

Abstract
BACKGROUND:
Phenylketonuria (PKU) is an inborn error of amino acid metabolism that results from a deficiency of phenylalanine hydroxylase (PAH). According to PAH database, exons 6 and 7 and their flanking introns of PAH gene contain the greatest number of mutant alleles. Therefore, as a preliminary study, nucleotide sequence analysis of exons 6 and 7 of the PAH gene has been performed in 25 PKU patients whose ancestors lived in Kermanshah province of Iran. To date, there has been no mutation data describing the genotypes of the PKU disease in this Kurdish ethnic region background.
MATERIALS AND METHODS:
Twenty-five patients (aged between 2 and 23 years) participated in this study. The DNA fragments containing two exons of the PAH gene [6 and 7] and their exon-flanking intronic sequences were amplified and sequenced.
RESULTS:
The total of detected mutations were R261X (8%), R176X (4%), R243Q (4%), R243X (2%) and R261Q (2%), as they accounted for 20% of all mutant alleles in this study. The identified polymorphisms are: IVS5 -54 G > A (22%), Q232Q (8%) and V245V (4%). All of the detected mutations in this study are related to CpG dinucleotides in the PAH gene sequence.
CONCLUSION:
The frequency of R261X, the most common mutation in our study, in Iranian population is <5%. Furthermore, there is no report of detection of R176X and R243Q in Isfahan and Azeri Turkish populations. These findings confirm the common Mediterranean mutations in this local population, although with more or lower frequencies than those reported in other related studies in Iran. Therefore, it may be necessary to study the PAH gene mutations in other provinces of Iran separately.
Keywords: Iran, kermanshah, mutation detection, phenylalanine hydroxylase, phenylketonuria
Introduction
Phenylketonuria (PKU), the most prevalent disorder of amino acid metabolism, is a widespread autosomal-recessive hereditary disease with a frequency of 1/10000 in Caucasians[1] and a variable incidence in other ethnics and populations.[2] In Iran, PKU is estimated to be about 1.6 in 10000.[3] The deficiency of the phenylalanine hydroxylase (PAH; MIM# 261600) enzyme leads to elevated phenylalanine (Phe) levels in the blood and various tissues including the brain and if left untreated, causes mental retardation.[4] This enzyme has three structural domains consisting of an N-terminal regulatory domain, a catalytic domain, and a C-terminal tetramerization domain.[5]
Occurred mutations at the PAH gene, encoding the PAH enzyme, is the most reason of the disease with over than 600 known mutations to date (http://data.mch.mcgill.ca/pahdb_new/). This gene spans about 90 kb on chromosome 12q and comprises 13 exons and 12 intervening sequences (IVS). The mutation profile of the PAH gene is spreads throughout the entire structural domains and shows enormous diversity.[6] The severity of the disease is also diverse from mild hyperphenylalaninemia (MHP) to classical PKU, which is characterized by pretreatment blood phenylalanine levels or dietary tolerance.[7]
Kermanshah province with the Kurdish ethnic background is located in the Central West portion of Iran, with about two million populations, bordering to the West with Iraq and to the North, East and South with the provinces of Kurdistan, Hamadan, Lorestan and Ilam.[8] In spite of some reports regarding the PKU disease among different ethnics in Iran,[9–11] there has been no mutation data describing the genotypes of the PKU disease in Kermanshah province.
Determining the relationship between genotype and phenotype would provide very useful information for planning dietary and therapeutic strategies. Therefore, we analyzed exons 6 and 7 of the PAH gene in 25 PKU patients born in Kermanshah province and compared them with those of other related studies in Iran.
Materials and Methods
Patients
From 2010 to 2011, 25 unrelated families who had an affected child with PKU were referred to the Medical Genetics Laboratory of the Reference Laboratory in Kermanshah; detailed questionnaires, including clinical and family history, were collected. The PKU patients, 15 males and 10 females (aged between 2 and 23 years) originated from Kermanshah province. They were selected from the database-records at the Imam Reza hospital in Kermanshah city. The primary diagnosis of these patients had based on clinical criteria and laboratory findings (detection of elevated Phenylalanine (Phe) levels in blood samples by using HPLC). According to the plasma phenylalanine concentration prior to phenylalanine restriction diet, they were classified to classical PKU, moderate PKU, or MHP with the level of 1200 μM or more; 600-1200 μM; and less than 600 μM, respectively. Urinary pterin analysis was performed to exclude BH4 deficiencies. Informed consent for DNA analysis was obtained from the patient or the patients’ family (in case of most patients). Consanguinity among parents was proven in 23 (92%) cases.
Methods
Genomic DNA was extracted using QIAamp DNA Mini Kit (Qiagen, USA) after collection of blood samples (5 ml) from patients and parents. The DNA fragments containing two exons of the PAH gene [6 and 7] and their exon-flanking intronic sequences were amplified by polymerase chain reaction (PCR) using the primers as showed in Table 1. PCR conditions were as follows: initial denaturation95°C, 5 min: each cycle: denaturation 94°C, 30 sec, annealing 57°C, 30 sec, elongation 72°C, 45 sec, 30 cycles: Final elongation: 72°C, 7 min. The agarose gel was stained with ethidium bromide for visualizing the fragment migration.
Table 1.
Oligonucleotide amplification primers and PCR conditions

Sequence Analysis
Samples were analyzed by direct sequencing of exons 6 and 7 of the PAH gene in an ABI-3130 DNA analyzer (Applied Biosystems, USA). PCR products were purified by using QIAquick PCR purification kit. Then, sequencing samples were precipitated with Ethanol-Sodium Acetate precipitation and were used for cycle sequencing. After Sequencing, the data were analyzed using DNA sequencing analysis v 5.2 software.
Results
Nucleotide sequence analysis of exons 6 and 7 of the PAH gene revealed 5 different mutations on 10 of the 50 mutant alleles (diagnostic efficiency of 20%) [Table 2].
Table 2.
The PAH mutations identifified as the results of the study
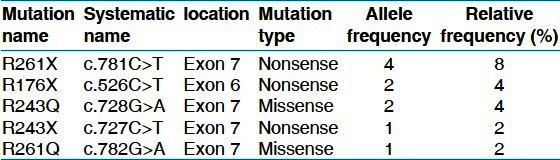
These included two missense mutations (6%) and three nonsense mutations (14%). The most frequent mutation was R261X (8%), accounted for four of all mutant alleles. The remaining four mutations were R176X, R243Q, R243X and R261Q representing 4, 4, 2 and 2%, respectively.
Among the seven patients with identified PAH gene mutations, three of them were homozygous (1 case of R243Q and 2 cases of R261X) and four of them showed only one mutant allele (two cases ofR176X and one case each of R243X and R261Q).
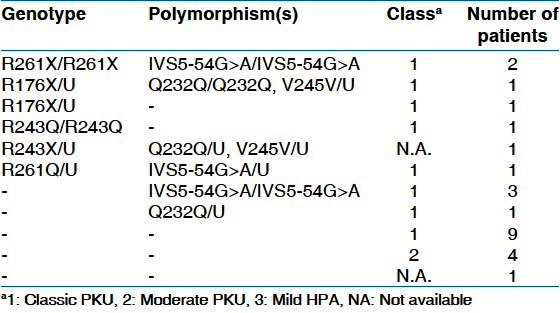
The identified polymorphysms are: IVS5 -54 G > A (intron 5), Q232Q (exon 6, CAA > CAG) and V245V (exon 7, GTG > GTA), with the frequencies of 22%, 8% and 4%, respectively [Table 3]. Genotypes of 25 PKU patients from this cohort are shown in Table 4.
Table 3.
The PAH polymorphisms identifified in the current study

Table 4.
Genotypes for mutations and polymorphisms of the PAH gene in 25 Kurdish PKU patients
Discussion
PKU is caused by the deficiency of PAH, an enzyme responsible for converting the amino acid phenylalanine into tyrosine in the liver, and leads to increase of phenylalanine in the blood. This accumulation is toxic to brain tissue and if untreated, infants with PKU develop mental retardation.[4]
In Kermanshah, Kurds are the prominent ethnic group that has lived in this area from the first millennium before the present age. Kermanshah has been known as the gate of Asia with the important Silk Road passing through this area.[12] This is the first report about the DNA lesions in the PAH gene causing PKU in Kermanshah province with the Kurdish ethnic background.
According to PAH database, exons 6 and 7 and their flanking introns contain the greatest number of mutant alleles. All of the detected mutations in this study are related to CpGdinucleotides in the PAH gene sequence. The CpG dinucleotide is often located at hotspots for pathogenic mutation in coding DNA. Deamination of the 5-methylcytosine at these sites leads to the conversion of Arg to Gln and Arg to stop codon (X).
R261X, the most common mutation in our study, is also detected in Iran[10,11] and some countries such as Croatia,[2] Italy,[13] Brazil,[14] Germany,[2] Korea,[15] Lithuania[16] and Portugal[17] [Table 5]. This mutation as well as R176X and R243X are nonsense mutations that lead to a premature step in translation.
Table 5.
Comparison of frequencies of detected mutations in this study with other studies in Iran and some other countries.
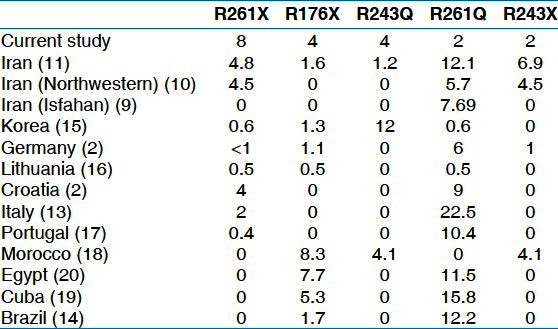
R243Q andR176X were seen with the frequency of 4% in this study. R243Q is a common mutation in the Southeast Asia countries.[15] R176X has previously been reported as the second major PKU-causing mutation in Moroccan[18] and in relatively frequencies in Cuban[19] and Egyptian[20] populations. The frequencies of these mutations in Iranian population is <2%.[11] Furthermore, there is no report of detection of these two mutations in Isfahan[9] and Azeri Turkish (northwestern of Iran)[10] populations [Table 5].
Two mutation remaining, R261Q and R243X are major mutations in Iranian population but were observed with the frequencies of 2% in this study. R261Q has previously been reported in relatively high frequencies in various populations, primarily in the Mediterranean and southern Europe. This mutation is a common mutation in the world and the second common mutation in Turkey.[21]
The neutral polymorphisms IVS5 -54 G > A (intron 5), Q232Q (exon 6, CAA > CAG) and V245V (exon 7, GTG > GTA), which does not lead to amino acid substitution, with the frequencies of 22%, 8% and 4%, respectively, was identified.
Conclusion
As seen on achieved results, the common Mediterranean mutations were detected in this local population, although with different frequencies than those reported in other related studies in Iran. Therefore, it may be necessary to study the PAH gene mutations in other provinces in Iran separately. Moreover, obtaining the full spectrum of mutations of this gene in Kermanshah province PKU patients needs to analysis of 11 remaining exons.[1–5,8–13]
Acknowledgements
We thank the patients and their family members for cooperation. This study was supported by a grant from Kermanshah University of Medical Sciences. The Vice Chancellor for Research at Kermanshah University of Medical Sciences in Iran has provided a grant to support this study.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Bickel H, Bachmann C, Beckers R, Brandt NJ, Clayton BE, Corrado HG, et al. Neonatal mass screening for metabolic. Eur J Pediatr. 1981;137:133–9. [Google Scholar]
- 2.Zschocke J. Phenylketonuria mutations in Europe. Hum Mutat. 2003;21:345–56. doi: 10.1002/humu.10192. [DOI] [PubMed] [Google Scholar]
- 3.Habib A, Fallahzadeh MH, Kazeroni H, Ganjkarimi AH. Incidence of phenylketonuria in southern Iran. Iranian J Med Sci. 2010;35:137–9. [Google Scholar]
- 4.Surtees R, Blau N. The neurochemistry of phenylketonuria. Eur J Pediatr. 2000;159:109–13. doi: 10.1007/pl00014370. [DOI] [PubMed] [Google Scholar]
- 5.Williams RA, Mamotte CD, Burnett JR. Phenylketonuria: An inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29:31–41. [PMC free article] [PubMed] [Google Scholar]
- 6.Scriver CR, Hurtubise M, Konecki D, Phommarinh M, Prevost L, Erlandsen H, et al. PAHdb: What a locus-specific knowledgebase can do? Hum Mutat. 2003;21:333–44. doi: 10.1002/humu.10200. [DOI] [PubMed] [Google Scholar]
- 7.Guldberg P, Rey F, Zschocke J, Romano V, François B, Michiels L, et al. A European multicenter study of phenylalanine hydroxylase deficiency: Classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am J Hum Genet. 1998;63:71–9. doi: 10.1086/301920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghotbi N, Tsukatani T. Evaluation of the national health policy of thalassemia screening in the Islamic Republic of Iran. East Mediterr Health J. 2005;11:308–18. [PubMed] [Google Scholar]
- 9.Vallian S, Barahimi E, Moeini H. Phenylketonuria in Iranian population: A study in institutions for mentally retarded in Isfahan. Mutat Res. 2003;526:45–52. doi: 10.1016/s0027-5107(03)00015-0. [DOI] [PubMed] [Google Scholar]
- 10.Bonyadi M, Omrani O, Moghanjoghi SM, Shiva S. Mutations of the phenylalanine hydroxylase gene in Iranian Azeri Turkish patients with phenylketonuria. Genet Test Mol Biomarkers. 2010;14:233–5. doi: 10.1089/gtmb.2009.0153. [DOI] [PubMed] [Google Scholar]
- 11.Zare-Karizi Sh, Hosseini-Mazinani SM, Khazaei-Koohpar Z, Seifati SM, Shahsavan-Behboodi B, Akbari MT, et al. Mutation spectrum of phenylketonuria in Iranian population. Mol Genet Metab. 2011;102:29–32. doi: 10.1016/j.ymgme.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Soltani MA. Tehran: Soha Press; 1999. Historical geography and comprehensive history of Kermanshah. [Google Scholar]
- 13.Guzzetta V, Bonapace G, Dianzani I, Parenti G, Lecora M, Giannattasio S, et al. Phenylketonuria in Italy: Distinct distribution pattern of three mutations of the phenylalanine hydroxylase gene. J Inherit Metab Dis. 1997;20:619–24. doi: 10.1023/a:1005315106604. [DOI] [PubMed] [Google Scholar]
- 14.Acosta A, Silva W, Jr, Carvalho T, Gomes M, Zago M. Mutations of the phenylalanine hydroxylase (PAH) gene in Brazilian patients with phenylketonuria. Hum Mutat. 2001;17:122–30. doi: 10.1002/1098-1004(200102)17:2<122::AID-HUMU4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 15.Lee DH, Koo SK, Lee KS, Yeon YJ, Oh HJ, Kim SW, et al. The molecular basis of phenylketonuria in Koreans. J Hum Genet. 2004;49:617–21. doi: 10.1007/s10038-004-0197-5. [DOI] [PubMed] [Google Scholar]
- 16.Kasnauskiene J, Giannattasio S, Lattanzio P, Cimbalistiene L, Kucinskas V. The molecular basis of phenylketonuria in Lithuania. Hum Mutat. 2003;21:398–402. doi: 10.1002/humu.9113. [DOI] [PubMed] [Google Scholar]
- 17.Rivera I, Leandro P, Lichter-Konecki U, Tavares de Almeida I, Lechner MC. Population genetics of hyperphenylalaninaemia resulting from phenylalanine hydroxylase deficiency in Portugal. J Med Genet. 1998;35:301–4. doi: 10.1136/jmg.35.4.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahri S, Desviat LR, Pérez B, Leal F, Ugarte M, Chabraoui L. Mutation analysis of phenylketonuria patients from Morocco: High prevalence of mutation G352fsdelG and detection of a novel mutation p.K85X. Clin Biochem. 2010;43:76–81. doi: 10.1016/j.clinbiochem.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 19.Desviat LR, Pérez B, Gutierrez E, Sánchez A, Barrios B, Ugarte M. Molecular basis of phenylketonuria in Cuba. Hum Mutat. 2001;18:252–5. doi: 10.1002/humu.1183. [DOI] [PubMed] [Google Scholar]
- 20.Effat L, Kuzmin A, Kasem N, Meguid NA, Kotb S, Eisensmith RC, et al. Haplotypes and mutations of the PAH locus in Egyptian families with PKU. Eur J Hum Genet. 1999;7:259–62. doi: 10.1038/sj.ejhg.5200270. [DOI] [PubMed] [Google Scholar]
- 21.Ozgûç M, Ozalp I, Coçkun T, Yilmaz E, Erdem H, Ayter S. Mutation analysis in Turkish phenylketonuria patients. J Med Genet. 1993;30:129–30. doi: 10.1136/jmg.30.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]