

Abstract
Dehydroepiandrosterone-sulfate (DHEAS) is a hormone produced by the adrenal gland and is a precursor for both androgens and estrogens. Atherosclerosis is a well characterized inflammatory disease, but little is known about the role of DHEAS in vascular inflammation. We hypothesize that DHEAS can reduce inflammation in vascular endothelial cells and the mechanism involves the peroxisome proliferator-activated receptor α (PPARα), thereby inhibiting transcription factors involved in endothelial cell inflammation. To test our hypothesis, aortic endothelial cells were pretreated for 48 hours with DHEAS, then with TNF-α. TNF-α-induced upregulation of the expression of inflammatory genes interleukin (IL)-8 and intracellular adhesion molecule (ICAM)-1 was attenuated by incubation with DHEAS. DHEAS inhibited the TNF-α-induced surface expression of vascular cell adhesion molecule (VCAM)-1. This effect was abolished by the addition of MK866, a PPARα inhibitor, indicating that PPARα is involved in the mechanism of this inhibition. The addition of the aromatase inhibitor letrozole had no effect on the inhibition of TNF-α-induced VCAM-1 expression by DHEAS. Treatment of endothelial cells with DHEAS dramatically inhibited the TNF-α-induced activation of NF-κB, an inflammatory transcription factor, and increased protein levels of the NF-κB inhibitor, IκB-α. These results signify the ability of DHEAS to directly inhibit the inflammatory process and show a potential direct effect of DHEAS on vascular inflammation that has implications for the development of atherosclerotic cardiovascular disease.
Keywords: Dehydroepiandrosterone sulfate, VCAM, PPAR, endothelial cell, inflammation
1. Introduction
Dehydroepiandrosterone-sulfate (DHEAS) and its precursor dehydroepiandrosterone (DHEA) are the most abundant circulating steroids in the human adult (Baulieu et al., 1965; Orentreich et al., 1984). While both are secreted by the adrenal gland, synthesis of DHEAS is not under the same tight control by adrenocorticotropic hormone (ACTH) as DHEA (Hornsby, 1995). The main function of these two hormones has been thought to be as precursors for androgens and estrogens in peripheral tissues (Alexandersen et al., 1996), (Figure 1) but they are also weak androgens and can play a role in pubertal changes. Circulating levels of the two hormones are 3 ng/ml (∼30 nM) for DHEA, whereas DHEAS circulates at 400 times that level, 1.2 ug/ml (∼10uM) (Alexandersen et al., 1996; Celec and Starka, 2003; Hornsby 1995). The dramatic decline in levels with age has led many investigators to hypothesize that DHEA and DHEAS could contribute to many diseases seen with aging, including diabetes mellitus, obesity, cancer, and atherosclerosis (Porsova-Dutoit et al., 2000).
Figure 1.

A schematic representation of where DHEA and DHEA-sulfate (DHEAS) fit into the steroid biosynthetic pathway. DHEA is a major precursor to androstenedione, which is the only precursor to both the androgens and estrogens in the steroid pathway in humans. DHEAS is the major circulating form, but is converted to DHEA by a sulfotransferase in tissue.
Many epidemiological studies of DHEA and DHEAS (DHEA(S)) as they affect vascular disease have shown that there is an inverse relationship between death from cardiovascular complications and DHEA(S) levels in men (Barrett-Connor et al., 1986; LaCroix et al., 1992). Others have shown no relationship (Contoreggi et al., 1990; Newcomer et al., 1994) or a negative one (Barrett-Connor et al., 1986). The data are further complicated by gender differences: often, there is no or a negative correlation between circulating DHEA(S) and cardiovascular events in women (Johannes et al., 1999; Khaw, 1996). Therefore, the epidemiological data on beneficial effects of DHEA(S) on cardiovascular health are controversial.
Animal studies in rodents and rabbits have shown a decisive benefit of DHEA and DHEAS on vascular disease. Oral administration of DHEA to cholesterol-fed rabbits resulted in a 50% reduction in plaque size (Gordon et al., 1988). Additional studies have shown that DHEA(S) have lipid lowering effects in both rodents and rabbits (Lea-Currie et al., 1997; Regelson and Kalimi, 1994) and protect against vascular reperfusion injury (Aragno et al., 2000; Ayhan et al., 2003). Together, the data demonstrate an atheroprotective effect of DHEA(S) in rodent models.
In vitro data also suggest that DHEA(S) are beneficial in preventing cardiovascular disease. They have been shown to relax vascular tissue through a calcium-related mechanism, inhibit smooth muscle cell proliferation, and inhibit the accumulation of cholesterol ester in macrophage cells (Barbagallo et al., 1995; Taniguchi et al., 1996; Williams et al., 2002). Thus, in vitro studies reveal a protective effect of DHEA(S) against the pathological mechanisms involved in atherosclerosis.
Among the earliest steps in the pathogenesis of atherosclerosis, endothelial cell injury and inflammation have been shown to be key mechanisms. Inflammatory mediators interact with the vascular endothelium to cause increased permeability, increased expression of adhesion molecules and chemoattractants, and leukocyte recruitment. Disrupted endothelial barrier function and increased recruitment and transmigration of activated leukocytes are hallmarks of atherosclerotic cardiovascular disease. In addition, inflammatory activation of the endothelium has been shown to decrease its vasomodulatory ability and antithrombotic properties, contributing to the pathogenesis of atherosclerosis (Ross, 1999). This study was designed to test the hypothesis that DHEAS can prevent endothelial cell injury and inflammation, key early events in the development of atherosclerosis. Peroxisome proliferator-activated receptor α (PPARα) has been shown to antagonize the nuclear factor-κB (NF-κB signaling pathway involved in the vascular inflammation of atherosclerosis (Delerive et al., 1999; Rival et al., 2002). We were interested in determining whether PPARα signaling is involved in the potential anti-inflammatory effects of DHEAS. Our studies show that DHEAS attenuates the inflammatory processes implicated in the development of atherosclerosis by a PPARα-mediated mechanism and is actively involved in the regulation of NF-κB.
2. Materials and Methods
2.1 Materials
All media and supplements were obtained from Cascade Biologics (Portland, Oregon). DHEAS was obtained from Sigma (St Louis, MO). TNF-α was obtained from Roche (Indianapolis, IN). Fluorescently conjugated antibodies and Cytofix/Cytoperm™ were obtained from BD Biosciences (San Diego, CA). Letrozole was a kind gift from Dr. Al Conley (University of California, Davis, CA). MK866 was obtained from Calbiochem (San Diego, CA). ICI 182,780 was from Tocris Cookson (Ellisville, MO). All cell culture plates were supplied by Fisher (Houston, TX). Protein assay was obtained from BioRad (Hercules, CA). Nuclear extraction kits, electrophoretic mobility kits and X-ray film were obtained from Pierce Biotechnology (Rockford, IL). NF-κB consensus sequence was obtained from Panomics (Redwood City, CA). Nylon membrane was provided by Pall (East Hills, NY).
2.2 Cell Culture
Human aortic endothelial cells (HAECs) were obtained from Cascade Biologics. Cells were grown in Medium 200 supplemented with 1 ug/ml hydrocortisone, 10 ng/ml human epidermal growth factor, 3 ng/ml fibroblast growth factor, 10 ug/ml heparin, 2% fetal bovine serum, 100 U/ml penicillin G, 100 ug/ml streptomycin sulfate, and 0.25 ug/ml amphotericin B. Cells were maintained in a 37 °C humidified incubator with a 5% CO2 and 95% air environment. Medium was changed every 2-3 days until desired confluency (80%) was reached. Cells were used between passages 5-7 for all experiments.
2.3 Cell Viability
Cells were grown in 6-well cell culture plates to 80% confluency (∼106 cells) then treated with increasing concentrations (0.1 uM -1 mM) of DHEAS for 48 hrs. Cells then were harvested by trypsinization, and counted on a hemocytometer using the trypan blue exclusion assay. All dead cells stain blue, while live cells do not stain and are refractile.
2.4 Flow Cytometry
Cells were grown in 6-well cell culture plates to 80% confluency then treated with 1 mM DHEAS for 48 hrs in the presence or absence of either letrozole (100nm), MK866 (1 uM) or ICI 182,780 (10nM) and then stimulated with TNF-α (25 ng/ml). DHEAS was dissolved in DMSO before being added to media, and cells not treated with DHEAS were exposed to equivalent concentrations of DMSO only. Cells were stained according to BD Biosciences cell surface antigen staining protocol with anti-VCAM-1 fluorescent conjugated monoclonal antibody. Briefly, cells were washed with cold PBS and harvested by trypsinization. After centrifugation cell pellets were washed with staining buffer (1X PBS with 1% v/v heat inactivated fetal calf serum and 0.09% w/v sodium azide), then incubated with fluorescent antibody for 30 min at 4°C. After staining, cell pellets were washed again with staining buffer and fixed with Cytofix/Cytoperm™ for 20 min at 4°C. Cells were read on a BD FACSCalibur flow cytometer (San Jose, CA).
2.5 Electrophoretic Mobility Shift Assays (EMSA)
EMSAs were performed according to the protocol previously described (Kota et al., 2005). Briefly, nuclear extracts were obtained using the Pierce NE-PER nuclear extraction kit according to the manufacturer’s instructions. Protein concentrations were determined using the Bio Rad DC Protein Assay (Hercules, CA). EMSAs were performed using double-stranded oligonucleotides (Santa Cruz Biotechnology, Santa Cruz, CA) for the consensus binding sites of transcription factors. The binding reactions consisted of 25 ug of nuclear protein and 20 fmol of the 32P-labeled oligonucleotide probe. Oligonucleotides were labeled (20 ng/ul) with 5 units of T4 polynucleotide kinase, and 3 ul of [γ-32P]dATP at 37°C for 45 min. End-labeled oligonucleotides were separated from unincorporated nucleotides by using chromaspin-10 columns (Clontech, Mountain Valley, CA). The double-stranded NF-κB consensus sequence, 5′-GATCCAGAGGGGACTTTCCGAGT-3′ was used in the reaction. The binding reactions (20 ul) were carried out for 20 min in binding buffer consisting of 20 mM HEPES, pH 7.9, 50mM KCl, 0.5 mM dithiothreitol, 0.1 mM EDTA, 2 ug of salmon sperm DNA, 10% glycerol, 5 mM MgCl2, 3 ug of poly(dI-dC). To check the specificity of the protein-DNA interactions, the supershift reaction was carried out by adding 1 ug of polyclonal antibody to NF-κB (p50 or p65 subunit) to the binding reaction. The reaction products were separated in a 5% polyacrylamide gel and analyzed following the autoradiography of the dried gel.
2.6 Quantitative Real-Time PCR
Total RNA was obtained using TRIZOL reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Cells were washed 1X with cold 1X PBS before addition of TRIZOL. Total RNA was quantified by absorbance at 260 and 280 nm, and 5 ug of RNA was used to prepare cDNA with the Superscript II RNase H-reverse transcriptase (Invitrogen, Carlsbad, CA) by following the manufacturer’s instructions. Primers were designed using Primer Express (Applied Biosystems, Foster City, CA) and synthesized by Operon Technologies (Alameda, CA). The primer sequences are as follows: IL-8 (sense, 5′-GTGTAAACATGACTTCCAAGCTGG-3′; antisense, 5′-GCACCTTCACACAGAGCTGC-3′), ICAM-1 (sense, 5′-CAGAAGAAGTGGCCCTCCATAG-3′; antisense, 5′-GGGCCTTTGTGTTTTGATGCTA-3′), and GAPDH (sense, 5′-CACCAACTGCTTAGCACCCC-3′; antisense, 5′-TGGTCATGAGTCCTTCCACG-3′). Sample cDNAs were diluted 1:10 and analyzed in duplicate on a 96-well plate using the SYBR Green I Master Mix (Applied Biosystems, Foster City, CA). Forward and reverse primers were added at a concentration of 600 nM for each reaction well. Detection of gene transcript levels was performed using the GeneAmp 7900 HT system (Applied Biosystems, Foster City, CA). The thermal cycling program was as follows: thermal activation for 10 min at 95°C and 40 cycles of PCR (melting for 15 seconds at 95°C, followed by annealing/extension for 1 minute at 60°C). The control sample was diluted 1:10, 1:20, 1:40, 1:80, 1:160 and used to construct a standard curve for each gene. PCR product formation was verified by analysis of product heat-dissociation curves and amplification plots, and product quantification values were taken from the linear region of the PCR curve. Gene expression levels were normalized to GAPDH levels for each sample.
2.7 Western Blots
Western blots were performed according to Sambrook and Russell (2001). Briefly, HAECs were incubated with increasing doses of DHEAS for 48 hours prior to cellular lysate collection. Cells were trypsinized and washed with 1X PBS before lysates were collected using the Pierce NE-PER nuclear extraction kit according to the manufacturer’s instructions. Lysates were mixed with 2X sample buffer and boiled for 3 minutes then run on a 10% SDS-PAGE gel and proteins transferred to nitrocellulose membrane. The membrane was blocked for one hour with 5% Blotto in Tris-buffered saline with 0.1% Tween 20 (TBST). For detection of IκB-α protein expression, the membrane was incubated with rabbit anti-human Iκ-B (Santa Cruz Biotechnology, C-21, Santa Cruz, CA) diluted 1:8000 in TBST overnight at 4°C. The membrane was then incubated with goat anti-rabbit peroxidase conjugated secondary antibody (Vector Labs, Burlingame, CA) diluted 1:10,000 in TBST for one hour. For detection of β-actin, the mouse monoclonal anti-human β-actin (Sigma, clone AC-15) was used as the primary antibody, and a sheep anti-mouse peroxidase conjugate was the secondary antibody (Amersham Biosciences, Pittsburgh, PA). Protein bands were detected using ECL Western Blotting Detection Reagents (Amersham Biosciences, Pittsburgh, PA) according to the manufacturer’s instructions. The membrane was exposed to film for 1-2 min then developed on a Konica Model SRX-101A film processor.
2.8 Statistical Analyses
All values are expressed as the mean ± SEM. Statistical analyses between multiple comparisons were performed using one-way ANOVA followed by a Bonferroni adjustment. A value of P < 0.05 was considered significant. Experiments were performed at least 2 times, with a maximum of 5 times.
3. Results
3.1 Viability of human aortic endothelial cells exposed to DHEAS in culture
HAECS were not detrimentally affected by exposure to increasing concentrations of DHEAS for 48 hrs (Table 1). Cell viability was determined by trypan blue exclusion assay and expressed as a percentage of cells alive after treatment.
TABLE 1.
Percentage of cells surviving after 48 hrs of treatment with increasing amounts of DHEAS
| DHEAS | % cells alive |
|---|---|
| 0 (vehicle) | 98.86 ± 0.65 |
| 1 uM | 97.92 ± 0 .48 |
| 10 uM | 95.56 ± 1.12 |
| 100 uM | 98.50 ± 0.87 |
| 1 mM | 97.61 ± 0.93 |
3.2 Dose-dependent effect of DHEAS on the expression of TNF-α-induced vascular cell adhesion molecule (VCAM)-1 expression in human aortic endothelial cells
To determine the effective dose of DHEAS and establish a dose response curve to DHEAS, cells were pretreated with increasing doses of DHEAS (0.1 uM-1 mM). These treatments resulted in a decreased percentage of cells expressing VCAM-1 after stimulation by TNF-α (Figure 2). No VCAM-1 expression was detected without stimulation with TNF-α. The highest dose of DHEAS, 1 mM, significantly decreased the percentage of cells expressing VCAM-1 by 83% ± 7.11% (only 10% of cells expressing VCAM-1) when compared to cells receiving no DHEAS treatment and stimulated with TNF-α. The lower doses of DHEAS decreased VCAM-1 expression in a dose dependent manner. DHEAS alone did not have any effect on the lack of VCAM-1 expression in HAECs not stimulated by TNF-α.
Figure 2.

Endothelial cells expressing the inflammatory marker VCAM-1 in the presence or absence of DHEAS over a range of concentrations. VCAM-1 expression was analyzed by flow cytometry. Endothelial cells were cultured in 6-well plates in the presence of DHEAS at varying concentrations for 48 hrs then stimulated with 25 ng/ml TNF-α. Cells not exposed to DHEAS were treated with vehicle only (DMSO, 0.7%). All cells were stimulated with 25 ng/ml TNF-α for 4 hrs. *P < 0.05 denotes significant difference when compared to no treatment with DHEAS.
3.3 Effect of DHEAS on the expression of TNF-α-induced inflammatory gene expression in human aortic endothelial cells
We assessed whether DHEAS had any effect on the expression of inflammatory genes IL-8 and ICAM-1 using quantitative RT-PCR analysis (Figure 3). HAECs were incubated with DHEAS for 48 hrs, followed by stimulation with TNF-used to prepare cDNA for qRT-PCR reactions. Both genes showed upregulation with TNF-α treatment alone, which was significantly attenuated by pre-incubation with DHEAS. With DHEAS, the level of IL-8 gene expression was reduced nearly 5-fold, while ICAM-1 expression was reduced 2-fold compared to TNF treatment alone. Cells treated with vehicle only (DMSO) showed minimal inflammatory gene expression.
Figure 3.

Quantitative real-time polymerase chain reaction (RT-PCR) analysis of gene expression in HAECs exposed to either TNF-α or DHEAS plus TNF-α. Endothelial cells were incubated in the presence of DHEAS (1 mM) for 48 hrs and then stimulated with 25 ng/ml TNF-α. Cells not exposed to DHEAS were treated with vehicle only (DMSO, 0.7%). A and B show the IL-8/GAPDH (A) and ICAM-1/GAPDH (B) expression ratios. Data are expressed as mean ± SEM. *P < 0.05 denotes significant difference when compared to TNF-α treatment alone.
3.4 Effect of an aromatase inhibitor on TNF-α-induced VCAM-1 expression in the presence or absence of DHEAS
To determine whether the conversion of DHEAS to estradiol or another estrogen played a role in the observed effect of DHEAS on VCAM-1 expression stimulated by TNF-α, an aromatase inhibitor was employed in the studies (Figure 4). Inhibition of aromatase prevents the conversion of DHEAS, an upstream precursor, to any aromatized product such as estradiol. The aromatase inhibitor letrozole did not have any significant effect on the DHEAS-reduced expression of VCAM-1 in HAECs after stimulation with TNF-α when compared to cells pretreated with vehicle only. DHEAS significantly reduced VCAM-1 expression such that only 5.5% of cells expressed the marker, and with the addition of letrozole that did not change, with 5.1% of cells expressing VCAM-1. Cells exposed to DHEAS and letrozole still exhibited the same dramatic decrease in VCAM-1 expression when stimulated with TNF-α.
Figure 4.

Percentage of endothelial cells expressing the inflammatory marker VCAM-1 in the presence or absence of DHEAS and/or letrozole, an aromatase inhibitor. The addition of letrozole did not alter the inhibition of VCAM-1 expression by DHEAS. Endothelial cells were cultured in the presence of DHEAS and in the presence or absence of letrozole (100nM) for 48 hrs, and then stimulated with 25 ng/ml TNF-α for 4 hours. Cells not exposed to DHEAS were treated with vehicle only (DMSO, 0.7%). *P < 0.05 denotes significant difference when compared to TNF-α treatment alone.
3.5 Effect of the estrogen receptor inhibitor ICI 182,780 on TNF-α-induced VCAM-1 expression in the presence or absence of DHEAS
Even though an aromatase inhibitor had no effect on the DHEAS inhibition of TNF-α-induced VCAM-1 expression, it is still possible that the effect is mediated through estrogen receptors. To determine whether estrogen receptors were playing a role in this DHEAS result, we used an estrogen receptor antagonist, ICI 182,780, which irreversibly binds the estrogen receptor and prevents downstream signaling. ICI 182,780 was incubated with HAECs at a concentration of 10 nM in the presence or absence of 1 mM DHEAS, for 48 hrs prior to stimulation with TNF-α (25 ng/ml) for 4 hrs. Figure 5 shows the results of ICI 182,780 on VCAM-1 expression. The presence of ICI 182,780 did not have any effect on the DHEAS-mediated inhibition of VCAM-1 expression in HAECs stimulated with TNF-α. In the presence of DHEAS, VCAM-1 expression was reduced to 6.6%. With treatment of both DHEAS and ICI 182,780 the expression of VCAM-1 was 11.4%, not significantly different than with treatment by DHEAS alone.
Figure 5.

Percentage of endothelial cells expressing the inflammatory marker VCAM-1 in the presence or absence of DHEAS and/or ICI 182,780, an estrogen receptor antagonist. Endothelial cells were cultured in the presence of DHEAS and in the presence or absence of ICI 182,780 (10nM) for 48 hrs, and then stimulated with 25 ng/ml TNF-α for 4 hours. Cells not exposed to DHEAS were treated with vehicle only (DMSO, 0.7%). *P < 0.05 denotes significant difference when compared to TNF-α treatment alone.
3.6 Effect of a PPARα inhibitor on TNF-α-induced VCAM-1 expression
The PPARα inhibitor, MK866, was used in the studies to determine whether the mechanism by which DHEAS inhibits VCAM-1 expression is through PPARα activation. Figure 6 shows that the addition of MK866 partially restored the expression of VCAM-1 (26% of cells expressing VCAM-1) by HAECs when compared to TNF-α stimulation alone (51% of cells expressing VCAM-1), whereas DHEAS treatment significantly reduced VCAM-1 expression (7.1% of cells). MK866 by itself had no effect on the TNF-α-induced expression of VCAM-1 in HAECS with 47% of the cells expressing VCAM-1. This indicates that PPARs are involved but that other mediators may play a role in DHEAS inhibition of VCAM-1 expression.
Figure 6.

Percentage of endothelial cells expressing the inflammatory marker VCAM-1 in the presence or absence of DHEAS and/or MK866, a PPARα inhibitor. Endothelial cells were cultured in the presence of DHEAS and in the presence of absence of MK866 at varying concentrations for 48 hrs, and then stimulated with TNF-α (25 ng/ml) for 4 hours. Cells not exposed to DHEAS were treated with vehicle only (DMSO, 0.7%). *P < 0.05 denotes significant difference when compared to TNF-α treatment alone.
3.7 NF-κB activation is inhibited by DHEAS in human aortic endothelial cells stimulated with TNF-α
We assessed whether DHEAS mediates its effects on endothelium by activation of NF-κB using electrophoretic mobility shift assays (EMSAs) (Figure 7). EMSA analysis showed the TNF-α-induced activation of NF-κB activity (lane 3). DHEAS (1mM) markedly inhibited NF-κB activity (lane 4) as compared to cells which were pretreated with vehicle only for 48 hrs prior to stimulation with TNF-α (lane 3). The specificity of NF-κB was determined by incubation with specific antibodies. The addition of p50 antibody abolished the middle and slow migrating bands, indicating the presence of p50 specificity. The addition of p65 antibody abolished the presence of the slow migrating band, indicating the presence of the p50/p65 heterodimer. Vehicle treated unstimulated control cells (lane 1) showed no constitutive NF-κB activity. The results demonstrate that incubation with DHEAS suppressed TNF-α-induced NF-κB activation, as indicated by the diminished presence of both the p50/p50 and p50/p65 dimers in EMSA analysis.
Figure 7.
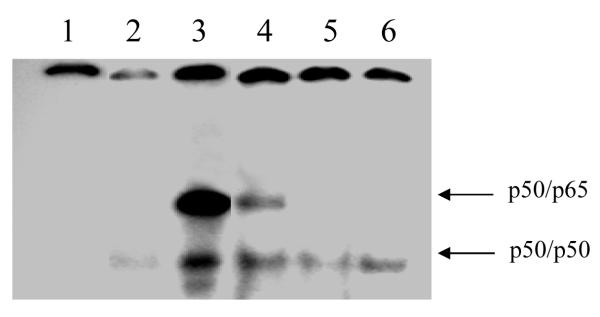
Electrophoretic mobility shift assay showing activity of transcription factor NF-κB. Endothelial cells were stimulated with TNF-α (25 ng/ml) for 4 hrs after pretreatment (48 hrs) with DHEAS (1 mM) or vehicle. EMSAs were carried out using nuclear protein extracted from cells incubated with various treatments. Lane 1: Free probe; Lane 2: Vehicle (DMSO)-treated cells; Lane 3: Vehicle-treated cells stimulated with TNF-α; Lane 4: Cells treated with DHEAS (1mM) for 48 hrs prior to stimulation with TNF-α; Lane 5: Cells treated with DHEAS (1mM) for 48 hrs prior to stimulation with TNF-α and supershifted with the p50 subunit of NF-κB antibody; Lane 6: Cells treated with DHEAS (1mM) for 48 hrs prior to stimulation with TNF-α and supershifted with the p65 subunit of NF-κB antibody. This image is representative of 3 different assays.
3.8 Dose-dependent effect of DHEAS on expression of IkB-α in human aortic endothelial cells
We determined whether DHEAS had any effect on the level of IκB-α, the primary inhibitor of NF-κB activation, using Western blotting techniques (Figure 8). HAECs were incubated with increasing concentration of DHEAS for 48 hrs prior to isolation of cellular lysates. Cellular lysates were then probed for IκB-α protein levels. There were increasing amounts of IκB-α protein that correlated with increasing concentrations of DHEAS (1-1000 uM DHEAS). Cells treated with vehicle only (DMSO) had little IκB-α detectable in the cellular lysate.
Figure 8.

Western blot showing protein expression of IκB-α in cellular lysates of HAECS exposed to increasing doses of DHEAS. Endothelial cells were incubated with increasing doses of DHEAS for 48 hrs prior to collection of cellular lysates. Cells receiving no DHEAS exposure were incubated with vehicle (DMSO) only. The lower blot shows β-actin levels as a loading control. This image is representative of 3 different blots.
4. Discussion
To our knowledge, this is the first report of DHEAS inhibiting VCAM-1 expression, a marker of endothelial cell injury, by inhibiting NF-κB activity. Our work showed that the addition of a PPARα inhibitor, MK866, partially restored the expression of VCAM-1. DHEAS also increased IκB-α protein level in a dose-dependent manner. These actions demonstrate a significant role for DHEAS in the prevention of inflammatory processes in the endothelium, and suggest that DHEAS has direct effects, and is not working by conversion to a downstream metabolite.
The effect of DHEA(S) on any cellular activity might be through their conversion to downstream end products, specifically estrogens (Haning et al., 1991). It has been well documented that estrogens are capable of inhibiting NF-κB in a variety of tissues (Sharma et al., 2001; Shaul, 2003; Wen et al., 2004). No specific intracellular receptors for DHEA(S) have been identified, even though a recent report has provided evidence of a cell surface receptor for DHEA in bovine aortic endothelial cells and an intracellular receptor in murine lymphocytes (Liu and Dillon, 2002; Liu and Dillon, 2004; Meikle et al., 1992). Controversy exists as to whether human endothelial cells have the ability to convert DHEAS to estrogens (Harada et al., 1999; Neve et al., 2000). Aromatase is the enzyme that makes the final conversion of the C-19 steroid testosterone to the C-18 steroid estradiol (Figure 1). The presence of letrozole inhibits that conversion. If it is the conversion of DHEAS to estrogen that inhibits TNF-α-induced VCAM-1 expression, then the presence of letrozole would prevent estrogen-mediated reduction in VCAM-1 expression. In our study, letrozole had no effect on the striking inhibition of VCAM-1 expression by DHEAS, indicating that conversion of DHEAS to estradiol is not playing a role in the mechanism of inhibition.
DHEAS can also be converted to other metabolites, including androstenediol, androstenedione, androsterone and 3α,17β-diol (Dufort et al., 2001; Provost et al., 2000), some of which have been reported to have an affinity for the estrogen receptor (Adams et al., 1981). We incubated DHEAS in the presence or absence of an effective estrogen receptor antagonist ICI 182,780. The addition of the antagonist had very little effect on the ability of DHEAS to inhibit TNF-α-induced VCAM-1 expression. Together, these results suggest that DHEAS has primary actions to reduce endothelial cell inflammatory marker expression; conversion to the downstream metabolite estradiol and involvement of the estrogen receptor are not necessary.
DHEAS appears to be mediating anti-inflammatory effects through the activation of the peroxisome proliferator-activated receptor (PPAR). Previously, it was shown in studies employing a PPARα deficient mouse that DHEAS did not activate PPARα responsive genes, as it did in wild-type mice (Peters et al., 1996). Other studies have shown that PPARα is involved in atherosclerotic disease by reducing tissue factor expression in monocytes, inhibiting expression of inflammatory cytokines and cell adhesion molecules in both macrophages and endothelial cells, and inhibiting the proliferation of vascular smooth muscle cells (Marx, 2002; Neve et al., 2000). These findings demonstrate an anti-inflammatory function of PPARα in vascular and immune tissues, which is strengthened by the ability of PPARα to inhibit the activation of nuclear factor-κB (NF-κB) (Marx et al., 1998; Marx et al., 1999; Vanden Berghe et al., 2003). In our study we employed the use of a PPARα inhibitor, MK866, and observed that addition of the PPARα inhibitor partially restored the ability of TNF-α to induce VCAM-1 expression in cells that had been pretreated with DHEAS and then activated with TNF-α. However, since it did not fully restore expression, we speculate that PPARα activation is not the only mechanism by which DHEAS can inhibit inflammation in aortic endothelial cells.
Nuclear factor κB (NF-κB) is known to be the primary transcription factor responsible for inflammation. Electrophoretic mobility assays were used in this project to determine if DHEAS could inhibit activation of NF-κB. As expected from the previously obtained data, DHEAS did inhibit the activation of NF-κB. We assessed the ability of DHEAS to increase IκB-α protein levels in the same cells. Our data demonstrate that DHEAS is capable of increasing IκB-α protein levels in a dose dependent manner. The increase in IκB-α results in the inhibition of NF-κB activation. However, previous studies have shown that DHEAS can inhibit the ability of reactive oxygen species to stimulate degradation of the IκB-α protein (Iwasaki et al., 2004). Previous studies have also shown that DHEAS is intimately involved in the oxidative balance in the cell (Mastrocola et al., 2003). DHEAS can upregulate the action of endothelial cell nitric-oxide synthase (eNOS), which has an anti-inflammatory effect in endothelial cells (Hayashi et al., 2000; Liu and Dillon, 2004). Activation of eNOS results in the production of nitric oxide (NO), a potent vasodilator, and evidence shows that a lack of NO in endothelial cells results in an increase in proliferation of vascular smooth muscle cells, and an increase in platelet aggregation and adherence (Simoncini et al., 2000).
The ability of the weak androgens such as DHEA and DHEAS to affect multiple pathways may also be playing a role in the anti-inflammatory effects of these hormones. DHEA(S) can affect the mitogen-activated protein kinase (MAPK) pathway in human vascular smooth muscle cells by inhibiting ERK-1 phosphorylation events (Vanden Berghe et al., 2003), can reduce baseline levels of c-fos mRNA in osteoblastic cells (Bodine et al., 1995) and can regulate the enzyme catalase, a specific inhibitor of NF-κB (Poynter and Daynes, 1998). These key molecular studies illustrate a potential role for DHEAS in regulating endothelial cell function through novel signal transduction mechanisms and redox reactions.
At a physiological concentration, DHEAS (10 uM) caused a 21% reduction in VCAM-1 expression that was not significantly different from HAECs treated with TNF-α only. However, a dose-response effect of DHEAS on VCAM-1 expression was clearly detected. DHEAS levels vary greatly between individuals and may go higher than 10 uM in some individuals (Celec and Starka, 2003). No studies have yet measured normal tissue levels of DHEA or DHEAS. Based on our dose-response experiments, we used these concentrations of DHEAS to maximize whatever effects we might see in the experimental assays to examine the mechanisms of DHEAS effect.
Our studies show a dose-response of VCAM-1 expression and IκB-α protein levels to DHEAS concentration, indicating that DHEAS is having a specific effect, rather than a generalized or artifactual effect. The data presented here offer preliminary information about the role of androgens in vascular physiology. Our studies point towards a potential cellular role for adrenal androgens in vascular biology and open the door for testing other hypotheses regarding how weak androgens affect vascular physiology and pathophysiology. Additional studies should be undertaken to further elucidate the exact mechanisms of how DHEAS affects the endothelium as well as other vascular tissues.
Acknowledgements
We would like to thank Drs. Bill Lasley and Al Conley (U.C. Davis) for their help in completing this study. We also thank Dr. Tom Famula (U.C. Davis) for his help in statistical analysis. This work was supported by the National Institutes of Health grant HL55667 and by a grant from the Richard A. & Nora Eccles Harrison Endowed Chair.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams J, Garcia M, Rochefort H. Estrogenic effects of physiological concentrations of 5-androstene-3 beta, 17 beta-diol and its metabolism in MCF7 human breast cancer cells. Cancer Res. 1981;41(11 Pt 1):4720–6. [PubMed] [Google Scholar]
- Alexandersen P, Haarbo J, Christiansen C. The relationship of natural androgens to coronary heart disease in males: a review. Atherosclerosis. 1996;125:1–13. doi: 10.1016/0021-9150(96)05864-9. [DOI] [PubMed] [Google Scholar]
- Aragno M, Parola S, Brignardello E, Mauro A, Tamagno E, Manti R, Danni O, Boccuzzi G. Dehydroepiandrosterone prevents oxidative injury induced by transient ischemia/reperfusion in the brain of diabetic rats. Diabetes. 2000;49:1924–1931. doi: 10.2337/diabetes.49.11.1924. [DOI] [PubMed] [Google Scholar]
- Ayhan S, Tugay C, Norton S, Araneo B, Siemionow M. Dehydroepiandrosterone protects the microcirculation of muscle flaps from ischemia-reperfusion injury by reducing the expression of adhesion molecules. Plast Reconstr Surg. 2003;111:2286–2294. doi: 10.1097/01.PRS.0000060242.85268.8F. [DOI] [PubMed] [Google Scholar]
- Barbagallo M, Shan J, Pang PK, Resnick LM. Effects of dehydroepiandrosterone sulfate on cellular calcium responsiveness and vascular contractility. Hypertension. 1995;26:1065–1069. doi: 10.1161/01.hyp.26.6.1065. [DOI] [PubMed] [Google Scholar]
- Barrett-Connor E, Khaw KT, Yen SSC. A prospective study of dehydroepiandrosterone sulfate, mortality, and cardiovascular disease. N Engl J Med. 1986;315:1519–1524. doi: 10.1056/NEJM198612113152405. [DOI] [PubMed] [Google Scholar]
- Baulieu EE, Corp’echot C, Dray F, Emiliozzi R, Lebeau MC, Mauvais-Jarvis P, Robel P. An adrenal-secreted “androgen”: Dehydroepiandrosterone Sulfate, its metabolism and a tentative generalization on the metabolism of other steroid conjugates in man. Recent Prog Horm Res. 1965;21:411–500. [PubMed] [Google Scholar]
- Bodine PV, Riggs BL, Spelsberg TC. Regulation of c-fos expression and TGF-beta production by gonadal and adrenal androgens in normal human osteoblastic cells. J Steroid Biochem Mol Biol. 1995;52:149–158. doi: 10.1016/0960-0760(94)00165-i. [DOI] [PubMed] [Google Scholar]
- Celec P, Starka L. Dehydroepiandrosterone - is the fountain of youth drying out? Physiol Res. 2003;52(4):397–407. [PubMed] [Google Scholar]
- Contoreggi CS, Backman MR, Anders R, Muller CD, Lakatta EG, Fleg JL, Harman SM. Plasma levels of estradiol, testosterone, and DHEAS do not predict risk of coronary artery disease in men. J Androl. 1990;11:460–470. [PubMed] [Google Scholar]
- Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, Fruchart JC, Tedgui A, Haegeman G, Staels B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999;274(45):32048–32054. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- Dufort I, Soucy P, Lacoste L, Luu-The V. Comparative biosynthetic pathway of androstenol and androgens. J Steroid Biochem Mol Biol. 2001;77(4-5):223–7. doi: 10.1016/s0960-0760(01)00057-7. [DOI] [PubMed] [Google Scholar]
- Gordon GB, Bush DE, Weisman HF. Reduction of atherosclerosis by administration of dehydroepiandrosterone. A study in the hypercholesterolemic New Zealand white rabbit with aortic intimal injury. J Clin Invest. 1988;82:712–720. doi: 10.1172/JCI113652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haning RV, Jr, Carlson IH, Flood CA, Hackett RJ, Longcope C. Metabolism of dehydroepiandrosterone sulfate (DS) in normal women and women with high DS concentrations. J Clin Endocrinol Metab. 1991;73:1210–1215. doi: 10.1210/jcem-73-6-1210. [DOI] [PubMed] [Google Scholar]
- Harada N, Sasano H, Murakami H, Ohkuma T, Nagura H, Takagi Y. Localized expression of aromatase in human vascular tissues. Circ Res. 1999;84:1285–91. doi: 10.1161/01.res.84.11.1285. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Esaki T, Muto E, Kano H, Asai Y, Thakur NK, Sumi D, Jayachandran M, Iguchi A. Dehydroepiandrosterone retards atherosclerosis formation through its conversion to estrogen: the possible role of nitric oxide. Arterioscler Thromb Vasc Biol. 2000;20:782–792. doi: 10.1161/01.atv.20.3.782. [DOI] [PubMed] [Google Scholar]
- Hornsby PJ. Biosynthesis of DHEAS by the human adrenal cortex and its age-related decline. Ann N Y Acad Sci. 1995;774:29–46. doi: 10.1111/j.1749-6632.1995.tb17370.x. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y, Asai M, Yoshida M, Nigawara T, Kambayashi M, Nakashima N. Dehydroepiandrosterone-sulfate inhibits nuclear factor-kappaB-dependent transcription in hepatocytes, possibly through antioxidant effect. J Clin Endocrinol Metab. 2004 Jul;89(7):3449–54. doi: 10.1210/jc.2003-031441. [DOI] [PubMed] [Google Scholar]
- Johannes CB, Stellato RK, Feldman HA, Longcope C, McKinlay JB. Relation of dehydroepiandrosterone and dehydroepiandrosterone sulfate with cardiovascular disease risk factors in women: longitudinal results from the Massachusetts Women’s Health Study. J Clin Epidemiol. 1999 Feb;52(2):95–103. doi: 10.1016/s0895-4356(98)00144-9. [DOI] [PubMed] [Google Scholar]
- Khaw KT. Dehydroepiandrosterone, dehydroepiandrosterone sulphate and cardiovascular disease. J Endocrinol. 1996;150(Suppl):S149–S153. [PubMed] [Google Scholar]
- Kota RS, Ramana CV, Tenorio FA, Enelow RI, Rutledge JC. Differential effects of lipoprotein lipase on tumor necrosis factor-α and interferon-γ-mediated gene expression in human endothelial cells. J Biol Chem. 2005;280:31076–31084. doi: 10.1074/jbc.M412189200. [DOI] [PubMed] [Google Scholar]
- LaCroix AZ, Yano K, Reed DM. Dehydroepiandrosterone sulfate, incidence of myocardial infarction, and extent of atherosclerosis in men. Circulation. 1992;86:1529–1535. doi: 10.1161/01.cir.86.5.1529. [DOI] [PubMed] [Google Scholar]
- Lea-Currie YR, Wu SM, McIntosh MK. Effects of acute administration of dehydroepiandrosterone-sulfate on adipose tissue mass and cellularity in male rats. Int J Obes Relat Metab Disord. 1997;21:147–154. doi: 10.1038/sj.ijo.0800382. [DOI] [PubMed] [Google Scholar]
- Liu D, Dillon JS. Dehydroepiandrosterone activates endothelial cell nitric-oxide synthase by a specific plasma membrane receptor coupled to Galpha(i2,3) J Biol Chem. 2002;277:21379–21388. doi: 10.1074/jbc.M200491200. [DOI] [PubMed] [Google Scholar]
- Liu D, Dillon JS. Dehydroepiandrosterone stimulates nitric oxide release in vascular endothelial cells: evidence for a cell surface receptor. Steroids. 2004 Apr;69(4):279–89. doi: 10.1016/j.steroids.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Marx N, Schonbeck U, Lazar MA, Libby P, Plutzky J. Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells. Circ Res. 1998;83:1097–1103. doi: 10.1161/01.res.83.11.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999;99:3125–3131. doi: 10.1161/01.cir.99.24.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx N. Peroxisome proliferator-activated receptor gamma and atherosclerosis. Curr Hypertens Rep. 2002;4:71–77. doi: 10.1007/s11906-002-0056-8. [DOI] [PubMed] [Google Scholar]
- Mastrocola R, Aragno M, Betteto S, Brignardello E, Catalano MG, Danni O, Boccuzzi G. Pro-oxidant effect of dehydroepiandrosterone in rats is mediated by PPAR activation. Life Sci. 2003;73:289–299. doi: 10.1016/s0024-3205(03)00287-x. [DOI] [PubMed] [Google Scholar]
- Meikle AW, Dorchuck RW, Araneo BA, Stringham JD, Evans TG, Spruance SL, Daynes RA. The presence of a dehydroepiandrosterone-specific receptor binding complex in murine T cells. J Steroid Biochem Mol Biol. 1992;42:293–304. doi: 10.1016/0960-0760(92)90132-3. [DOI] [PubMed] [Google Scholar]
- Neve BP, Fruchart JC, Staels B. Role of the peroxisome proliferator-activated receptors (PPAR) in atherosclerosis. Biochem Pharmacol. 2000;60:1245–1250. doi: 10.1016/s0006-2952(00)00430-5. [DOI] [PubMed] [Google Scholar]
- Newcomer LM, Manson JE, Barbieri RL, Hennekens CH, Stampfer MJ. Dehydroepiandrosterone sulfate and the risk of myocardial infarction in US male physicians: a prospective study. Am J Epidemiol. 1994;140:870–875. doi: 10.1093/oxfordjournals.aje.a117175. [DOI] [PubMed] [Google Scholar]
- Orentreich N, Brind JL, Rizer RL, Vogelman JH. Age changes and sex differences in serum dehydroepiandrosterone sulfate concentrations throughout adulthood. J Clin Endocrinol Metab. 1984;59(3):551–555. doi: 10.1210/jcem-59-3-551. [DOI] [PubMed] [Google Scholar]
- Peters JM, Zhou YC, Ram PA, Lee SS, Gonzalez FJ, Waxman DJ. Peroxisome proliferator-activated receptor alpha required for gene induction by dehydroepiandrosterone-3 beta-sulfate. Mol Pharmacol. 1996;50:67–74. [PubMed] [Google Scholar]
- Porsova-Dutoit I, Sulcova J, Starka L. Do DHEA/DHEAS play a role in coronary heart disease? Physiol. Res. 2000;49(Suppl. 1):S43–S56. [PubMed] [Google Scholar]
- Poynter ME, Daynes RA. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J Biol Chem. 1998;273:32833–32841. doi: 10.1074/jbc.273.49.32833. [DOI] [PubMed] [Google Scholar]
- Provost PR, Blomquist CH, Godin C, Huang XF, Flamand N, Luu-The V, Nadeau D, Tremblay Y. Androgen formation and metabolism in the pulmonary epithelial cell line A549: expression of 17beta-hydroxysteroid dehydrogenase type 5 and 3alpha-hydroxysteroid dehydrogenase type 3. Endocrinology. 2000;141(8):2786–94. doi: 10.1210/endo.141.8.7589. [DOI] [PubMed] [Google Scholar]
- Regelson W, Kalimi M. Dehydroepiandrosterone (DHEA)--the multifunctional steroid. II. Effects on the CNS, cell proliferation, metabolic and vascular, clinical and other effects. Mechanism of action? Ann N Y Acad Sci. 1994;719:564–575. doi: 10.1111/j.1749-6632.1994.tb56860.x. [DOI] [PubMed] [Google Scholar]
- Rival Y, Beneteau N, Taillandier T, Pezet M, Dupont-Passelaigue E, Patoisseau JF, Junquero D, Colpaert FC, Delhon A. PPARalpha and PPARdelta activators inhibit cytokine-induced nuclear translocation of NF-kappaB and expression of VCAM-1-1 in EAhy926 endothelial cells. Eur J Pharmacol. 2002;435(2-3):143–151. doi: 10.1016/s0014-2999(01)01589-8. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis — an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Third Edition Cold Spring Harbor; 2001. [Google Scholar]
- Sharma RV, Gurjar MV, Bhalla RC. Selected contribution: estrogen receptor-alpha gene transfer inhibits proliferation and NF-kappaB activation in VSM cells from female rats. J Appl Physiol. 2001;91:2400–2406. doi: 10.1152/jappl.2001.91.5.2400. discussion 2389-2390. [DOI] [PubMed] [Google Scholar]
- Shaul PW. Endothelial nitric oxide synthase, caveolae and the development of atherosclerosis. J Physiol. 2003;547:21–33. doi: 10.1113/jphysiol.2002.031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoncini T, Maffei S, Basta G, Barsacchi G, Genazzani AR, Liao JK, De Caterina R. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ Res. 2000;87:19–25. doi: 10.1161/01.res.87.1.19. [DOI] [PubMed] [Google Scholar]
- Taniguchi S, Yanase T, Kobayashi K, Takayanagi R, Nawata H. Dehydroepiandrosterone markedly inhibits the accumulation of cholesteryl ester in mouse macrophage J774-1 cells. Atherosclerosis. 1996;126:143–154. doi: 10.1016/0021-9150(96)05902-3. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe W, Vermeulen L, Delerive P, De Bosscher K, Staels B, Haegeman G. A paradigm for gene regulation: inflammation, NF-kappaB and PPAR. Adv Exp Med Biol. 2003;544:181–96. doi: 10.1007/978-1-4419-9072-3_22. [DOI] [PubMed] [Google Scholar]
- Wen Y, Yang S, Liu R, Perez E, Yi KD, Koulen P, Simpkins JW. Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain Res. 2004;1008:147–154. doi: 10.1016/j.brainres.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Williams MR, Ling S, Dawood T, Hashimura K, Dai A, Li H, Liu JP, Funder JW, Sudhir K, Komesaroff PA. Dehydroepiandrosterone inhibits human vascular smooth muscle cell proliferation independent of ARs and ERs. J Clin Endocrinol Metab. 2002;87:176–181. doi: 10.1210/jcem.87.1.8161. [DOI] [PubMed] [Google Scholar]