

Abstract
The delivery of therapeutics via nanoscaled vehicles for solid cancer treatment can be enhanced by the incorporation of a targeting capability. Here, we describe a new method for assembling a targeted nanoparticle that utilizes the reversible covalent complexation between boronic acids and diols to achieve a targeted nanoparticle for the delivery of anti-cancer drug, camptothecin (CPT). CPT is conjugated to a biocompatible, hydrophilic copolymer of mucic acid and PEG (MAP). When this polymer-drug conjugate is placed in water, it self-assembles into MAP-CPT nanoparticles of ca. 30 nm (diameter) and slightly negative zeta potential. The antibody Herceptin is attached to a boronic acid via a polyethylene glycol (PEG) spacer, and this boronic acid-containing targeting moiety is complexed with the diol-containing MAP to form a targeted MAP-CPT nanoparticle. The addition of Herceptin targeting agent to the MAP-CPT nanoparticles yields targeted MAP-CPT nanoparticles with increased nanoparticle size to ca. 40 nm (diameter). The main mechanisms of CPT release from MAP-CPT nanoparticles are found by in vitro analysis to be hydrolysis and nanoparticle disruption by fat. Cellular uptake of nanoparticles is enhanced by 70% compared to non-targeted version by the incorporation of a single Herceptin antibody targeting agent per nanoparticle. This single Herceptin antibody targeted MAP-CPT nanoparticle system carries ca. 60 CPT molecules per nanoparticle and shows prolonged plasma circulation with an elimination half-life of 21.2 h and AUC value of 2766 μg.h/ml at a 10 mg CPT/kg tail vein injection in mice.
Introduction
Nanoparticles are finding application for the delivery of a broad range of therapeutic and imaging agents, some of which are currently being investigated in human clinical trials (1, 2). When nanoparticles are employed for the delivery of therapeutics to solid cancers, they rely upon the enhanced permeability and retention (EPR) effect to accumulate in the tumors (3). After reaching the tumor site by the EPR effect, nanoparticles containing targeting agents that can engage cancer cell surface receptors are shown to achieve uptake into cancer cells in amounts exceeding those of non-targeted nanoparticles with similar size and surface charge (4–6).
Our research group has been involved in translating two polymer-based nanoparticles from the laboratory to the clinic. The first, CRLX101 (previously known as IT-101) is a ca. 30 nm (diameter), non-targeted nanoparticle composed of a cyclodextrin-polymer conjugate of the anti-cancer drug camptothecin (CPT) (7). The second, CALAA-01, is a ca. 70 nm (diameter) targeted (human transferrin protein) nanoparticle comprised of a cyclodextrin-polymer and siRNA (8, 9). The method of assembling the targeting moiety onto the CALAA-01 nanoparticle involves the formation of inclusion complexes of adamantane with the cyclodextrins in the polymer (8). Since the method of assembling CRLX101 utilizes inclusion complex formation between the cyclodextrins in the polymer and the conjugated CPT, the targeting methodology employed with CALAA-01 is not amenable for CRLX101. Because of this issue and others that were encountered as these nanoparticles were translated to the clinic, we have been exploring new ways of assembling targeted nanoparticles that could carry any type of payload. Here, we describe a methodology to create targeted nanoparticles that involves the use of boronic acid-diol complexes.
Boronic acids have been used: (i) in a variety of pharmaceutical agents, (ii) in creating devices to sense sugars and (iii) in compounds to create drug delivery materials and devices (10). For example, the complexation of boronic acids with diols has recently been employed to assemble cross-linked micelles (11, 12). Here, we demonstrate a new method for the assembly of targeted nanoparticles that employs boronic acid complexation with diols. That is, nanoparticles are assembled using polymers that have sugars in their backbone, e.g., mucic acid, to provide a repeating unit that contains vicinal diols. Nanoparticle targeting agents are conjugated with flexible spacing groups, e.g., polyethylene glycol (PEG), and terminated with a boronic acid. These targeting moieties are then assembled onto the surface of the nanoparticle via the reversible, covalent bonding between the boronic acid and the surface vicinal diols of the polymer (Scheme 1). This nanoparticle system can have numerous functionalities that allow for the assembly with a wide variety of therapeutic and imaging agents (13). For example, small hydrophobic drug molecules like CPT can be conjugated to the polymer backbone and self-assembled into nanoparticles.
Scheme 1.
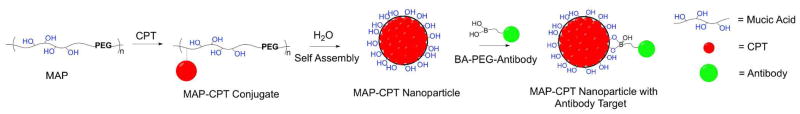
Targeted MAP-CPT nanoparticle delivery system. MAP, copolymer of mucic acid and polyethylene glycol; CPT, camptothecin; BA, boronic acid.
In this study, we report on an antibody (Herceptin) targeted, polymer-based nanoparticle system for the delivery of CPT that utilizes the complexation of boronic acids on the antibody targeting moiety and vicinal diols on the polymer for assembly of the targeted nanoparticle. CPT was conjugated onto a biocompatible hydrophilic copolymer backbone of mucic acid and PEG (MAP). This conjugate, when placed in water self-assembled into nanoparticles. Addition of the antibody targeting agent was achieved by exploiting the complexation between boronic acids in the targeting moiety and the mucic acid repeats in the polymer (Scheme 1).
Experimental Procedures
General
All commercially purchased reagents and solvents including anhydrous and HPLC grades were used without further purification. Boc-L-aspartic acid 4-benzyl 1-(N-hydroxysuccinimide) ester (Boc-Asp(OBzl)-OSu) was purchased from Bachem (Bubendorf, Switzerland). Disuccinimidyl propionate poly(ethylene glycol) (diSPA-PEG, MW 3400) was purchased from SunBio (South Korea). Campthothecin was obtained from Boehringer Ingelheim (Ingelheim, Germany). 3-carboxylphenylboronic acid and 3-carboxy 5-nitrophenylboronic acid were purchased from Alfa Aesar (Ward Hill, MA). Amine polyethylene glycol carboxylic acid (NH2-PEG-CO2H, MW 5000) was purchased from JenKem Technology USA (Allen, TX). Herceptin® (Trastuzumab) was obtained from Dr. Y. Yen at the City of Hope (Duarte, CA). BT-474, MCF-7, SKBR-3 and MDA-MB-231 breast cancer cell lines were purchased from American Type Culture Collection (Manassas, VA). Female BALB/c mice were purchased from Jackson Laboratory (Bar Habor, ME). Mice plasma was processed from fresh blood collected from female BALB/c mice. All other reagents were purchased from Sigma (St Louis, MO).
1H, 13C and 19F NMR spectra were recorded on either a 500 MHz or 600 MHz spectrometer (Inova, Varian). Mass spectrometry was carried out using a LCQ ion trap mass spectrometer (Finnigan, Thermo) by direct infusion electrospray ionization or MALDI-TOF mass spectrometer (Voyager DE™-PRO, Applied Biosystems).
Concentrations of unconjugated CPT and polymer-bound CPT were analyzed on a HPLC system (Agilent) equipped with a fluorescence detector (excitation 370 nm, emission 440 nm) and a reverse phase column (Synergi 4 μm Hydro-RP 80 Å, Phenomenex) complete with guard column. 50% acetonitrile, 50% potassium phosphate buffer (10 mM, pH 4) was used as the eluent at a flow rate of 0.5 ml/min.
Absorbance and other fluorescence measurements were taken on a microplate reader (Infinite 200, Tecan).
Preparation and Characterization of MAP-CPT Nanoparticles
1. Mucic acid dimethyl ester
120 ml of methanol and 0.4 ml of concentrated sulfuric acid were added to 5 g (22.8 mmol) of mucic acid. This mixture was refluxed at 85 °C overnight under constant stirring. Solids were filtered, washed with methanol, recrystallized and dried under vacuum overnight to yield 8 g (71%) of mucic acid dimethyl ester. 1H NMR ((CD3)2SO) δ 4.88–4.91 (d, 2H), 4.78–4.81 (m, 2H), 4.28–4.31 (d, 2H), 3.77–3.78 (d, 2H), 3.63 (s, 6H). ESI/MS (m/z): 261.0 [M+Na]+.
2. Protected mucic acid diamine
60 ml of methanol, 5.25 g (32.7 mmol) of N-Boc-ethylenediamine and 5.7 ml (32.7 mmol) of N, N-diisopropylethylamine (DIPEA) were added to 3 g (12.6 mmol) of mucic acid dimethyl ester. This reaction mixture was refluxed at 85 °C overnight under constant stirring. Solids were filtered, washed with methanol, recrystallized and dried under vacuum to give 4.65 g (75%) of product. 1H NMR ((CD3)2SO) δ 7.72 (t, 2H), 6.82 (t, 2H), 5.13–5.15 (d, 2H), 4.35–4.38 (d, 2H), 4.08–4.11 (d, 2H), 3.78–3.80 (d, 2H), 2.95–3.15 (m, 8H), 1.36 (s, 18). ESI/MS (m/z): 517.1 [M+Na]+.
3. Mucic acid diamine
8 g (16.2 mmol) of protected mucic acid diamine was transferred to a flask containing 3 N HCl in methanol (160 ml) and reacted for 6 h at room temperature under constant stirring. Precipitate was filtered, washed with methanol and vacuum dried overnight to yield 5.69 g (96%) of mucic acid diamine. 1H NMR ((CD3)2SO) δ 7.93–7.99 (m, 8H), 5.31–5.34 (d, 2H), 4.57 (d, 2H), 4.10–4.17 (d, 2H), 3.82 (d, 2H), 3.32–3.38 (m, 4H), 2.82–2.86 (m, 4H). ESI/MS (m/z): 295.1 [M+H]+.
4. Mucic acid di(Asp(OBzl)-Boc)
9.27 g (22.1 mmol) of Boc-Asp(OBzl)-OSu in 75 ml of acetonitrile and 2.88 ml (35.8 mmol) of pyridine was added to 2.7 g (7.35 mmol) of mucic acid diamine dissolved in 45 ml water. This reaction mixture was refluxed at 60 °C overnight under constant stirring. Acetonitrile was removed by rotavap and resulting precipitate was recrystallized from hot water and dried under vacuum to yield 2.03 g (31%) mucic acid di(Asp(OBzl)-Boc). 1H NMR ((CD3)2SO) δ 7.93 (t, 2H), 7.75 (t, 2H), 7.29–7.35 (m, 10H), 7.05 (d, 2H), 5.11 (t, 2H), 5.06 (s, 4H), 4.34–4.39 (d, 2H), 4.25–4.30 (d, 2H), 4.10–4.12 (d, 2H), 3.77–3.79 (d, 2H), 3.11–3.15 (m, 8H), 2.53–2.78 (m, 4H), 1.36 (s, 18H). ESI/MS (m/z): 927.4 [M+Na]+.
5. Mucic acid di(Asp(OBzl)-amine)
6 ml (80.8 mmol) of trifluoroacetic acid was slowly added to 2 g (2.21 mmol) of mucic acid di(Asp(OBzl)-Boc) dissolved in 18 ml of dichloromethane on an ice bath. The reaction was allowed to proceed for 4 h in ice bath. Solvent was removed, precipitate recrystallized with tetrahydrofuran and dried on vacuum to yield 1.74 g (84%) of product. 1H NMR ((CD3)2SO) δ 8.46 (t, 2H), 8.21 (s, 6H), 7.79 (t, 2H), 7.32–7.38 (m, 10H), 5.19 (t, 2H), 5.14 (s, 4H), 4.41 (s, 2H), 4.11–4.14 (d, 2H), 4.02–4.06 (d, 2H), 3.81 (s, 2H), 3.11–3.23 (m, 8H), 2.79–3.02 (m, 4H). ESI/MS (m/z): 705.4 [M+H]+.
6. Mucic acid di(Asp-amine)
511 mg (13.5 mmol) of 20 w % palladium hydroxide on carbon was transferred to 1.26 g (1.35 mmol) of mucic acid di(Asp(OBzl)-amine) dissolved in 50 ml of methanol. Reaction vessel was sealed and vented with argon. Hydrogen gas was added via double-layered balloon for 24 h. Catalyst was filtered away, solvent removed and precipitate dried on vacuum. This was then reconstituted in water, filtered with a 0.2 μm membrane filter (Acrodisc) and lyophilized to yield 711 mg (70%) of product. 1H NMR ((CD3)2SO) δ 8.44 (t, 2H), 8.19 (broad, 6H), 7.80 (t, 2H), 5.22 (t, 2H), 4.48 (s, 2H), 4.12 (s, 2H), 3.94–3.99 (m, 2H), 3.80 (s, 2H), 3.11–3.23 (m, 8H), 2.70–2.85 (m, 4H). ESI/MS (m/z): 525.2 [M+H]+.
7. MAP (Mucic Acid containing Polymer)
111 mg of mucic acid di(Asp-amine) (0.15 mM) and 500 mg of diSPA-PEG3.4kDa (0.15 mM) were dried under vacuum for 6 h. 3.5 ml of anhydrous dimethyl sulfoxide was added under argon to dissolve the reactants. 82 μl (0.47 mM) of anhydrous DIPEA was then added under argon and reaction was allowed to proceed for 40 h at room temperature under constant stirring. Reaction mixture was then dialyzed with 10 kDa MWCO membrane filter device (Amicon) 4 times, filtered through a 0.2 μm membrane filter (Acrodisc) and lyophilized to yield 466 mg (81%) MAP. GPC (for medium MAP): Mn = 55.5 kDa, Mw = 75.3 kDa, Mw/Mn = 1.36. 1H NMR ((CD3)2SO, for medium MAP) δ 8.07 (d, 1H), 8.04 (d, 1H), 7.83 (t, 1H), 7.77 (t, 1H), 7.69 (t, 2H), 4.43 (td, 2H), 4.10 (d, 2H), 3.75 (ddt, 2H), 3.55 (t, 4.3H), 3.42–3.52 (PEG peak, 330H), 3.05–3.16 (m, 8H), 2.39–2.53 (m, 4H), 2.34 (t, 4.3H). 13C NMR (D2O, for medium MAP) δ 175.7, 174.9, 174.0, 172.9, 66.5, 69.6, 70.6, 50.6, 38.8, 38.3, 36.5, 35.7.
8. Absolute molecular weight determination of MAP
The absolute determination of molecular weights does not require standards for calibration. The values were determined on a GPC system equipped with an Agilent pump, degasser and autosampler, double gel permeation columns (PL aquagel-OH 40 8 μm 300 × 7.5 mm, Polymer Laboratory) in series connected to a multi-angle laser light scattering (MALS) detector (DAWN HELEOS, Wyatt) and a refractive index (RI) detector (Optilab rEX, Wyatt). PBS (1x, 0.02 w/v % sodium azide, pH 7.4) was used as the eluent at a flow rate of 0.7 ml/min. Specific refractive increment, dn/dc was measured by batch injection of various concentrations of MAP dissolved in PBS into Optilab rEX at 0.2 ml/min.
9. Molecular weight control of MAP
Molecular weight of MAP was controlled by the amount of tertiary base, N, N-diisopropylethylamine (DIPEA) added to the reaction. After dissolving equal molar ratios of mucic acid di(Asp-amine) and diSPA-PEG (3.4 kDa) in anhydrous dimethyl sulfoxide, different equivalents (1.1, 1.6 and 2) of anhydrous DIPEA to amine groups on mucic acid di(Asp-amine) were added. The reactions were then carried out and the polymers processed as per the methods described in section 7 (vide supra). Three polymers of differing molecular weights were prepared and denoted as short, medium and long.
10. 20-O-glycinylcamptothecin trifluoroacetic acid salt (CPT-glyTFA)
20-O-glycinylcamptothecin trifluoroacetic acid salt (CPT-glyTFA) was synthesized as previously reported (14).
11. MAP-CPT conjugate
403 mg (0.22 mM on -CO2H basis) of MAP was dissolved in 20 ml of anhydrous dimethyl sulfoxide. To this was added 167 mg (0.87 mM) of EDC and 75 mg (0.65 mM) of NHS dissolved in 6 ml of anhydrous dimethyl sulfoxide, 340 mg (0.65 mM) of 20-O-glycinylcamptothecin trifluoroacetic acid salt (CPT-glyTFA) dissolved in 6 ml of anhydrous dimethyl sulfoxide and 114 μl (0.65 mM) of anhydrous DIPEA under argon. After overnight reaction under constant stirring, the reaction mixture was dialyzed against dimethyl sulfoxide 3 times and water 2 times using 10 kDa MWCO membrane (Spectra/Por 7). Any insoluble material was removed by centrifugation and supernatant was filtered with a 0.2 μm filter (Acrodisc). A portion of this clear yellow solution was lyophilized for yield determination and percent CPT conjugation analysis. The remaining was formulated into 0.9 w/v% saline and stored at −20 °C.
12. CPT content in MAP-CPT
Weight percent of CPT in MAP-CPT was determined by the intrinsic fluorescence of CPT. MAP-CPT was dissolved in dimethyl sulfoxide at 10 mg/ml. This was diluted into 0.1 mg/ml with 1 N NaOH and incubated overnight. Fluorescence was measured at excitation 370 nm, emission 440 nm, and compared to that of known concentrations of CPT.
13. Nanoparticle composition
Nanoparticle compositions were analyzed as per the methods described in section 8 (vide supra) except gel permeation columns were bypassed. Dn/dc value of MAP-CPT nanoparticles was measured by batch injection of various concentrations of MAP-CPT nanoparticles dissolved in PBS into Optilab rEX at 0.2 ml/min. This allowed for the molecular weight determination of MAP-CPT nanoparticles. The number of MAP-CPT conjugates per nanoparticle was then calculated from this data.
14. Nanoparticle size and surface charge
Particle size and zeta potential measurements were determined by dynamic light scattering (DLS) using a ZetaPALS instrument (Brookhaven Instruments, Holtsville, NY). Effective hydrodynamic diameter was recorded and averaged from 10 runs at 1 min each. Zeta potential measurements were collected in 1 mM KCl solution for three sets of 10 runs at a target residual of 0.012, and the results were averaged.
15. Nanoparticle morphology
Nanoparticle morphology was visualized by cryo-electron microscopy (cryo-EM). Glow discharged Quantifoil holey carbon grid (SPI Supplies) was loaded into an automated climate-controlled plunge-freezer (Vitrobot, FEI) and 3 μl of sample was applied. Grid was blotted, drained, plunged into liquid ethane, transferred and stored in liquid nitrogen. Samples were visualized under cryogenic temperatures with transmission electron microscope (Tecnai T12, FEI) equipped with a cryo-specimen holder. Acceleration voltage was set at 120 kV.
Preparation and Characterization of Targeted MAP-CPT Nanoparticles
1. PBA-PEG-CO2H (3-amide-PEG(5kDa)-carboxylic acid phenylboronic acid)
To 200 mg (1.21 mM) of 3-carboxyphenylboronic acid dissolved in 5 ml of anhydrous tetrahydrofuran was added 18.7 μl (0.24 mM) of anhydrous dimethylformamide under argon. This reaction vessel was transferred into an ice bath and 195 μl (2.89 mM) of oxalyl chloride was slowly added under argon. Reaction was allowed to proceed under vent and constant stirring for 2 h at room temperature. Solvent and excess reagent were removed under vacuum. 37 mg (0.2 mM) of this dried acyl chloride compound was dissolved in 15 ml of anhydrous dichloromethane under argon. To this was added 500 mg (0.1 mM) of NH2-PEG(5kDa)-CO2H and 52 μl (0.3 mM) of dry DIPEA under argon. After overnight reaction under constant stirring, solvent was removed under vacuum and the dried product was reconstituted in 0.5 N HCl. This solution was passed through a 0.2 μm filter (Acrodisc) and dialyzed against water with a 3 kDa MWCO membrane filter device (Amicon) until constant pH was attained. The supernatant was then filtered with a 0.2 μm filter (Acrodisc) and lyophilized to yield 377 mg (73%) of PBA-PEG-CO2H. 1H NMR ((CD3)2SO) δ 12.52 (s, 1H), 8.39 (t, 1H), 8.22 (s, 1H), 8.14 (s, 2H), 7.88 (d, 1H), 7.82 (d, 1H), 7.39 (t, 1H), 3.99 (s, 2H), 3.35–3.62 (PEG peak). MALDI-TOF (m/z): 5600 g/mol (NH2-PEG5kDa-CO2H, 5400 g/mol).
2. nitroPBA-PEG-CO2H (3-amide-PEG(5kDa)-carboxylic acid, 5-nitrophenylboronic acid)
Synthesis of nitroPBA-PEG-CO2H was performed in the same way as for PBA-PEG-CO2H except the starting material was 3-carboxy 5-nitrophenylboronic acid. The reaction yielded 393 mg (76%) of nitroPBA-PEG-CO2H. 1H NMR ((CD3)2SO) δ 12.52 (s, 1H), 8.89 (t, 1H), 8.72 (s, 1H), 8.68 (s, 1H), 8.64 (s, 1H), 8.61 (s, 2H), 3.99 (s, 2H), 3.35–3.62 (PEG peak). MALDI-TOF (m/z): 5600 g/mol (NH2-PEG5kDa-CO2H, 5400 g/mol).
3. pKa determination for PBA
pKa of PBA was found by measuring the change in absorbance of PBA as it converted from trigonal (in low pH) to tetrahedral (in high pH) conformation. To a solution of 10−3 M PBA in 0.1 M PBS, pH 7.4 was titrated 1 N NaOH. pH was recorded and corresponding samples were removed for absorbance measurements at 268 nm.
4. Binding constant determination between PBA and MAP
A three component competitive assay involving fluorescent dye Alizarin Red S, PBA and MAP (diol containing) was used to determine the binding constant between PBA and MAP. This technique was developed by Springsteen and Wang (15). Briefly, the association constant Keq1 for Alizarin Red S-PBA complex was determined by changes in fluorescence as PBA was titrated into Alizarin Red S. MAP was then titrated into a solution containing Alizarin Red S and PBA. The addition of the MAP perturbed the first equilibrium due to MAP competing with Alizarin Red S for binding to PBA. Changes in fluorescence were measured and the association constant Keq between PBA and MAP was then determined. Measurements were performed in 0.1 M PBS at pH 7.4.
5. Herceptin-PEG-nitroPBA (Herceptin-PEG(5kDa)-5-nitrophenylboronic acid)
36 mg (6.4 μM) of nitroPBA-PEG-CO2H, 12.3 mg (64 μM) of EDC and 11.1 mg (96 μM) of NHS were dissolved in 2.4 ml of 0.1 M MES buffer, pH 6.0. This mixture was reacted for 15 min on a rotating shaker at room temperature. Excess reactants were dialyzed away with 3 kDa MWCO membrane filter device (Amicon). This activated carboxylic acid PEG compound was added to 20 mg (0.14 mM) of Herceptin in 0.1 M PBS, pH 7.2. Reaction was carried out on a rotating shaker at room temperature for 2 h and then dialyzed 4 times with 50 kDa MWCO membrane filter device (Amicon) against 1x PBS at pH 7.4. MALDI-TOF: average conjugation of 1 to 2 nitroPBA-PEG per antibody.
6. Formulation of targeted MAP-CPT nanoparticles
Herceptin-PEG-nitroPBA was added to the medium MAP-CPT nanoparticles to form targeted MAP-CPT nanoparticles at a ratio of one Herceptin per nanoparticle in 1x PBS, pH 7.4.
7. Particle size, surface charge and morphology
The methods used for the targeted nanoparticle were the same as for the non-targeted nanoparticles as described in Preparation and Characterization of MAP-CPT Nanoparticles sections 14 and 15 (vide supra).
Cellular Uptake Studies
24 well plates were seeded with either BT-474 (in RPMI-1640 medium supplemented with 10% fetal calf serum) or MCF-7 (in Dulbecco’s Modified Eagle medium supplemented with 10% fetal calf serum) (Cellgro, Manassas, VA) at 20,000 cells per well and kept at 37 °C in a humidified oven with 5% CO2. After 40 h, media were replaced with 0.3 ml of fresh media containing either medium MAP-CPT (40 μg CPT/ml), medium MAP-CPT with free Herceptin at 10 mg/ml, targeted MAP-CPT (40 μg CPT/ml) at varying targeting densities or targeted MAP-CPT with free Herceptin at 10 mg/ml. Transfection was conducted for 30 min at 37 °C. Formulations were then removed and cells washed twice with cold PBS. 200 μl of RIPA buffer was added per well for cell detachment and lysis. Lysed cells were then incubated at 4 °C for 15 min and centrifuged at 14,000 g for 10 min at 4 °C. A portion of the supernatant was used for protein quantification via BCA assay. To another portion was added an equal amount of 0.1 N NaOH, this was incubated at room temperature overnight and fluorescence was measured at 370/440 nm using known concentrations of MAP-CPT as standard.
In Vitro Release Studies
Release of CPT from short, medium, long MAP-CPT nanoparticles and targeted MAP-CPT nanoparticles were conducted at 0.32 mg CPT/ml in 1x PBS at pH 6.5, 7 or 7.4; BALB/c mice plasma, human plasma, and 1x PBS at pH 7.4 containing 3 mg/ml of low density lipoprotein (LDL), or 100 units/ml of Butyrylcholinesterase (BCHE) or a combination of LDL and BCHE.
Media pipetted into 96 well plates were incubated at 37 °C in a humidified oven for 2 h for equilibration. Formulations were mixed into the relevant media and placed back into the oven. Samples were taken out at predetermined time points and immediately frozen at −80 °C until time for analysis. For release in BALB/c mice plasma and in human plasma, incubation was carried out in a humidified oven at 37 °C with 5% CO2 to maintain the carbonic acid/bicarbonate buffer system, the major pH buffer system in plasma at physiological pH levels.
The amount of unconjugated CPT was determined by first mixing 10 μl of sample with 10 μl of 0.1 N HCl and incubating at room temperature for 30 min. 80 μl of methanol was then added and the mixture incubated at room temperature for 3 h for protein precipitation. This mixture was centrifuged at 14,000 g for 10 min at 4 °C, supernatant was filtered with a 0.45 μm filter (Millex-LH), diluted 20 folds with methanol and 10 μl of the resulting solution injected into HPLC. The peak area of the eluted CPT (at 7.8 min) was compared to that of control. To measure the total amount of CPT, 10 μl of sample was mixed with 6.5 μl of 0.1 N NaOH. This solution was incubated at room temperature for 1 h for CPT to be released from parent polymer. 10 μl of 0.1 N HCl was then added to convert the carboxylate CPT form to the lactone form. 73.5 μl of methanol was subsequently added and mixture incubated for 3 h at room temperature. The sample was then centrifuged and processed as above. Polymer-bound CPT concentration was determined from the difference between total CPT and unconjugated CPT concentrations.
Cytotoxicity Assays
Cells were kept at 37 °C in a humidified oven with 5% CO2. BT-474, MCF-7, SKBR-3 and MDA-MB-231 cell lines were incubated in RPMI-1640 medium, Dulbecco’s Modified Eagle medium, McCoy’s 5A Modified medium and Dulbecco’s Modified Eagle medium respectively (all supplemented with 10% fetal calf serum). 3,000 cells per well were plated into 96 well plates. After 24 h, media was removed and replaced with fresh media containing different concentrations of medium MAP, nitroPBA-PEG, CPT, medium MAP-CPT nanoparticles, targeted MAP-CPT nanoparticles or Herceptin. After 72 h of incubation, formulations were replaced with fresh media, and 20 μl of CellTiter 96® AQueous One Solution cell proliferation assay (Promega) was added per well. This was incubated for 2 h in a humidified oven at 37 °C with 5% CO2 before absorbance measurements at 490 nm. Wells containing untreated cells were used as controls.
Pharmacokinetic Studies
All animals were treated as per National Institute of Health Guidelines for Animal Care and approved by California Institute of Technology Institutional Animal Care and Use Committee. Formulations were administered via bolus tail vein injection into 12 – 16 weeks old female BALB/c mice. At predetermined time points, blood was collected via saphenous vein bleed with blood collection tubes (Microvette CB 300 EDTA, Sarstedt). Samples were immediately centrifuged at 10,000 g, 4 °C for 15 min and supernatant removed and stored at −80 °C until time for analysis. Analyses for unconjugated and polymer-bound CPT were as per the methods described in the section In vitro Release Studies (vide supra). Non-compartmental modeling software PK Solutions 2.0 by Summit Research Services (Montrose, CO) was used for pharmacokinetic data analysis.
Results and Discussions
Preparation and Characterization of MAP-CPT Nanoparticles
The synthesis methodology of the MAP-CPT conjugate and its assembly into MAP-CPT nanoparticles is outlined in Scheme 2. Mucic acid was modified to contain both amino and carboxyl groups to allow for polymerization with PEG (amine group) and for conjugation of CPT (carboxyl group), respectively. It was observed via 19F NMR that the mucic acid di(Asp-amine) formed was the trifluoroacetic acid salt by comparison to a standard (2-(trifluoromethyl)acetophenone). Therefore, for the polymerization reaction between mucic acid di(Asp-amine) and diSPA-PEG to proceed, a base was required for the deprotonation of -NH3+ group into the reactive -NH2 form. Here, the tertiary base N, N-diisopropylethylamine (DIPEA) was used. Supplementary Fig. 1 shows representative NMR data for the MAP polymers. The polymer end groups were determined to be -PEG-CO2H by 1H NMR comparison of PEG and mucic acid di(Asp-amine) peak integrals. The molecular weight of MAP can be controlled by the amount of DIPEA added per NH3+ group on mucic acid di(Asp-amine) (Table 1). As the amount of DIPEA was increased, an increase in viscosity of the reaction mixture was observed. Three polymers were prepared and their properties are listed in Table 1.
Scheme 2.

Synthesis of MAP-CPT nanoparticles.
Table 1.
Characterization of MAP, MAP-CPT conjugate, MAP-CPT nanoparticles and targeted MAP-CPT nanoparticles
| Short | Medium | Long | ||
|---|---|---|---|---|
| MAP | Base added (equivalent)a | 1.1 | 1.6 | 2 |
| MWb (kDa) | 20 | 65 | 102 | |
| Polydispersityc | 1.22 | 1.36 | 1.13 | |
| # repeat units (n) | 5–7 | 15–21 | 25–30 | |
|
| ||||
| MAP-CPT conjugate | MWb (kDa) | 22 | 75 | 114 |
| wt % CPT conjugated | 9.8 | 12.7 | 10.1 | |
|
| ||||
| MAP-CPT nanoparticle | # conjugates/particle | 2–3 | 2–3 | 2–3 |
| # CPT/particle | ~14 | ~60 | ~72 | |
| particle size (nm) | ~30 | ~30 | ~30 | |
| zeta potential (mV) | −1.3+/−0.6 | −0.5+/−0.5 | −0.8+/−0.5 | |
|
| ||||
| Targeted MAP-CPT nanoparticle | # Herceptin/particle | 1 | ||
| particle size (nm) | ~40 | |||
| zeta potential (mV) | −0.4+/−0.6 | |||
Equivalent amount of N, N-diisopropylethylamine (DIPEA) added per amine group in mucic acid di(Asp-amine).
MW, molecular weight determined as (Mw+Mn)/2; Mw, weight average molecular weight; Mn, number average molecular weight.
Polydispersity determined as Mw/Mn.
The absolute molecular weights of the different MAP samples were measured using multi-angle laser light scattering (MALS) and refractive index (RI) detections. A refractive index increment (dn/dc) value of 0.134 ml/g was found for the medium MAP. This dn/dc value along with MALS and RI data were used to determine the number average molecular weight (Mn), weight average molecular weight (Mw) and polydispersity (Mw/Mn) of the MAP samples (Table 1).
CPT modified at 20-OH position has been reported to show significant reduction in deactivation caused by ring opening of the lactone (16). We therefore used CPT modified at this site with a glycine linker, CPT-gly, as was done previously with IT-101 (14). The CPT-gly was conjugated onto MAP to form MAP-CPT conjugate by standard EDC coupling technique (Scheme 2). The amount of CPT conjugated onto the three MAP polymers is reported in Table 1.
Upon addition of water, the MAP-CPT conjugates self-assemble into nanoparticles since CPT is very soluble in dimethyl sulfoxide but not soluble to any significant amount in water. To determine the number of MAP-CPT conjugates per MAP-CPT nanoparticle, dn/dc values of MAP-CPT nanoparticles were found, and the nanoparticles were characterized by MALS and RI detection. From these data, the molecular weight of each MAP-CPT nanoparticle was determined. Since the molecular weights of MAP-CPT conjugates were known, the MAP-CPT nanoparticles were found to be composed of 2 to 3 MAP-CPT conjugates. This finding along with the observation that all MAP-CPT nanoparticles have approximately the same size (ca. 30 nm in diameter) suggests that the nanoparticles have different packing densities.
MAP-CPT nanoparticles have an average particle size of ca. 30 nm (by DLS measurements) and a slightly negative surface charge (by Zeta Potential measurements). The negative surface charge is presumably due to the presence of unconjugated carboxylic acid groups on the MAP backbone. Cryo-EM was used to visualize the medium MAP-CPT nanoparticles in their native environment. Spherical nanoparticles with a narrow size range of 20–40 nm are observed (Fig. 1A).
Figure 1.

Cryo-EM images of (A) medium MAP-CPT nanoparticles, (B) targeted MAP-CPT nanoparticles
Preparation and Characterization of Targeted MAP-CPT Nanoparticles
To prepare a targeted nanoparticle, the covalent, reversible binding property between boronic acids and MAP (diol containing) was used. The binding constant between phenylboronic acid (PBA) and MAP is low at ca. 20 M−1. To increase the binding constant, PBAs with electron withdrawing groups on the phenyl ring were employed to increase the acidity of the boron atom and thus elevate the binding constant with MAP. Commercially available 3-carboxyPBA and 3-carboxy 5-nitroPBA were converted into acyl chlorides using oxalyl chloride (Scheme 3). These acyl chloride species were then reacted with NH2-PEG-CO2H to form PBA-PEG-CO2H and nitroPBA-PEG-CO2H, respectively (Scheme 3). The addition of bases DMF and DIPEA were required for the synthesis reactions to proceed. However, the presence of these bases resulted in tetrahedral adduct formations with acidic PBA. To remove these adducts, work up in 0.5 N HCl with subsequent equilibration to neutral pH by dialysis against water was carried out.
Scheme 3.
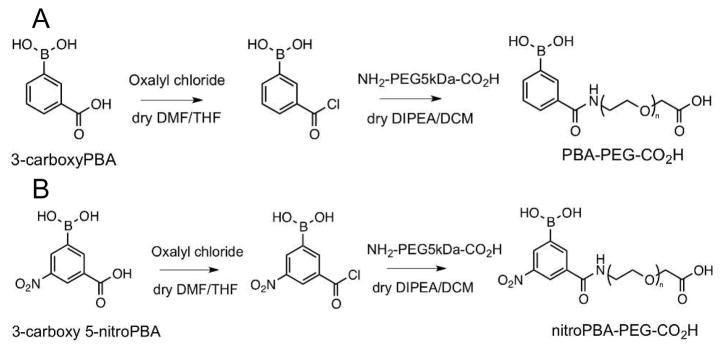
Synthesis of (A) PBA-PEG-CO2H and (B) nitroPBA-PEG-CO2H.
pKa values of the modified PBAs were determined by absorbance changes due to conformational change of PBA from trigonal to tetrahedral form as the pH was increased (Table 2). PBA with electron withdrawing groups resulted in lower pKa’s, with nitroPBA-PEG-CO2H having the lowest pKa of 6.8. Thus, at physiological pH, most of the PBA was present in the reactive anionic tetrahedral form. Binding constants between PBA and MAP were found by competitive binding with Alizarin Red S in 0.1 M PBS, pH 7.4. Because of the low pKa value and high binding constant with MAP observed for nitroPBA-PEG-CO2H, it was chosen as the linker between Herceptin and MAP-CPT nanoparticles.
Table 2.
pKa values and binding constants of various PBAs (phenylboronic acids) to MAP
| pKa | Binding Constant with MAP (M−1) | |
|---|---|---|
| PBA | 8.8 | 20 |
| PBA-PEG-CO2H | 8.3 | 520 |
| nitroPBA-PEG-CO2H | 6.8 | 1420 |
The conjugation reaction between Herceptin and nitroPBA-PEG-CO2H proceeded via EDC coupling (Scheme 4). An average of 1 to 2 PEGs were attached per Herceptin antibody.
Scheme 4.
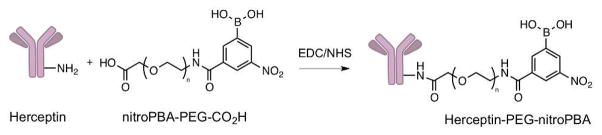
Synthesis of Herceptin-PEG-nitroPBA.
An average size of ca. 40 nm was observed for targeted MAP-CPT nanoparticles by DLS and cryo-EM (Table 1 and Fig. 1B). This increase in ca. 10 nm from the non-targeted nanoparticle suggests the attachment of Herceptin to the nanoparticle (the amount was ca. one Herceptin antibody per nanoparticle). Indeed, from cryo-EM images, the targeted nanoparticles were not spherical but appeared to have protrusions indicating the attachment of antibodies. The surface charge of targeted MAP-CPT nanoparticles was slightly higher than MAP-CPT nanoparticles due to the presence of a positively charged Herceptin at pH 7.4 (pI of Herceptin, 9.2 (17)).
Cellular Uptake Studies
BT-474, a HER2 overexpressing human breast cancer cell line, was used to examine the cellular uptake of Herceptin targeted MAP-CPT nanoparticles versus the non-targeted nanoparticles. MCF-7, a HER2 negative breast cancer cell line, was used as a negative control. It was observed that one Herceptin-PEG-nitroPBA per nanoparticle was sufficient to achieve ca. 70% greater uptake in BT-474 cell line compared to the non-targeted nanoparticle (Fig. 2A). Uptake of the targeted MAP-CPT in BT-474 cells showed inhibition in the presence of free Herceptin (Fig. 2B). In contrast, uptake of MAP-CPT nanoparticles in MCF-7 exhibited no dependence on targeting or on presence of free Herceptin (Fig. 2B). These data indicate that there is receptor mediated uptake in the BT-474 cells by the targeted MAP-CPT nanoparticles via engagement of the HER2 receptor. Cellular uptake of both the targeted and non-targeted MAP-CPT nanoparticles also occurred via nonspecific fluid phase endocytosis.
Figure 2.

Cellular uptake of (A) targeted MAP-CPT nanoparticles at increasing ratio of Herceptin-PEG-nitroPBA to nanoparticle in BT-474, (B) medium MAP-CPT nanoparticles or targeted MAP-CPT nanoparticles with or without free Herceptin (10 mg/ml) in BT-474 and MCF-7 cell lines.
In Vitro Release Studies
Release of CPT from the short, medium and long MAP-CPT nanoparticles and from the targeted MAP-CPT nanoparticles all exhibited first-order kinetics. Release half-lives of CPT from the short, medium and long MAP-CPT nanoparticles were very similar in all conditions tested, while longer half-lives for the targeted MAP-CPT were observed (Table 3). A strong dependence of release rate with pH was observed. As the pH increased from 6.5 to 7.4, the release half-lives reduced from 338 h to 58 h for the short, medium and long MAP-CPT nanoparticles. These data indicate that hydrolysis plays an important role in the release of CPT. A release half-life of 59 h was observed in BALB/c mice plasma, while a significantly lower half-life of 38 h was obtained in human plasma for the short, medium and long MAP-CPT nanoparticles. It is known that mice plasma contains more esterase activity than human plasma, while human plasma contains more “fat” than mouse plasma. Therefore, to understand the differences in release rates between human and mice plasma, the contributions of esterase and fat to CPT release rates were individually tested. Butyrylcholinesterase (BCHE) is present in both mice and human plasma and was used to test esterase contribution to the release rate. Low density lipoprotein (LDL) was chosen to test the contribution of “fat” to the release rate. These components were constituted in PBS (pH 7.4). It was found that the presence of BCHE did not affect release rate (half-life of 61 h compared to 58 h in PBS, pH 7.4, for the short, medium and long MAP-CPT nanoparticles). However, the addition of LDL dramatically increased the release rates. This effect is likely due to nanoparticle disruption by competing hydrophobic interactions. Therefore, nanoparticle disruption by the presence of “fat”, and the subsequent CPT cleavage by hydrolysis appears to be another main mechanism of CPT release from MAP-CPT nanoparticles. In vitro release from the Herceptin targeted MAP-CPT nanoparticles shows longer half-lives than the non-targeted versions. It is possible that the presence of Herceptin with a pI of 9.2 [17] increased the stability of the negatively charged nanoparticles by electrostatic interactions, and thus shielded the nanoparticles from some of the competing hydrophobic interactions.
Table 3.
In vitro release half-lives (t1/2) of CPT from short, medium and long MAP-CPT nanoparticles and from targeted MAP-CPT nanoparticles in various media
| PBS | t1/2 (h) | pH 6.5 | pH 7 | pH 7.4 |
|---|---|---|---|---|
| Short, medium, long MAP-CPT | 338 | 178 | 58 | |
| Targeted MAP-CPT | 396 | 204 | 78 | |
|
| ||||
| Plasma | t1/2 (h) | Mice | Human | |
|
| ||||
| Short, medium, long MAP-CPT | 59 | 38 | ||
| Targeted MAP-CPT | 63 | 46 | ||
|
| ||||
| PBS pH 7.4 | t1/2 (h) | LDLa | BCHEb | LDL + BCHEc |
|
| ||||
| Short, medium, long MAP-CPT | 45 | 61 | 44 | |
| Targeted MAP-CPT | 62 | 76 | 62 | |
Abbreviations: PBS, phosphate buffered saline; LDL, low density lipoprotein; BCHE, Butyrylcholinesterase.
PBS, pH 7.4, containing 3 mg/ml LDL.
PBS, pH 7.4, containing 100 units/ml BCHE.
PBS, pH 7.4, containing 3 mg/ml LDL and 100 units/ml BCHE.
Cytotoxicity Assays
In vitro cytotoxicities of medium MAP, nitroPBA-PEG, CPT, medium MAP-CPT nanoparticles, targeted MAP-CPT nanoparticles and Herceptin were evaluated in two HER2+ breast cancer cell lines (BT-474, SKBR-3) and two HER2- breast cancer cell lines (MCF-7, MDA-MB-231) (Table 4).
Table 4.
IC50 values of medium MAP, nitroPBA-PEG, CPT, medium MAP-CPT nanoparticles, targeted MAP-CPT nanoparticles and Herceptin for a range of breast cancer cell lines
| MCF-7 | MDA-MB-231 | SKBR-3 | BT-474 | ||
|---|---|---|---|---|---|
| HER2 expression | − | − | + | + | |
|
| |||||
| IC50 (μM) | Medium MAP | >500 | >500 | >500 | >500 |
| nitroPBA-PEG | >1000 | >1000 | >1000 | >1000 | |
| CPT | 0.3 | 0.1 | 0.03 | 4 | |
| Medium MAP-CPT | 0.5 | 0.6 | 0.2 | 40 | |
| Targeted MAP-CPT | 0.5 | 0.6 | 0.1 | 6 | |
| Herceptin | no effect | no effect | no valuea | no valuea | |
no value, IC50 value was not obtained over the concentration range of 0.001–0.5 μM.
Medium MAP and nitroPBA-PEG gave IC50 values of above 500 μM and 1000 μM (highest concentrations tested), respectively, indicating minimal toxicity. IC50 concentrations of CPT, medium MAP-CPT nanoparticles and targeted MAP-CPT nanoparticles were based on the content of the CPT. It is noted that CPT was released gradually from medium MAP-CPT nanoparticles and targeted MAP-CPT nanoparticles (see In Vitro Release Studies). Additionally, nanoparticle uptake by cells results in long release times due to the acidic nature of the endosomes. These factors contribute to the observed higher IC50 values for the medium MAP-CPT nanoparticles and targeted MAP-CPT nanoparticles compared to CPT (Table 4). In HER2+ cell lines, the targeted MAP-CPT nanoparticles gave lower IC50 values compared to MAP-CPT alone, while in HER2- cell lines, targeting did not affect the IC50. BT-474 was the most resistant cell line to CPT, and consequently to medium MAP-CPT nanoparticles. The addition of Herceptin at concentrations of 0.001–0.5 μM to BT-474 or SKBR-3 cells resulted in a constant cell viability of ca. 60% as compared to a no treatment control. The lack of a true IC50 value over this concentration range for these cell lines has been observed previously (18). There were no effects observed in HER2- cell lines at Herceptin concentrations of up to 0.5 μM.
Pharmacokinetic Studies
The nanoparticles displayed a biphasic profile with a fast redistribution phase (α) and a long elimination phase (β). The elimination phase for the medium MAP-CPT nanoparticles was prolonged with a half-live of 16.6 h and a high AUC value of 2298 μg.h/ml. In contrast, mice injected with CPT alone at 10 mg/kg showed fast clearance, with AUC of only 1.6 μg.h/ml (19). Targeting of the medium MAP-CPT nanoparticles affected the pharmacokinetic profile by increasing the redistribution phase and prolonging the elimination phase to 21.2 h with a high AUC value of 2766 μg.h/ml. The amount of unbound CPT in plasma for both nanoparticles was low at all time points. The PK data are similar to those of CRLX101 (7) that is currently being investigated in a number of Phase II clinical trials.
Summary
We report here a new methodology for creating targeted nanoparticles that uses boronic acid-diol complexation as a way to assemble the targeting agents onto the polymer-based nanoparticles. The specific example of assembling an antibody targeted nanoparticle delivery system for the small hydrophobic drug, CPT, was used to demonstrate the assembly method. The targeted nanoparticle was composed of a biocompatible copolymer of mucic acid and PEG (MAP) onto which the anti-cancer drug CPT was conjugated. Targeting was achieved by attaching boronic acids to the targeting moiety of choice, in this case Herceptin antibody, and the subsequent complexation via boronic acid-diol formation with the mucic acid repeats in the MAP polymer. A ca. 40 nm nanoparticle size and a slightly negative surface charge allowed for its long circulation in vivo with an elimination half-life of 21.2 h and an AUC value of 2766 μg.h/ml at a 10 mg/kg CPT injection in mice. This extended circulation will be helpful in exploiting the EPR effect for localization in solid tumors. Nanoparticle cellular uptake was enhanced by 70% via targeting by receptor mediated uptake compared to the non-targeted version. This single antibody targeted system contained ca. 60 CPT molecules per nanoparticle. We have recently completed the initial in vivo testing of the nanoparticles described in this work in nude mice bearing BT-474 tumors. The targeted nanoparticles were able to show complete tumor regression while the non-targeted versions did not. These antitumor results along with further PK and biodistribution data will be reported elsewhere (20).
Supplementary Material
Acknowledgments
We thank Alasdair McDowall, David VanderVelde and Leonard Medrano for assistance with cryo-EM imaging, NMR spectroscopy and cell culture preparation respectively. This project was financially supported by the National Cancer Institute Grant CA 151819.
References
- 1.Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nature Rev Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 2.Kamaly N, Xiao ZY, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Society Rev. 2012;41:2971–3010. doi: 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Controlled Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 4.Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong KL, Nielsen UB, Marks JD, Benz CC, Park JW. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732–6740. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- 5.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci USA. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi CHJ, Alabi CA, Webster P, Davis ME. Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles. Proc Natl Acad Sci USA. 2010;107:1235–1240. doi: 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis ME. Design and development of IT-101, a cyclodextrin-containing polymer conjugate of camptothecin. Adv Drug Delivery Rev. 2009;61:1189–1192. doi: 10.1016/j.addr.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6:659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 9.Davis ME, Zuckerman JE, Choi CHJ, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–U140. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang WQ, Gao XM, Wang BH. Boronic acid compounds as potential pharmaceutical agents. Med Res Rev. 2003;23:346–368. doi: 10.1002/med.10043. [DOI] [PubMed] [Google Scholar]
- 11.Chen WX, Cheng YF, Wang BH. Dual-responsive boronate crosslinked micelles for targeted drug delivery. Angew Chem Int Edit. 2012;51:5293–5295. doi: 10.1002/anie.201201179. [DOI] [PubMed] [Google Scholar]
- 12.Li YP, Xiao WW, Xiao K, Berti L, Luo JT, Tseng HP, Fung G, Lam KS. Well-defined, reversible boronate crosslinked nanocarriers for targeted drug delivery in response to acidic pH values and cis-diols. Angew Chem Int Edit. 2012;51:2864–2869. doi: 10.1002/anie.201107144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alabi CA, Davis ME. 20100040556 US Patent Appl.
- 14.Cheng JJ, Khin KT, Jensen GS, Liu AJ, Davis ME. Synthesis of linear, beta-cyclodextrin-based polymers and their camptothecin conjugates. Bioconjugate Chem. 2003;14:1007–1017. doi: 10.1021/bc0340924. [DOI] [PubMed] [Google Scholar]
- 15.Springsteen G, Wang BH. A detailed examination of boronic acid-diol complexation. Tetrahedron. 2002;58:5291–5300. [Google Scholar]
- 16.Zhao H, Lee C, Sai PK, Choe YH, Boro M, Pendri A, Guan SY, Greenwald RB. 20-O-acylcamptothecin derivatives: Evidence for lactone stabilization. J Org Chem. 2000;65:4601–4606. doi: 10.1021/jo000221n. [DOI] [PubMed] [Google Scholar]
- 17.Wiig H, Gyenge CC, Tenstad O. The interstitial distribution of macromolecules in rat tumours is influenced by the negatively charged matrix components. J Physiology (London) 2005;567:557–567. doi: 10.1113/jphysiol.2005.089615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillips GDL, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blättler WA, Lambert JM, Chari RVJ, Lutz RJ, Wong WLT, Jacobson FS, Koeppen H, Schwall RH, Kenkare-Mitra SR, Sliwkowski MX. Targeting HER2-positive breast cancer with Trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68:9280–9290. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 19.Supko JG, Malspeis L. Pharmacokinetics of the 9-amino and 10,11-methylenedioxy derivatives of camptothecin in mice. Cancer Res. 1993;53:3062–3069. [PubMed] [Google Scholar]
- 20.Han H, Davis ME. Single antibody, targeted nanoparticle delivery of camptothecin. 2013. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.