

Abstract
Aims
Inward remodelling of the resistance vasculature is predictive of hypertension and life-threatening cardiovascular events. We hypothesize that the contractile mechanisms responsible for maintaining a reduced diameter over time in response to prolonged stimulation with vasoconstrictor agonists are in part responsible for the initial stages of the remodelling process. Here we investigated the role of vascular smooth muscle (VSM) actin polymerization on agonist-induced vasoconstriction and development of inward remodelling.
Methods and results
Experiments were conducted in Sprague–Dawley rat resistance vessels isolated from the cremaster and mesentery. Within blood vessels, actin dynamics of VSM were monitored by confocal microscopy after introduction of fluorescent actin monomers through electroporation and by differential centrifugation to probe globular (G) and filamentous (F) actin content. Results indicated that 4 h of agonist-dependent vasoconstriction induced inward remodelling and caused significant actin polymerization, elevating the F-/total-actin ratio. Inhibition of actin polymerization prevented vessels from maintaining prolonged vasoconstriction and developing inward remodelling. Activation of the small GTPases Rho/Rac/Cdc42 also increased the F-/total-actin ratio and induced inward remodelling, while inhibition of Rho kinase or Rac-1 prevented inward remodelling. Disruption of the actin cytoskeleton reversed the inward remodelling caused by prolonged vasoconstriction, but did not affect the passive diameter of freshly isolated vessels.
Conclusion
These results indicate that vasoconstriction-induced inward remodelling is in part caused by the polymerization of actin within VSM cells through activation of small GTPases.
Keywords: Microcirculation, Hypertension, Cytoskeletal remodelling, Rho-kinase, Rac-1
1. Introduction
Hypertension seriously afflicts humanity. In 2006, the World Health Organization estimated that by the year 2030 almost 23 million people will die from hypertension-related cardiovascular diseases, mainly from heart disease and stroke.1 The pathophysiology of hypertension is still largely not understood, with 95% of it considered essential. However, the most common structural change observed in resistance arteries of individuals with essential hypertension is inward eutrophic remodelling.2–4 Moreover, the presence of inward eutrophic remodelling has the best predictive value for subsequent life-threatening cardiovascular events in both normotensive and hypertensive individuals.5,6
Inward euthrophic remodelling is characterized by a reduced passive luminal diameter without a change in cross-sectional area of the vascular wall.4,7 It is postulated that, in hypertension, the main stimulus inducing this remodelling is increased vascular resistance secondary to prolonged vasoconstriction.6,8 Therefore, we hypothesized that mechanisms linked to vascular smooth muscle (VSM) contraction during prolonged vasoconstriction are involved in the initial stages of inward remodelling.9
Traditionally, vasoconstriction has been ascribed to a process in which stimuli cause a rise in intracellular calcium that activates myosin light-chain kinase with the subsequent cycling of actomyosin cross-bridges in VSM.9 However, recent evidence indicates that VSM contraction is also associated with rapid actin polymerization and VSM cytoskeletal remodelling.10,11 Furthermore, experimental results12 suggest that cytoskeletal remodelling is time-dependent, with actin polymerization increasing over time as vasoconstriction persist. Prolonged vasoconstriction has also been associated with the repositioning of VSM cells within the vascular wall of resistance vessels,13 a process that should require cytoskeletal remodelling and the formation of new cellular adhesions.14 In the light of the aforementioned evidence, we investigated whether prolonged vasoconstriction of small resistance arteries resulted in VSM actin polymerization that triggered inward eutrophic remodelling.
2. Methods
2.1. Animals and vessels
All animal procedures complied with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011), and were approved by the Animal Care and Use committee at the University of Missouri-Columbia. Male Sprague–Dawley rats (150–250 g) were anaesthetized with pentobarbital sodium (100 mg/kg). Anaesthesia was confirmed by loss of spinal nociceptive reflexes. First-order segments of the cremaster-feed arterioles were isolated and cannulated as described previously.15 Isolated arterioles were electroporated with Actin-Alexa-488, cannulated, and pressurized in an isolation chamber, and subsequently examined for Actin-Alexa-488-dependent fluorescence with a confocal microscope. Euthanasia was induced by pneumothorax and exsanguination while under anaesthesia.
2.2. Experimental protocols
To determine the effects of prolonged vasoconstriction on actin dynamics, we exposed electroporated arterioles to norepinephrine (NE, 10−5.5 M) + angiotensin-II (Ang-II, 10−7 M) for 4 h and measured the fluorescence intensity of Actin-Alexa-488 within individual VSM cells over time. We also determined the effects of preventing actin polymerization on Actin-Alexa-488-dependent fluorescence using electroporated arterioles exposed to cytochalasin-D (500 nM) for 20 min and then to NE + Ang-II while in the presence of cytochalasin-D for 4 h. Confocal images were taken before the addition of the vasoconstrictor agonists, every hour during exposure to NE + Ang-II, and immediately after washing off the agonists. In additional experiments, arterioles not electroporated were first dilated with 10−4 M adenosine and then exposed to calcium-free physiological saline solution (PSS) to obtain maximal passive diameter. Subsequently, vessels were allowed to regain tone in PSS with calcium and then exposed to 500 nM cytochalasin-D for 20 min followed by a 4 h exposure to NE + Ang-II. After this, vessels were washed with PSS for 30 min and then exposed to adenosine followed by calcium-free PSS. In these experiments, vascular diameter was continuously recorded using video microscopy.16
In vitro experiments were also conducted to determine the effects of Actin-Alexa-488 polymerization on its fluorescence intensity. Experiments were conducted in mini-well slides placed on a confocal microscope using the same objective and excitation/emission parameters as when imaging arterioles. Actin-Alexa-488 (1 µg/µL) was exposed to actin polymerization or control buffer, and images were taken every 30 s for 10 min. Subsequently, mycalolide-B (2 μM) was added and images taken every 30 s for 10 min.
To determine the effects of Rho/Rac/Cdc42 activation on actin polymerization and inward remodelling, arterioles were exposed to 1 µg/mL CN04 for 4 h. As in the previous protocols, a maximal passive diameter was obtained before and after the prolonged exposure to CN04. Actin polymerization was measured as the ratio of F-/total-actin by differential ultracentrifugation using a commercially available kit17 in rat mesenteric arterioles exposed to NE + Ang-II, jasplakinolide, CN04 (1 µg/mL), or vehicle control for 4 h. To determine the effects of Rho-associated protein kinase (ROCK) or Rac-1 inhibition on vasoconstriction-induced inward remodelling, isolated cremaster arterioles were exposed to 10 µM Y27632 or 100 µM NSC23766 for 20 min prior to and during a 4 h exposure to NE + Ang-II. As described above, the maximal passive diameter was obtained before and after the prolonged exposure to agonists.
2.3. Immunohistochemistry
To determine the effects of cytochalasin-D and mycalolide-B on the structure of VSM stress fibres, cannulated and pressurized arterioles were exposed to cytochalasin-D (10 µM), mycalolide-B (2 µM), or vehicle control for 1 h and then fixed with 4% paraformaldehyde. Vessels were incubated with a combination of phalloidin, anti-tubulin antibodies, and 4′,6-diamidino-2-phenylindole (DAPI) in order to stain stress fibres, microtubules, and nuclei, respectively. Vessels were washed and imaged on a confocal microscope.
2.4. Statistics
Data are presented as means ± SE. Statistical comparisons were made using paired or unpaired t-tests, or one-way ANOVA followed by Tukey's test for multiple comparisons as appropriate. Values of P ≤ 0.05 were considered significant. (See extended Methods in Supplementary material online.)
3. Results
3.1. Prolonged exposure to NE + Ang-II decreased Actin-Alexa-488-fluorescence within VSM cells of electroporated arterioles
As an approach to quantify changes in actin polymerization, we measured the changes in fluorescence intensity of VSM cells over time in arterioles electroporated with Actin-Alexa-488 (Figure 1A). We found that the fluorescence intensity was inversely proportional to the degree of actin polymerization. Cellular fluorescence intensity decreased 42% after 4 h of exposure to NE + Ang-II. After removing the vasoconstrictor agonists, the fluorescence intensity increased by 9%. In comparison, cells in control vessels not exposed to NE + Ang-II experienced no significant change in fluorescence intensity over time (Figure 1B). In arterioles treated with cytochalasin-D to inhibit actin polymerization, exposure to NE + Ang-II for 4 h caused only a 16% decrease in fluorescence intensity (Figure 1B). Consequently, the slope of the regression line representing the reduction in fluorescence intensity in arterioles exposed for 4 h to NE + Ang-II was significantly greater than that of control arterioles or vessels exposed to NE + Ang-II in the presence of cytochalasin-D (P ≤ 0.05) (Figure 1B).
Figure 1.
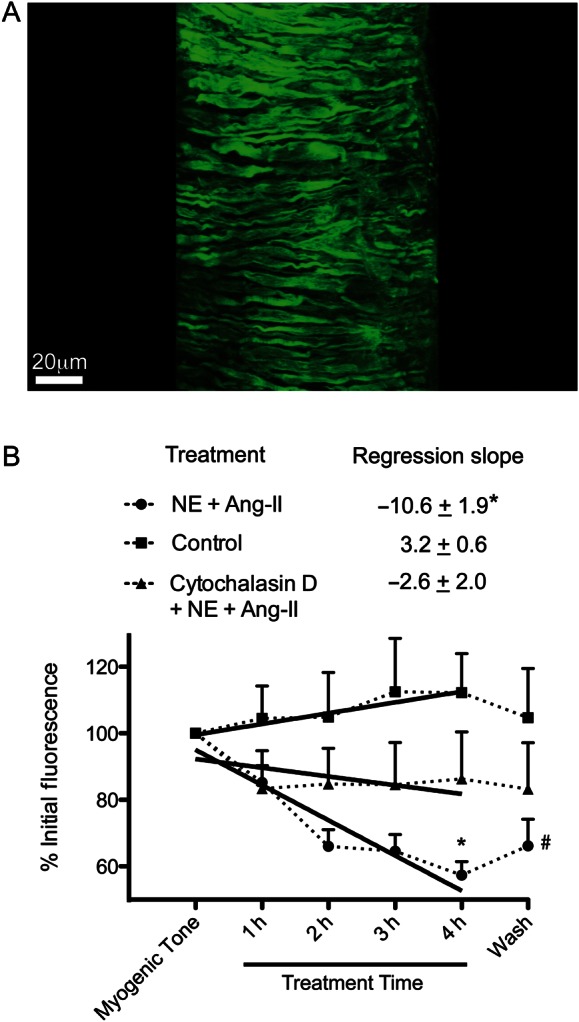
Prolonged (4 h) exposure of arterioles to NE (10−5.5 M) + Ang-II (10−7 M) results in a significant decrease in Actin-Alexa-488 fluorescent intensity compared with control vessels with myogenic tone. (A) Representative image of an arteriole showing the fluorescence associated with Actin-Alexa-488 within VSM cells. The image is a three-dimensional reconstruction of confocal Z-stacks of a vessel electroporated with Actin-Alexa-488. The reconstructed image was segmented to eliminate the adventitial extracellular matrix fluorescence and allow for visualization of the Actin-Alexa-488 within individual VSM cells. (B) Change in Actin-Alexa-488 fluorescence intensities over time within VSM cells of arterioles exposed to either control conditions (n = 6), cytochalasin-D (500 nM) + NE + Ang-II (n = 8), or NE + Ang-II (n = 9). Fluorescence intensities were obtained from a minimum of two cells per vessel from raw images taken with exactly the same excitation, emission, and detection parameters. Fluorescence intensities were normalized to the initial level of fluorescence detected in the first image taken with the vessel having spontaneous myogenic tone. The regression slopes (mean ± SE) represent the change in fluorescence intensities from the initial level at myogenic tone to that after 4 h of incubation under each condition. The regression slope of vessels exposed to NE + Ang-II is significantly different from that of the other conditions (*P ≤ 0.05). In the line graph, the overall fluorescence intensity (mean ± SE) was significantly reduced in NE + Ang-II-exposed arterioles compared with the other conditions at 4 h of incubation (*P ≤ 0.05) and remained reduced compared with control 30 min after the removal of all agonists (Wash, #P ≤ 0.05).
3.2. Polymerization of Actin-Alexa-488 in vitro decreases its fluorescence
To confirm that the changes in Actin-Alexa-488-dependent fluorescence were related to actin polymerization, we performed a series of experiments in which Actin-Alexa-488 was polymerized in vitro. Exposure of Actin-Alexa-488 to general actin buffer (control) caused an overall 29% reduction in fluorescence after 10 min due to dilution effects (Figure 2A). In comparison, Actin-Alexa-488 exposed for 10 min to actin polymerization buffer resulted in an overall 61% reduction in fluorescence intensity (P ≤ 0.05 vs. control) (Figure 2B). Subsequent addition of mycalolide-B caused a further reduction in fluorescence in actin exposed to control buffer and a fluorescence increase in actin previously exposed to polymerization buffer (Figure 2).
Figure 2.

In vitro polymerization decreases the fluorescence intensity of Actin-Alexa-488. (A) Florescence intensities of Actin-Alexa-488 in solution before and after the addition of control buffer for 10 min followed by 2 μM mycalolide-B for 10 min. (B) Fluorescence intensities of Actin-Alexa-488 in solution before and after the addition of actin polymerization buffer for 10 min followed by 2 μM mycalolide-B for 10 min. All data are means ± SE. *P ≤ 0.05 vs. control.
3.3. Cytochalasin-D caused the vasoconstriction induced by NE + Ang-II to wane over time
Exposure of arterioles to cytochalasin-D (20 min) caused a paradoxical tendency to increase the level of tone without preventing the initial (within 5 min) vasoconstriction induced by exposure to NE + Ang-II (Figure 3A). Thereafter, cytochalasin-D prevented NE + Ang-II from maintaining vasoconstriction over time. Vessels dilated to diameters approaching maximal passive diameter despite the presence of NE + Ang-II for 4 h. The removal of cytochalasin-D and the vasoconstrictors by washing the vessels for 30 min caused a small but insignificant reduction in diameter. Subsequent exposure to adenosine and calcium-free solution confirmed that the vessels did not remodel (Figure 3A). Vessels treated with only cytochalasin-D lost myogenic tone over time, but at a lesser rate and smaller extent than the loss of constriction associated with exposure to NE + Ang-II (Figure 3B). In comparison, vessels exposed 4 h to NE + Ang-II without cytochalasin-D remained constricted throughout exposure to the agonists and developed inward remodelling characterized by a reduced passive luminal diameter and an increased wall to lumen ratio (Figure 3C and see Supplementary material online, Figure S2).
Figure 3.

Exposure of isolated arterioles to cytochalasin-D causes the constriction evoked by NE + Ang-II to subside over time and prevents inward remodelling. (A) Arterioles incubated (20 min) with cytochalasin-D (500 nM) and then exposed (4 h) to NE (10−5.5 M) + Ang-II (10−7 M) in the presence of cytochalasin-D (n = 6). Before and after the incubation with cytochalasin-D and NE + Ang-II, arterioles were allowed to develop spontaneous myogenic tone, and exposed to Adenosine (Ado, 10−4 M), and then to calcium-free solution. (B) Arterioles were incubated (4 h, 20 min) with cytochalasin-D (500 nM, n = 6). Before and after the incubation with cytochalasin-D, arterioles were allowed to develop spontaneous myogenic tone, and exposed to Adenosine (Ado, 10−4 M), and then to calcium-free solution. (C) Arterioles were exposed (4 h) to only NE + Ang-II (n = 8). Before and after the incubation with NE + Ang-II, arterioles were allowed to develop spontaneous myogenic tone, and exposed to Adenosine (Ado, 10−4 M), and then to calcium-free solution. Data are means ± SE of the per cent maximal diameter obtained under calcium-free conditions before incubation with the agonists. *P ≤ 0.05 vs. the passive diameter (calcium-free) obtained before exposure to NE + Ang-II.
3.4. Actin depolymerization reversed the inward remodelling caused by 4 h of vasoconstriction
Consistent with a previous report,18 our current data indicated that prolonged exposure to agonist-induced vasoconstriction promoted actin polymerization. To determine whether polymerized actin played a role in the initial stages of the inward remodelling process, remodelled arterioles were treated 1 h with actin-depolymerizing agents under calcium-free passive conditions. As shown in Figure 4A, arterioles exposed 4 h to NE + Ang-II underwent inward remodelling with a reduction in the passive diameter of 9.07 ± 0.36 μm. Exposure of these remodelled arterioles to mycalolide-B reversed the inward remodelling by 76% (Figure 4B). In comparison, remodelled arterioles maintained 1 h under calcium-free conditions with vehicle control or cytochalasin-D had no significant changes in the diameter. Immunofluorescence showed that compared with controls, arterioles exposed to cytochalasin-D had only a partial and patchy disruption of VSM actin fibres, whereas arterioles exposed to mycalolide-B had an almost complete disruption of the actin cytoskeleton (Figure 4C). Freshly isolated arterioles unexposed to NE + Ang-II showed no changes in passive diameter when exposed for 1 h to actin-depolymerizing agents (data not shown).
Figure 4.

Actin depolymerization reverses the inward remodelling caused by 4 h of exposure to NE + Ang-II. (A) Arterioles exposed (4 h) to NE (10−5.5 M) + Ang-II (10−7 M). Before and after the 4 h incubation with NE + Ang-II, arterioles were allowed to develop spontaneous myogenic tone, and were exposed to adenosine (Ado, 10−4 M), and then to calcium-free solution. After 5 min under the second exposure to calcium-free conditions, arterioles were treated (1 h) with vehicle control (n = 8), cytochalasin-D (10 μM, n = 9), or mycalolide-B (2 μM, n = 11). Data are means ± SE of the per cent maximal diameter obtained under calcium-free conditions before the 4 h incubation with NE + Ang-II. *P ≤ 0.05 vs. control or cytochalasin-D. (B) Change in diameter caused by exposure to vehicle control, cytochalasin-D, or mycalolide-B is expressed as a per cent change from the reduction in passive diameter following the second exposure to calcium-free conditions. Data are means ± SE. *P ≤ 0.05 vs. control or cytochalasin-D. (C) Three-dimensional reconstruction images of arterioles stained with DAPI (red) to visualize nuclei, phalloidin (green) to visualize F-actin, and in the left panel anti-tubulin antibody (blue) to visualize microtubules. Left, control vessels were incubated with vehicle control for 1 h. Middle, arterioles were exposed to 10 μM cytochalasin-D for 1 h. Right, arterioles were exposed (1 h) to 2 μM mycalolide-B and show an almost complete disruption of the actin cytoskeleton. Microtubules stained (blue) show that cytoskeletal structures other than F-actin remained intact.
3.5. Prolonged vasoconstriction or Rho/Rac/Cdc42 activation increased F-/total-actin in arterioles
To explore the mechanisms associated with prolonged vasoconstriction-induced actin polymerization and inward remodelling, we first corroborated that arteriolar exposure to NE + Ang-II resulted in a nearly 10% increase in F-/total-actin using a differential centrifugation assay (Figure 5). Exposure to jasplakinolide for 4 h increased the F-/total-actin nearly 20% vs. control (Figure 5). As exposure to vasoconstrictor agonists activates cytoskeletal remodelling pathways and the small GTP-binding proteins Rho, Rac, and Cdc42,19 we examined the effects of the Rho/Rac/Cdc42 activator-I, CN04, on isolated arterioles. Exposure for 4 h to CN04 induced a 14.8% increase in F-/total-actin compared with controls (Figure 6A) and caused an overall constriction of 13% (data not shown). Subsequent exposure to calcium-free solution revealed that arterioles exposed to CN04 had a small but significantly reduced (6.2%) passive diameter compared with control arterioles unexposed to CN04 (Figure 6B). Arterioles exposed 4 h to jasplakinolide showed no signs of inward remodelling (data not shown).
Figure 5.

F-actin content is increased in vessels exposed to NE + Ang-II for 4 h. The Top panel shows a representative immunoblot of actin separated by differential ultracentrifugation into G-(monomeric) and F-(filamentous) actin portions from control arterioles and those exposed to NE (10−5.5 M) + Ang-II (10−7 M) or Jasplakinolide (Jas, 100 nM) for 4 h. The Lower panel shows the F-/total-actin ratios normalized to those of control as obtained from the densitometric analysis of the actin bands from the immunoblots (n = 5 independent experiments for each group). Data are means ± SE. *P ≤ 0.05 vs. control.
Figure 6.

Activation of Rho/Rac/Cdc42 increases actin polymerization and evokes inward remodelling. (A) Upper panel: a representative immunoblot of actin separated by differential ultracentrifugation into G- and F-actin portions from control arterioles and those exposed (4 h) to CN04 (1 µg/mL). Lower panel: the F-/total-actin ratios normalized to those of control as obtained from the densitometric analysis of the actin bands from the immunoblots (n = 10 independent experiments for each group). Data are means ± SE. *P ≤ 0.05 vs. control. (B) The bars represent the maximal passive diameter of arterioles kept under control conditions (60 mmHg intraluminal pressure at 34.5°C) or exposed (4 h) to CN04 (1 µg/mL) (n = 7/group). Data are means ± SE of the per cent maximal diameter obtained under calcium-free conditions before the 4 h incubation period. *P ≤ 0.05 vs. control. (C) Arterioles incubated (20 min) with the ROCK inhibitor Y27632 (10 µM) and then exposed (4 h) to NE (10−5.5 M) + Ang-II (10−7 M) in the presence of Y27632 (n = 9). Before and after the incubation with Y27632 and NE + Ang-II, arterioles were allowed to develop spontaneous myogenic tone, and exposed to Adenosine (Ado, 10−4 M), followed by calcium-free solution. Data are means ± SE of the per cent maximal diameter obtained under calcium-free conditions before the incubation with Y27632. (D) Arterioles incubated (20 min) with the Rac-1 inhibitor NSC23766 (100 µM) and then exposed (4 h) to NE + Ang-II in the presence of NSC23766 (n = 11). Before and after the incubation with NSC23766 and NE + Ang-II, arterioles were allowed to develop spontaneous myogenic tone, and exposed to Adenosine (Ado, 10−4 M), and then to calcium-free solution. Data are means ± SE of the per cent maximal diameter obtained under calcium-free conditions before the incubation with NSC23766.
3.6. Inhibition of ROCK or Rac-1 prevented vasoconstriction-induced inward remodelling
During agonist-induced vasoconstriction, Rho-mediated ROCK activation causes calcium sensitization of contractile proteins and the phosphorylation of LIM domain kinase1 (LIMK1), which translates into cofilin inactivation, actin polymerization, and actin filament stabilization.15 To determine the role of ROCK on vasoconstriction and inward remodelling, we incubated arterioles with the ROCK inhibitor Y27632 (10 µM) 20 min before and during exposure (4 h) to NE + Ang-II. The initial 20 min exposure to Y27632 resulted in a nearly 19% dilation from myogenic tone (Figure 6C). Y27632 did not prevent NE + Ang-II from inducing an initial constriction, but prevented NE + Ang-II from maintaining vasoconstriction over time (Figure 6C). Removal of NE + Ang-II and Y27632 for 30 min caused further dilation, and exposure to adenosine 10−4 M and calcium-free solution showed that these vessels did not remodel (Figure 6C). In comparison, inhibition of Rac-1 with NSC23766 (100 µM) caused an initial reduction in vascular diameter, did not prevent NE + Ang-II from maintaining vasoconstriction for 4 h, but prevented these vessels from remodelling (Figure 6D).
4. Discussion
The primary finding of this study was that prolonged exposure of isolated arterioles to vasoconstrictor agonists induced VSM actin polymerization, which in turn was, in part, responsible for the development of inward remodelling in these vessels.
First, in isolated arterioles electroporated with Actin-Alexa-488, we observed that actin-dependent fluorescence diminished over time in response to prolonged agonist-induced vasoconstriction. Control arterioles not exposed to the agonists did not show significant changes in fluorescence. Therefore, we associated the process of agonist-induced vasoconstriction with reduction in G-actin-dependent fluorescence and interpreted the decrease in fluorescence as increased actin polymerization. It is likely that the Actin-Alexa-488 electroporated into arterioles had not reached an equilibrium between monomeric and fibrillar actin at the beginning of each experiment. Therefore, the fact that vasodilation did not fully restore fluorescence intensity does not necessarily mean that a quantitatively different depolymerization occurred between the constriction and relaxation events. In vitro, we confirmed that the reduction in Actin-Alexa-488 fluorescence was indeed associated with actin polymerization. The overall fluorescence of Actin-Alexa-488 decreased upon exposure to actin polymerization buffer. Subsequent exposure to the actin depolymerization agent mycalolide-B20 caused an increase in fluorescence. In contrast, exposure of Actin-Alexa-488 to control buffer followed by mycalolide-B caused only slight reductions in fluorescence due to dilution effects. The significant reduction in fluorescence caused by actin polymerization is likely the result of fluorescence quenching caused by the nucleation of actin monomers to form F-actin.
To determine the effects of inhibiting actin polymerization on prolonged vasoconstriction and inward remodelling, we treated isolated arterioles with cytochalasin-D before and during a prolonged exposure to NE + Ang-II. Cytochalasin-D binds to the barbed end of actin filaments and inhibits the association of new actin subunits. Cytochalasin-D at 500 nM inhibits actin polymerization without affecting the initial phase of agonist-induced vasoconstriction.21–23 In accordance, cytochalasin-D did not prevent the initial contractile response of vessels exposed to NE + Ang-II, but resulted in their inability to maintain constriction over time. The addition of cytochalasin-D in the absence of NE + Ang-II also caused the loss of myogenic tone over time, a result consistent with those of other laboratories indicating that myogenic constriction requires actin polymerization.24–26 After 4 h of exposure to NE + Ang-II with cytochalasin-D, vessels dilated and showed no signs of inward remodelling. Similar results were observed when latrunculin, an agent that binds actin monomers and inhibits actin polymerization, was used (data not shown). These results suggest that VSM actin polymerization is needed for the basal maintenance of myogenic tone and for maintenance of agonist-induced vasoconstriction as well as for the development of vasoconstriction-induced inward remodelling in arterioles.
The bimodal response of vessels to NE + Ang-II in the presence of cytochalasin-D suggests that two mechanisms are involved in the onset and maintenance of constriction during prolonged exposure to agonists. The mechanism likely responsible for the initial response to NE + Ang-II is mainly based on the calcium-dependent actomyosin interaction and cross bridge cycling controlled through the phosphorylation of myosin light chain. Our results indicate that actin polymerization does not affect this mechanism, as vessels had similar acute constrictions to NE + Ang-II in the presence or absence of cytochalasin-D. The second mechanism involved in maintaining vasoconstriction appears contingent on actin polymerization, as it was sensitive to the presence of cytochalasin-D or latrunculin.
Actin polymerization has been shown to be critical in the acute phase of smooth muscle contraction in tracheal tissues.11,27,28 Through processes involving the phosphorylation of paxillin, the activation of small GTP-binding proteins, and conformational changes within focal adhesion sites,29–31 contractile agonists increase the incorporation of G-actin into F-actin stress fibres and stiffen the cytoskeleton. Studies in blood vessels also indicate that inhibition of actin polymerization blocks the development or maintenance of full contractile force in arteries.12,21–23,25,26,32–34 However, a number of studies suggest that in blood vessels inhibition of actin polymerization interferes with agonist-induced vasoconstriction in a time-dependent manner.12,23,33 In those studies, cytochalasin used at low concentrations did not affect the maximal acute (within seconds) constriction of arteries. In addition, the phosphorylation of paxillin and the formation of F-actin fibres occurred during the sustained phase of vasoconstriction, between 10 and 20 min after the initial exposure to vasoconstrictor stimuli. At that time actin polymerization strengthened and stiffened the cytoskeleton, stiffening the cell over time and increasing the mechanical efficiency of the cell for the transmission of force.12 This cellular stiffening or ‘cellular solidification’ occurs concomitantly to paxillin phosphorylation and F-actin formation. We have previously shown that intracellular calcium during prolonged vasoconstriction is reduced to pre-constriction levels at timeframes similar to those reported for the increase in paxillin phosphorylation and F-actin formation.35 Results from the present study demonstrate, for the first time, that actin polymerization continues, as vessels remain exposed to vasoconstrictor agonists for 4 h. They also indicate that actin remains polymerized under passive conditions after the removal of the vasoconstrictor agonists and in the presence of adenosine and calcium-free PSS. This is supported by our observation that mycalolide-B reversed vasoconstriction-induced inward remodelling (Figure 4). To corroborate that mycalolide-B indeed disrupted the actin cytoskeleton; we determined that 1 h exposure with the toxin almost completely destroyed all actin fibres detectable by phalloidin staining in isolated arterioles. In comparison, incubation with vehicle or cytochalasin-D (10 μM) for 1 h did not significantly disrupt actin fibres and failed to reverse vasoconstriction-induced inward remodelling.
To induce actin cytoskeletal remodelling through mechanisms consistent with those activated by vasoconstrictor agents, we exposed isolated arterioles to CN04, an activator of the small Rho family GTPases Rho/Rac/Cdc42. These small GTPases have been shown to be key regulators of the actin cytoskeleton in diverse cell lines.36–38 For example, Rho activation results in the formation of stress fibres as well as in calcium sensitization through pathways involving its downstream target ROCK. In comparison, Rac regulates the formation of lamellipodia and membrane ruffles, and its activation leads to the reorganization of the actin cytoskeleton.39,40 Cdc42, the third GTPase activated by CN04, is required for the extension of filopodia, and its activation during agonist-induced contraction of tracheal smooth muscle has been shown to regulate active tension development and actin polymerization.30 In the present study, 4 h exposure of resistance vessels to CN04 resulted in constriction and inward remodelling. Vessels treated with CN04 also had greater F-/total-actin ratios than controls. In comparison, exposure of arterioles to jasplakinolide failed to induce inward remodelling (data not shown) in spite of increasing the ratio of F-/total-actin. Jasplakinolide induces actin polymerization and the stabilization of actin fibres in part through the incorporation of G-actin into disordered amorphous masses of F-actin, which sequesters monomeric actin and makes it unavailable for the remodelling of cellular stress fibres.41 This may explain the inability of jasplakinolide to induce inward remodelling under our experimental conditions. Moreover, it suggests that stabilization of stress fibres is not enough to induce inward remodelling. Vessels appear to require the specific cytoskeletal remodelling induced by activation of the small Rho family of GTPases.
To begin exploring the role of these GTPases on the remodelling process, we determined the effect of ROCK or Rac-1 inhibition on vasoconstriction-induced inward remodelling. Exposure of arterioles to Y27632 or NSC23766 prevented the NE + Ang-II-induced inward remodelling. However, the mechanisms involved in preventing inward remodelling by these inhibitors may or may not be related to their capacity to affect actin polymerization pathways. Y27632 is an inhibitor of ROCK, which is implicated in calcium sensitization as well as in actin polymerization pathways. In cerebral resistance arteries, ROCK predominantly affects actin polymerization rather than calcium sensitization.42 Therefore, it is likely that ROCK inhibition in the present study prevented inward remodelling by inhibiting actin polymerization, but this remains to be experimentally corroborated. Consistent with our results, in vivo inhibition of the Rho-ROCK pathway has been shown to ameliorate hypertension and vascular remodelling.43–45
In comparison with ROCK, Rac-1 is involved in actin polymerization pathways responsible for the formation of lamellipodia, as well as the production of superoxide as part of the NAD(P)H oxidase enzymes.46,47 Similar to our previous findings that NAD(P)H oxidase inhibition with apocynin prevented the development of inward remodelling without affecting agonist-induced vasoconstriction,48 NSC23766 also prevented inward remodelling without affecting the constriction caused by 4 h of exposure to NE + Ang-II. As inhibition of actin polymerization prevented NE + Ang-II from maintaining vasoconstriction over time, these results suggest that Rac-1 inhibition prevents inward remodelling by inhibiting NAD(P)H oxidase superoxide production rather than by inhibiting lamellipodia formation. Consistent with these findings, transgenic overexpression of Rac-1 in VSM cells of mice caused hypertension dependent on the overproduction of superoxide anion.49
In conclusion, results from the present study indicate that actin polymerization induced by prolonged vasoconstriction is in part responsible for the development of arteriolar inward eutrophic remodelling. Our results also suggest that actin polymerization participating in the initial stages of the remodelling process is not a mere stabilization of stress fibres, but likely a specific type of actin polymerization induced by the activation of small Rho family GTPases, in particular RhoA and its downstream effector ROCK. Further studies are required to determine the specific characteristics of the actin structures formed and their temporal or permanent role on reducing the passive diameter of the remodelled vessels.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by the National Institutes of Health(HL088105 to L.A.M-L., P01HL095486 to G.A.M.) and the American Heart Association(0530031N to L.A.M-L.).
Supplementary Material
References
- 1.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heagerty AM, Aalkjaer C, Bund SJ, Korsgaard N, Mulvany MJ. Small artery structure in hypertension: dual processes of remodeling and growth. Hypertension. 1993;21:391–397. doi: 10.1161/01.hyp.21.4.391. [DOI] [PubMed] [Google Scholar]
- 3.Heagerty AM, Heerkens EH, Izzard AS. Small artery structure and function in hypertension. J Cell Mol Med. 2010;14:1037–1043. doi: 10.1111/j.1582-4934.2010.01080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Intengan HD, Schiffrin EL. Structure and mechanical properties of resistance arteries in hypertension: role of adhesion molecules and extracellular matrix determinants. Hypertension. 2000;36:312–318. doi: 10.1161/01.hyp.36.3.312. [DOI] [PubMed] [Google Scholar]
- 5.Rizzoni D, Porteri E, Boari GEM, De Ciuceis C, Sleiman I, Muiesan ML, et al. Prognostic significance of small-artery structure in hypertension. Circulation. 2003;108:2230–2235. doi: 10.1161/01.CIR.0000095031.51492.C5. [DOI] [PubMed] [Google Scholar]
- 6.Mathiassen ON, Buus NH, Sihm I, Thybo NK, Mørn B, Schroeder AP, et al. Small artery structure is an independent predictor of cardiovascular events in essential hypertension. J Hypertens. 2007;25:1021–1026. doi: 10.1097/HJH.0b013e32805bf8ed. [DOI] [PubMed] [Google Scholar]
- 7.Mulvany MJ, Baumbach GL, Aalkjaer C, Heagerty AM, Korsgaard N, Schiffrin EL, et al. Vascular remodeling. Hypertension. 1996;28:505–506. [PubMed] [Google Scholar]
- 8.Bakker EN, van der Meulen ET, van den Berg BM, Everts V, Spaan JA, VanBavel E. Inward remodeling follows chronic vasoconstriction in isolated resistance arteries. J Vasc Res. 2002;39:12–20. doi: 10.1159/000048989. [DOI] [PubMed] [Google Scholar]
- 9.Martinez-Lemus LA, Hill MA, Meininger GA. The plastic nature of the vascular wall: a continuum of remodeling events contributing to control of arteriolar diameter and structure. Physiology (Bethesda) 2009;24:45–57. doi: 10.1152/physiol.00029.2008. [DOI] [PubMed] [Google Scholar]
- 10.Kim HR, Appel S, Vetterkind S, Gangopadhyay SS, Morgan KG. Smooth muscle signalling pathways in health and disease: contractility in Health and DiseaseReview Series. J Cell Mol Med. 2008;12:2165–2180. doi: 10.1111/j.1582-4934.2008.00552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehta D, Gunst SJ. Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol. 1999;519(Pt 3):829–840. doi: 10.1111/j.1469-7793.1999.0829n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rembold CM, Tejani AD, Ripley ML, Han S. Paxillin phosphorylation, actin polymerization, noise temperature, and the sustained phase of swine carotid artery contraction. Am J Physiol Cell Physiol. 2007;293:C993–C1002. doi: 10.1152/ajpcell.00090.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Lemus LA, Hill MA, Bolz SS, Pohl U, Meininger GA. Acute mechanoadaptation of vascular smooth muscle cells in response to continuous arteriolar vasoconstriction: implications for functional remodeling. FASEB J. 2004;18:708–710. doi: 10.1096/fj.03-0634fje. [DOI] [PubMed] [Google Scholar]
- 14.Hong Z, Sun Z, Li Z, Mesquitta WT, Trzeciakowski JP, Meininger GA. Coordination of fibronectin adhesion with contraction and relaxation in microvascular smooth muscle. Cardiovasc Res. 2012;96:73–80. doi: 10.1093/cvr/cvs239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 16.Martinez-Lemus LA. Persistent agonist-induced vasoconstriction is not required for angiotensin II to mediate inward remodeling of isolated arterioles with myogenic tone. J Vasc Res. 2008;45:211–221. doi: 10.1159/000112513. [DOI] [PubMed] [Google Scholar]
- 17.Kim HR, Gallant C, Leavis PC, Gunst SJ, Morgan KG. Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent. Am J Physiol Cell Physiol. 2008;295:C768–C778. doi: 10.1152/ajpcell.00174.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turner CE, Pietras KM, Taylor DS, Molloy CJ. Angiotensin II stimulation of rapid paxillin tyrosine phosphorylation correlates with the formation of focal adhesions in rat aortic smooth muscle cells. J Cell Sci. 1995;108:333–342. doi: 10.1242/jcs.108.1.333. [DOI] [PubMed] [Google Scholar]
- 19.Puetz S, Lubomirov LT, Pfitzer G. Regulation of smooth muscle contraction by small GTPases. Physiology (Bethesda) 2009;24:342–356. doi: 10.1152/physiol.00023.2009. [DOI] [PubMed] [Google Scholar]
- 20.Saito S, Watabe S, Ozaki H, Fusetani N, Karaki H. Mycalolide B, a novel actin depolymerizing agent. J Biol Chem. 1994;269:29710–29714. [PubMed] [Google Scholar]
- 21.Adler KB, Krill J, Alberghini TV, Evans JN. Effect of cytochalasin D on smooth muscle contraction. Cell Motil. 1983;3:545–551. doi: 10.1002/cm.970030521. [DOI] [PubMed] [Google Scholar]
- 22.Shaw L, Ahmed S, Austin C, Taggart MJ. Inhibitors of actin filament polymerisation attenuate force but not global intracellular calcium in isolated pressurised resistance arteries. J Vasc Res. 2003;40:1–10. doi: 10.1159/000068940. [DOI] [PubMed] [Google Scholar]
- 23.Wright G, Hurn E. Cytochalasin inhibition of slow tension increase in rat aortic rings. Am J Physiol. 1994;267:H1437–H1446. doi: 10.1152/ajpheart.1994.267.4.H1437. [DOI] [PubMed] [Google Scholar]
- 24.Flavahan NA, Bailey SR, Flavahan WA, Mitra S, Flavahan S. Imaging remodeling of the actin cytoskeleton in vascular smooth muscle cells after mechanosensitive arteriolar constriction. Am J Physiol Heart Circ Physiol. 2005;288:H660–H669. doi: 10.1152/ajpheart.00608.2004. [DOI] [PubMed] [Google Scholar]
- 25.Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. 2002;16:72–76. doi: 10.1096/cj.01-0104hyp. [DOI] [PubMed] [Google Scholar]
- 26.Cipolla MJ, Osol G. Vascular smooth muscle actin cytoskeleton in cerebral artery forced dilatation. Stroke. 1998;29:1223–1228. doi: 10.1161/01.str.29.6.1223. [DOI] [PubMed] [Google Scholar]
- 27.Jones KA, Perkins WJ, Lorenz RR, Prakash YS, Sieck GC, Warner DO. F-actin stabilization increases tension cost during contraction of permeabilized airway smooth muscle in dogs. J Physiol. 1999;519(Pt 2):527–538. doi: 10.1111/j.1469-7793.1999.0527m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Wu Y, Wu C, Gunst SJ. Integrin-linked kinase regulates N-WASp-mediated actin polymerization and tension development in tracheal smooth muscle. J Biol Chem. 2007;282:34568–34580. doi: 10.1074/jbc.M704966200. [DOI] [PubMed] [Google Scholar]
- 29.Tang D, Mehta D, Gunst SJ. Mechanosensitive tyrosine phosphorylation of paxillin and focal adhesion kinase in tracheal smooth muscle. Am J Physiol. 1999;276:C250–C258. doi: 10.1152/ajpcell.1999.276.1.C250. [DOI] [PubMed] [Google Scholar]
- 30.Tang DD, Gunst SJ. The small GTPase Cdc42 regulates actin polymerization and tension development during contractile stimulation of smooth muscle. J Biol Chem. 2004;279:51722–51728. doi: 10.1074/jbc.M408351200. [DOI] [PubMed] [Google Scholar]
- 31.Zhang W, Gunst SJ. Interactions of airway smooth muscle cells with their tissue matrix: implications for contraction. Proc Am Thorac Soc. 2008;5:32–39. doi: 10.1513/pats.200704-048VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X, Pavlish K, Zhang HY, Benoit JN. Effects of chronic portal hypertension on agonist-induced actin polymerization in small mesenteric arteries. Am J Physiol Heart Circ Physiol. 2006;290:H1915–H1921. doi: 10.1152/ajpheart.00643.2005. [DOI] [PubMed] [Google Scholar]
- 33.Ohanian V, Gatfield K, Ohanian J. Role of the actin cytoskeleton in G-protein-coupled receptor activation of PYK2 and paxillin in vascular smooth muscle. Hypertension. 2005;46:93–99. doi: 10.1161/01.HYP.0000167990.82235.3c. [DOI] [PubMed] [Google Scholar]
- 34.Saito SY, Hori M, Ozaki H, Karaki H. Cytochalasin D inhibits smooth muscle contraction by directly inhibiting contractile apparatus. J Smooth Muscle Res. 1996;32:51–60. doi: 10.1540/jsmr.32.51. [DOI] [PubMed] [Google Scholar]
- 35.Hill MA, Potocnik SJ, Martinez-Lemus LA, Meininger GA. Delayed arteriolar relaxation after prolonged agonist exposure: functional remodeling involving tyrosine phosphorylation. Am J Physiol Heart Circ Physiol. 2003;285:H849–H856. doi: 10.1152/ajpheart.00986.2002. [DOI] [PubMed] [Google Scholar]
- 36.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 37.Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family Gtpases in Mammalian cells. Annu Rev Biochem. 1999;68:459–486. doi: 10.1146/annurev.biochem.68.1.459. [DOI] [PubMed] [Google Scholar]
- 38.Ridley AJ. Rho family proteins and regulation of the actin cytoskeleton. Prog Mol Subcell Biol. 1999;22:1–22. doi: 10.1007/978-3-642-58591-3_1. [DOI] [PubMed] [Google Scholar]
- 39.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 40.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 41.Bubb MR, Spector I, Beyer BB, Fosen KM. Effects of Jasplakinolide on the kinetics of actin polymerization. J Biol Chem. 2000;275:5163–5170. doi: 10.1074/jbc.275.7.5163. [DOI] [PubMed] [Google Scholar]
- 42.Corteling RL, Brett SE, Yin H, Zheng X-L, Walsh MP, Welsh DG. The functional consequence of RhoA knockdown by RNA interference in rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2007;293:H440–H447. doi: 10.1152/ajpheart.01374.2006. [DOI] [PubMed] [Google Scholar]
- 43.Xu EZ, Kantores C, Ivanovska J, Engelberts D, Kavanagh BP, McNamara PJ, et al. Rescue treatment with a Rho-kinase inhibitor normalizes right ventricular function and reverses remodeling in juvenile rats with chronic pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2010;299:H1854–H1864. doi: 10.1152/ajpheart.00595.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi N, Nakano S, Mita S, Kobayashi T, Honda T, Tsubokou Y, et al. Involvement of Rho-kinase pathway for angiotensin II-induced plasminogen activator inhibitor-1 gene expression and cardiovascular remodeling in hypertensive rats. J Pharmacol Exp Ther. 2002;301:459–466. doi: 10.1124/jpet.301.2.459. [DOI] [PubMed] [Google Scholar]
- 45.Takeshima H, Kobayashi N, Koguchi W, Ishikawa M, Sugiyama F, Ishimitsu T. Cardioprotective effect of a combination of Rho-kinase inhibitor and p38 MAPK inhibitor on cardiovascular remodeling and oxidative stress in Dahl rats. J Atheroscler Thromb. 2012;19:326–336. doi: 10.5551/jat.11114. [DOI] [PubMed] [Google Scholar]
- 46.Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008;275:3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 47.Merajver SD, Usmani SZ. Multifaceted role of Rho proteins in angiogenesis. J Mammary Gland Biol Neoplasia. 2005;10:291–298. doi: 10.1007/s10911-006-9002-8. [DOI] [PubMed] [Google Scholar]
- 48.Martinez-Lemus LA, Zhao G, Galinanes EL, Boone M. Inward remodeling of resistance arteries requires reactive oxygen species-dependent activation of matrix metalloproteinases. Am J Physiol Heart Circ Physiol. 2011;300:H2005–H2015. doi: 10.1152/ajpheart.01066.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hassanain HH, Gregg D, Marcelo ML, Zweier JL, Souza HP, Selvakumar B, et al. Hypertension caused by transgenic overexpression of Rac1. Antioxid Redox Signal. 2007;9:91–100. doi: 10.1089/ars.2007.9.91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.