

Abstract
Significance
Myofibroblasts are responsible for wound closure that occurs in healed acute wounds. However, their actions can result in disfiguring scar contractures, compromised organ function, and a tumor promoting stroma. Understanding the mechanisms regulating their contractile machinery, gene expression, and lifespan is essential to develop new therapies to control their function.
Recent Advances
Mechanical stress and transforming growth factor beta-1 (TGF-β1) regulate myofibroblast differentiation from mesenchymal progenitors. As these precursor cells differentiate, they assemble a contractile apparatus to generate the force used to contract wounds. The mechanisms by which mechanical stress promote expression of contractile genes through the TGF-β1 and serum response factor pathways and offer therapeutic targets to limit myofibroblast function are being elucidated.
Critical Issues
Emerging evidence suggests that the integration of mechanical cues with intracellular signaling pathways is critical to myofibroblast function via its effects on gene expression, cellular contraction, and paracrine signaling with neighboring cells. In addition, while apoptosis is clearly one pathway that can limit myofibroblast lifespan, recent data suggest that pathogenic myofibroblasts can become senescent and adopt a more beneficial phenotype, or may revert to a quiescent state, thereby limiting their function.
Future Directions
Given the important role that myofibroblasts play in pathologies as disparate as cutaneous scarring, organ fibrosis, and tumor progression, knowledge gained in the areas of intracellular signaling networks, mechanical signal transduction, extracellular matrix biology, and cell fate will support efforts to develop new therapies with a wide impact.

Livingston Van De Water, PhD
Scope
In normal acute wounds, myofibroblasts are transiently present and orchestrate time limited and spatially restricted scarring. However, when myofibroblasts persist at sites of pathogenic scarring, organ fibrosis, or within the tumor stroma, exuberant deposition and contraction of extracellular matrix (ECM) occur. Here, we will review data on the cellular mechanisms that govern myofibroblast function and persistence, focusing on recent evidence that these cells respond to mechanical signals and are the mechanically active cells responsible for wound contraction.
Translational Relevance
Given their central role in scarring and fibrosis, and the tumor microenvironment, myofibroblasts are important therapeutic targets. To succeed in developing therapies to limit myofibroblast function, we must understand the extracellular inputs and intracellular signaling networks that govern myofibroblast function, in vitro and in vivo. A recent important development has been the recognition that myofibroblasts are mechanically responsive cells and that mechanical forces influence important determinants of myofibroblast formation, function, and fate, such as growth factor activation extracellularly and transcription factor regulation intracellularly.
Clinical Relevance
Scarring and fibrosis, when taken together as a clinical entity, are responsible for a remarkably large disease burden, which by some estimates is some 45% of all chronic diseases in the western world.1 Excessive scarring following cutaneous injury results in conditions such as hypertrophic scars (HTS), burn contractures, and keloids; while palmar fibromatosis (Dupuytren's disease) results in contracture and scarring of the palmar fascia in the absence of an initiating injury. Myofibroblasts are central determinants of the course of fibrosis and there are currently no therapies that are effective in preventing or reversing myofibroblast function.
Discussion of Findings and Relevant Literature
Myofibroblasts: what is good and what is bad?
What are they and where are they?
Normal connective tissues fibroblasts are embedded in a collagen-rich ECM and maintain tissue homeostasis by synthesizing interstitial collagens, proteoglycans, and adhesive non-collagenous proteins and by exerting mild contractive force on this ECM. These resting connective tissue fibroblasts are also shielded from “routine” external mechanical perturbations in the skin by the collagen-rich ECM that they have assembled.2 Very low levels of growth factors, cytokines, and blood plasma proteins percolate through this microenvironment and this rather placid setting maintains the fibroblast in a quiescent state.
Normal dermal fibroblasts experience dramatic changes in their microenvironment after injury. These include changes in the complement of growth factors and cytokines, alterations in the mechanical microenvironment and importantly, conversion of the formerly collagen-rich dermal ECM into one that is predominantly comprised of fibrin and fibronectin. These conditions are thought to be required for conversion of the fibroblast through an intermediate (“proto-myofibroblast”) and into a differentiated myofibroblast.2 Myofibroblasts express smooth muscle alpha-actin (SM α-actin), the actin isoform found in vascular SM cells, larger adhesion sites (termed “focal adhesions”), and pronounced actin-myosin containing stress fibers.3–5 These components are obvious when myofibroblasts are differentiated on a rigid, planar surface (e.g., coverslip or culture dish) but take on a different morphology when myofibroblasts are enmeshed in a collagen gel (Fig. 1). Compared with fibroblasts, myofibroblasts express increased amounts of Type I and Type III collagen, proteoglycans, specific forms of fibronectin, and a plethora of proteins including contractile proteins, growth factors, cytokines, matricellular proteins, and proteins that regulate the cell cycle and cell fate.6
Figure 1.

Morphology of the myofibroblast. (A, B) Cartoons depicting the morphology and cytoskeletal components of a fibroblast (A) versus a myofibroblast (B). For more detail, see text. (C, D) Immunofluorescence images showing the stress fiber organization (red-phalloidin staining) and focal adhesion proteins (A, green-Hic-5 immunostaining; B, green-vinculin immunostaining) of a myofibroblast on a glass coverslip (C) or in a collagen gel (D). Scale bars: (C) 50 μm; (D) 40 μm.
Recent studies have also demonstrated that in addition to normal connective tissue fibroblasts, there are other sources of myofibroblast progenitors. These progenitor cell types include a disintegrin and metalloproteinase-12 (ADAM-12)-positive perivascular cells, fibrocytes, and cells derived from an epithelial–mesenchymal transition (EMT).7–9 While it is clear that these cells have the potential to form myofibroblasts, their total contribution to the myofibroblast population that forms in an acute wound is unclear.
Although for the purposes of this review we will focus on the myofibroblast phenotype in cutaneous wounds, pathogenic scars, organ fibrosis, and tumor stroma, it is important to note that cells with features of myofibroblasts are also found in some developing tissues and specialized normal adult tissues (Table 1).10–21 Their role in these normal tissue settings is not clear, but given the prominence of the contractile apparatus in myofibroblasts this likely involves a mechanical component. An important concept that has become increasingly clear is that under some circumstances myofibroblasts are “good,” as in normal acute wounds and some normal tissues and under pathogenic settings myofibroblasts are “bad,” depositing and contracting excessive scar and elaborating growth factors and cytokines that perpetuate the pathology.
Table 1.
Fibroblastic cells of normal organs with myofibroblastic features
| Localization | Reference |
|---|---|
| Uterine submucosa | Glasser and Julian (1986)10 |
| Reticular cells of lymph notes and spleen | Toccanier-Pelte et al. (1987)11 |
| Intestinal pericryptal cells | Sappino et al. (1989)12 |
| Intestinal villous core | Kaye et al. (1968)13 |
| Testicular stroma | Skalli et al. (1986)14 |
| Theca externa of the ovary | Czernobilsky et al. (1989)15 |
| Periodontal ligament | Beertsen et al. (1974)16 |
| Adrenal-gland capsule | Bressler (1973)17 |
| Hepatic perisinusoidal cells | Yokoi et al. (1984)18 |
| Lung septa | Kapanci et al. (1992)19 |
| Bone-marrow stroma | Charbord et al. (1990)20 |
| Capillary and venular pericytes | Lindahl and Betsholtz (1998)21 |
Functions at the acute wound site
During normal acute wound healing, the myofibroblast dramatically upregulates collagen and fibronectin deposition over an interval of ∼7–14 days in the rodent models commonly employed (Fig. 2).22–25 However, the duration of myofibroblasts varies depending on the size and type of cutaneous wound, and the animal species.2,26 Importantly, exuberant expression and deposition of a collagen-rich ECM is a prominent feature of scarring and fibrosis, which is closely associated with the presence of SM α-actin–positive myofibroblasts.27
Figure 2.
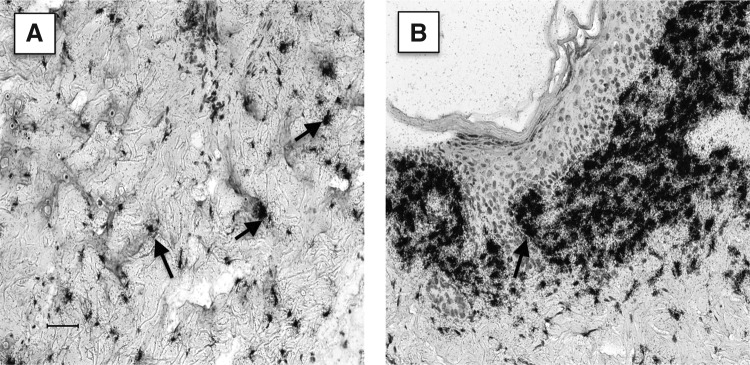
Type I collagen mRNA expression in normal dermis versus wound tissue. Paraffin sections of normal rat skin (A) or an 8-day wound (B) were reacted with an 35S-labeled transcribed RNA probe reactive with Type I Collagen. Note that normal dermal fibroblasts (arrows) in the reticular dermis (A) are modestly labeled with the probe while fibroblasts in the wound (B) are heavily labeled. Photographic emulsion was exposed for the same length of time. Scale bar: 50 μm.
Guillio Gabbiani identified wound fibroblasts as the prominent cell type likely to exert contractive force in wounds and in ex vivo granulation tissue experiments.28,29 The SM α-actin isoform has been reported to mediate the increased intracellular tension observed in myofibroblasts enabling these cells to exert increased force on the ECM, thereby serving to remodel the wound matrix.30 However, SM α-actin is not strictly required for wound contraction because mice lacking this actin isoform close wounds normally, potentially by the compensatory function of other muscle actin isoforms.31 Importantly, in healing acute wounds, the action of the myofibroblast results in imperfect healing that optimizes the need to rapidly repair defects in skin or other organs thereby maximizing rapid recovery of function. However, this comes at the expense of mechanical integrity, because the scar does not restore the connective tissue to a mechanical resilience akin to the original tissue.2 This is certainly important in the skin and critically important in the heart following a myocardial infarction. The scar that heals myocardial infarctions restores the cardiac architecture, albeit imperfectly.32,33
Impact of persistent myofibroblast function: contractures and excessive scarring
While myofibroblast function is temporally and spatially limited in normal acute wounds, this is not true of the pathogenic scarring observed in HTS, burn contractures, and keloids. The abnormal appearance and persistent myofibroblast is now a well-established feature of fibrotic lesions in skin disorders including Dupuytren's disease (palmar contracture) and scleroderma in skin in addition to organs that undergo fibrotic reactions, including lung, liver, and kidney.2,34–36 This prodigious ECM deposition and excessive contraction of the wound matrix is thought to be the result of feed-forward loops in myofibroblasts in fibrotic lesions and pathogenic scars (Figs. 3 and 4).
Figure 3.
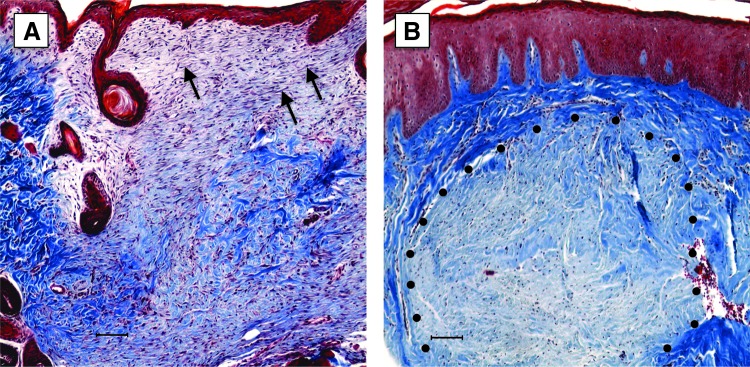
Trichrome stain of normal rodent wound and human HTS. Paraffin sections of a normal rat 14 day wound (A) or human HTS (B) were stained with Massons Trichrome. Note that red color that predominates in the wound bed (A) represents a cell-rich and less pronounced ECM. The deep blue color in the HTS is a result of prominent collagen deposition. Also, the cells within the wound (A, arrow) are linearly arrayed, while in the HTS they are organized into a nodule (dotted line). Scale bars: 100 μm. ECM, extracellular matrix; HTS, hypertrophic scar.
Figure 4.
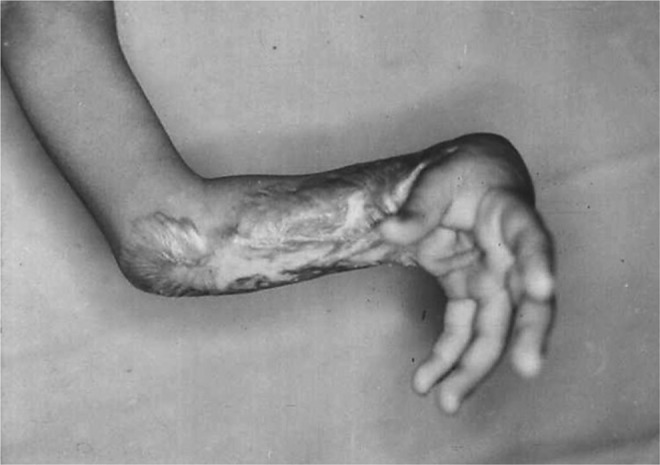
Burn injury resulting in scar contracture across the wrist. Existence of myofibroblasts following a burn injury can deposit excessive ECM and contract the resulting scar leading to a pathologic contracture. As observed in this individual, the wrist is held in permanent flexion by the contracture that resulted from a burn injury. (Courtesy of Dr. Jaysheela Mudera, University College, London. Reprinted with permission from Tomasek et al.2)
Chronic wounds and myofibroblasts
While myofibroblast persistence is characteristic of pathogenic scarring and organ fibrosis, there appears to be a paucity of myofibroblasts in chronic wounds such as diabetic ulcers and venous stasis ulcers, in which robust granulation tissue is not evident.37 The compromised granulation tissue formation that is characteristic of these wounds is likely the consequence of persistent bacterial biofilm formation, reduced epidermal barrier function, impaired growth factor production, and compromised angiogenesis in the wound bed.38–40 The deficiency in angiogenesis is, in turn, thought to result in a chronically hypoxic microenvironment within these chronic wounds. These conditions of prolonged low oxygen tension have recently been shown to decrease myofibroblast formation and wound contraction in a rodent experimental model.41 Importantly, expression of SM α-actin is markedly reduced, in vitro, when myofibroblasts are subjected to hypoxic conditions (2% O2).42 This is an important and understudied area of myofibroblast biology owing in part to the fact that investigators have traditionally cultured cells in the lab under conditions of ambient oxygen tension (21% O2), which are hyperoxic conditions for most cells including wound fibroblasts and do not reflect the ischemic wound microenvironment.43 Importantly, the paucity of myofibroblasts in chronic wounds may be secondary to the impaired vasculature. Indeed, recent fate-mapping experiments show that ADAM-12–positive perivascular cells are an important source of collagen producing myofibroblasts following injury.7 A failure to generate ADAM-12–positive precursors may be due to the compromised vasculature and could contribute to the deficiency of myofibroblasts observed in chronic wounds.
Myofibroblasts in tumor stroma
Clinicians and pathologists have known that certain types of tumors are distinctly hard on palpation (“desmoplastic”) and may in some instances contract neighboring tissues to the point of retracting skin visibly.34 These “scirrhous” tumors result from an abundant fibrous stroma, which is now known to be attributable to the myofibroblasts' deposition and contraction of a collagen-rich ECM by cancer-associated fibroblasts (CAFs, also termed tumor-associated fibroblasts [TAFs]).44–46 Importantly, CAFs are heterogeneous and include FSP-1–positive cells that are SM α-actin–negative or positive.46 These myofibroblast functions, while aiding diagnosis by palpation, also underlie an important feature because this tissue stiffness has recently been shown to support tumor progression that in turn promotes further myofibroblast differentiation.47,48 CAFs play a central role in this “feed-forward” loop, which is a recurring theme in fibrotic settings and provide important paracrine signals to tumor cells.36,49 The concept that the microenvironment of tumors and wounds share important features was originally proposed by Dvorak and has served as an important impetus for recent work in this area.50,51
In summary, myofibroblasts or related “activated” fibroblasts are present in certain normal tissues and transiently in acute wounds where they deposit ECM and exert functionally appropriate mechanical tension on the tissue microenvironment thereby promoting homeostatic mechanisms. However, the extracellular and intracellular network of signals supporting this homeostasis can be disrupted in a spectrum of pathologies. As a result, feed-forward loops are established that can lead to inappropriate mechanical and paracrine signals that perpetuate pathologies such as contractures, tumor growth, and fibrosis. We will now take a closer look at the mechanisms that support the genesis and longevity of myofibroblasts.
Myofibroblasts and their formation, function, and fate during wound healing
There have been a number of excellent reviews in recent years that emphasize different aspects of the signals that induce myofibroblast differentiation, including the role of inflammatory cells,1 mechanical signals,36,52 the ECM,6 and the pro-fibrotic growth factor, transforming growth factor beta-1 (TGF-β1).2,35,36,53 We will highlight those aspects that we believe are important determinants of myofibroblast differentiation, function, and fate.
The proto-myofibroblast
In early granulation, tissue fibroblasts adopt a phenotype characterized by increased bundles of actin–myosin stress fibers and more prominent focal adhesions structures.54,55 Typically, these cells, termed proto-myofibroblasts, do not express SM α-actin.2 Although we are currently limited by the lack of specific cell surface markers with which we can isolate and characterize the proto-myofibroblast, this group of cells provides a mechanistic intermediate that is useful in understanding the process through which myofibroblasts differentiate and potentially regress.
When dermal fibroblasts are explanted into a culture dish in the presence of growth medium containing serum, a large proportion of the cells display obvious focal adhesions and prominent stress fibers and express “cellular” fibronectins (FNs), but most cells under these conditions are SM α-actin–negative proto-myofibroblasts.2 Early in situ hybridization studies demonstrated that the FN mRNAs that are present in uninjured dermal fibroblasts are largely devoid of alternatively spliced segments, termed EDA (or EIIIA) and EDB (EIIIB).24 When these cells are explanted and cultivated in vitro they undergo a change in their FN mRNA pattern of alternative splicing and include the EDA and EDB domains in a form of FN termed “cellular” FN.22,25,56
What are the conditions that fibroblasts encounter after injury and how might this initiate their transformation to proto-myofibroblasts? The fibroblastic precursor cells residing in the collagen-rich dermal ECM closely adjacent to the wound are abruptly confronted with a dramatic change in signals from the ECM, growth factors, and mechanical features of the ECM.57 The provisional matrix that is established in the first few minutes following injury results from hemorrhage of severed blood vessels and then by extravasation of plasma proteins over several days after injury. Extravasation is actively mediated by vascular endothelial growth factor, a potent vascular permeability factor.58,59 Extravasated plasma proteins are thought to maintain the provisional matrix during the inflammatory and early granulation tissue phases of wound healing.57 This provisional matrix is initially highly mechanically compliant with a Young's modulus of <1,000 Pa (defined in Table 2)52,60 and contains a complex mixture of growth factors, including platelet-derived growth factor (PDGF) that is thought to be one important factor that promotes fibroblast migration into the granulation tissue.61,62 Importantly, dramatic changes in inflammatory state mediated in part by immune cells also impact the microenvironment during this interval.1 One important determinant, recently identified, is the switch in macrophage “class” from the M1 (inflammatory) phenotype to the M2 (resolution) phenotype.63
Table 2.
Definition of terms in biomechanics
| Tension is a pulling force tending to stretch or elongate. It is the reaction force exerted by a stretched string (or similar object) on the objects that stretch it. Tension also develops within a material when it is stretched beyond its slack length and begins to resist elongation. Tension is the magnitude of a force and is measured in newtons (N). | |
| Compression is the opposite of tension; a force tending to shorten or compress. | |
| Stress is a measure of the area-normalized internal pressure forces acting within a deformable material. It represents the average force per unit area of a surface within the material on which the internal forces act. It is measured in units of pressure, that is, force per unit area typically expressed in SI units of pascals (Pa); (1 Pa=1 N/m2). | |
| Strain is a normalized measure of deformation representing the ratio of the change in length to the original length of a material; it is dimensionless measure and is typically expressed as a decimal fraction or a percentage. | |
| Elasticity describes the ability of materials to return to their original shape after applied stresses are removed. Young's modulus is a measure of the stiffness of an elastic material. It is the ratio of stress over strain, measured units of force per unit area per strain, typically expressed in pascals. It is an intrinsic property of the material, unaffected by specimen geometry. | |
| Stiffness describes the extent to which an object resists deformation in response to an applied force. Stiffness is a property of a structure; elastic modulus is a property of the constituent material. Stiffness is a measure of resistance offered by an elastic body to deformation and is measured in units of force per change in length (newtons/m). It is an extrinsic property of the elastic body, influenced by both the material and specimen geometry. | |
| Compliance is the inverse of stiffness. | |
| Young's modulus for wounded tissues: | |
| Fibrin clot | 10–1,000 Pa |
| 7 day rat wound granulation tissue | ∼18 kPa |
| 8 day rat wound granulation tissue | ∼25 kPa |
| 9 day rat wound granulation tissue | ∼30 kPa |
| 12 day rat wound granulation tissue | ∼50 kPa |
| Young's modulus for cellular events | |
| Formation of proto-myofibroblast (stress fibers) | 3–5 kPa |
| Activation of TGF-β1 from latent ECM complex | 5–9 kPa |
| Formation of myofibroblast (stress fibers with SM α-actin) | 16–20 kPa |
Fibroblasts migrating in the provisional matrix of the early granulation tissue exhibit increased F-actin staining and EDA-positive FN staining a few days ahead of the appearance of SM α-actin–positive cells and resemble proto-myofibroblasts.26 Although the exact mechanisms through which this proto-myofibroblast phenotype is acquired are unclear, it is currently thought that this is caused by the increased stiffness induced by tractional forces accompanying fibroblast migration within the fibrin-rich provisional matrix and newly made collagen.2,5,52,64 This view is consistent with the well-known fact that fibroblasts can reorganize collagen gels and increase gel stiffness in vitro.61 Focal adhesion size increases with increasing stiffness and this is accompanied by clustering of integrins within focal adhesions and increases in prominent actin-rich stress fibers in a process that is driven by Rho GTPase.65 The expression of SM α-actin and its incorporation into actin-containing stress fibers does not occur until a threshold stiffness is sensed by the proto-myofibroblast.5 However, measuring bulk changes in wound mechanical features is difficult, and it is generally accepted that the actual stiffness “felt” by individual fibroblasts and proto-myofibroblast within the wound microenvironment is unknown. Additionally, how changes in growth factors, the ECM including FN isoforms and collagen–fibrin mixtures at changing compliances govern proto-myofibroblast maturation is currently unclear, but it is an area for additional research because it offers opportunities to therapeutically target the progression to fully differentiated myofibroblasts (reviewed recently by Klingberg et al.).66
Differentiation of myofibroblasts
As indicated above, differentiated myofibroblasts are critical cellular elements that deposit scars in normal acute wounds and in pathogenic settings such as fibrotic lesions and tumors. Accordingly, the mechanisms promoting their differentiation have been intensively studied and there are a number of excellent reviews on this subject.2,35,36,52,53 The key factors that have been identified in the process of myofibroblast differentiation include TGF-β1, sufficient mechanical stiffness, and the presence of specific isoforms of FN (Fig. 5).5,25,64,67
Figure 5.

Model of myofibroblast differentiation and wound contraction. (A) In normal tissues, fibroblasts are shielded from “routine” external mechanical perturbations in the skin by the collagen-rich ECM that they have assembled, such that the organization of a contractile cytoskeleton is not stimulated (light pink area of dermis). Following a full-thickness dermal injury, the wound is filled with a provisional matrix comprised of fibrin and fibronectin and a complement of newly released growth factors and cytokines. Fibroblasts, along with blood vessels, are stimulated to migrate into this pro-migration microenvironment and over time replace the provisional matrix with an ECM comprised of collagen and cellular FNs to form granulation tissue. (B) Tractional forces accompanying fibroblast migration are responsible for local areas of increased stiffness in newly made collagen. Focal adhesion assembly is increased with increasing stiffness and this is accompanied by clustering of integrins within focal adhesions and increased stress fiber assembly resulting in fibroblast acquisition of the proto-myofibroblast phenotype. Tensional forces and growth factors stimulate proto-myofibroblasts to secrete transforming growth factor-beta1 (TGF-β1) and increased levels of ED-A FN. (C) In response to TGF-β1, a threshold of mechanical stiffness and the presence of specific isoforms of FN proto-myofibroblasts differentiate into myofibroblasts. Feed-forward pathways, including TGF-β1, the mechanical environment, actin dynamics, SRF/MRTF transcriptional activation, and Hic-5, are responsible for myofibroblast function and persistence. At the same time, differentiated myofibroblasts deposit collagen and other ECM components, and produce proteases. This complex process of remodeling results in shortening of the collagen matrix with corresponding wound closure. (D) During normal acute wound healing, these feed-forward mechanisms are temporally limited and myofibroblast numbers diminish as a result of apoptosis and/or senescence. (E) In pathological situations, such as HTS formation, these feed-forward pathways presumably persist allowing for continued myofibroblast presence resulting in continued ECM deposition and remodeling. In conclusion, myofibroblasts, far from being a “bad” cell type, are functionally essential cells. It is their dysregulation that is the cause of tissue dysfunction. (Modified from Fig. 9, Tomasek et al.2). FN, fibronectins; SRF, serum response factor; MRTFA/B, myocardin-related transcription factor A/B.
TGF-β1 is released from cells in a large latent complex (LLC) that is comprised of TGF-β1, a latency-associated peptide (LAP) and a latent TGF-β1–binding protein (LTBP-1). LTBP-1 is capable of binding to ECM proteins including FN, vitronectin, and fibrillin and in doing so anchors the LLC in the ECM where it is thought to serve as a ready reservoir of latent TGF-β1.68 Importantly, TGF-β1 can be liberated from the LLC by a number of mechanisms that include proteolysis of peptide sequences within LAP and/or LTBP-1 by matrix metalloproteinases (MMPs)-9 and serine proteases including plasmin.68 These proteases are particularly relevant to wound healing.69,70 For example, active MMP-9 can be docked at the cell surface, where it can efficiently activate latent TGF-β1.71
Interactions between the LLC and thrombospondin 1 (TSP-1) also are known to promote release from and activation of TGF-β1.72 TSP-1 is a member of a group of “matricellular proteins” that bind within the ECM but have regulatory roles rather than structural roles.73 Active TGF-β1 release from the LLC can be induced by a short synthetic peptide derived from TSP-1 and the interaction is thought to occur via the LAP within the latent complex.72 Recent data have also revealed an important mechanically induced mechanism that promotes TGF-β1 activation.74,75 In brief, several integrins (αvβ5, αvβ6, αvβ3, and αvβ8) have been shown to interact with LAP within the latent complex-ECM.74,76 These integrins, coupled to a contractile cytoskeleton, tug on the latent complex, and it is thought that in doing so induce a conformation change in the latent complex resulting in the release of TGF-β1. Importantly, as shown by Wipff et al., this requires a threshold level of mechanical stiffness (Young's modulus >5,000 Pa) in the ECM that resists the tugging by myofibroblast cytoskeleton, thereby opening up the latent complex.75
Despite this clear link between mechanical stress and TGF-β1 activation, the addition of exogenous active TGF-β1 to fibroblasts in a compliant mechanical environment is not sufficient to promote the differentiation of myofibroblasts.5,67 On the other hand, blockade of active TGF-β1 signaling in a stiff mechanical environment blocks differentiation.2,30,64 Hence, while active TGF-β1 is required, there must be other important mechanically sensitive components within the signaling networks controlling myofibroblast differentiation. Indeed, application of increased mechanical stress to healing wounds promotes more rapid myofibroblast formation and, conversely, loss of mechanical stress in a granulation tissue model results in a loss of SM α-actin from myofibroblasts.26 When studied in culture, myofibroblasts elaborate large focal adhesions and prominent stress fibers, and when a threshold focal adhesion size is attained the cells gain expression of SM α-actin.4,5 Under conditions of low extracellular stiffness, focal adhesions are small; while on stiff substrates, focal adhesions and stress fibers grow.77,78 Importantly, Rho GTPase–dependent networks are central to these adhesion maturation processes.65 In addition, ECM proteins, focal adhesion complexes, and SM α-actin all serve as mechanical transducers in cells.79
The mechanically sensitive processes discussed above have important implications for the pathogenesis of exuberant scar formation. When splints are applied to wounds for long intervals a HTS phenotype is observed in mice, presumably by increasing mechanical stress and promoting myofibroblast differentiation, but also by impacting inflammatory cells.80,81 Similarly, the increasing stiffness of the tumor microenvironment contributed by the tumor stroma ECM promotes myofibroblast differentiation.60 This apparent feed-forward mechanism promotes myofibroblast persistence by activating TGF-β1, increasing Rho activation, cytoskeletal stiffening and stress fiber stabilization, FN and collagen expression and deposition that in turn increase the stiffness of the microenvironment. Importantly, work from Tschumperlin's group has identified a mechanically sensitive threshold at which lung fibroblasts cultured on substrata at or above ∼1kPa resulted in increased proliferation and collagen synthesis, and decreased apoptosis, protease gene expression, cyclooxygenase-2 expression and prostaglandin E2 (PGE2) production.82 PGE2 is an inhibitor of the fibrotic response and these data support a model in which increased matrix stiffening enables a positive feedback loop that promotes fibrosis.
Many intriguing unknowns remain for future study. When fibroblasts are dispersed into culture on rigid surfaces (e.g., glass or plastic) in the presence of exogenous active TGF-β1, why does it typically take 3–5 days for differentiation into myofibroblasts? In the absence of exogenous TGF-β1, why do some fibroblasts isolated from some locations and species display a low percentage of cells expressing SM α-actin, while fibroblasts from other locations or species display a high percentage of cells expressing SM α-actin? Why does culturing fibroblasts at low density promote myofibroblast formation?83 Why do only a proportion of fibroblasts form myofibroblasts, even in the presence of exogenous TGF-β1? Intriguingly, when fibroblasts or myofibroblasts are grown under these conditions and then cloned to single cells, the cloned population has a similar percentage of cells expressing SM α-actin suggesting cell autonomous processes are engaged.84*
Function of myofibroblasts
Myofibroblasts in wounds, fibrotic lesions, and the tumor microenvironment deposit the collagen-rich ECM that is then remodeled by contractile forces and by ECM matrix modifying proteins, including proteases and matricelluar proteins.2,36,85 Myofibroblasts also are central elements in an intricate network of paracrine signaling between epithelial cells—wound keratinocytes and carcinoma cells in tumors—and inflammatory cells and endothelial cells.45,62,86,87
Scar contractures are irreversible and occur in part as the result of reversible contraction by myofibroblasts. And yet, the process of scar contracture is distinct from muscle contraction. How is this possible? As with muscle, myofibroblast contraction is driven by the interaction between actin and myosin and is reversible (Fig. 6). In myofibroblasts, myosin II and actin are within stress fibers linked to focal adhesions, thereby providing functional continuity between the intracellular contractile force and the ECM. When fibroblasts are cultured in collagen gels they contract the gels. When cytochalasin D (a drug that induces disruption of filamentous actin) is added early in the time course, the collagen relaxes. However, if cytochalasin D is added at later times relaxation is not complete, suggesting the presence of a residual matrix tension.88,89 The collagen in this model and, by extension the wound ECM, is thought to undergo a progressive shortening in a “ratchet mechanism.”2,61,66,89 Rather than collagen fibers sliding past each other, the collagen matrix is interconnected into a network that is stabilized by covalent and non-covalent interactions. The interconnectedness of the wound collagen mesh (or network) amplifies the relatively small-scale single cell contraction (on the order of tens of microns) into large-scale contracture through a network effect within the structure of the wound ECM.90 As the myofibroblast contracts this matrix the contraction is stabilized by the aforementioned associated ECM proteins and reversibility becomes limited leading to a contracture.2,61,89 This has important therapeutic implications for fibrotic lesions suggesting that it may be productive to target mechanisms that stabilize the wound ECM rather than the myofibroblasts themselves. One such promising target is the enzyme, lysyl oxidase, which cross-links collagen fibrils.91
Figure 6.
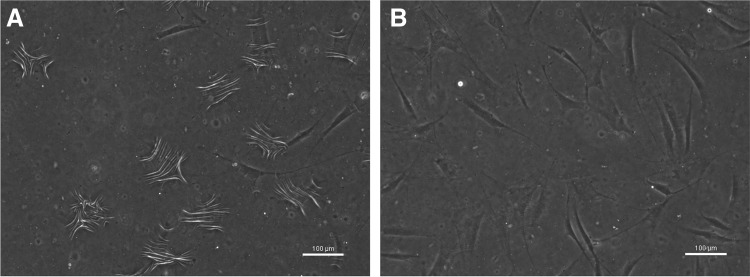
Dermal fibroblasts generate contractile force sufficient to deform silicone substrates when stimulated with serum. Normal primary dermal fibroblasts were cultured on a deformable silicone substrate (PDMS). Serum stimulation is sufficient to generate wrinkling (A); wrinkling is lost upon removal of serum (B), demonstrating reversibility of contractile forces. Cells are viewed under phase microscopy. Scale bars: 100 μm.
Myofibroblast fate and persistence in fibrosis (apoptosis, reversion, and senescence)
Although the thrust of research in the areas of scarring and fibrosis is to develop new therapies for these diseases, scarring has also been used for decorative purposes in some cultures, particularly continental Africa, to denote social standing or to mark significant milestones in one's life (Fig. 7).92 Although the practice is fading in traditional tribal cultures its use is increasing in western cultures. Whether traditional or modern, scars are made with incisions followed by repeated application of irritants such as charcoal in traditional cultures and mineral or herbal irritants.93 The resulting decorative scars resemble HTS or keloids.
Figure 7.
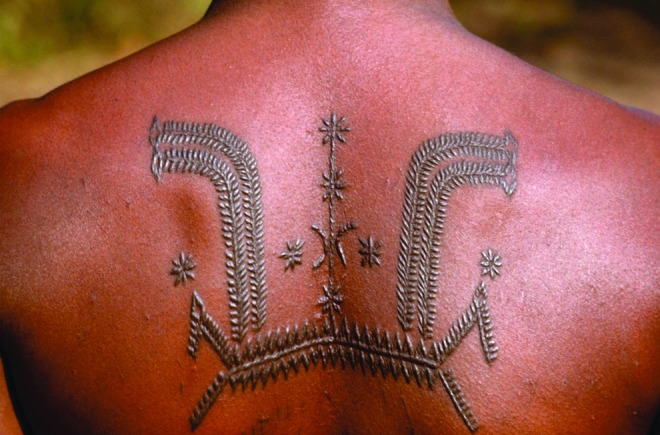
Decorative scarring. Ornate welts of raised scar tissue are a mark of beauty on the back of an individual from Mozambique. (Photo by Volkmar K. Wentzel/National Geographic Stock; all rights reserved.92) To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
The mechanisms promoting persistent myofibroblast function, whether pathogenic or decorative, are not well understood but likely involve feed-forward loops (Fig. 8). The presence of persistent inflammation—which could result from chronic irritants—and prolonged production and activation of TGF-β1 are important components as are the aforementioned feed-forward mechanisms resulting from increased mechanical stiffness.1,36 Moreover, HTS myofibroblasts elaborate an autocrine, positive feedback loop in which latent TGF-β1 is produced.94 A focal adhesion protein, termed Hic-5 (also called TGFB1I1) and a homologue of paxillin, has been shown to be necessary and sufficient for the production of latent TGF-β1 and maintenance of the myofibroblast phenotype.95 Genetically silencing Hic-5 reduces the cardinal features of myofibroblasts including SM α-actin, focal adhesion size, Collagen I (alpha 2) production, and collagen contraction.95 Some of these functions (i.e., Collagen I and latent TGF-β1 production) require Hic-5 and others (e.g., SM α-actin expression and collagen contraction) are secondary to Hic-5–dependent regulation of TGF-β1 production. The Hic-5–dependent production of latent TGF-β1 could coordinately work with the contraction dependent activation of TGF-β1.75
Figure 8.

Feed-forward pathways that support myofibroblast persistence. Active TGF-β1 induces many pathways but we highlight here Rho-dependent processes that include increases (light gray arrows left) in actin polymerization, dissociation of MRTF-A from G-actin, and translocation of MRTF-A to the nucleus where with SRF induces cytoskeletal genes, some focal adhesion proteins, including Hic-5 (black arrow, left), and Type I collagen synthesis (black arrow, right). The result is increased (light gray arrows, right) collagen synthesis and deposition, increased contractile force (intracellular tension) via the actin-myosin II containing stress fibers. SM α-actin promotes increased contractile force that in turn supports focal adhesion growth and stress fiber assembly (dark gray arrow, right). Hic-5 has been shown to be necessary for latent TGF-β1production and mechanical contraction promotes latent TGF-β1 activation (dark gray arrows, left). SM α-actin, smooth muscle alpha-actin.
Hic-5 also has been shown to be both necessary and sufficient for the TGF-β1 regulated cell cycle in normal dermal fibroblasts and hypertrophic scar myofibroblasts.96 Hic-5, unlike paxillin, is induced by TGF-β1 and remains constitutively high in hypertrophic scar myofibroblasts in the absence of exogenous TGF-β1.96 Inhibiting Hic-5 expression in pathogenic myofibroblasts in culture—by addition of either TGF-β1 antagonists or Hic-5 siRNA—reduces Hic-5 levels and relieves the inhibition of proliferation by downregulating the expression of p21cip1, a CDK inhibitor that governs the G1/S transition of the cell cycle.94,96 Hic-5, again differing from its homologue paxillin, translocates to and can be retained in the nucleus under conditions of high reactive oxygen species and serves as a transcriptional coregulator of a number of genes, including p21cip1.97–100 Hic-5 is expressed by myofibroblasts in prostate tumor stroma, HTS, and Dupuytren's disease.101,†
During normal acute wound healing, the TGF-β–dependent feed-forward mechanisms are likely temporally limited and SM α-actin–positive myofibroblast numbers diminish over time. It has been known for nearly 20 years that myofibroblast numbers are reduced in normal wounds and the proportion of apoptotic fibroblasts increase as the proliferative, granulation tissue phase of wound healing wanes.102 The mechanisms that dictate when and which myofibroblasts undergo apoptosis are not clear, although several important leads have been found recently. Thannickal's group has discovered a mechanism in which TGF-β1 elicits an apoptosis-resistant phenotype through separate but converging pathways. One pathway transduces cell adhesion signals through a SMAD3 and focal adhesion kinase (FAK)–dependent pathway; the other pathway utilizes a p38 MAPK and PI3 kinase/Akt–dependent signaling.103–106 These pathways converge to support TGF-β1–dependent resistance to apoptosis and anoikis, a form of programmed cell death induced by loss of adhesion-dependent signaling.
Recent data have suggested mechanisms other than apoptosis may regulate myofibroblast persistence. The CCN family of matricellular proteins include CCN1 (Cy61), CCN2 (CTGF), and CCN3 (NOV) and have been shown to regulate myofibroblast fate.107,108 The CCN proteins have binding sites that interact with ECM proteins, including FN, decorin, perlecan, and vitronectin, in addition to integrins α5β1, α6β1, αvβ3, and αvβ5. CCN proteins through integrins and other receptors can synergize with intracellular signaling pathways that involve TNF-α, NADPH oxidase 1, Jun kinase, and p38 and, depending upon which, can promote apoptosis or myofibroblast senescence. For example, CCN1 via its binding to α6β1 integrins and proteoglycans can induce reactive oxygen species in cells via NADPH oxidase 1 and induce a p53 and Rb protein–dependent senescence.107 Induction of the senescent state in myofibroblasts results in a decrease in the wound ECM and fibrosis, in part because of decreased collagen production and increased matrix-degrading enzymes.108 Indeed, mice engineered to express a defective CCN1 exhibit exaggerated fibrosis in the skin.107 Increasing the production of CCN1 therapeutically to induce the senescent state in myofibroblasts holds promise as a means to limit fibrosis.
Although washing away or blocking the action of TGF-β1 in myofibroblast cultures is sufficient to reverse the phenotype in vitro,2,64 until recently the reversibility of the myofibroblast phenotype has not been demonstrated in animals. Using a carbon tetrachloride model of liver fibrosis model and a fluorescent protein fate mapping approach, Kisseleva et al. demonstrated that when carbon tetrachloride is removed and fibrosis subsides, ∼50% of the myofibroblasts do not undergo apoptosis.109 Instead, the myofibroblast reverts to a phenotype that resembles an activated form of the quiescent hepatic stellate cell, the myofibroblast progenitor cell in liver. Earlier work had shown, in agreement with the studies mentioned above, that myofibroblasts in fibrotic liver also can undergo apoptosis and/or senescence.110 Taken together, these studies provide new avenues to therapeutically blunt myofibroblast life span and functional state. More work is needed to determine whether the pathways governing the fate of myofibroblasts depend upon the fibrotic setting or are universally applicable to disease as it occurs in different organ systems and tumors.
Impact of myofibroblast intracellular tension and contraction on function
Research over the past decade has begun to illuminate the subtle interplay between mechanical signals, growth factors, ECM, and their roles in dictating myofibroblast function. We will now expand on what is known about how myofibroblasts sense alterations in the mechanical microenvironment and translate these into changes in gene expression and contraction.
How myofibroblasts sense and adapt to changes in the mechanical microenvironment
It now appears that ECM, the integrins that bind these proteins, focal adhesions, and the cytoskeleton form a linear “train” of functional modules that respond to mechanical stress.111–113 Importantly, although these structures appear static when viewed in the microscope, it has also become clear that many—but likely not all—of the molecular components within each module (e.g., ECM, focal adhesions, etc.) undergo dynamic assembly and disassembly and rapid associations with other proteins.79
ECM and integrins
Because plasma and cellular FNs are prominent in healing wounds at different times after injury, FNs serve as particularly relevant models for ECM proteins that are responsive to mechanical forces. This adhesive glycoprotein is comprised of FN Type III repeats, which have been found through both simulations and laboratory measurements to undergo reversible unfolding when force is applied.114 Individual FN molecules bind to integrins and then are assembled into fibrils in a process that is mechanically regulated. Tugging on FN by integrins on the cell opens up cryptic sites within FN (notably in a segment called FN III-1) that facilitate the addition of more FN molecules and assembly into a fibrillar array.115,116 Recent studies have described other domains within FN that appear to be “cryptic.”117 These additional domains may also release activities when subjected to mechanical tension, although proof of this is currently lacking. FN serves as a useful model, but there are many other ECM components within the wound that modulate myofibroblast function (reviewed by Klingberg et al.).66
The structure of integrins has been worked out in molecular detail and has yielded a wealth of data as a foundation for mechanistic studies (see also the article by Leask, this issue, p. XXX).118–121 Although the details are beyond the scope of this article, several important concepts should be mentioned. Integrins undergo a conformation change that results in a transition from an inactive (bent) to an active (upright) form with potential intermediate conformations also possible. When in the active conformation, integrin complexes at adhesions sites can transduce to the cell a “sense” of the extracellular tension. This sensing mechanism involves increases in the apparent affinity of integrins for ECM molecules and the binding of associated proteins (e.g., talin).122 Increased extracellular rigidity imposed on regions of the cell, strengthens the linkage between integrins and the ECM resulting in “adhesion strengthening.”123–125 This apparent increased affinity in ligand binding by integrins is reinforced by recruitment of talin1 that unfolds under mechanical force, thereby recruiting vinculin and engaging the actin cytoskeleton resulting in enlarged focal adhesions that serve to keep the force per unit area constant and support increased force generation.126–128 Maturation of focal adhesions into the larger macromolecular structures is characteristic of myofibroblasts.4,5,52,94 Recently, an engineered vinculin “tensiometer” has enabled measurements of force generated at the level of a single vinculin molecule.129 Vinculin is a key element in the link between the ECM, integrins, and the cytoskeleton because it both stabilizes focal adhesions under force and registers increases and decreases in force as they occur within the focal adhesion. While focal adhesions are prominent and intensively studied mechanical signal transducers, it is now clear that stretch-sensitive ion channels also mediate fibroblast and myofibroblast responses.122,130
Focal adhesions and cell contraction
“Supermature” focal adhesions are a distinctive feature of myofibroblasts on mechanically stiff surfaces and reflect the intimate link between focal adhesions and the cytoskeleton.4,5,94 Focal adhesion maturation is blocked by contractility inhibitors (e.g., Rho kinase [ROCK] and myosin II inhibitors) and the growth of focal adhesions controls tension-dependent recruitment of SM α-actin.4,5,65 Focal adhesions are complexes of hundreds of molecules—in addition to integrins, talin, and vinculin discussed above—including receptors, scaffolding proteins, kinases, and phosphatases that interact combinatorially, serving as signaling centers that activate networks of “downstream” kinases regulating cell functions.111 Recent data have highlighted several of these components as potentially important in governing or contributing to the myofibroblast phenotype. FAK undergoes conformation changes induced by mechanical stress that relieve the inhibition of its kinase activity by the internal FERM domain.131 Recently, control mice or mice in which fibroblasts were null for FAK were tested with incisional wounds under prolonged mechanical stress, conditions that in normal mice exhibit a “hypertrophic scar” phenotype. FAK null mice did not elaborate this pathologic scarring and importantly, this was mediated by markedly reduced levels of gene products, notably monocyte chemoattractant protein 1 (MCP-1) that mediates recruitment of macrophages among other functions.81 Indeed, there were many other changes observed on cDNA arrays underscoring the capacity of FAK to govern paracrine functions during wound healing. Additional support for a central role for FAK in the myofibroblast phenotype was obtained in scleroderma fibroblasts knocked down for FAK or treated with a FAK phosphorylation inhibitor.132
Paxillin and Hic-5 are closely related focal adhesion proteins that serve as scaffolds for signaling and have important, but reciprocal functions in regulating tumor cell migration.133 There is a competitive relationship between Hic-5 and paxillin with the former promoting mesenchymal-like movement and the latter ameboid movement.133 In two-dimensional cultures both paxillin and Hic-5 localize to focal adhesions, however, paxillin is associated with active vinculin in relatively immature focal complexes at the cell periphery in a process governed by Rac, while Hic-5 associates with active vinculin in maturing focal adhesions driven by Rho GTPase.134 When Hic-5 is knocked down in pathogenic myofibroblasts, collagen contraction is dramatically reduced, potentially because of the loss of mature focal adhesions.95 These studies suggest that Hic-5 may have important roles in regulating the maturation of focal adhesions, and the generation of tension in contracting myofibroblasts and potentially mechanical activation of TGF-β1 (Fig. 8).
Mechanoregulation of myofibroblast genes
The serum response factor (SRF) pathway is now known to serve as a central regulator of this process linking the assembly of actin to gene transcription. SRF is constitutively present in cells, but by itself is a weak transcription factor.135,136 The myocardin-related transcription factor (MRTF) family includes myocardin that is constitutively present in the nucleus of SM cells but also MRTF-A that associates with the G-actin monomers and is transcriptionally inactive when complexed with G-actin.137 When actin polymerization occurs G-actin dissociates from MRTF-A as it becomes incorporated into growing actin filaments. Upon its disassociation from G-actin, MRTF-A translocation to, and retention in the nucleus is favored. Once nuclear, MRTF-A docks with SRF on promoters to regulate the expression of up to 100 genes.138 Interestingly, many of the contractile genes regulated by MRTF-A, which are conditionally expressed in myofibroblasts, including SM α-actin and Hic-5, are constitutively expressed in SM cells and are regulated by myocardin.139 The differential utilization of SRF cofactors may elaborate a key difference in the way in which mechanical environment impinges upon contractile gene expression in myofibroblasts, whereas these genes in constitutively contractile cell types such as SM cells may be less mechanically influenced.
Crider et al. recently reported that MRTF-A/B, SRF cofactors, are required for differentiation of myofibroblasts and for establishing the contractile phenotype in myofibroblasts.140 They observed that the expression of SM α-actin and SM γ-actin, SM22-α, h1-calponin, and vinculin required MRTF-A/B and that overexpression of MRTF-A was sufficient to drive upregulation in the absence of TGF-β1. Knocking down MRTF-A/B also resulted in the loss of super-mature focal adhesions and a reduction in contractile force generation.
Given the effects of ECM stiffness on actin polymerization and as a consequence MRTF-A/B localization, these findings fit well with earlier data showing that the compliance of collagen gels was an important factor in regulating SM α-actin expression.64,67 When collagen-coated magnetite beads were pulled with a magnet, after binding to osteoblastic cells, the SM α-actin promoter activity was increased in a mechanism that required an intact SRF promoter element (“CarG box”).141 Huang et al. reported that induction of SM α-actin was dependent upon environmental stiffness and required activation of the Rho-ROCK pathway and MRTF-A interactions with the promoter.142 Indeed, MRTF-A translocation to the nucleus, a critical regulatory step in activating the SRF-MRTF-A pathway has been reported to be dependent upon a critical stiffness when 3T3 cells are cultured in collagen gels of various compliances.143 These findings are likely to have important relevance for the fibrotic process. Mice that are null for MRTF-A show reductions in the expression of contractile proteins, including SM α-actin and SM-22 alpha and interstitial collagens and reductions in scarring and fibrosis in a model of myocardial infarction.144 Importantly, other pathways likely counterbalance the SRF-MRTF-A–dependent induction of the contractile phenotype. For example, during EMT, TGF-β–dependent activation of SMAD3 results in inhibition of MRTF-A stimulating activity at the CArG elements in the SM α-actin promoter.145 This inhibition by SMAD3 can be tempered by β-catenin.146
Taken together, these findings support a model in which MRTF-A is central to establishing and maintaining the myofibroblast phenotype in response to mechanical tension (Figs. 8 and 9).140 Interestingly, targeting the SRF-MRTF-A “CarGome” pathway may be more effective in reducing myofibroblast function than targeting SM α-actin itself.
Figure 9.

Mechanical environment regulates transcriptional activity of MRTF-A via actin dynamics. Stress fibers and filamentous actin (F-actin) are stabilized under high intracellular tension resulting in the liberation of MRTF-A from globular actin (G-actin). Free to shuttle to the nucleus from the cytoplasm, MRTF-A drives expression of contractile genes promoting further increases in intracellular tension. (Adapted from Crider et al.140) To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Future Developments of Interest
We now briefly discuss potential approaches for therapy in which myofibroblast function and longevity are targets. The reader is referred to other articles in this issue that address additional therapeutic approaches for fibrosis and wound contractures (see also the articles in this issue by Levinson, p. 149, and by Sharpe and Martin, p. 167).147,148
Nonhealing wounds
In nonhealing wounds the formation of myofibroblasts is compromised, owing in part to the hypoxic conditions in chronic wounds. Because proper, but temporally limited, myofibroblast function is necessary for normal acute wound healing, therapeutic approaches should include steps to reverse the chronic hypoxic state.38–43 Promising data indicate that promoting a suitable mechanical microenvironment in the context of an ECM scaffold and stem cells will be beneficial.149 This ECM scaffold would likely include Type III collagen—the predominant collagen in granulation tissue—and a mixture of proteoglycans and alternatively spliced cellular FNs. In addition, a source of active TGF-β1 is necessary and could be provided by encoding “constitutively active” TGF-β1 in an adenoviral vector, thereby providing a transient transduction for temporally limited expression. Alteration of the mechanical environment may also assist in healing of chronic wounds; negative-pressure wound therapy using a vacuum dressing to apply frequency-dependent loading on the wound and surrounding tissue has been demonstrated, in certain studies, to assist with chronic wound closure.150,151 The underlying cellular mechanisms for this response are unclear and deserve further study. In addition, given what we know about the pronounced inhibition of healing caused by persistent biofilms, it will be important to treat these to make an environment permissive for robust healing.
Pathological scarring and contractures
Presently therapies to reverse preexisting scarring and fibrotic lesions in which myofibroblasts have matured, reverted, or disappeared must be deferred until sophisticated methods for remodeling collagen-rich scars are developed. However, the goal of modulating myofibroblast function, differentiation, or lifespan in developing scars seems more attainable currently. One promising therapeutic approach may be to target wounds with decorin, a small leucine-rich proteoglycan that is reduced in pathogenic scarring, including HTS.152 Decorin has a high affinity for interstitial collagen and null mice show defects in collagen fibrillogenesis and exhibit skin fragility.153 Intriguingly, decorin may therefore have important effects on the ability of collagen fibers to transmit mechanical signals. Moreover, decorin tightly interacts with TGF-β, sequestering it and thereby downmodulating TGF-β–dependent wound processes and decorin null mice exhibit enhanced liver fibrosis.154,155 One recent novel approach is to use targeting peptides to deliver decorin to the wound microenvironment.156 Another approach may be to promote myofibroblast apoptosis or senescence to force these cells out of the feed-forward loops that maintain their pathologic function. Certain matricellular proteins (i.e., CCN1) promote the ability of fibroblasts to attain a senescent phenotype and this “senescence-associated secretory phenotype” is one in which upregulation of ECM degrading enzymes and downregulation of the ECM genes occurs.108 It may also be possible to promote apoptosis of myofibroblasts by targeting anti-apoptotic genes in these cells or by modulating the protective effects of the p53 and Rb pathways, although specificity may be a concern. Increased intracellular tension is a characteristic of the differentiated myofibroblast, but evidence from a variety of experiments suggest that relaxing environmental tension may reverse the formation or prominence of stress fibers within cells and potentially induce fibroblast apoptosis.157
Given the role of mechanical tension in promoting and maintaining the feed-forward loops (Fig. 8) that sustain myofibroblasts, reducing tension in the tissues surrounding the wound could provide an important therapeutic tool (see the article by Wong et al., this issue, p. 185).158 This approach could have the potential to blunt ongoing TGF-β1 activation, which as we have discussed is a mechanically sensitive process for the myofibroblast.36 Because there is a clear link between focal adhesion size and the growth of stress fibers, targeting components within focal adhesions may also provide a means to modulate stress fiber formation and SM α-actin incorporation.5 Conversely, blocking SM α-actin incorporation into stress fibers with an amino terminal peptide has been shown to reduce intracellular tension.159–161 Importantly, one outcome of reducing intracellular tension (perhaps by modulating wound stiffness) would be to reverse the steady state polymerization of G-actin into F-actin, thereby reducing or blocking the transcriptional activity of the MRTF-A/B–SRF pathways regulating contractile gene expression.140 Because Rho GTPase and its “downstream” effector, ROCK, are important components that promote actin polymerization, directly targeting ROCK may prove to be an important therapeutic avenue. Recently, a ROCK inhibitor (Y-27632) was used to reduce granulation tissue contraction, in situ.162 There are currently ROCK inhibitors in clinical trials (e.g., Fasudil) for cardiovascular diseases.163 However, because Rho GTPase and ROCK are critical components in pathways that regulate fundamental cell functions, targeting them may have serious side effects. Finally, an alternative approach to reduce the tension within the scar may be to target the ECM, potentially by modulating FN function or by antagonizing lysyl oxidase, the enzymes that cross-link collagen tropocollagen and fibrils, thereby affecting collagen fiber structure.164
Take-Home Messages.
Myofibroblasts are the cell type responsible for wound closure in normal acute wounds and when persist can result in fibrosis and tissue contracture.
While plentiful in normal acute wounds, myofibroblast numbers are depleted in chronic wounds.
Myofibroblasts differentiate from normally quiescent fibroblasts through a process that requires active TGF-β1, ECM proteins, and mechanical stiffness.
The myofibroblast response to these extrinsic signals is coupled by the cell to intrinsic intracellular mechanisms involving actin and myosin structures. These are intricately linked to key processes such as contraction—the motive force in wound contraction—and gene expression that endows the cell with contractile and ECM proteins.
The mechanisms that govern myofibroblast disappearance during normal acute wounds but persistence in fibrosis are poorly understood.
Myofibroblast persistence is regulated by “feed-forward” pathways that integrate the mechanical environment, extracellular growth factor activation and signaling, and intracellular tension and gene expression together.
Uncovering the cellular processes that regulate myofibroblast contraction, gene expression and long life spans at sites of injury will provide important new avenues for therapies to modulate the robust scarring and wound contracture that affect patients with a wide range of these pathologies.
In summary, recent work has revealed a great deal about the myofibroblast and its roles in governing the normal wound healing response and its pathological role in cases where myofibroblasts are not present or are overly active. This growing foundation of information will provide us with important new “nodes” within the network of signals that govern myofibroblast function. These nodes will likely provide the means to target myofibroblasts therapeutically with more specificity then may be possible by blocking “pleiotropic” growth factors like TGF-β1.
Abbreviations and Acronyms
- ADAM 12
a disintegrin and metalloproteinase 12
- CAF
carcinoma-associated fibroblasts (also sometimes termed TAF=tumor-associated fibroblasts)
- CCN
family of cytokines; name is acronym derived from three of the family members, C for CTGF, C for Cyr-61, and N for Nov
- ECM
extracellular matrix
- EMT
epithelial to mesenchymal transition
- FAK
focal adhesion kinase
- FERM
F for 4.1 protein, E for ezrin, R for radixin, and M for moesin
- FN
fibronectin
- Hic-5
hydrogen peroxide-inducible clone-5 (also called TGFB1I1)
- HTS
hypertrophic scar
- LAP
latency-associated peptide
- LLC
large latent complex
- LTBP-1
latent TGF-β1–binding protein
- MCP-1
monocyte chemoattractant protein-1
- MMP
matrix metalloproteinase
- MRTFA/B
myocardin-related transcription factor A/B
- p38 MAPK
p38 MAP kinase
- PDGF
platelet-derived growth factor
- PGE2
prostaglandin E2
- ROCK
Rho kinase
- SM α-actin
smooth muscle alpha-actin
- SRF
serum response factor
- TGF-β1
transforming growth factor beta-1
- TSP-1
thrombospondin 1
- VEGF
vascular endothelial growth factor
Footnotes
Tomasek JJ: Clonal analysis of Dupuytren's myofibroblasts. University of Oklahoma Health Sciences Center, Oklahoma City, OK, 2005 (unpublished).
Van De Water L and Tomasek JJ: The TGF-β1–inducible focal adhesion protein Hic-5 (TGFB1I1) is expressed in myofibroblasts in Dupuytren's disease. Albany Medical College, Albany, NY, and University of Oklahoma Health Sciences Center, Oklahoma City, OK, 2012 (unpublished).
Acknowledgments
The authors are grateful to Dr. David Corr, a biomedical engineer at Rensselaer Polytechnic Institute (Troy, NY), for advice on biomechanics, and to Debbie Moran (Albany Medical College) for her careful help in preparing this article.
Funding Sources
Research reported in this publication was supported by the National Institute of General Medical Science of the National Institutes of Health under award numbers R01GM056442 (to L.V.D.W.) and R01GM060651 (to J.J.T.), and by the American Recovery and Reinvestment Act (ARRA) funds to L.V.D.W. from the National Institute of General Medical Sciences, National Institutes of Health.
Author Disclosure and Ghostwriting
No competing financial interests exist. The content of this article was written entirely by the author(s) listed. No ghostwriters were used to write this article.
About the Authors
Livingston Van De Water is a Professor and Scott Varney a graduate student in the Center for Cell Biology and Cancer Research at Albany Medical College. James J. Tomasek is the David Ross Boyd Professor of Cell Biology and the Dean of the Graduate College at the University of Oklahoma Health Sciences Center. Both groups work on the mechanisms regulating the function of myofibroblasts in scarring and fibrosis.
References
- 1.Wynn TA. Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomasek JJ. Gabbiani G. Hinz B. Chaponnier C. Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 3.Darby I. Skalli O. Gabbiani G. α-Smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest. 1990;63:21. [PubMed] [Google Scholar]
- 4.Dugina V. Fontao L. Chaponnier C. Vasiliev J. Gabbiani G. Focal adhesion features during myofibroblastic differentiation are controlled by intracellular and extracellular factors. J Cell Sci. 2001;114:3285. doi: 10.1242/jcs.114.18.3285. [DOI] [PubMed] [Google Scholar]
- 5.Goffin JM. Pittet P. Csucs G. Lussi JW. Meister J-J. Hinz B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J Cell Biol. 2006;172:259. doi: 10.1083/jcb.200506179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vedrenne N. Coulomb B. Danigo A. Bonté F. Desmoulière A. The complex dialogue between (myo)fibroblasts and the extracellular matrix during skin repair processes and ageing. Pathol Biol. 2012;60:20. doi: 10.1016/j.patbio.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Dulauroy S. Di Carlo SE. Langa F. Eberl G. Peduto L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. 2012;18:1262. doi: 10.1038/nm.2848. [DOI] [PubMed] [Google Scholar]
- 8.Quan TE. Cowper SE. Bucala R. The role of circulating fibrocytes in fibrosis. Curr Rheumatol Rep. 2006;8:145. doi: 10.1007/s11926-006-0055-x. [DOI] [PubMed] [Google Scholar]
- 9.Kalluri R. Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glasser SR. Julian J. Intermediate filament protein as a marker of uterine stromal cell decidualization. Biol Reprod. 1986;35:463. doi: 10.1095/biolreprod35.2.463. [DOI] [PubMed] [Google Scholar]
- 11.Toccanier-Pelte M-F. Skalli O. Kapanci Y. Gabbiani G. Characterization of stromal cells with myoid features in lymph nodes and spleen in normal and pathological conditions. Am J Pathol. 1987;129:109. [PMC free article] [PubMed] [Google Scholar]
- 12.Sappino AP. Dietrich PY. Skalli O. Widgren S. Gabbiani G. Colonic pericryptal fibroblasts. Differentiation pattern in embryonic and phenotypie modulation in epithelial proliferative lesions. Virchows Arch A Pathol Anat Histopathol. 1989;415:551. doi: 10.1007/BF00718649. [DOI] [PubMed] [Google Scholar]
- 13.Kaye GI. Lane N. Pascal PR. Colonic pericryptal fibroblast sheath: replication, migration, and cytodifferentiation of a mesenchymal cell system in adult tissue. II. Fine structure aspects of normal rabbit and human colon. Gastroenterology. 1968;54:852. [PubMed] [Google Scholar]
- 14.Skalli O. Ropraz P. Trzeciak A. Benzonana G. Gillessen D. Gabbiani G. A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol. 1986;103:2787. doi: 10.1083/jcb.103.6.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Czernobilsky B. Shezen E. Lifschitz-Mercer B. Fogel M. Luzon A. Jacob N. Skalli O. Gabbiani G. Alpha smooth muscle actin (α-SM actin) in normal human ovaries, in ovarian stromal hyperplasia and in ovarian neoplasms. Virchows Arch B Cell Pathol Incl Mol Pathol. 1989;57:55. doi: 10.1007/BF02899065. [DOI] [PubMed] [Google Scholar]
- 16.Beertsen W. Everts V. van den Hooff A. Fine structure of fibroblasts in the periodontal ligament of the rat incisor and their possible role in tooth eruption. Arch Oral Biol. 1974;19:1087. doi: 10.1016/0003-9969(74)90235-0. [DOI] [PubMed] [Google Scholar]
- 17.Bressler RS. Myoid cells in the capsule of the adrenal gland and in monolayers derived from cultured adrenal capsules. Anat Rec. 1973;177:525. doi: 10.1002/ar.1091770406. [DOI] [PubMed] [Google Scholar]
- 18.Yokoi Y. Namihisa T. Kuroda H. Komatsu I. Miyasaki A. Watanabe S. Usui K. Immunocytochemical detection of desmin in fat-storing cells (Ito cells) Hepatology. 1984;4:709. doi: 10.1002/hep.1840040425. [DOI] [PubMed] [Google Scholar]
- 19.Kapanci Y. Ribaux C. Chaponnier C. Gabbiani G. Cytoskeletal features of alveolar myofibroblasts and pericytes in normal human and rat lung. J Histochem Cytochem. 1992;40:1955. doi: 10.1177/40.12.1333502. [DOI] [PubMed] [Google Scholar]
- 20.Charbord P. Lerat H. Newton I. Tamayo E. Gown AM. Singer JW. Herve P. The cytoskeleton of stromal cells from human bone marrow cultures resembles that of cultured smooth muscle cells. Exp Hematol. 1990;18:276. [PubMed] [Google Scholar]
- 21.Lindahl P. Betsholtz C. Not all myofibroblasts are alike: revisiting the role of PDGF-A and PDGF-B targeted mice. Curr Opin Neprhol Hypertens. 1998;7:21. doi: 10.1097/00041552-199801000-00004. [DOI] [PubMed] [Google Scholar]
- 22.Brown LF. Dubin D. Lavigne L. Logan B. Dvorak HF. Van De Water L. Macrophages and fibroblasts express embryonic fibronectins during cutaneous wound healing. Am J Pathol. 1993;142:793. [PMC free article] [PubMed] [Google Scholar]
- 23.Desmouliere A. Gabbiani G. The role of the myofibroblast in wound healing, fibrocontrative diseases. In: Clark RAF, editor. The Molecular and Cellular Biology of Wound Repair. New York: Plenum Press; 1988. pp. 391–423. [Google Scholar]
- 24.ffrench-Constant C. Van De Water L. Dvorak HF. Hynes RO. Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J Cell Biol. 1989;109:903. doi: 10.1083/jcb.109.2.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serini G. Bochaton-Piallat ML. Ropraz P. Geinoz A. Borsi L. Zardi L. Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142:873. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinz B. Mastrangelo D. Iselin CE. Chaponnier C. Gabbiani G. Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am J Pathol. 2001;159:1009. doi: 10.1016/S0002-9440(10)61776-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang K. Rekhter MD. Gordon D. Phan SH. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am J Pathol. 1994;145:114. [PMC free article] [PubMed] [Google Scholar]
- 28.Gabbiani G. Hirschel BJ. Ryan GB. Statkov PR. Majno G. Granulation tissue as a contractile organ. A study of structure and function. J Exp Med. 1972;135:719. doi: 10.1084/jem.135.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 30.Hinz B. Celetta G. Tomasek JJ. Gabbiani G. Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomasek J. Haaksma C. Schwartz R. Howard E. Whole animal knockout of smooth muscle alpha-actin does not alter excisional wound healing or the fibroblast-to-myofibroblast transition. Wound Repair Regen. 2013;21:166. doi: 10.1111/wrr.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coen M. Gabbiani G. Bochaton-Piallat ML. Myofibroblast-mediated adventitial remodeling: an underestimated player in arterial pathology. Arterioscler Thromb Vasc Biol. 2011;31:2391. doi: 10.1161/ATVBAHA.111.231548. [DOI] [PubMed] [Google Scholar]
- 33.van den Borne SW. Diez J. Blankesteijn WM. Verjans J. Hofstra L. Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 34.Hinz B. Gabbiani G. Seemayer T. Schurch W. Myofibroblast. In: Mills SE, editor. Histology for Pathologists. 4th. Philadelphia, PA: Lippincott, Williams and Wilkins; 2012. pp. 131–178. [Google Scholar]
- 35.Hinz B. Phan SH. Thannickal VJ. Galli A. Bochaton-Piallat M-L. Gabbiani G. The myofibroblast. Am J Pathol. 2007;170:1807. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hinz B. Phan SH. Thannickal VJ. Prunotto M. Desmouliere A. Varga J. De Wever O. Mareel M. Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olerud JE. Odland GF. Burgess EM. Wyss CR. Fisher LD. Matsen FA. A model for the study of wounds in normal elderly adults and patients with peripheral vascular disease or diabetes mellitus. J Surg Res. 1995;59:349. doi: 10.1006/jsre.1995.1175. [DOI] [PubMed] [Google Scholar]
- 38.Brem H. Tomic-Canic M. Cellular and molecular basis of wound healing in diabetes. J Clin Invest. 2007;117:1219. doi: 10.1172/JCI32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mustoe T. Understanding chronic wounds: a unifying hypothesis on their pathogenesis and implications for therapy. Am J Surg. 2004;187:65S. doi: 10.1016/S0002-9610(03)00306-4. [DOI] [PubMed] [Google Scholar]
- 40.Mustoe T. O'Shaughnessy K. Kloeters O. Chronic wound pathogenesis and current treatment strategies: a unifying hypothesis. Plast Reconstr Surg. 2006;117:35S. doi: 10.1097/01.prs.0000225431.63010.1b. [DOI] [PubMed] [Google Scholar]
- 41.Alizadeh N. Pepper MS. Modarressi A. Alfo K. Schlaudraff K. Montandon D. Gabbiani G. Bochaton-Piallat M-L. Pittet B. Persistent ischemia impairs myofibroblast development in wound granulation tissue: a new model of delayed wound healing. Wound Repair Regen. 2007;15:809. doi: 10.1111/j.1524-475X.2007.00312.x. [DOI] [PubMed] [Google Scholar]
- 42.Modarressi A. Pietramaggiori G. Godbout C. Vigato E. Pittet B. Hinz B. Hypoxia impairs skin myofibroblast differentiation and function. J Invest Dermatol. 2010;130:2818. doi: 10.1038/jid.2010.224. [DOI] [PubMed] [Google Scholar]
- 43.Sen CK. Roy S. Oxygenation state as a driver of myofibroblast differentiation and wound contraction: hypoxia impairs wound closure. J Invest Dermatol. 2010;130:2701. doi: 10.1038/jid.2010.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orimo A. Weinberg RA. Heterogeneity of stromal fibroblasts in tumors. Cancer Biol Ther. 2007;6:618. doi: 10.4161/cbt.6.4.4255. [DOI] [PubMed] [Google Scholar]
- 45.Otranto M. Sarrazy V. Bonte F. Hinz B. Gabbiani G. Desmouliere A. The role of the myofibroblast in tumor stroma remodeling. Cell Adh Migr. 2012;6:203. doi: 10.4161/cam.20377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sugimoto H. Mundel TM. Kieran MW. Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006;5:1640. doi: 10.4161/cbt.5.12.3354. [DOI] [PubMed] [Google Scholar]
- 47.Butcher DT. Alliston T. Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9:108. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paszek MJ. Zahir N. Johnson KR. Lakins JN. Rozenberg GI. Gefen A. Reinhart-King CA. Margulies SS. Dembo M. Boettiger D. Hammer DA. Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 49.Orimo A. Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 50.Dvorak HF. Tumors: wounds that do not heal. N Engl J Med. 1986;315:1650. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 51.Schafer M. Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9:628. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- 52.Hinz B. The myofibroblast paradigm for a mechanically active cell. J Biomechanics. 2010;43:146. doi: 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 53.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 54.Gabbiani G. Ryan GB. Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971;27:549. doi: 10.1007/BF02147594. [DOI] [PubMed] [Google Scholar]
- 55.Singer II. Kawka DW. Kazazis DM. Clark RA. In vivo co-distribution of fibronectin and actin fibers in granulation tissue: immunofluorescence and electron microscope studies of the fibronexus at the myofibroblast surface. J Cell Biol. 1984;98:2091. doi: 10.1083/jcb.98.6.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh P. Reimer CL. Peters JH. Stepp MA. Hynes RO. Van De Water L. The spatial and temporal expression patterns of integrin alpha9beta1 and one of its ligands, the EIIIA segment of fibronectin, in cutaneous wound healing. J Invest Dermatol. 2004;123:1176. doi: 10.1111/j.0022-202X.2004.23485.x. [DOI] [PubMed] [Google Scholar]
- 57.Clark RAF. Wound repair: overview, general considerations. In: Clark RAF, editor. The Molecular and Cellular Biology of Wound Repair. New York: Plenum Press; 1988. pp. 3–50. [Google Scholar]
- 58.Brown LF. Yeo KT. Berse B. Yeo TK. Senger DR. Dvorak HF. Van De Water L. Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J Exp Med. 1992;176:1375. doi: 10.1084/jem.176.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dvorak H. Vascular permeability to plasma, plasma proteins, and cells: an update. Curr Opin Hematol. 2010;17:225. doi: 10.1097/MOH.0b013e3283386638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu H. Mouw JK. Weaver VM. Forcing form and function: biomechanical regulation of tumor evolution. Trends Cell Biol. 2011;21:47. doi: 10.1016/j.tcb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grinnell F. Petroll WM. Cell motility and mechanics in three-dimensional collagen matrices. Annu Rev Cell Dev Biol. 2010;26:335. doi: 10.1146/annurev.cellbio.042308.113318. [DOI] [PubMed] [Google Scholar]
- 62.Werner S. Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83:835. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 63.Lucas T. Waisman A. Ranjan R. Roes J. Krieg T. Muller W. Roers A. Eming SA. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184:3964. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 64.Vaughan MB. Howard EW. Tomasek JJ. Transforming growth factor-β1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. 2000;257:180. doi: 10.1006/excr.2000.4869. [DOI] [PubMed] [Google Scholar]
- 65.Chrzanowska-Wodnicka M. Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klingberg F. Hinz B. White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol. 2013;229:298. doi: 10.1002/path.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arora PD. Narani N. McCulloch CA. The compliance of collagen gels regulates transforming growth factor-beta induction of alpha-smooth muscle actin in fibroblasts. Am J Pathol. 1999;154:871. doi: 10.1016/s0002-9440(10)65334-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Annes JP. Making sense of latent TGFβ activation. J Cell Sci. 2003;116:217. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 69.Jenkins G. The role of proteases in transforming growth factor-β activation. Int J Biochem Cell Biol. 2008;40:1068. doi: 10.1016/j.biocel.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 70.Stamenkovic I. Extracellular matrix remodelling: the role of matrix metalloproteinases. J Pathol. 2003;200:448. doi: 10.1002/path.1400. [DOI] [PubMed] [Google Scholar]
- 71.Yu Q. Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163. [PMC free article] [PubMed] [Google Scholar]
- 72.Sweetwyne M. Murphy-Ullrich J. Thrombospondin1 in tissue repair and fibrosis: TGF-β-dependent and independent mechanisms. Matrix Biol. 2012;31:178. doi: 10.1016/j.matbio.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wipff P. Hinz B. Integrins and the activation of latent transforming growth factor β1—an intimate relationship. Eur J Cell Biol. 2008;87:601. doi: 10.1016/j.ejcb.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 75.Wipff PJ. Rifkin DB. Meister JJ. Hinz B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J Cell Biol. 2007;179:1311. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sheppard D. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev. 2005;24:395. doi: 10.1007/s10555-005-5131-6. [DOI] [PubMed] [Google Scholar]
- 77.Bershadsky AD. Balaban NQ. Geiger B. Adhesion-dependent cell mechanosensitivity. Annu Rev Cell Dev Biol. 2003;19:677. doi: 10.1146/annurev.cellbio.19.111301.153011. [DOI] [PubMed] [Google Scholar]
- 78.Pelham RJ. Wang Yl. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci USA. 1997;94:13661. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoffman BD. Grashoff C. Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475:316. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aarabi S. Bhatt KA. Shi Y. Paterno J. Chang EI. Loh SA. Holmes JW. Longaker MT. Yee H. Gurtner GC. Mechanical load initiates hypertrophic scar formation through decreased cellular apoptosis. FASEB J. 2007;21:3250. doi: 10.1096/fj.07-8218com. [DOI] [PubMed] [Google Scholar]
- 81.Wong VW. Rustad KC. Akaishi S. Sorkin M. Glotzbach JP. Januszyk M. Nelson ER. Levi K. Paterno J. Vial IN. Kuang AA. Longaker MT. Gurtner GC. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat Med. 2011;18:148. doi: 10.1038/nm.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu F. Mih JD. Shea BS. Kho AT. Sharif AS. Tager AM. Tschumperlin DJ. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190:693–706. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Masur SK. Dewal HS. Dinh TT. Erenburg I. Petridou S. Myofibroblasts differentiate from fibroblasts when plated at low density. Proc Natl Acad Sci USA. 1996;93:4219. doi: 10.1073/pnas.93.9.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Desmouliere A. Rubbia-Brandt L. Abdiu A. Walz T. Macieira-Coelho A. Gabbiani G. a-smooth muscle actin is expressed in a subpopulation of cultured and cloned fibroblasts and is modulated by gamma-interferon. Exp Cell Res. 1992;201:64. doi: 10.1016/0014-4827(92)90348-c. [DOI] [PubMed] [Google Scholar]
- 85.Yates CC. Bodnar R. Wells A. Matrix control of scarring. Cell Mol Life Sci. 2011;68:1871. doi: 10.1007/s00018-011-0663-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martin P. Wound healing—aiming for perfect skin regeneration. Science. 1997;276:75. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- 87.Werner S. Krieg T. Smola H. Keratinocyte—fibroblast interactions in wound healing. J Invest Dermatol. 2007;127:998. doi: 10.1038/sj.jid.5700786. [DOI] [PubMed] [Google Scholar]
- 88.Guidry C. Grinnell F. Studies on the mechanism of hydrated collagen gel reorganization by human skin fibroblasts. J Cell Sci. 1985;79:67. doi: 10.1242/jcs.79.1.67. [DOI] [PubMed] [Google Scholar]
- 89.Marenzana M. Wilson-Jones N. Mudera V. Brown R. The origins and regulation of tissue tension: Identification of collagen tension-fixation process in vitro. Exp Cell Res. 2006;312:423. doi: 10.1016/j.yexcr.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 90.Sawhney RK. Howard J. Slow local movements of collagen fibers by fibroblasts drive the rapid global self-organization of collagen gels. J Cell Biol. 2002;157:1083. doi: 10.1083/jcb.200203069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Levental KR. Yu H. Kass L. Lakins JN. Egeblad M. Erler JT. Fong SFT. Csiszar K. Giaccia A. Weninger W. Yamauchi M. Gasser DL. Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wentzel VK. National Geographic Society; Ornate welts of raised scar tissue are a mark of beauty. stock picture ID: 615801. [Google Scholar]
- 93.Karamanoukian R. Ukatu C. Lee E. Hyman J. Sundine M. Kobayashi M. Evans GRD. Aesthetic skin branding: a novel form of body art with adverse clinical sequela. J Burn Care Res. 2006;27:108. doi: 10.1097/01.bcr.0000191958.51354.cd. [DOI] [PubMed] [Google Scholar]
- 94.Dabiri G. Campaner A. Morgan J. Van De Water L. A TGF-β1-dependent autocrine loop regulates the structure of focal adhesions in hypertrophic scar fibroblasts. J Invest Dermatol. 2006;126:963. doi: 10.1038/sj.jid.5700187. [DOI] [PubMed] [Google Scholar]
- 95.Dabiri G. Tumbarello DA. Turner CE. Van De Water L. Hic-5 promotes the hypertrophic scar myofibroblast phenotype by regulating the TGF-β1 autocrine loop. J Invest Dermatol. 2008;128:2518. doi: 10.1038/jid.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dabiri G. Tumbarello DA. Turnder CE. Van De Water L. TGF-β1 slows the growth of pathogenic myofibroblasts through a mechanism requiring the focal adhesion protein, Hic-5. J Invest Dermatol. 2008;128:280. doi: 10.1038/sj.jid.5700975. [DOI] [PubMed] [Google Scholar]
- 97.Heitzer MD. DeFranco DB. Hic-5, an adaptor-like nuclear receptor coactivator. Nucl Recept Signal. 2006;4:e019. doi: 10.1621/nrs.04019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim-Kaneyama J-R. Lei X-F. Arita S. Miyauchi A. Miyazaki T. Miyazaki A. Hydrogen peroxide-inducible clone 5 (hic-5) as a potential therapeutic target for vascular and other disorders. J Atheroscler Thromb. 2012;19:601. doi: 10.5551/jat.10736. [DOI] [PubMed] [Google Scholar]
- 99.Mori K. Hamanaka H. Oshima Y. Araki Y. Ishikawa F. Nose K. Shibanuma M. A HIC-5- and KLF4-dependent mechanism transactivates p21Cip1 in response to anchorage loss. J Biol Chem. 2012;287:38854. doi: 10.1074/jbc.M112.377721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shibanuma M. Mori K. Nose K. HIC-5: A mobile molecular scaffold regulating the anchorage dependence of cell growth. Int J Cell Biol. 2012;2012:1. doi: 10.1155/2012/426138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li P. Yu X. Ge K. Melamed J. Roeder R. Wang Z. Heterogeneous expression and functions of androgen receptor co-factors in primary prostate cancer. Am J Pathol. 2010;161:1467. doi: 10.1016/S0002-9440(10)64422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Desmouliere A. Redard M. Darby I. Gabbiani G. Apoptosis medidats the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56. [PMC free article] [PubMed] [Google Scholar]
- 103.Hecker L. Thannickal VJ. Nonresolving fibrotic disorders: idiopathic pulmonary fibrosis as a paradigm of impaired tissue regeneration. Am J Med Sci. 2011;341:431. doi: 10.1097/MAJ.0b013e31821a9d66. [DOI] [PubMed] [Google Scholar]
- 104.Horowitz JC. Lee DY. Waghray M. Keshamouni VG. Thomas PE. Zhang H. Cui Z. Thannickal VJ. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. 2003;279:1359. doi: 10.1074/jbc.M306248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Horowitz JC. Rogers DS. Sharma V. Vittal R. White ES. Cui Z. Thannickal VJ. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal. 2007;19:761. doi: 10.1016/j.cellsig.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Thannickal VJ. Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. 2006;3:350. doi: 10.1513/pats.200601-001TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jun J. Lau L. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jun J-I. Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov. 2011;10:945. doi: 10.1038/nrd3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kisseleva T. Cong M. Paik Y. Scholten D. Jiang C. Benner C. Iwaisako K. Moore-Morris T. Scott B. Tsukamoto H. Evans SM. Dillmann W. Glass CK. Brenner DA. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci USA. 2012;109:9448. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Krizhanovsky V. Yon M. Dickins RA. Hearn S. Simon J. Miething C. Yee H. Zender L. Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Geiger B. Yamada KM. Molecular architecture and function of matrix adhesions. Cold Spring Harbor Persp Biol. 2011;3:a005033. doi: 10.1101/cshperspect.a005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Orr A. Helmke B. Blackman B. Schwartz M. Mechanisms of mechanotransduction. Dev Cell. 2006;10:11. doi: 10.1016/j.devcel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 113.Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harbor Perspect Biol. 2010;2:a005066. doi: 10.1101/cshperspect.a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vogel V. Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol. 2006;7:265. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- 115.Gao M. Craig D. Lequin O. Campbell I. Vogel V. Schulten K. Structure and functional significance of mechanically unfolded fibronectin type III1 intermediates. Proc Natl Acad Sci USA. 2003;100:14784. doi: 10.1073/pnas.2334390100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhong C. Chrzanowska-Wodnicka M. Brown J. Shaub A. Belkin AM. Burridge K. Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J Cell Biol. 1998;141:539. doi: 10.1083/jcb.141.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schenk S. Quaranta V. Tales from the crypt[ic] sites of the extracellular matrix. Trends Cell Biol. 2003;13:366. doi: 10.1016/s0962-8924(03)00129-6. [DOI] [PubMed] [Google Scholar]
- 118.Arnaout M. Goodman S. Xiong J. Structure and mechanics of integrin-based cell adhesion. Curr Opin Cell Biol. 2007;19:495. doi: 10.1016/j.ceb.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Campbell ID. Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harbor Perspect Biol. 2011;3:a004994. doi: 10.1101/cshperspect.a004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Luo B. Carman C. Springer T. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Leask A. Integrin β1: a mechanosignaling sensor essential for connective tissue deposition by fibroblasts. Adv Wound Care. 2013;2 doi: 10.1089/wound.2012.0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Moore SW. Roca-Cusachs P. Sheetz MP. Stretchy proteins on stretchy substrates: the important elements of integrin-mediated rigidity sensing. Dev Cell. 2010;19:194. doi: 10.1016/j.devcel.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Balaban NQ. Schwarz US. Riveline D. Goichberg P. Tzur G. Sabanay I. Mahalu D. Safran S. Bershadsky A. Addadi L. Geiger B. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat Cell Biol. 2001;3:466. doi: 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
- 124.Choquet D. Felsenfeld DP. Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 1997;88:39. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- 125.Riveline D. Zamir E. Balaban NQ. Schwarz US. Ishizaki T. Narumiya S. Kam Z. Geiger B. Bershadsky AD. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153:1175. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.del Rio A. Perez-Jimenez R. Liu R. Roca-Cusachs P. Fernandez JM. Sheetz MP. Stretching single talin rod molecules activates vinculin binding. Science. 2009;323:638. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jiang G. Giannone G. Critchley DR. Fukumoto E. Sheetz MP. Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature. 2003;424:334. doi: 10.1038/nature01805. [DOI] [PubMed] [Google Scholar]
- 128.Papagrigoriou E. Gingras AR. Barsukov IL. Bate N. Fillingham IJ. Patel B. Frank R. Ziegler WH. Roberts GC. Critchley DR. Emsley J. Activation of a vinculin-binding site in the talin rod involves rearrangement of a five-helix bundle. EMBO J. 2004;23:2942. doi: 10.1038/sj.emboj.7600285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Grashoff C. Hoffman BD. Brenner MD. Zhou R. Parsons M. Yang MT. McLean MA. Sligar SG. Chen CS. Ha T. Schwartz MA. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466:263. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Castella LF. Gabbiani G. McCulloch CA. Hinz B. Regulation of myofibroblast activities: calcium pulls some strings behind the scene. Exp Cell Res. 2010;316:2390. doi: 10.1016/j.yexcr.2010.04.033. [DOI] [PubMed] [Google Scholar]
- 131.Tomar A. Schlaepfer DD. Focal adhesion kinase: switching between GAPs and GEFs in the regulation of cell motility. Curr Opin Cell Biol. 2009;21:676. doi: 10.1016/j.ceb.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mimura Y. Ihn H. Jinnin M. Asano Y. Yamane K. Tamaki K. Constitutive phosphorylation of focal adhesion kinase is involved in the myofibroblast differentiation of scleroderma fibroblasts. J Invest Dermatol. 2005;124:886. doi: 10.1111/j.0022-202X.2005.23701.x. [DOI] [PubMed] [Google Scholar]
- 133.Deakin NO. Turner CE. Distinct roles for paxillin and Hic-5 in regulating breast cancer cell morphology, invasion, and metastasis. Mol Biol Cell. 2011;22:327. doi: 10.1091/mbc.e10-09-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Deakin NO. Ballestrem C. Turner CE. Paxillin and Hic-5 interaction with vinculin is differentially regulated by Rac1 and RhoA. PLoS One. 2012;7:e37990. doi: 10.1371/journal.pone.0037990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Miano JM. Long X. Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol. 2007;292:C70. doi: 10.1152/ajpcell.00386.2006. [DOI] [PubMed] [Google Scholar]
- 136.Posern G. Treisman R. Actin’ together: serum response factor, its cofactors and the link to signal transduction. Trends in Cell Biol. 2006;16:588. doi: 10.1016/j.tcb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 137.Miralles F. Posern G. Zaromytidou A. Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 138.Sun Q. Chen G. Streb JW. Long X. Yang Y. Stoeckert CJ. Miano JM. Defining the mammalian CArGome. Genome Res. 2006;16:197. doi: 10.1101/gr.4108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang X. Hu G. Betts C. Harmon EY. Keller RS. Van De Water L. Zhou J. Transforming growth factor-β1-induced transcript 1 protein, a novel marker for smooth muscle contractile phenotype, is regulated by serum response factor/myocardin protein. J Biol Chem. 2011;286:41589. doi: 10.1074/jbc.M111.250878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Crider BJ. Risinger GM. Haaksma CJ. Howard EW. Tomasek JJ. Myocardin-related transcription factors A and B are key regulators of TGF-b1-induced fibroblast to myofibroblast differentiation. J Invest Dermatol. 2011;131:2378. doi: 10.1038/jid.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang J. Su M. Fan J. Seth A. McCulloch CA. Transcriptional regulation of a contractile gene by mechanical forces applied through integrins in osteoblasts. J Biol Chem. 2002;277:22889. doi: 10.1074/jbc.M203130200. [DOI] [PubMed] [Google Scholar]
- 142.Huang X. Yang N. Fiore VF. Barker TH. Sun Y. Morris SW. Ding Q. Thannickal VJ. Zhou Y. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. 2012;47:340. doi: 10.1165/rcmb.2012-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.McGee KM. Vartiainen MK. Khaw PT. Treisman R. Bailly M. Nuclear transport of the serum response factor coactivator MRTF-A is downregulated at tensional homeostasis. EMBO Rep. 2011;12:963. doi: 10.1038/embor.2011.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Small EM. Thatcher JE. Sutherland LB. Kinoshita H. Gerard RD. Richardson JA. DiMaio JM. Sadek H. Kuwahara K. Olson EN. Myocardin-related transcription factor-A controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Masszi A. Speight P. Charbonney E. Lodyga M. Nakano H. Szaszi K. Kapus A. Fate-determining mechanisms in epithelial-myofibroblast transition: major inhibitory role for Smad3. J Cell Biol. 2010;188:383. doi: 10.1083/jcb.200906155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Charbonney E. Speight P. Masszi A. Nakano H. Kapus A. beta-catenin and Smad3 regulate the activity and stability of myocardin-related transcription factor during epithelial-myofibroblast transition. Mol Biol Cell. 2011;22:4472. doi: 10.1091/mbc.E11-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Levinson H. A paradigm of fibroblast activation and dermal wound contraction to guide the development of therapies for chronic wounds and pathologic scars. Adv Wound Care. 2013;2:149. doi: 10.1089/wound.2012.0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sharpe JR. Martin Y. Strategies demonstrating efficacy in reducing wound contraction in vivo. Adv Wound Care. 2013;2:167. doi: 10.1089/wound.2012.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wong VW. Gurtner GC. Tissue engineering for the management of chronic wounds: current concepts and future perspectives. Exp Dermatol. 2012;21:729. doi: 10.1111/j.1600-0625.2012.01542.x. [DOI] [PubMed] [Google Scholar]
- 150.Lancerotto L. Bayer LR. Orgill DP. Mechanisms of action of microdeformational wound therapy. Semin Cell Dev Biol. 2012;23:987. doi: 10.1016/j.semcdb.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 151.Shirakawa M. Isseroff RR. Topical negative pressure devices: use for enhancement of healing chronic wounds. Arch Dermatol. 2005;141:1449. doi: 10.1001/archderm.141.11.1449. [DOI] [PubMed] [Google Scholar]
- 152.Honardoust D. Varkey M. Hori K. Ding J. Shankowsky HA. Tredget EE. Small leucine-rich proteoglycans, decorin and fibromodulin, are reduced in postburn hypertrophic scar. Wound Repair Regen. 2011;19:368. doi: 10.1111/j.1524-475X.2011.00677.x. [DOI] [PubMed] [Google Scholar]
- 153.Danielson KG. Baribault H. Holmes DF. Graham H. Kadler KE. Iozzo RV. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol. 1997;136:729. doi: 10.1083/jcb.136.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Baghy K. Dezso K. Laszlo V. Fullar A. Peterfia B. Paku S. Nagy P. Schaff Z. Iozzo RV. Kovalszky I. Ablation of the decorin gene enhances experimental hepatic fibrosis and impairs hepatic healing in mice. Lab Invest. 2011;91:439. doi: 10.1038/labinvest.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Neill T. Schaefer L. Iozzo RV. Decorin: a guardian from the matrix. Am J Pathol. 2012;181:380. doi: 10.1016/j.ajpath.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jarvinen TAH. Design of target-seeking antifibrotic compounds. Methods Enzymol. 2012;509:243. doi: 10.1016/B978-0-12-391858-1.00013-7. [DOI] [PubMed] [Google Scholar]
- 157.Grinnell F. Zhu M. Carlson MA. Abrams JM. Release of mechanical tension triggers apoptosis of human fibroblasts in a model of regressing granulation tissue. Exp Cell Res. 1999;248:608. doi: 10.1006/excr.1999.4440. [DOI] [PubMed] [Google Scholar]
- 158.Wong VW. Beasley B. Zepeda J. Dauskardt RH. Yock PG. Longaker MT. Gurtner GC. A mechanomodulatory device to minimize incisional scar formation. Adv Wound Care. 2013;2:185. doi: 10.1089/wound.2012.0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chaponnier C. Goethals M. Janmey PA. Gabbiani F. Gabbiani G. Vandekerckhove J. The specific NH2-terminal sequence Ac-EEED of a-smooth muscle actin plays a role in polymerization in vitro and in vivo. J Cell Biol. 1995;130:887. doi: 10.1083/jcb.130.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Clement S. Hinz B. Dugina V. Gabbiani G. Chapponnier C. The N-terminal Ac-EEED sequence plays a role in -smooth-muscle actin incorporation into stress fibers. J Cell Sci. 2005;118:1395. doi: 10.1242/jcs.01732. [DOI] [PubMed] [Google Scholar]
- 161.Hinz B. The NH2-terminal peptide of alpha-smooth muscle actin inhibits force generation by the myofibroblast in vitro and in vivo. J Cell Biol. 2002;157:657. doi: 10.1083/jcb.200201049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tomasek J. Vaughan M. Kropp B. Gabbiani G. Martin M. Haaksma C. Hinz B. Contraction of myofibroblasts in granulation tissue is dependent on Rho/Rho kinase/myosin light chain phosphatase activity. Wound Repair Regen. 2006;14:313. doi: 10.1111/j.1743-6109.2006.00126.x. [DOI] [PubMed] [Google Scholar]
- 163.Satoh K. Fukumoto Y. Shimokawa H. Rho-kinase: important new therapeutic target in cardiovascular diseases. Am J Physiol Heart Circ Physiol. 2011;301:H287. doi: 10.1152/ajpheart.00327.2011. [DOI] [PubMed] [Google Scholar]
- 164.Barry-Hamilton V. Spangler R. Marshall D. McCauley S. Rodriguez HM. Oyasu M. Mikels A. Vaysberg M. Ghermazien H. Wai C. Garcia CA. Velayo AC. Jorgensen B. Biermann D. Tsai D. Green J. Zaffryar-Eilot S. Holzer A. Ogg S. Thai D. Neufeld G. Van Vlasselaer P. Smith V. Allosteric inhibition of lysyl oxidase–like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16:1005. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]