest and apoptosis in cells with activated MAPK pathway,24 we examined the effect of MEK inhibitors on the romidepsin-resistant sublines. Cells were treated for 48 hours with the MEK inhibitors AZD6244, AS703026, and PD0325901. Exquisite sensitivity was noted in the resistant cells, with high levels of annexin staining evident after treatment with as little as 1 nM PD0325901 or 50 nM AZD6244 or AS702036 (Figure 3A; data from a representative experiment is shown in supplemental Figure 1). Minimal apoptosis was observed in the parental cells, even following 500 nM MEK inhibitor. ERK dephosphorylation confirmed inhibition of MEK kinase activity in the DpVp50 (Figure 3B) and DpP75 (Figure 3C) cells, following 48-hour treatment with PD0325901 or AZD6244, respectively. The increased apoptosis observed by flow cytometry after treatment with MEK inhibitors was confirmed by increased PARP cleavage (Figure 3B-C). Romidepsin-resistant cells were not sensitive to a 48-hour treatment with insulin receptor inhibitors (15 µM OSI-906 or 25 µM AG1024) or a PI3K inhibitor (5 µM LY294002) (data not shown).
Figure 3.

Romidepsin-resistant cells exhibit exquisite sensitivity to MEK inhibitors and reduction of Bim plays a role in resistance. (A) HuT78 parental, DpVp50, and DpP75 cells were incubated with the noted concentrations of the MEK inhibitors AS703026, PD0325901, and AZD6244 for 48 hours. Cells were then harvested and incubated with anti-annexin V antibody and propidium iodide as outlined in “Materials and methods.” Results are from at least 3 independent experiments. C, control. (B) DpVp50 or (C) DpP75 cells were treated with increasing concentrations of PD0325901 or AZD6244, respectively, for 48 hours. Cells were then harvested and whole-cell lysates were subjected to PAGE, transferred to nitrocellulose membranes, and probed with antibodies against PARP, cleaved PARP (c-PARP), phosphorylated ERK (p-ERK), total ERK, and GAPDH. The experiment was repeated three times. (D) HuT78 cells were treated with 25 ng/mL romidepsin (Dp) for 48 hours, and percent annexin-positive cells were determined as outlined in (A). DpVp50 cells were treated for 48 hours with 25 ng/mL romidepsin alone (Dp) or with 1, 3, or 5 nM PD0325901 (PD), 20 µM OSI-906 (OSI), or 10 µM LY294002 (LY) alone or in combination with 25 ng/mL romidepsin (Dp). Percent annexin-positive cells were determined as in (A). Bars denote the average percentage of cells that stained positively for annexin. Whole-cell lysates were prepared from HuT78 and the romidepsin-resistant lines, subjected to PAGE, and transferred to a nitrocellulose membrane that was probed for the apoptosis-associated proteins (E) Bim as well as (F) Bax, Bid, Bak, Bcl-xL, and Mcl-1. GAPDH served as a loading control. The romidepsin-resistant sublines (G) DpVp50 and (H) DpP75 were treated with the MEK inhibitors AZD6244 and PD0329501, respectively, for 48 hours after which whole-cell lysates were subjected to PAGE and transferred to nitrocellulose. HuT78 parental cells were included as a positive control for Bim in (G). Blots were subsequently probed for PARP, cleaved PARP (c-PARP), Bim, and GAPDH. C, control.
Next, we investigated whether the resistant cells could be resensitized by combining romidepsin with MEK (PD0325901), PI3K (LY294002), or insulin receptor (OSI-906) inhibitors (Figure 3D). The combination experiments were difficult to perform with the MEK inhibitor, given the marked sensitivity noted above. Both parental and resistant cells were treated for 48 hours with 25 ng/mL romidepsin. As expected, significant apoptosis followed romidepsin exposure in parental cells (P < .001) but not in the DpVp50 cells. Addition of MEK inhibitor induced cytotoxicity alone in the resistant cells (P < .002 for all concentrations compared with control). When romidepsin was combined with 1 or 3 nM PD0325901, a statistically significant increase in apoptosis was noted compared with treatment with MEK inhibitor alone (P = .022 and P = .048, respectively). Only a nonsignificant small increase in apoptosis was observed when either LY294002 or OSI-906 was combined with romidepsin.
Bim expression plays a role in apoptosis mediated by romidepsin and MEK inhibitors
In cell line models, activation of the MAPK pathway has been shown to lead to repression of the proapoptotic protein Bim. ERK directly phosphorylates Bim, leading to ubiquitination and degradation by the proteasome.25,26 MEK inhibitors prevent phosphorylation of Bim by ERK, thereby facilitating increased Bim expression and apoptosis.27 Immunoblot analysis showed readily detectable Bim expression in HuT78 cells but reduced Bim expression in the resistant sublines (Figure 3E).
Expression levels of other members of the Bcl-2 family were also assessed (Figure 3F). In drug-sensitive HuT78 cells, high Bim and high levels of prosurvival Mcl-1 were noted. In the resistant sublines, Mcl-1 levels were decreased compared with parental cells; this appeared to be due to loss of pSTAT3, since treatment of HuT78 cells with a STAT3 inhibitor resulted in decreased Mcl-1 (data not shown). Bax levels were similar in all cell lines and Bak levels were slightly elevated in the resistant lines. The resistant cells also expressed increased levels of the prosurvival protein Bcl-xL. Enforced expression of Bcl-xL has been shown to prevent romidepsin-mediated apoptosis in cell line models.28
To determine whether re-expression of Bim correlated with apoptosis in the romidepsin-resistant cells, we examined Bim and cleaved PARP in MEK inhibitor–treated cells. Treatment of DpVp50 (Figure 3G) and DpP75 (Figure 3H) cells with the MEK inhibitors AZD6244 and PD0329501, respectively, for 48 hours restored the level of Bim in the resistant sublines back to parental levels, even at concentrations as low as 1 nM (Figure 3H). Re-expression of Bim was accompanied by increased cell death as shown by increased PARP cleavage—in agreement with the increased annexin V levels measured by flow cytometry shown in Figure 3A.
We next targeted Bim expression in the DpP75 cells with an shRNA to its encoding gene BCL2L11. DpP75 cells stably transduced with empty vector, vector encoding a nontargeting shRNA, or vector containing the shRNA to BCL2L11 were exposed to 250 nM AZD6244 or PD0325901 for 48 hours. The expected induction of Bim and cleaved PARP were notably diminished after MEK inhibitor treatment in cells transduced with the shRNA to BCL2L11 (Figure 4A). Stable transfectants were then exposed to 25 ng/mL romidepsin and 250 nM AZD6244 or PD0325901 for 48 hours and subsequently stained with annexin V and propidium iodide. MEK inhibitors resulted in significant cell death (P < .0001 compared with C or Dp for all 3 transductions), but significantly more cells survived in the BCL2L11 shRNA-transduced cells compared with cells transduced with empty vector or nontargeting shRNA (P < .002 for both AZD6244 and PD0325901; Figure 4B). Similar results were observed in HuT78 cells, in which Bim expression was markedly reduced in HuT78 cells transduced with shRNA to BCL2L11 compared with cells transduced with empty vector or nontargeting shRNA (Figure 4C), and these cells were also more resistant to 48-hour treatment with 1 or 10 ng/mL romidepsin (P < .03 compared with empty vector or nontargeting shRNA) (Figure 4D). These results suggest that Bim plays a role in both apoptosis mediated by MEK inhibitor in the resistant cells and apoptosis mediated by romidepsin in HuT78 cells.
Figure 4.

Characterization of resistance mechanisms to romidepsin. (A) DpP75 cells were transduced with lentiviral particles containing empty vector, a non-targeting shRNA, or an shRNA to BCL2L11 and selected with puromycin to generate stable cell lines. Cells were then incubated in 250 nM AZD6244 (AZD) or PD0325901 (PD) for 48 hours, after which whole-cell lysates were extracted and subjected to immunoblot analysis for Bim and cleaved PARP (c-PARP). GAPDH served as a loading control. (B) Cells from (A) were then treated with 25 ng romidepsin (Dp), 250 nM AZD6244 (AZD), or 250 nM PD0325901 (PD) for 48 hours, after which the cells were incubated with annexin V and PI, and percent live cells was determined. Results compiled from at least 5 independent experiments are shown. (C) HuT78 cells were transduced with lentiviral particles as outlined in (A), after which whole-cell lysates were extracted and subjected to immunoblot analysis for Bim and GAPDH. (D) Cells from (C) were then treated with 1 or 10 ng/mL romidepsin (Dp) for 48 hours after which the cells were incubated with annexin V and PI, and percent live cells was determined. Results compiled from at least 3 independent experiments are shown. (E) HuT78 and DpVp50 cells were incubated with 25 ng/mL romidepsin (Dp) or 1 or 2.5 µM ABT-737 for 48 hours, after which cells were stained with annexin and PI as in (A). Percent annexin-positive cells was determined; results from at least 3 experiments are shown. (F) RPMI-8226 cells were incubated with 25 ng/mL romidepsin alone (Dp) or with 250 nM AZD6244 (Dp AZD) or PD0325901 (Dp PD) for 6 hours, after which media was removed and cells were incubated with romidepsin-free medium alone or continuing with AZD6244 or PD0325901 for an additional 42 hours. As controls, cells were also incubated with 250 nM AZD6244 (AZD) or PD0325901 (PD) alone for 48 hours. Percent annexin positive cells was determined as in (A). Results from at least 4 independent experiments are shown. C, control.
To explore whether the increased Bcl-xL was important in the resistant lines, we incubated HuT78 and DpVp50 cells with the Bcl-2 mimetic ABT-737, which targets cells that rely on expression of prosurvival Bcl-2, Bcl-xL or Bcl-w; cells that rely on Mcl-1 expression are known to be resistant to ABT-737.29 Parental HuT78 cells were resistant to ABT-737 at 1 or 2.5 µM when treated for 48 hours, while ABT-737 induced apoptosis in the DpVp50 cells (Figure 4E). Thus, the romidepsin-selected sublines appear to rely on increased Bcl-xL expression along with the reduction in Bim expression. This contrasts with parental cells, which appear to rely predominantly on Mcl-1 expression.
Short-term romidepsin treatment combined with MEK inhibition is effective in a different hematologic malignancy model with MAPK pathway activation
Our results suggested that cell lines with activation of the MAPK pathway might exhibit inherent resistance to romidepsin treatment and might be sensitized to romidepsin by MEK inhibitors. To address this question, we examined KRAS-mutant RPMI-8226 myeloma cells. To more closely represent the clinical dosing of romidepsin, we treated cells with 25 ng/mL romidepsin for 6 hours followed by a 42-hour incubation in romidepsin-free medium and examined apoptosis via annexin staining (Figure 4F). Approximately 50% of cells were annexin positive with romidepsin alone; a significant increase in apoptosis was observed when combined with 250 nM MEK inhibitors AZD6244 or PD0325901 (P < .006) that alone had little activity. Thus, response to romidepsin treatment can be improved by concomitant treatment with MEK inhibitors in hematologic cancers with activation of the MAPK pathway.
Perturbation of genes controlled by the MAPK pathway and decreased Bim expression in samples from patients treated with romidepsin
Having observed that activation of the MAPK pathway afforded protection from romidepsin-induced cell death, we sought evidence of interaction with the pathway by examining expression of genes regulated by the MAPK pathway. RNA was isolated from PBMCs obtained from 7 patients with circulating CTCL cells collected before and after receiving romidepsin. From a microarray analysis of the samples (GEO with accession number: GSE45405), we sourced expression data for a series of 17 genes that have been shown to be under the control of the MAPK pathway (shown in >3 model systems; see supplemental Table 1 for annotated list). As shown in Figure 5A, the genes clustered into 2 distinct groups: 1 in which gene expression was suppressed at 4 hours and rebounded at 24 hours and 1 in which gene expression increased at 4 hours and then decreased at 24 hours. Quantitative PCR analysis for a selection of MAPK downregulated genes was confirmed in another subset of patients and contrasted with ABCB1 (Figure 5B), which we previously found upregulated in peripheral blood samples taken from patients treated with romidepsin.14 These results demonstrate that romidepsin alters expression of genes controlled by the MAPK pathway, a finding consistent with a role for perturbation of this pathway in romidepsin effect.
Figure 5.
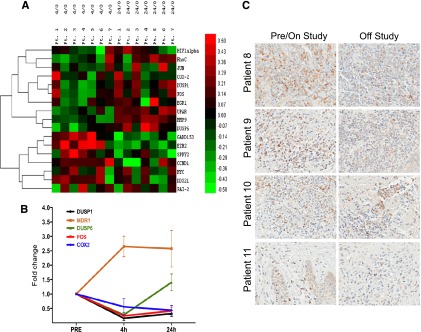
Examination of the MAPK pathway and Bim expression in patient samples. (A) Microarray analysis of total RNA isolated from samples of circulating tumor cells from 7 patients before romidepsin infusion (0 hours), at 4 hours after initiation of the infusion (4 hours), and 24 hours after initiation of the infusion (24 hours). An annotated list of genes that were shown to be under the control of the MAPK pathway was compiled from a series of studies and selected for analysis (supplemental Table 1). Ratios for all genes versus pretreatment values were log transformed and mean centered. An additional filter was applied to the untransformed ratios such that only genes with at least 1 ratio >2 were considered. A heatmap was then generated by using the centroid method for hierarchical clustering and Pearson correlation for the similarity metric. (B) Quantitative PCR analysis of selected MAPK-regulated genes as well as MDR-1 was performed in a subset of patients with CTCL treated with romidepsin. Genes observed to be decreased in the array in (A) were confirmed in a separate set of messenger RNA samples. (C) Bim expression as detected by immunohistochemistry in skin biopsies collected before or while on study (left column) and at disease progression (right column) from 4 patients treated with romidepsin. Magnification ×40.
We also examined Bim expression in skin biopsy samples to look for potential changes after romidepsin treatment. Samples were generally obtained before patients received the first dose of romidepsin, except in the case of patient 8 in whom the sample was obtained after 2 months on study and near the off-study date. As shown in Figure 5C, in patients 8 and 9, Bim expression was readily detectable initially but decreased in the off-study sample. Although tumor shrinkage was observed in patients 8 and 9, neither had a confirmed clinical response. Focal positivity was observed in patient 10, who had a sustained partial remission. Bim expression was low in both samples in patient 11, who experienced only disease progression.
Discussion
Despite the fact that two HDIs have gained approval for the treatment of CTCL and/or PTCL, knowledge of clinical mechanisms of resistance to this class of antiproliferative agents is limited.30 Although Pgp confers transporter-mediated resistance to romidepsin, this was not observed in the clinical samples studied.14 We thus focused on non-Pgp resistance mechanisms, evaluating HuT78 CTCL sublines independently selected in romidepsin in the presence of verapamil or valspodar to prevent the emergence of Pgp-mediated resistance.17 We probed potential resistance mechanisms in the romidepsin-resistant lines by using a high-throughput TaqMan-based quantitative RT-PCR assay,31 finding increased INSR expression as well as increased IR protein and phospho-MEK expression. The MAPK pathway activation evidently led to suppression of the proapoptotic protein Bim, a mechanism of resistance not previously described in romidepsin-selected cells.
Some earlier studies pointed to activation of the MAPK pathway as a potential mechanism of resistance to anticancer agents including romidepsin. In cell lines selected in the mutant BRAF inhibitor vemurafenib (PLX4032), N-RAS upregulation was linked to restoration of MAPK pathway activation and resistance.32 Similarly, erlotinib- or gefitinib-resistant lung cancer cells overexpress MET, leading to increased MEK phosphorylation.33 Transfection of A549 lung carcinoma cells with constitutively active MEK lowered the proapoptotic effects of romidepsin.15 Similarly, Ozaki et al suggested that combination of a MEK inhibitor with an HDI including romidepsin enhanced apoptosis in cells with a constitutively active ERK pathway.16 Our results extend these earlier observations and present evidence that perturbation of the MAPK pathway may lead to resistance to romidepsin-mediated apoptosis. Consistent with these observations are results in clinical samples that show inhibition of MAPK-controlled genes immediately after romidepsin treatment; other MAPK-controlled genes increase immediately after romidepsin treatment. These clinical results together with the HuT78 model suggest that romidepsin alters the basal regulation of the MAPK pathway in malignant T cells. Resistance emerges by upregulation or adaptation of the MAPK pathway to the romidepsin effect. Thus, combination of romidepsin with MEK inhibitors may increase the clinical efficacy of romidepsin in cancers with MAPK pathway activation. MEK inhibitors are currently being evaluated in clinical trials to treat lymphoma (NCT01278615). In separate studies, we have found that MEK inhibitors increase the sensitivity of solid tumor cell lines to romidepsin, suggesting that this mechanism of resistance identified in the HuT78 cell line may be a fundamental way that solid tumor cells evade sensitivity to romidepsin (manuscript in preparation; R.W.R., V.L.L., Julian C. Bahr, A.R.C., Z.Z., Alexandra Zimmer, and S.E.B.).
Bim, a proapoptotic protein that has been shown to interact with antiapoptotic proteins such as Bcl-2, Bcl-xL, and Mcl-1, is known to be transcriptionally regulated by the forkhead box transcription factor class O (FoxO), particularly FoxO3 and FoxO1, and posttranslationally regulated by the MAPK pathway.34 Activation of MAPK leads to phosphorylation of Bim by ERK, resulting in degradation via the proteasome.25,26 Low Bim expression has been shown to confer resistance to cytotoxics, including resistance to paclitaxel in transformed mouse kidney epithelial cells following phosphorylation and degradation of Bim by the RAS/MAPK pathway.35 Cell lines with low basal levels of Bim were shown to respond poorly to treatment with kinase inhibitors, and Bim levels in chemonaive tumor samples predicted clinical benefit from treatment with EGFR inhibitors.36 We detected decreased levels of Bim in romidepsin-resistant HuT78 cells compared with parental cells, suggesting that romidepsin may be less effective in cells with decreased Bim or in cells where romidepsin treatment does not increase levels of Bim. This latter theory is supported by Yang et al, who demonstrated decreased apoptosis in H1299 cells when treated with romidepsin because of a lack of FoxO1-mediated increase in Bim.37 Additionally, cells in which Bim has been deleted have been shown to be resistant to vorinostat treatment in a mouse model of B-cell lymphoma.38
In the clinic, HDIs have shown promising responses as a single agent only in certain hematologic malignancies.2 Romidepsin has significant and sustained single-agent activity in patients with refractory CTCL, with a 34% response rate and duration of response of 11 to 15 months.5 However, despite response to the therapy, resistance emerges in both PTCL and CTCL. Moreover, the effects of romidepsin seen in preclinical and in vitro models have not been translated to the clinic in solid tumors. Studies have shown that HDIs can synergize with a wide range of antitumor agents, and several HDIs, including romidepsin, are currently in clinical trials in combination with different anticancer agents.39 Undoubtedly, resistance to romidepsin will be multifactorial, but our observations of the role of activated MEK in limiting the activity of romidepsin suggest that future clinical trials combining MEK inhibitors with romidepsin may result in significant and sustained responses.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Donna Butcher and Dr Miriam R. Anver of the Histotechnology Laboratory at the Frederick National Laboratory for Cancer Research for the immunohistochemistry analyses.
This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research and by a Cooperative Research and Development Agreement with Celgene Corporation, as well as by NCI grant RO1 CA132098 (to L.C.S.). The Wistar Institute Genomics and Bioinformatics facilities are supported by National Institutes of Health, Cancer Center Support Grant P30 CA010815.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.R.C. performed research, collected data, analyzed and interpreted data, performed statistical analysis, and wrote the manuscript; R.W.R. designed and performed research, collected, analyzed, and interpreted data, performed statistical analysis. and wrote the manuscript; V.L.L. performed research; Z.Z. performed research and collected data; R.L.P. performed research and collected clinical trial data; J.-P.G. performed research and collected, analyzed, and interpreted data; A.V.K. analyzed gene expression data and contributed vital analytic tools; J.W. analyzed data and performed statistical analysis; L.C.S. performed research and collected gene expression data; M.M.G. contributed vital analytical tools; N.L.C. designed research; and S.E.B. designed research, analyzed and interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: This research was supported in part by a cooperative research and development agreement between the National Cancer Institute and Celgene Corporation (S.E.B.). The remaining authors declare no competing financial interests.
Correspondence: Susan E. Bates, National Cancer Institute, National Institutes of Health, 9000 Rockville Pike, Bldg 10, Room 12N226, Bethesda, MD 20892; e-mail: batess@helix.nih.gov.
References
- 1.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1(4):287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 2.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 3.Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res. 2009;15(12):3958–3969. doi: 10.1158/1078-0432.CCR-08-2785. [DOI] [PubMed] [Google Scholar]
- 4.Whittaker SJ, Demierre MF, Kim EJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(29):4485–4491. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 5.Piekarz RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piekarz RL, Frye R, Prince HM, et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011;117(22):5827–5834. doi: 10.1182/blood-2010-10-312603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piekarz RL, Robey R, Sandor V, et al. Inhibitor of histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case report. Blood. 2001;98(9):2865–2868. doi: 10.1182/blood.v98.9.2865. [DOI] [PubMed] [Google Scholar]
- 8.Furumai R, Matsuyama A, Kobashi N, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62(17):4916–4921. [PubMed] [Google Scholar]
- 9.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6(3):238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T, Okuhara M. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J Antibiot (Tokyo) 1994;47(3):301–310. doi: 10.7164/antibiotics.47.301. [DOI] [PubMed] [Google Scholar]
- 11.Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV, Bates SE. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer. 2000;83(6):817–825. doi: 10.1054/bjoc.2000.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee JS, Paull K, Alvarez M, et al. Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute drug screen. Mol Pharmacol. 1994;46(4):627–638. [PubMed] [Google Scholar]
- 13.Robey RW, Zhan Z, Piekarz RL, Kayastha GL, Fojo T, Bates SE. Increased MDR1 expression in normal and malignant peripheral blood mononuclear cells obtained from patients receiving depsipeptide (FR901228, FK228, NSC630176). Clin Cancer Res. 2006;12(5):1547–1555. doi: 10.1158/1078-0432.CCR-05-1423. [DOI] [PubMed] [Google Scholar]
- 14.Bates SE, Zhan Z, Steadman K, et al. Laboratory correlates for a phase II trial of romidepsin in cutaneous and peripheral T-cell lymphoma. Br J Haematol. 2010;148(2):256–267. doi: 10.1111/j.1365-2141.2009.07954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu XD, Wang SY, Chen GA, et al. Apoptosis induced by depsipeptide FK228 coincides with inhibition of survival signaling in lung cancer cells. Cancer J. 2007;13(2):105–113. doi: 10.1097/PPO.0b013e318046eedc. [DOI] [PubMed] [Google Scholar]
- 16.Ozaki K, Minoda A, Kishikawa F, Kohno M. Blockade of the ERK pathway markedly sensitizes tumor cells to HDAC inhibitor-induced cell death. Biochem Biophys Res Commun. 2006;339(4):1171–1177. doi: 10.1016/j.bbrc.2005.11.131. [DOI] [PubMed] [Google Scholar]
- 17.Piekarz RL, Robey RW, Zhan Z, et al. T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistance. Blood. 2004;103(12):4636–4643. doi: 10.1182/blood-2003-09-3068. [DOI] [PubMed] [Google Scholar]
- 18.Gillet JP, Wang J, Calcagno AM, et al. Clinical relevance of multidrug resistance gene expression in ovarian serous carcinoma effusions. Mol Pharm. 2011;8(6):2080–2088. doi: 10.1021/mp200240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamada H, Arakawa Y, Saito S, Agawa M, Kano Y, Horiguchi-Yamada J. Depsipeptide-resistant KU812 cells show reversible P-glycoprotein expression, hyper-acetylated histones, and modulated gene expression profile. Leuk Res. 2006;30(6):723–734. doi: 10.1016/j.leukres.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 21.Gillet JP, Gottesman MM. Advances in the molecular detection of ABC transporters involved in multidrug resistance in cancer. Curr Pharm Biotechnol. 2011;12(4):686–692. doi: 10.2174/138920111795163931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fantin VR, Loboda A, Paweletz CP, et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008;68(10):3785–3794. doi: 10.1158/0008-5472.CAN-07-6091. [DOI] [PubMed] [Google Scholar]
- 23.Belfiore A, Malaguarnera R. Insulin receptor and cancer. Endocr Relat Cancer. 2011;18(4):R125–R147. doi: 10.1530/ERC-11-0074. [DOI] [PubMed] [Google Scholar]
- 24.Rusconi P, Caiola E, Broggini M. RAS/RAF/MEK inhibitors in oncology. Curr Med Chem. 2012;19(8):1164–1176. doi: 10.2174/092986712799320510. [DOI] [PubMed] [Google Scholar]
- 25.Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22(43):6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 26.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278(21):18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 27.Meng J, Fang B, Liao Y, Chresta CM, Smith PD, Roth JA. Apoptosis induction by MEK inhibition in human lung cancer cells is mediated by Bim. PLoS ONE. 2010;5(9):e13026. doi: 10.1371/journal.pone.0013026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newbold A, Lindemann RK, Cluse LA, Whitecross KF, Dear AE, Johnstone RW. Characterisation of the novel apoptotic and therapeutic activities of the histone deacetylase inhibitor romidepsin. Mol Cancer Ther. 2008;7(5):1066–1079. doi: 10.1158/1535-7163.MCT-07-2256. [DOI] [PubMed] [Google Scholar]
- 29.van Delft MF, Wei AH, Mason KD, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fantin VR, Richon VM. Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin Cancer Res. 2007;13(24):7237–7242. doi: 10.1158/1078-0432.CCR-07-2114. [DOI] [PubMed] [Google Scholar]
- 31.Orina JN, Calcagno AM, Wu CP, et al. Evaluation of current methods used to analyze the expression profiles of ATP-binding cassette transporters yields an improved drug-discovery database. Mol Cancer Ther. 2009;8(7):2057–2066. doi: 10.1158/1535-7163.MCT-09-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104(52):20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillings AS, Balmanno K, Wiggins CM, Johnson M, Cook SJ. Apoptosis and autophagy: BIM as a mediator of tumour cell death in response to oncogene-targeted therapeutics. FEBS J. 2009;276(21):6050–6062. doi: 10.1111/j.1742-4658.2009.07329.x. [DOI] [PubMed] [Google Scholar]
- 35.Tan TT, Degenhardt K, Nelson DA, et al. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell. 2005;7(3):227–238. doi: 10.1016/j.ccr.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 36.Faber AC, Corcoran RB, Ebi H, et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1(4):352–365. doi: 10.1158/2159-8290.CD-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Zhao Y, Liao W, et al. Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia. 2009;11(4):313–324. doi: 10.1593/neo.81358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindemann RK, Newbold A, Whitecross KF, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci USA. 2007;104(19):8071–8076. doi: 10.1073/pnas.0702294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bots M, Johnstone RW. Rational combinations using HDAC inhibitors. Clin Cancer Res. 2009;15(12):3970–3977. doi: 10.1158/1078-0432.CCR-08-2786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.