

Key Points
Targeting APCs to enhance GVT.
CD8+ DCs are important for optimizing antitumor responses after allogeneic bone marrow transplantation.
Abstract
The graft-versus-tumor (GVT) effect after allogeneic hematopoietic cell transplantation (allo-HCT) represents an effective form of immunotherapy against many malignancies. Meaningful separation of the potentially curative GVT responses from graft-versus-host disease (GVHD), the most serious toxicity following T-cell replete allo-HCT, has been an elusive goal. GVHD is initiated by alloantigens, although both alloantigens and tumor-specific antigens (TSAs) initiate GVT responses. Emerging data have illuminated a role for antigen-presenting cells (APCs) in inducing alloantigen-specific responses. By using multiple clinically relevant murine models, we show that a specific subset of host-derived APCs—CD8+ dendritic cells (DCs)—enhances TSA responses and is required for optimal induction of GVT. Stimulation of TLR3, which among host hematopoietic APC subsets is predominantly expressed on CD8+ DCs, enhanced GVT without exacerbating GVHD. Thus, strategies that modulate host APC subsets without direct manipulation of donor T cells could augment GVT responses and enhance the efficacy of allo-HCT.
Introduction
Allogeneic hematopoietic cell transplantation (allo-HCT) is a curative treatment for malignant hematopoietic diseases.1 The curative potential of allo-HCT is a consequence of the graft-versus-tumor (GVT) responses.1 However, the beneficial GVT responses are tightly linked to the potentially life-threatening complications of graft-versus-host disease (GVHD), which has limited wider application of allo-HCT.2 Most patients who undergo allo-HCT receive hematopoietic stem cells and T cells from human leukocyte antigen–matched but genetically nonidentical donors.2 In these patients, the primary antigenic targets of donor T cells responsible for GVHD are the host minor histocompatibility alloantigens (miHAs).3-5 Alloantigens are also critical for GVT responses.3 In addition to the miHAs, donor T cells respond to tumor-specific antigens (TSAs) that are either virally encoded and/or mutated tumor antigens that may represent additional important targets for GVT responses without causing concomitant GVHD, although this remains to be definitively determined.3,4 Augmenting GVT responses through elucidation of relevant TSAs and T cells that specifically respond to them has been clinically challenging. The alloantigens derived primarily from the endogenously polymorphic peptides in the host target tissues—hematopoietic-derived and nonhematopoietic-derived antigen-presenting cells (APCs)—are presented directly either to donor CD8+ or CD4+ T cells by major histocompatibility complex (MHC) class I and class II molecules, respectively, and induce GVHD.3 Experimental evidence demonstrates an obligatory role for host hematopoietic APCs in GVT response.6-8 The role of different hematopoietic-derived APC subsets and their relative importance in graft-versus-leukemia (GVL) response is not known. By using several clinically relevant murine and tumor models, we have found that host CD8α+ dendritic cells (DCs) are required for optimal GVT response without aggravating GVHD.
Materials and methods
Mice
Female C57BL/6 (B6, H-2b, CD45.2+), C3H.sw (H-2b, CD45.2+; Jackson Laboratory, Bar Harbor, ME), B6 Ly5.2 (H-2b, CD45.1+), and BALB/c (H-2d) mice (Charles River Laboratories, Wilmington, MA) and B6 β2m−/− mice (H-2b, CD45.2+) were obtained from Taconic (Hudson, NY). Batf3−/− mice on a 129 background were provided by Kenneth Murphy (Washington University School of Medicine, St. Louis, MO) and were backcrossed by ≥ 6× generations onto B6 background at our facility. Tlr3−/− mice on a B6 background were obtained from Gary Luker (University of Michigan, Ann Arbor, MI).9 All animals were cared for under regulations reviewed and approved by the University Committee on Use and Care of Animals of the University of Michigan, based on University Laboratory Animal Medicine guidelines.
Generation of BM chimeras
Bone marrow (BM) chimeras were generated as described before.6,10,11 Briefly, B6 Ly5.2 wild-type (WT) animals were administered 11 Gy total-body irradiation (TBI; 137Cs source) and then injected intravenously with BM and whole spleen cells from WT B6 on day 1 or Tlr3−/− donor mice on day 0. Donor hematopoietic chimerism was confirmed by using the CD45.2 monoclonal antibody 3 to 4 months after BM transplantation (BMT; donor type >95.0%).
BMT
BMTs were performed as described previously.6,12-14 Briefly, splenic T cells from donors were enriched while the BM was depleted of T cells by autoMACS (Miltenyi Biotec, Bergisch Gladbach, Germany). WT B6 and Batf3−/− animals received 10 Gy (137Cs source) on day −1 and 0.5 × 106 CD8+ T cells along with 5 × 106 T-cell–depleted BM (TCD-BM) from either syngeneic B6 or allogeneic C3H.sw donors. For studies in which the recipients were BM chimeras, we induced GVHD 3 to 5 months after the generation of BM chimeras as described previously.6,12,13 Briefly, the chimeras received 9 Gy TBI on day −1 and were injected intravenously with 5 × 106 BM and 0.5 × 106 CD8+ T cells from either the syngeneic B6 or allogeneic C3H.sw donors on day 0. Survival was monitored daily, and recipients’ body weight and GVHD clinical scores were measured weekly.
Induction of leukemia and lymphoma
Tumors (MBL-2 or EL-4, H-2b) were introduced during BMT at two different doses as described previously.6 MBL-2 is a Moloney murine leukemia virus–induced T-cell lymphoma,15,16 and EL-4 is a chemically induced T-cell lymphoma.17 Both are of B6 origin (H-2b) and were extensively used as models of acute leukemia and lymphoma. We attributed death to tumor if tumor was present at necropsy and death and to GVHD only if no tumor was evident and there was histologic evidence of GVHD. We established the presence of tumor by macroscopic and microscopic visualization of tumor cells in liver and spleen and/or by hind-leg paralysis.18 Mice surviving beyond the observation period of BMT were euthanized for histologic evaluation to determine leukemia- and lymphoma-free survival.
Systemic and histopathologic analysis of GVHD
We monitored survival daily after BMT and assessed the degree of clinical GVHD weekly as described previously.19 Histopathologic analysis of liver and gastrointestinal tract (primary GVHD target organs) was performed as described20 by using a semiquantitative scoring system by a single pathologist (C.L.).20 After scoring, the codes were broken and data were compiled.
Splenic DC isolation
Splenic CD11c+ DCs were enriched by 2-step immunomagnetic bead enrichment. Briefly, spleens were minced and digested with collagenase D (Roche) for 25 to 40 minutes at 37°C. After incubation, cells were washed and preincubated with the rat anti-mouse FcR monoclonal antibody (mAb) 2.4G2 (eBioscience, San Diego, CA) for 15 minutes at 4°C to block nonspecific FcR binding of labeled antibodies. Next, cells were incubated with the APC-conjugated B220 and CD90.2 mAbs (BioLegend, San Diego, CA) for 10 minutes at 4°C and then incubated with anti-APC microbeads (Miltenyi Biotec) for 15 minutes. Single-cell suspensions were negatively isolated to deplete B cells, T cells, and plasmacytoid DCs by autoMACS (Miltenyi Biotec). The remaining negative populations were incubated with CD11c microbeads (Miltenyi Biotec) and then isolated with positive selection per the manufacturer’s protocol. The purity of the enriched CD11c+ DC preparations was >70%. In some experiments, splenic CD8+ DCs were isolated by using a CD8+ DC isolation kit (Miltenyi Biotec) per the manufacturer’s protocol, and the remaining negative populations were incubated with CD11c microbeads (Miltenyi Biotec) and then isolated with positive selection (these are CD8− DCs).
FACS analysis
Fluorescent-activated cell sorter (FACS) analyses were performed as described previously.12,13 Briefly, to analyze chimerism, splenocytes from transplanted mice were resuspended in FACS wash buffer (2% bovine serum albumin in phosphate-buffered saline) and stained with conjugated mAbs—fluorescein isothiocynate (FITC)–conjugated mAbs to mouse CD4, CD8, CD44, and CD45.2; phycoerythrin (PE)-conjugated mAbs to CD25, CD62L, CD69, CD80, CD86, PD-L1, and I-Ab; interferon gamma (IFN-γ); granzyme B; FoxP3; allophycocyanin-conjugated mAbs to CD45.1 and CD229.1; and PerCPcy5.5-conjugated CD25 (eBioscience). The procedure was performed as described previously.13 Cells were analyzed by using an FACS Vantage SE (Becton Dickinson, San Jose, CA) or C6 cytometer (BD Accuri cytometers; BD Biosciences, Ann Arbor, MI). For intracellular staining (FoxP3, IFN-γ, granzyme B, and CD107a), 2.0 to 3.0 × 106 cells per well were stained for cell surface markers, fixed, and permeabilized per the manufacturer’s protocol. IFN-γ and granzyme B production by donor T cells on day 14 were assessed following in vitro restimulation with 10 µg/mL CD3 and 1 ng/mL CD28 for 5 hours with brefeldin A (eBioscience). Cells underwent intracellular staining for cell surface markers and were fixed and permeabilized per the manufacturer’s protocol.
Cytokine enzyme-linked immunosorbent assay
Cytokines were measured in serum and culture supernatants by enzyme-linked immunosorbent assay with specific anti-mouse mAbs for IFN-γ and interleukin-17A (IL-17A; BioLegend) according to the manufacturer’s protocol and read at 450 nm by using a microplate reader (Model 3550; Bio-Rad Laboratories, Hercules, CA). All samples and standards were run in duplicate.
Analysis of MBL-2–specific cytotoxic T cells
Donor MBL-2–specific CD8+ T cells were analyzed on day14 after BMT with the immunodominant PE-conjugated peptide tetramer CCLCLTVFL epitope, a gag-encoded protein of Friend-Moloney-Rauscher retrovirus that is recognized in the context of H-2b.15,16 An H-2Db–restricted influenza peptide (DbPA, SSLENFRAYV) was used as negative control. Tetramers were made by the National Institutes of Health tetramer core facility (Atlanta, GA). Briefly, splenocytes from transplanted mice were resuspended in FACS wash buffer (2% bovine serum albumin in phosphate-buffered saline), stained with conjugated mAbs, incubated with PE-conjugated gag-specific tetramer, washed twice with FACS wash buffer, fixed with 1× BD FACS Lysing Solution (BD Biosciences), and analyzed by using a C6 cytometer (BD Biosciences).
Mixed DC and tumor reaction imaging
Splenic DCs (1 × 105) labeled with CellTracker Green 5-chloromethylfluorescein diacetate (CMFDA; Molecular Probes, Eugene, OR) were cocultured with 1 × 105 EL-4 cells stably transduced with mCherry in 6-well plates for 6 hours. Samples were imaged with an Olympus FluoView 1000 laser scanning confocal microscope using ×25 (XLPlan N ×25 MP; NA = 1.05) and ×60 (LUMPlanFLN ×60; NA = 1.00) water immersion objectives. The Green CMFDA and mCherry were excited with 488-nm and 559-nm laser lines, respectively. The green fluorescent signal was detected with a wavelength range of 490 to 590 nm, and the red fluorescent signal was detected with a wavelength range of 570 to 580 nm. All the fluorescent images (1024 × 1024 pixels; 12 bits, bitmap) were collected under identical photomultiplier tube high voltage (∼600V) and pixel integration time (12.5 μs/pixel).
Luciferase+ MBL-2 cell line
MBL-2 cells were transduced with a third-generation lentivirus coexpressing green fluorescent protein (GFP) and firefly luciferase.21-23 Briefly, 0.5 × 106 fresh MBL-2 cells were suspended in 1 mL of 10% Dulbecco’s modified Eagle medium (DMEM) and 8 μg/mL of polybrene in a 6-well plate. Cells were centrifuged at 2000 rpm at 32°C for 30 minutes. Cells were then cultured for 7 hours at 37°C. One milliliter of fresh media (total 2 mL) was added for overnight incubation. On day 2, MBL-2 cells were washed with fresh 10% DMEM and allowed to expand for 24 hours. Stably transduced cells were sorted (FACS Diva) for high expression of GFP and were recultured in vitro for 2 to 4 days with 10% DMEM.
Bioluminescence imaging
Bioluminescence imaging was performed with a cryogenically cooled charge-coupled device (CCD) camera (IVIS; Caliper, Hopkinton, MA). Acquisition and analysis of images were performed as previously described.23 All animals were imaged 10 minutes after being injected intraperitoneally with 100 μL (40 mg/mL) of firefly d-luciferin (Biosynth AG, Staad, Switzerland). Animals were imaged 30 seconds to 5 minutes, depending on the signal strength. All animals were maintained under isoflurane anesthesia in a 37°C heated environment.
In vivo treatment with polyI:C
For the in vivo experiments, the [B6→B6Ly5.2] and [Tlr3−/−→B6Ly5.2(F)] chimeras received 900 cGy TBI (137Cs source) on day −1 and were then transplanted on day 0 with 5 × 106 BM cells plus isolated 0.5 × 106 CD8+T cells from donor mice along with 2 × 104 MBL-2 cells. Polyinosinic:polycytidylic acid (polyI:C, 2.5 mg/kg; Amersham/GE Healthcare, Piscataway, NJ) or the control diluents were administered by intraperitoneal injection on days 0 and +1 after BMT.
Cross-presentation of tumor antigen assay
The CD8+ T cells isolated from B6 mice were stained with carboxyfluorescein succinimidyl ester (CFSE; Invitrogen, Eugene, OR). Then, 5 × 104 CFSE-labeled CD8+ T cells were incubated with 5 × 104 CD11c+ DCs from either WT B6 or Batf3−/− animals, 5 × 104 irradiated class I−/− splenocytes from β2m−/− animals, and MBL-2 tumor lysates in round-bottom 96-well plates for 4 days and were analyzed by using an Accuri C6 cytometer (BD Biosciences).
Statistical analysis
The Mann-Whitney U test was used for the statistical analysis of in vitro data and clinical scores. We plotted survival curves by using Kaplan-Meier estimates, and the Wilcoxon rank-sum test was used to analyze survival data. A P value < .05 was considered statistically significant.
Results
Host CD8+ DCs are dispensable for induction of GVHD across MHC-matched minor antigen disparate BMTs
We investigated the role of host DC subsets in an MHC-identical, multiple miHA-mismatched murine model, analogous to most human allo-HCTs. We first compared the severity of GVHD in C57BL/6 (B6, H-2b) WT mice and the basic leucine zipper transcription factor ATF-like3 (Batf3−/−) –deficient B6 animals (these are deficient only in CD8α+ DCs).9,24-26 They were lethally irradiated with 10 Gy on day −1 and were transplanted with 0.5 × 106 CD8+ T cells and 5 × 106 TCD-BM cells from either syngeneic B6 or allogeneic MHC-matched, miHA-mismatched C3H.sw donors.6 All of the syngeneic recipients survived with no signs of GVHD. By contrast, the allogeneic WT animals showed significantly greater mortality and clinical severity of GVHD than the syngeneic animals (Figure 1A-B). The allogeneic Batf3−/−B6-recipient animals showed mortality, clinical severity, and GVHD-specific histopathologic damage in the target organs similar to that of the WT allogeneic recipients (supplemental Tables 1 and 2). Donor CD8+ T-cell expansion, effector memory (CD44+CD62L−), central memory (CD44+CD62L+), naive (CD44−CD62L+), IFN-γ+, granzyme B+, CD4+CD25+FoxP3+ T cells and serum levels of IFN-γ and IL-17A were similar in both the allogeneic WT and Batf3−/− animals on days 14 (Figure 1C-I) and 21 (supplemental Figure 1A-B), demonstrating that the activation, expansion, and differentiation of alloreactive T cells was not altered in the absence of host CD8+ DC subsets.
Figure 1.

Host CD8+ DCs are dispensable for induction of allo-reactivity and GVHD severity. (A) Survival (n = 6 to 17 per group, pooled from 2 experiments) and (B) clinical GVHD score after allo-HCT (n = 6 to 17 per group, pooled from 2 experiments). (C) Donor CD45.2+CD8+ T cells (syngeneic) or CD229.1+CD8+ T cells (allogeneic) (n = 8 to 12 per group, pooled from 3 experiments) expansion in spleen on day 14. (D) The subpopulation of donor-type CD8+ T cells. Effector memory (EM; CD62L−CD44+), central memory (CM; CD62L+CD44+) and naive (CD62L+CD44−) CD8+ T cells were based on the expression of CD62L and CD44 (n = 6 to 8 per group, pooled from 2 experiments). (E) Donor-type IFN-γ+ and (F) granzyme B+ CD8+ T cells (n = 8 to 17 per group, pooled from 3 experiments) in spleen on day 14. (G) Donor-type CD4+CD25+Foxp3+ regulatory T cells on day 14 (n = 8 to 12 per group, pooled from 3 experiments). Serum levels of (H) IFN-γ and (I) IL-17A on day 14 (n = 8 to12 per group, pooled from 3 experiments). (C-I) The bars represent mean ± standard deviation. N.S. denotes no significant differences (P > .05).
Absence of host CD8+ DCs reduces antitumor responses after allogeneic BMT
Experimental evidence demonstrates a crucial role for APCs in GVT response.6,27 We next examined whether the GVT responses were distinct from GVHD in the absence of host CD8+ DCs.9 To simultaneously evaluate GVT responses, we infused 2 × 104 MBL-2 (H-2b) tumor cells at the time of transplantation. MBL-2 is a Moloney murine sarcoma virus/Moloney murine leukemia virus–induced T-cell lymphoma16,17 that is syngeneic to the host B6 mouse and is uniformly lethal at a dose of 5 × 103 cells in a syngeneic setting.6 MBL-2 cells express the immune-dominant TSA, the gag-encoded protein,16 in addition to the miHAs that can be recognized by the donor C3H.sw CD8+ T cells.6 All of the syngeneic animals showed no signs of GVHD and died of tumor burden, demonstrating that TSA responses alone are insufficient for elimination of the tumor cells in these models (Figure 2A). Although all of the allogeneic B6 animals showed signs of GVHD, only 45% of them died of tumor, thus demonstrating the generation of a significant GVT response and GVHD (Figure 2A). By contrast, even though the allogeneic Batf3−/− mice demonstrated significantly prolonged survival when compared with syngeneic animals and all showed signs of GVHD, 100% eventually died of tumor (P = .004; Figure 2A). Thus the allogeneic Batf3−/− hosts demonstrated significantly poor GVT response, despite showing similar GVHD severity when compared with WT animals. To further determine the magnitude of GVT response, we transplanted as above but with a 75% reduction in the tumor dose. Once again, 100% of the syngeneic animals demonstrated tumor without evidence of acute GVHD, while all of the allogeneic WT animals demonstrated GVHD, but only 20% of them had tumors at necropsy (Figure 2B). By contrast, despite all of the Batf3−/− hosts showing similar signs of GVHD, almost 90% of them showed tumors at necropsy (Figure 2B; P < .0001). Collectively, these data suggest that the allogeneic Batf3−/− animals showed significantly reduced GVT response, despite generating similar alloreactivity and GVHD, demonstrating the requirement of host CD8+ DCs for generating an effective GVT response.
Figure 2.
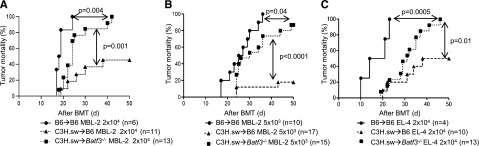
Absence of host CD8+ DCs reduces GVT responses. (A) Tumor mortality data of 2 × 104 MBL-2 cells (n = 6 to 11 per group, pooled from 2 experiments) or (B) 5 × 103 MBL-2 cells (n = 10 to 17 per group, pooled from 3 experiments). (C) Tumor mortality data of 2 × 104 EL-4 (n = 4 to 13 per group, pooled from 2 experiments).
To investigate for any potential intrinsic role of tumors in the altered GVT responses, we also used EL-4 leukemia cells in similar GVT response experiments. EL-4 is a B6 MHC class II negative/low T-cell leukemia/lymphoma (H-2b) and is thus syngeneic (H-2b) to B6 hosts and allogeneic to C3H.sw donors.18 Only the allogeneic Batf3−/− animals demonstrated reduced GVT responses against EL-4 tumors (Figure 2C).
Presence of host CD8+ DCs optimizes GVT responses
In order to evaluate the mechanisms of GVT effect induced by CD8+ DCs per se, we cotransferred either WT splenic CD8+ DCs or CD8− DCs to hosts at the time of allo-BMT. We used β2-microglobulin–deficient (B2m−/−) mice that are functionally incapable of efficient antigen presentation to donor CD8+ T cells on either endogenous host–derived hematopoietic or nonhematopoietic cells.6 The B2m−/− animals were irradiated and injected with TCD BM and 0.5 × 106 CD8+ T cells from C3H.sw donors. All recipient animals were injected with 2 × 104 MBL-2 tumor cells alone or along with 1 × 106 B6 splenic CD8+ or CD8− DCs derived from WT mice. To determine GVT-specific responses, we analyzed donor T-cell responses against immune-dominant TSA, the CCLCLTVFL epitope of gag protein16,17 which is present only in the tumor cells and not in the host tissues. The frequency (2.34% ± 0.67%; n = 6) and absolute number (0.08 ± 0.04 × 106) of gag-tetramer+CD8+ T cells from CD8+ DC-transferred animals were significantly higher than in those animals that received either no DC transfer (1.30% ± 0.52%; P = .26; 0.007 ± 0.002 × 106; P = .04; n = 6) or those animals that received CD8− DCs (1.01% ± 0.32%; P = .04; 0.01 ± 0.0004 × 106; P = .02; n = 6) on day 14 after allo-HCT (Figure 3A-B). We also found that absolute tumor cell numbers were decreased in CD8+ DC-transferred animals (Figure 3C). These data demonstrated that the GVL effect was dependent on the CD8+ DCs.
Figure 3.

CD8+ DCs are required for optimal GVT effect. Host CD8+ DCs are able to increase GVT effect in allo-HCT. β2m−/− animals received 10 Gy on day −1 and 0.5 × 106 CD8+ T cells along with 5 × 106 TCD-BM from allogeneic C3H.sw donors; syngeneic 2 × 104 MBL-2 tumor cells and 1 × 106 splenic CD8+ DCs or CD8− DCs were added at the time of BMT. MBL-2–specific CD8+ T cells were analyzed by using gag-tetramer staining. (A) The representative histogram of gag expression in CD229.1+CD8+ on day 14 after allo-HCT. The percentage of gag positivity in allogeneic β2MG−/− with host-type CD8+ DCs was 1.8. (B) The absolute number of donor-derived gag-tetramer+CD8+ T cells (n = 6 per group). (C) Absolute number of infiltrated GFP+MBL-2 tumor cells in spleen (n = 6 per group). (D) Impaired GVL response is the intrinsic effect of the absence of Batf3 on DCs. β2m−/− animals received 10 Gy on day −1 and 0.5 × 106 CD8+ T cells along with 5 × 106 TCD-BM from either syngeneic B6 or allogeneic C3H.sw donors; syngeneic MBL-2 tumor cells and 2 × 106 splenic DCs from either B6 WT or Batf3−/− mice were added at the time of BMT. Survival (n = 7 to 11 per group, pooled from 2 experiments). (E) Splenic DC phenotype. The frequency of CD8+ DCs and the expression of costimulation molecules of CD11c+ cells were analyzed from WT B6 and the naive Batf3−/− animals that were not transplanted (n = 3 to 5 per group, pooled from 2 experiments). The bars represent mean ± standard deviation. (F) Tumor mortality data of 2 × 104 MBL-2 cells (n = 9 to 22 per group, pooled from 3 experiments) in the 10× generations Batf3−/− hosts.
Because the purity of transferred DCs was ∼70%, it is possible that some additional host-type stem cells or precursors were transferred into these animals that could affect host-derived hematopoietic recovery in the allogeneic animals. However, all of the allogeneic animals demonstrated similar levels of donor chimerism at 21 days after BMT (>90%), regardless of whether they received DCs. Nonetheless, these experiments cannot definitively rule out the possibility of the potential impact of host hematopoietic recovery.
Next, to further confirm whether the reduction in GVT response in Batf3−/−recipients is due only to a cell-autonomous defect in the host hematopoietic APCs (absence of CD8α+ DCs), we performed DC add-back experiments from Batf3−/− and WT B6 animals into B2m−/− animals by using an experimental design similar to the one described above. The B2m−/− recipient animals were injected with 2 × 104 MBL-2 tumor cells alone or with 2 × 106 spleen DCs derived from WT or Batf3−/− mice. All syngeneic animals died of tumor (Figure 3D). Similarly, all (100%) of the B2m−/− allogeneic animals that did not receive any host-type DCs also died of tumor, although a significant number of the B2m−/−allogeneic animals that received WT B6 spleen DCs demonstrated tumor-free survival, indicating the generation of a GVT response. The phenotype of the transferred DCs from WT and Batf3−/− animals was analyzed. As shown in Figure 3D, the animals that received either WT or Batf3−/− cells demonstrated similar expression of costimulatory molecules and were distinct only in the numbers of CD8+ DCs, suggesting that the impact on TSA responses was not due to impact of maturation of the transplanted DCs. Thus, consistent with previous observations, tumor cells (which express class I) by themselves were not directly capable of sufficiently activating the allogeneic or TSA CD8+ T cells to induce a robust GVT response in the absence of functional host hematopoietic-derived APCs.6 However, all of the B2m−/− allogeneic animals that received Batf3−/− spleen DCs demonstrated tumor at necropsy (P = .03; Figure 3D), demonstrating the generation of an ineffective GVT response despite the presence of all other DC subsets except the CD8+ DCs.
The above experiments were performed with Batf3−/− animals that were backcrossed 6× onto B6 background. These animals showed very few to no CD8+ DCs (supplemental Figure 2C). However, when we backcrossed the animals 10× onto B6 background, we observed that animals showed higher, but still significantly reduced, numbers of CD8+ DCs in comparison with WT animals, consistent with a recent article suggesting development of a Batf3 independent manner.28 To further examine whether the emergence of CD8+ DCs modulated GVL response, we performed additional experiments with 10× Batf3−/− animals and compared the results with those from WT recipients. As shown in Figure 3F, these animals still demonstrated reduced tumor elimination compared with WT animals (50% vs 10% tumor-induced mortality; P = .02). However, the tumor elimination was superior to that observed with 6× Batf3−/− animals (50% vs 80% tumor-induced mortality; Figure 3D,F). Tumor elimination after allo-BMT is maximal in WT mice and reduced in 10× Batf−/− but is minimal in 6× Batf−/− recipients; this correlated with the presence and numbers of CD8+ DCs and suggests that optimal GVL responses are dependent on host CD8+ DCs. Alternatively, it is possible that the CD8+ DCs that develop in the absence of Batf3−/− might be functionally different from those that develop in WT animals, but this remains to be tested.
CD8+ DCs are required for induction of TSA responses
We next determined the mechanisms for the dependence of GVT responses on the host CD8+ DCs. Because GVHD is driven by alloantigens and was similar in WT and Batf3−/− recipients after MHC-matched BMT, we reasoned that the differential effect on GVT response might be from lack of efficient presentation of TSAs in the absence of host CD8+ DCs following allo-HCT.6,29,30
We first analyzed whether there was a difference in the ability of the DCs to take up tumor cells in the presence of or absence of Batf3. We cultured both spleen CD8+ DCs and CD8− DCs that were freshly isolated and stained them with Cell Tracker Green CMFDA (Molecular Probes) from WT B6 with mCherry-transduced EL-4 tumor cells. As shown in Figure 4A, although CD8− DCs are known to be the dominant population responsible for uptake of exogenous antigen, CD8+ DCs were able to take up more tumors than CD8− DCs (34.1% ± 6.1% vs 13.6% ± 3.5%; P = .01) in this assay. This assay, however, does not rule out the potential impact of trogocytosis.29 Next, we directly evaluated the ability of the WT and Batf3−/− DCs for functional presentation of tumors to CD8+ T cells. The WT or Batf3−/− splenocytes were enriched for CD11c, cultured with irradiated MHC class I−/− splenocytes and CFSE-labeled CD45.1+ CD8+ T cells, and pulsed with MBL-2 tumor cells.9 The WT DCs induced significant CD8+ T-cell proliferation (supplemental Figure 2A-B). To directly demonstrate in vivo the relevance of the presentation capacity of tumors by the WT DCs but not by the Batf3−/− in the presentation and generation of an effective GVT response, we analyzed the generation of TSA CD8+ T-cell responses following allo-HCT. We analyzed T-cell responses to the immune-dominant TSA—the CCLCLTVFL epitope of gag protein16,17—which is present only in the tumor cells and not in the host tissues. The frequency (0.46% ± 0.11%; n = 11) and absolute number (2.52 ± 0.77 × 104) of gag-tetramer+CD8+ T cells of Batf3−/− animals were significantly less than in WT animals (1.2% ± 0.26%; P = .02; 6.62 ± 1.72 × 104: P = .04; n = 11) on day 14 after allo-HCT (Figure 4C-D), demonstrating the lack of an efficient generation of TSA T-cell response in the absence of host CD8+ DCs. Collectively, these data demonstrate that presentation of nonallogeneic TSAs or tumor-specific miHAs, along with alloantigens by the host CD8+ DC subsets to donor T cells is required to elicit an effective GVT response.
Figure 4.

CD8+ DCs are required for cross-presentation of TSAs and generation of TSA donor T-cell responses. (A) The representative confocal microscopy images revealed efficient B6 WT CD8+ DC uptaking tumor cells. DCs and tumor cells are shown labeled with green (Cell Tracker Green CMFDA) and red (mCherry) fluorescent protein, respectively. Yellow arrows represent overlap of both fluorescent signals. (B) The percentage of engulfed DCs per high-power field (HPF) in either CD8+ DCs or CD8− DCs (n = 7, pooled from 2 experiments). (C) Representative data from donor-derived Gag-specific CD8+ T cells of either allogeneic WT B6 animals (upper panels) or Batf3−/− animals (lower panels) on day 14 after allo-BMT. Negative control hemagglutinin-tetramer staining (left) and Gag-tetramer staining (right). (D) The frequency and absolute number of donor-type Gag+CD8+ T cells (n = 7 to 11, pooled from 3 experiments). The bars represent mean ± standard deviation.
TLR3 expression on host APCs is required for the generation of an effective GVT response
Toll-like receptor 3 (TLR3) is critical for presentation of viral double-stranded RNA.24,31,32 Among the DC subsets, TLR3 is predominantly expressed on CD8+ DC subsets. Therefore, we next used Tlr3−/− mice as hosts to correlate the relevance of host CD8+ DCs on GVHD and GVT response following allo-HCT. B6 WT and Tlr3−/− animals were lethally irradiated and transplanted with CD8+ T cells and TCD-BM from either B6 or C3H.sw donors along with 2 × 104 MBL-2 tumor cells. The allogeneic WT B6 mice demonstrated GVHD, and only 10% of them had tumors at necropsy (supplemental Figure 3A). By contrast, 70% of the allogeneic Tlr3−/− B6 animals had tumors at necropsy, despite demonstrating similar severity of GVHD (P = .01; supplemental Figure 3A). TLR3 is primarily, but not exclusively, expressed on CD8α+ DC subsets.31,32 To analyze the impact of TLR3 deficiency on only host hematopoietic-derived APCs without the confounding effect of its deficiency on the host nonhematopoietic cells, we generated BM chimeras. WT B6 Ly5.2 animals were lethally irradiated and infused with BM and splenocytes from syngeneic Ly5.1 WT B6 or Tlr3−/− donors to generate the [WT-B6→B6Ly5.2] and [Tlr3−/−B6→B6Ly5.2] chimeras. These animals were used as recipients 4 months later in allo-BMT. They were lethally irradiated and transplanted with 0.5 × 106 CD8+ T cells along with 5 × 106 TCD-BM from either syngeneic B6 or allogeneic C3H.sw donors. The [WT-B6→B6Ly5.2] and the [Tlr3−/−B6→B6Ly5.2] animals showed similar survival, histopathologic and clinical severity of GVHD (Figure 5A-C; supplemental Figure 3B). Donor T-cell expansion, donor IFN-γ+ T cells, donor granzyme B+ T cells, serum levels of IFN-γ, IL-17A, and GVHD-specific histopathology were equivalent in the allogeneic [Tlr3−/−B6→B6Ly5.2] animals compared with the allogeneic [WT-B6→B6Ly5.2] animals (Figure 5D-F; supplemental Figure 3C-D).
Figure 5.

TLR3 expression on host APCs is critical for GVT response but not for GVHD. (A) Survival of the [WT-B6→B6Ly5.2] and the [Tlr3−/−B6→B6Ly5.2] animals (n = 6 to 12 per group, pooled from 2 experiments). Histopathologic GVHD scores of (B) gut (small and large intestine) and (C) liver on day 14 after allo-HCT (n = 6 to 9 per group, pooled from 2 experiments). (D) Donor CD45.2+CD8+ T cell (syngeneic) or CD229.1+CD8+ T cell (allogeneic) (n = 6 to 9 per group, pooled from 2 experiments) expansion in spleen on day 14. (E) Donor-type IFN-γ+ and (F) granzyme B+ CD8+ T cells (n = 6 to 9 per group, pooled from 2 experiments) in spleen on day 14. (B-F) The bars represent mean ± standard deviation. (G) Tumor mortality data for the [WT-B6→ B6Ly5.2] and the [Tlr3−/−B6→B6Ly5.2] animals with 2 × 104 MBL-2 tumor cells (n = 7 to 19 per group, pooled from 3 experiments). (H) Representative photos from the bioluminescence study on day 21. (I) Tumor mortality data for 5 × 103 MBL mice (n = 6 to 10 per group, pooled from 2 experiments). (J) Tumor mortality data for MHC-mismatched BMT model (BALB/c→[WT-B6→B6Ly5.2] or [Tlr3−/−B6→B6Ly5.2] animals with 2 × 104 MBL-2 cells (n = 3 to 13 per group, pooled from 2 experiments). N.S., not significant.
When [WT-B6→B6Ly5.2] and [Tlr3−/−B6→B6Ly5.2] chimeras were transplanted as above, along with 2 × 104 MBL-2 tumor cells, a majority of the allogeneic [WT-B6→B6 Ly5.2] animals eliminated tumor (only 20% had tumor at necropsy). By contrast, 87.5% of allogeneic [Tlr3−/−→B6Ly5.2] animals had tumor at necropsy (P < .0001; Figure 5G). The data were also confirmed by using bioluminescence imaging, which clearly demonstrated the progression of tumor growth in the allogeneic [Tlr3−/−→B6Ly5.2] animals but not in the allogeneic [WT-B6→B6 Ly5.2] animals (Figure 5H; supplemental Figure 3E).
To analyze the loss of magnitude of GVT response, the tumor dose was decreased by 75% reduction of the above dose (5 × 103 MBL-2 tumor cells). All (100%) of the syngeneic animals died with tumors. By contrast, 10% of the allogeneic [WT-B6→B6Ly5.2] animals died with tumor, and 62.5% of the [Tlr3−/−→B6Ly5.2] animals died with tumors (P = .03; Figure 5I). We also confirmed the decreased GVT responses by using a different MHC-mismatched BALB/c→[WT-B6→B6Ly5.2] or [Tlr3−/−B6→B6Ly5.2] model (P = .01; Figure 5J).
TLR3 stimulation enhances GVT response without exacerbating GVHD
To confirm the specificity of TLR3 and to determine whether enhancing cross-presentation can augment GVT responses, we treated the [WT-B6→B6Ly5.2] and [Tlr3−/−B6→B6Ly5.2] chimeras with either 50 μg polyI:C (a TLR-3–specific ligand shown to promote cross-presentation)32 per mouse (day 0 and 1) or with diluent control with or without the MBL-2 tumor cells after allo-HCT. Treatment with polyI:C did not alter GVHD severity in either the [WT-B6→B6Ly5.2] or [Tlr3−/−B6→B6Ly5.2] allogeneic animals, demonstrating no clinically significant enhancement of alloreaction. However, it significantly reduced tumor-induced mortality in the allogeneic [WT-B6→B6Ly5.2] animals (50% vs 9.3%; P < .01) but did not significantly alter tumor-induced death in the [Tlr3−/−→B6Ly5.2] animals (100% vs 93%; P = not significant; Figure 6A) and was confirmed with bioluminescence imaging (Figure 6B). These data suggest that (1) enhancement of TSA-only responses (in syngeneic animals with polyI:C) alone was insufficient to eliminate tumor and (2) enhancement of TSA responses only in the presence of alloantigen-driven responses is required for optimal tumor elimination. Thus, collectively, they demonstrate that GVT responses can be maximized without aggravating GVHD by enhancing TLR3 responses in the host hematopoietic-derived APCs.
Figure 6.
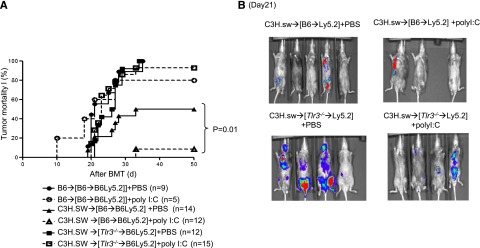
Stimulation of TLR3 on host hematopoietic APCs enhances GVT response. BMT and tumor induction were performed in [WT-B6→B6Ly5.2] and [Tlr3−/−B6→B6Ly5.2] animals as described in “Materials and methods.” The recipient animals received TLR3 stimulant polyI:C or the diluent control and were analyzed for GVHD and tumor-free survival. (A) Tumor mortality data for [WT-B6→B6Ly5.2] and [Tlr3−/−B6→B6Ly5.2] animals with 2 × 104 MBL-2 cells (n = 5 to 14 per group, pooled from 2 experiments). (B) Representative photos from bioluminescence study at day 21 after allo-BMT.
Discussion
The GVT effect after allo-HCT represents a potent form of immunotherapy for hematologic malignancies. Both clinical and experimental data have shown that donor cytotoxic T lymphocytes are critical for GVT.3 Unfortunately, T cells that are specific for tumor alloantigens are also shared by the host tissues, and this largely explains the tight linkage of GVT response and GVHD.3 Although tumor cells also express immunogenic TSAs, it has been challenging to identify and harness T cells specific against TSAs before clinical transplantation. Thus, an effective separation of GVT response from GVHD is the central problem in the field of allo-HCT.33 We show that under certain conditions, targeting specific host APC subsets might be a novel and viable strategy for separating GVT response from GVHD. In multiple tumor and allo-HCT models, we demonstrate an important role for host CD8+ DCs in the activation of responses specific to TSAs but not to alloantigens, thus inducing a robust GVT response without significantly aggravating GVHD.
Alloantigens are expressed by all host APC subsets, and these antigens are presented by them directly to the donor CD8+ T cells. The host leukemias/tumors likewise express alloantigens, but also additionally express TSAs. However, even when they express APC-like features, they likely have undergone a process of immune editing, making them poor direct stimulators of an effective T-cell response through a variety of immune-suppressive mechanisms.34 Therefore, to generate an optimal GVT response by the donor T cells, the TSAs are preferentially presented on host hematopoietic-derived subsets of professional APCs—the CD8+ DCs. Nonetheless, despite the inferior tumor elimination, the allorecipients that lacked CD8+ DCs still demonstrated an increase in tumor-free survival (Figure 2) when compared with the syngeneic animals. In addition, absence of an alloantigen response despite polyI:C stimulation in the syngeneic animals (Figure 6) also did not lead to an efficient tumor elimination. These data are consistent with the notion that TSA-driven antitumor responses only are not sufficient in these models and also that alloantigen-driven antitumor responses only mediate some GVT responses.4 Collectively, they show that direct alloantigen-driven responses are required for GVT response.6 By contrast, the presence of both TSA and alloantigen-induced responses (Figures 1, 4, 5, and 6) is required for the most efficient GVT response. It is therefore possible that anti-alloantigen (miHA) responses enhance anti-TSA responses. However, our data do not formally rule out the possibility that C3H.sw T cells might simply be more responsive than B6 T cells against certain TSAs.
Our data are consistent with the data that CD8α+ DCs are critical for presentation of tumor and viral antigens and now extend those observations to the context of alloresponses.9,32,35 Although CD8α+ DCs usually compose between 5% and 15% of the murine spleen DC compartment and are not observed in humans, a recent study demonstrated that BDCA3 (CD141)+ conventional DCs in humans are equivalent of murine CD8α+ DCs,36-40 suggesting that it may be possible to enhance GVT response in humans by enhancing the function of these DC subsets.
Most CD8α+ DCs highly express TLR3,41 and we show that TLR3 expression on host hematopoietic APCs is also important for inducing optimal GVT response. The experiments with polyI:C demonstrated that an effective immune adjuvant can be used to generate a more robust GVT response after allo-HCT. Although the specific use of polyI:C may be limited to virally induced tumors/leukemias,32 it is important to note that the immune-dominant miHAs that drive GVH responses in this model system are known. However, that is not the case in most clinical BMTs. Therefore, in experimental or clinical instances in which endogenous viruses contribute to immune-dominant miHAs that drive GVH responses, stimulation of TLR3 might also lead to enhanced GVHD along with enhanced GVT response. Our observations, nonetheless, underscore the principle of enhancing antigen presentation on a subset of host APCs as a strategy for increasing the efficacy of allo-HCT.
It is also possible that in the chimera experiments, some host APC subsets are radio-resistant and therefore might persist.42 But any residual WT host APCs in these chimeras were clearly not sufficient to induce effective GVT responses. Our tumor models also may not accurately reflect all aspects of spontaneously occurring malignancies.43 However, these tumors possess all the characteristic hallmarks of malignancy and have been useful in elucidating effector mechanisms of immunotherapy and in uncovering underlying biological principles of allo-HCT.43,44 In addition, the rapid kinetics of the tumor-induced mortality in these models suggest that our observations are most applicable to acute leukemia and other aggressive hematologic malignancies rather than indolent malignant diseases. Furthermore, the rapidity of GVT responses in our model system and the lack of availability of knockout (Batf3 and β2MG) mice and relevant tumors in other strain backgrounds have precluded careful and direct analyses of the role of donor-derived CD8+ DCs. Clinically, our findings might therefore be particularly relevant to high-risk acute leukemias that are likely to relapse despite clinical GVHD.
When taken in light of recent observations that host DCs are not critical for induction of GVHD, our results suggest that either enhancing the numbers and/or modulating the function of the specific host hematopoietic APC subsets might be an effective way to enhance GVT responses without substantially impacting GVHD.10,11,45,46 Collectively, our findings have implications for clinical allo-HCT and suggest that strategies targeting specific host APC subsets could maximize GVT responses without concomitantly exacerbating GVHD.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health: AI075284, HL090775, and CA143379 (P.R.); and R01CA136553, R01CA136829, and P50CA093990 (G.L.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: T.T. designed and performed research, analyzed data, and wrote the paper; Y.S., G.L., and J.L. performed research and analyzed data; K.E.L., I.T., and R.E. performed research; C.L. performed research and analyzed data; N.M., C.M., and E.N. performed research; S.C. provided intellectual input; K.M.M. provided intellectual input and contributed new reagents and mice; and P.R. conceived the project, designed experiments, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Pavan Reddy, 3312 Comprehensive Cancer Center, 1500 East Medical Center Dr, Ann Arbor, MI 48109-0942; e-mail; reddypr@umich.edu.
References
- 1.Appelbaum FR. Haematopoietic cell transplantation as immunotherapy. Nature. 2001;411(6835):385–389. doi: 10.1038/35077251. [DOI] [PubMed] [Google Scholar]
- 2.Welniak LA, Blazar BR, Murphy WJ. Immunobiology of allogeneic hematopoietic stem cell transplantation. Annu Rev Immunol. 2007;25:139–170. doi: 10.1146/annurev.immunol.25.022106.141606. [DOI] [PubMed] [Google Scholar]
- 3.Bleakley M, Riddell SR. Molecules and mechanisms of the graft-versus-leukaemia effect. Nat Rev Cancer. 2004;4(5):371–380. doi: 10.1038/nrc1365. [DOI] [PubMed] [Google Scholar]
- 4.Vincent K, Roy DC, Perreault C. Next-generation leukemia immunotherapy. Blood. 2011;118(11):2951–2959. doi: 10.1182/blood-2011-04-350868. [DOI] [PubMed] [Google Scholar]
- 5.van den Brink MR, Burakoff SJ. Cytolytic pathways in haematopoietic stem-cell transplantation. Nat Rev Immunol. 2002;2(4):273–281. doi: 10.1038/nri775. [DOI] [PubMed] [Google Scholar]
- 6.Reddy P, Maeda Y, Liu C, Krijanovski OI, Korngold R, Ferrara JL. A crucial role for antigen-presenting cells and alloantigen expression in graft-versus-leukemia responses. Nat Med. 2005;11(11):1244–1249. doi: 10.1038/nm1309. [DOI] [PubMed] [Google Scholar]
- 7.Mapara MY, Kim YM, Wang SP, Bronson R, Sachs DH, Sykes M. Donor lymphocyte infusions mediate superior graft-versus-leukemia effects in mixed compared to fully allogeneic chimeras: a critical role for host antigen-presenting cells. Blood. 2002;100(5):1903–1909. doi: 10.1182/blood-2002-01-0023. [DOI] [PubMed] [Google Scholar]
- 8.Billiau AD, Fevery S, Rutgeerts O, Landuyt W, Waer M. Crucial role of timing of donor lymphocyte infusion in generating dissociated graft-versus-host and graft-versus-leukemia responses in mice receiving allogeneic bone marrow transplants. Blood. 2002;100(5):1894–1902. doi: 10.1182/blood-2002-02-0419. [DOI] [PubMed] [Google Scholar]
- 9.Hildner K, Edelson BT, Purtha WE, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toubai T, Tawara I, Sun Y, et al. Article I. Induction of acute GVHD by sex-mismatched H-Y antigens in the absence of functional radio-sensitive host hematopoietic-derived antigen-presenting cells. Blood. 2012;119(16):18:3844-3853. doi: 10.1182/blood-2011-10-384057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shlomchik WD, Couzens MS, Tang CB, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285(5426):412–415. doi: 10.1126/science.285.5426.412. [DOI] [PubMed] [Google Scholar]
- 12.Reddy P, Sun Y, Toubai T, et al. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J Clin Invest. 2008;118(7):2562–2573. doi: 10.1172/JCI34712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toubai T, Sun Y, Tawara I, et al. Ikaros-Notch axis in host hematopoietic cells regulates experimental graft-versus-host disease. Blood. 2011;118(1):192–204. doi: 10.1182/blood-2010-12-324616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teshima T, Ordemann R, Reddy P, Gagin S, Liu C, Cooke KR, Ferrara JL. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. 2002;8(6):575–581. doi: 10.1038/nm0602-575. [DOI] [PubMed] [Google Scholar]
- 15.Chen W, Qin H, Chesebro B, Cheever MA. Identification of a gag-encoded cytotoxic T-lymphocyte epitope from FBL-3 leukemia shared by Friend, Moloney, and Rauscher murine leukemia virus-induced tumors. J Virol. 1996;70(11):7773–7782. doi: 10.1128/jvi.70.11.7773-7782.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Facchinetti A, Dalla Santa S, Mezzalira S, Rosato A, Biasi G. A large number of T lymphocytes recognize Moloney-murine leukemia virus-induced antigens, but a few mediate long-lasting tumor immunosurveillance. J Immunol. 2005;174(9):5398–5406. doi: 10.4049/jimmunol.174.9.5398. [DOI] [PubMed] [Google Scholar]
- 17.van Hall T, van Bergen J, van Veelen PA, et al. Identification of a novel tumor-specific CTL epitope presented by RMA, EL-4, and MBL-2 lymphomas reveals their common origin. J Immunol. 2000;165(2):869–877. doi: 10.4049/jimmunol.165.2.869. [DOI] [PubMed] [Google Scholar]
- 18.Reddy P, Maeda Y, Hotary K, Liu C, Reznikov LL, Dinarello CA, Ferrara JL. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc Natl Acad Sci USA. 2004;101(11):3921–3926. doi: 10.1073/pnas.0400380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J, Jr, Crawford JM, Ferrara JL. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88(8):3230–3239. [PubMed] [Google Scholar]
- 20.Hill GR, Cooke KR, Teshima T, et al. Interleukin-11 promotes T cell polarization and prevents acute graft-versus-host disease after allogeneic bone marrow transplantation. J Clin Invest. 1998;102(1):115–123. doi: 10.1172/JCI3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith MC, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, Luker GD. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64(23):8604–8612. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- 22.Contag CH, Jenkins D, Contag PR, Negrin RS. Use of reporter genes for optical measurements of neoplastic disease in vivo. Neoplasia. 2000;2(1-2):41–52. doi: 10.1038/sj.neo.7900079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duran-Struuck R, Tawara I, Lowler K, et al. A novel role for the semaphorin Sema4D in the induction of allo-responses. Biol Blood Marrow Transplant. 2007;13(11):1294–1303. doi: 10.1016/j.bbmt.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Satpathy AT, Murphy KM, Kc W. Transcription factor networks in dendritic cell development. Semin Immunol. 2011;23(5):388–397. doi: 10.1016/j.smim.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mashayekhi M, Sandau MM, Dunay IR, et al. CD8α(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity. 2011;35(2):249–259. doi: 10.1016/j.immuni.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edelson BT, Bradstreet TR, Hildner K, et al. CD8α(+) dendritic cells are an obligate cellular entry point for productive infection by Listeria monocytogenes. Immunity. 2011;35(2):236–248. doi: 10.1016/j.immuni.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chakraverty R, Sykes M. The role of antigen-presenting cells in triggering graft-versus-host disease and graft-versus-leukemia. Blood. 2007;110(1):9–17. doi: 10.1182/blood-2006-12-022038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tussiwand R, Lee WL, Murphy TL, et al. Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature. 2012;490(7421):502–507. doi: 10.1038/nature11531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amigorena S, Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr Opin Immunol. 2010;22(1):109–117. doi: 10.1016/j.coi.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 30.Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10(6):403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- 31.Miyake T, Kumagai Y, Kato H, et al. Poly I:C-induced activation of NK cells by CD8 alpha+ dendritic cells via the IPS-1 and TRIF-dependent pathways. J Immunol. 2009;183(4):2522–2528. doi: 10.4049/jimmunol.0901500. [DOI] [PubMed] [Google Scholar]
- 32.Schulz O, Diebold SS, Chen M, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433(7028):887–892. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 33.Appelbaum FR. Hematopoietic-cell transplantation at 50. N Engl J Med. 2007;357(15):1472–1475. doi: 10.1056/NEJMp078166. [DOI] [PubMed] [Google Scholar]
- 34.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21(2):137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 35.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8alpha+ dendritic cells. J Exp Med. 2011;208(10):2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lauterbach H, Bathke B, Gilles S, et al. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J Exp Med. 2010;207(12):2703–2717. doi: 10.1084/jem.20092720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poulin LF, Salio M, Griessinger E, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med. 2010;207(6):1261–1271. doi: 10.1084/jem.20092618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bachem A, Güttler S, Hartung E, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med. 2010;207(6):1273–1281. doi: 10.1084/jem.20100348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crozat K, Guiton R, Contreras V, et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med. 2010;207(6):1283–1292. doi: 10.1084/jem.20100223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jongbloed SL, Kassianos AJ, McDonald KJ, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010;207(6):1247–1260. doi: 10.1084/jem.20092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edwards AD, Diebold SS, Slack EM, et al. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol. 2003;33(4):827–833. doi: 10.1002/eji.200323797. [DOI] [PubMed] [Google Scholar]
- 42.Merad M, Hoffmann P, Ranheim E, et al. Depletion of host Langerhans cells before transplantation of donor alloreactive T cells prevents skin graft-versus-host disease. Nat Med. 2004;10(5):510–517. doi: 10.1038/nm1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reddy P, Negrin R, Hill GR. Mouse models of bone marrow transplantation. Biol Blood Marrow Transplant. 2008;14(1 Suppl 1):129–135. doi: 10.1016/j.bbmt.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 45.Koyama M, Kuns RD, Olver SD, et al. Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat Med. 2011;18(1):135–142. doi: 10.1038/nm.2597. [DOI] [PubMed] [Google Scholar]
- 46.Li H, Demetris AJ, McNiff J, et al. Profound depletion of host conventional dendritic cells, plasmacytoid dendritic cells, and B cells does not prevent graft-versus-host disease induction. J Immunol. 2012;188(8):3804–3811. doi: 10.4049/jimmunol.1102795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.