

Abstract
Rearrangements of the mixed-lineage leukemia (MLL) gene occur predominately in pediatric leukemia cases and are generally predictors of a poor prognosis. These chromosomal rearrangements result in fusion of the protein MLL to one of more than 60 protein partners. MLL fusions are potent inducers of leukemia through activation of oncogene expression; therefore, targeting this transcriptional activation function may arrest MLL-rearranged (MLL-R) leukemia. Leukemic cell lines harboring the most common fusion protein, MLL-AF4, require the direct interaction of AF4 with the transcription factor AF9 to survive and self-renew; disrupting this interaction with a cell-penetrating AF4-derived peptide results in cell death, suggesting that the AF4-AF9 interaction could be a viable target for a novel MLL-R leukemia therapy. Here we describe the use of AlphaScreen technology to develop a high-throughput screening (HTS) assay to detect nonpeptidic inhibitors of AF4-AF9 binding. The assay is economical, requiring only low nanomolar concentrations of biotinylated AF4-derived peptide and FLAG-tagged AF9 in low-volume 384-well plates. A Z′-factor of 0.71 and a signal-to-background ratio of 21.3 showed the assay to be robust, and sensitivity to inhibition was demonstrated with competing AF4-derived peptides. Two pilot screens comprising 5,680 compounds served as validation for HTS at Nemours and the Broad Institute. Assay artifacts were excluded using a counterscreen comprising a biotinylated FLAG peptide. This is the first reported HTS-compatible assay to identify compounds that inhibit a key binding interaction of an MLL fusion partner, and the results presented here demonstrate suitability for screening large chemical libraries in high-density, low-volume plate formats.
Introduction
As part of the Nemours Center for Childhood Cancer Research, the High-Throughput Screening and Drug Discovery Lab is focused on the discovery of novel chemical probes to explore the ways in which the biology of pediatric cancer differs from that of adult malignancy; our ultimate goal is to develop targeted therapeutics for these devastating diseases. In the past 30 years, few new anticancer agents have been approved for use in pediatric patients, and survival in very high-risk patient groups remains poor despite intensive cytotoxic therapies. Rationally designed drugs have benefited adults, but with the exception of BCR-ABL-positive leukemias, targeted therapies have so far done little to improve pediatric outcomes.1 Recently, research into the mechanistic basis for pediatric cancer has begun to uncover defined molecular targets, raising the possibility of treatment by specifically designed drugs.
A major focus of our pediatric cancer research is leukemia resulting from specific chromosomal rearrangement of the mixed-lineage leukemia (MLL) gene. MLL-rearranged (MLL-R) leukemia is a particularly aggressive cancer that occurs predominately in children. MLL rearrangement is found in 2–8% of pediatric acute lymphoblastic leukemia (ALL) and 10–20% of acute myeloid leukemia (AML), including up to 80% of infant ALL and therapy-related secondary AML. While other types of childhood acute leukemia boast event-free survival rates approaching 80–90%, the prognosis for children suffering from MLL-R leukemia is particularly dismal.2 For infants in particular, conventional chemotherapy regimens often fail, leading to a five-year event-free survival rate of only 34%.2,3 Thus, specifically targeted novel therapies are urgently needed.
MLL is a large multidomain protein commonly expressed in hematopoietic cells. In normal cells, MLL, a histone methyltransferase, is responsible for methylating the lysine 4 residue on histone 3 (H3K4) and regulating the expression of genes that have a crucial role in the development of the hematopoietic system. In leukemic cells, chromosomal rearrangements lead to loss of the C-terminal methyltransferase domain of MLL, giving rise to oncogenic fusion proteins comprising the N-terminus of MLL linked to one of more than 60 partner proteins (Fig. 1A).4 MLL fusions are potent inducers of leukemia; through mechanisms that are still under investigation, the fusion proteins cause abnormal expression of the MLL target genes, notably the homeobox (HOX) genes.5–7 The HOX genes regulate normal hematopoietic cell differentiation and must be appropriately induced and suppressed at different stages in blood development to ensure normal hematopoiesis. Overexpression of the oncogene HOXA9 in particular is repeatedly found in MLL-R leukemia. A loss of the control of gene expression at a critical stage in hematopoietic development blocks differentiation of hematopoietic cell progenitors, which acquire the capacity for unlimited self-renewal, leading to malignant transformation.2,7
Fig. 1.

MLL-AF4–AF9 binding interaction. (A) Schematic of MLL wild type, MLL fusion, AF4, and AF9. The truncation of MLL in MLL fusions results in the loss of the plant homology domain (PHD), transactivation domain (TAD), and Su-var3-enhancer of zeste (SET) domain (which is responsible for methyltransferase activity); all 3 are important in transcriptional activation. In contrast, MLL fusions retain the AT-hook and CXXC domains of MLL and consequently DNA-binding activity. The AF4 fusion to MLL comprises ∼850 amino acids from the carboxy terminus, including an AF9 interaction domain. Chromosomal breakpoints for cell lines carrying the t(4;11) translocation are indicated. The ANC1 homology domain (AHD) of AF9 binds multiple transcription factors, including AF4. Figure not drawn to scale. (B) AlphaScreen assay to screen for inhibitors of AF4-AF9 binding. Binding of a biotinylated AF4 peptide to FLAG-tagged AF9 protein is detected by the addition of streptavidin-coated donor beads and anti-FLAG-coated acceptor beads. If peptide and protein are bound, laser excitation of the donor beads results in singlet oxygen (1O2) transfer to the acceptor beads and light emission. If a small-molecule inhibitor disrupts the peptide–protein binding, singlet oxygen transfer fails to occur due to the increased distance between the donor and acceptor beads. Thus, inhibitor binding is detectable by a decrease in light emission. MLL, mixed-lineage leukemia.
Recent studies have revealed that MLL fusions affect gene expression by recruiting a complex of proteins, including several transcription factors and the histone methyltransferase DOT1L, which regulate the activity of RNA polymerase II during transcriptional elongation.2,8,9 Therefore, disruption of one or more of the key protein–protein interactions within the transcriptional elongation complex may block MLL-R leukemia and restore normal hematopoietic differentiation. Although numerous fusion partners for MLL have been discovered, five transcription factors account for 80% of MLL fusions. MLL-AF4 is the most common fusion; in infants, it alone accounts for half of the leukemia cases, and is associated with the worst prognosis.10,11 We decided to focus our initial probe and drug discovery efforts on MLL-AF4 due to its importance in high-risk pediatric leukemia and based on published work validating the interaction of MLL-AF4 and the transcription factor AF9 as a potentially important target. Hemenway and coworkers found that the direct interaction between AF4 and the transcription factor AF9 is required for proliferation and survival of leukemic cell lines harboring the MLL-AF4 fusion.12,13 Yeast two-hybrid assays identified a 12-amino-acid sequence in AF4 that binds to the C-terminus of AF9. They also reported that a 10-amino acid peptide sequence derived from the AF9-binding site of AF4 was sufficient to inhibit binding of AF9 to AF4 with a single-digit nanomolar half-maximal inhibition concentration (IC50) potency in an enzyme-linked immunosorbent assay. Moreover, a cell-permeable penetratin-containing peptide (penetratin-LWVKIDLDLLSRV) was shown by fluorescence microscopy to disrupt intracellular AF4-AF9 binding. This cell-penetrating peptide caused leukemia cell lines harboring the MLL-AF4 fusion to undergo cell death; it was not toxic to normal hematopoietic cells.13,14 Further studies demonstrated synergism between the AF9-binding peptide and conventional chemotherapeutic agents in the selective killing of leukemia cells containing MLL-AF4.14,15
The peptide work of Hemenway and coworkers demonstrates that targeting the AF4-AF9 interaction could be a viable therapeutic strategy against leukemias harboring MLL-AF4 fusions and provides proof of principle for our small-molecule drug discovery efforts. The relatively small size of the peptide that inhibits the AF4-AF9 binding interaction suggests that it should be possible to identify small nonpeptidic AF9 antagonists.16 To this end, we have designed a high-throughput screening (HTS) assay for the use at Nemours and transfer to the Broad Institute for screening of the Molecular Libraries Small Molecule Repository (MLSMR) collection to identify compounds that disrupt the binding interaction between AF9 and AF4. Herein, we describe the development of a method that uses AlphaScreen™ (Perkin Elmer, Waltham, MA) to measure binding between full-length AF9 and an AF4-derived peptide. Further, we validate its suitability for large-scale HTS and report assay performance in 2 pilot screens comprising a total of 5,680 compounds.
Materials and Methods
Reagents
Potassium phosphate monobasic, potassium phosphate dibasic, sodium chloride (NaCl), and Tween-20 were obtained from Fisher Chemicals (Waltham, MA). Phosphate-buffered saline (PBS; pH 7.4) was made up to a final concentration of 1.47 mM potassium phosphate monobasic, 4.3 mM sodium phosphate dibasic, 2.7 mM potassium chloride, and 137 mM NaCl. Gray, 384-well AlphaPlates SW, 7.5% bovine serum albumin (BSA), and AlphaScreen FLAG detection kit (Catalog no. 6760613C) containing anti-FLAG-coated acceptor beads and streptavidin donor beads were purchased from Perkin Elmer. Biotin-FLAG peptide used in the counterscreen was from Perkin Elmer or Biomatik (Wilmington, DE; sequence: Biotin-GGSGGSGGSGGSGGSGGDYKDDDDK). The N-terminal biotinylated AF4 27-mer (residues 748–773), representing the AF9-binding region of AF4 (Fig. 1A), and nonbiotinylated peptides (Table 1) were from Biomatik. We chose to use these peptides to replicate the results found by Srinivasan et al.13 The AF9 protein possessing a C-terminal FLAG tag was custom-expressed and purified by Origene (Rockville, MD). Corning 3095 microplate sealing tape was obtained from Fisher Scientific (Morris Plains, NJ).
Table 1.
AF4 Peptide Sequences
| Peptide | Sequencea |
|---|---|
| Biotin-AF4 27-merb | Biotin-LSPLRDTPPPQSLMVKITLDLLSRIPQ-OH |
| AF4 27-mer | NH2-LSPLRDTPPPQSLMVKITLDLLSRIPQ-OH |
| 8-merc | NH2-WVKIDLDL-OH |
| 10-mer | NH2-LWVKIDLDLL-OH |
| 12-mer | NH2-LWVKIDLDLLSR-OH |
| Inactive 12-merd | NH2-LWEKSDLDLLSR-OH |
aResidues key to AF9 binding are in bold type.
bThe biotinylated and nonbiotinylated AF4 27-mer peptides are derived from residues 747–774 of AF4.
cThe 8, 10, and 12-mer peptides are derived from the homolog FMR2, which is closely related to AF4 and shares the AF9 interaction domain. We chose to use these peptides to replicate the results found by Srinivasan et al.13
dUnderline indicates substituted amino acids.
Compound Libraries
The Spectrum Collection compound library containing 2,000 U.S. Food and Drug Administration–approved drugs and bioactive natural products at 10 mM concentration in dimethyl sulfoxide (DMSO) was obtained from MicroSource, Inc. (Gaylordsville, CT).17 The library was reformatted into polypropylene V-bottom 384-well microplates obtained from Greiner BioOne (Catalog no. 781280; Monroe, NC) at 4 mM in DMSO.
The Natural Products Library (NPL) and Natural Derivatives Library (NDL) were obtained from TimTec (Newark, DE).18 The NPL contained 680 natural products, and the NDL contained 3,000 synthetic compounds whose design is based on natural product–derived scaffolds. The compounds from these libraries at 10 mM in DMSO were reformatted from 96- to 384-well plates and diluted to 2 mM with DMSO in polypropylene round-bottom plates from Nalge Nunc International (Catalog no. 264573; Rochester, NY).
Assay Development and Optimization
For initial assay development, we performed a cross-titration of AF9-FLAG and biotin-AF4 27-mer. AF9-FLAG (3 μL) and biotin-AF4 27-mer (3 μL) were added to the wells containing 4 μL water/0.01% Tween-20. Final concentrations were 0–100 nM AF9-FLAG and biotin-AF4 27-mer in 10 μL of 1× PBS (pH 7.4) containing 0.1% BSA, and 0.01% Tween-20. The resulting mixtures were incubated for 90 min at room temperature. Anti-FLAG acceptor beads and streptavidin-conjugated donor beads (120 μg/mL each) were then added simultaneously in 2 μL of assay buffer and incubated for 60 min. All incubations after bead addition were protected from light. AlphaScreen signals were read on a Perkin Elmer Envision Multilabel Plate Reader 2104 equipped with the AlphaScreen optical detection module using standard settings (excitation: 680 nm; emission: 570 nm). For all subsequent experiments, the following assay conditions were used: 2.4 nM AF9-FLAG and 2.4 nM biotin-AF4 27-mer in 10 μL assay buffer, followed after 90 min by the addition of 2 μL of 120 μg/mL donor and acceptor beads, to give a final bead concentration of 20 μg/mL in 12 μL.
Once optimal concentrations of peptide and AF9 were determined, two timecourse experiments were performed. The kinetics of the interaction between AF9 and biotin-AF4 27-mer were assessed by preincubation of biotin-AF4 27-mer and AF9 together (2.4 nM each) in 10 μL assay buffer for 0, 15, 30, 60, 90, 120, 180, and 240 min. Donor and acceptor beads (120 μg/mL) in 2 μL of assay buffer were then added simultaneously, the mixture incubated for an additional 60 min, and AlphaScreen signals read. The time course for bead capture was determined by preincubating biotin-AF4 27-mer and AF9-FLAG for 90 min in 10 μL assay buffer, and varying the incubation time after bead addition. Donor and acceptor beads (120 μg/mL) were added simultaneously in 2 μL of assay buffer, the mixture incubated for 0, 30, 60, 90, or 120 min, and then AlphaScreen signals read.
Optimization of the amount of AlphaScreen beads to reduce the cost per well while maintaining adequate standard deviation, Z′-factor and signal-to-background ratio (S/B) were also performed. Dilutions of AF9 and biotin-AF4 27-mer in 10 μL assay buffer were incubated for 90 min, after which varying amounts of beads (20, 15, 10, 12, 8, or 5 μg/mL final each) were added in 2 μL assay buffer. The mixture was incubated for 60 min, after which AlphaScreen signals were read.
The order of addition was also tested in four sequences: (A) biotin-AF4 27-mer preincubated for 90 min with AF9-FLAG, followed by the addition of a mixture of both beads and additional 60-min incubation; (B) biotin-AF4 27-mer preincubated with AF9-FLAG for 90 min, followed by addition of acceptor beads and 60-min incubation, then addition of donor beads, and a final 60-min incubation before reading; (C) AF9-FLAG preincubated with beads for 60 min, followed by addition of biotin-AF4-27mer, and a second incubation of 90 min; and (D) biotin-AF4 27-mer, AF9-FLAG, and beads added simultaneously and incubated for 90 min before reading (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/adt).
DMSO compatibility was evaluated by adding biotin-AF4 27-mer (in 3 μL) and AF9-FLAG (in 3 μL) to wells containing increasing concentrations of DMSO in 4 μL water/0.01% Tween-20. Biotin-AF4 27-mer and AF9-FLAG were incubated for 90 min at room temperature, followed by a 60-min bead incubation. The AF4-AF9 binding signal in the absence of DMSO (100%) was compared to the signal generated in the presence of DMSO. Final concentrations in the 12-μL assay were 2 nM biotin-AF4 27-mer, 2 nM AF9-FLAG, and 20 μg/mL each donor and acceptor beads in the assay buffer.
Saturation Binding of Biotin-AF4 27-mer to AF9-FLAG
AF9-FLAG (in 3 μL assay buffer) was added to wells containing 7 μL of a series of concentrations of biotin-AF4 27-mer in assay buffer. Final concentrations of AF9-FLAG and biotin-AF4 27-mer were 0.12 nM and 0.06–12 nM (in 10 μL), respectively. Background wells contained 0.12 nM AF9-FLAG, increasing concentrations of biotin-AF4 27-mer, and 300 nM nonbiotinylated AF4 27-mer. This concentration of nonbiotinylated AF4 27-mer is sufficient to completely block the interaction of biotin-AF4 27-mer with AF9-FLAG, ensuring that the background wells measure only nonspecific binding and background counts. The binding partners were incubated for 90 min at room temperature, followed by the addition of anti-FLAG acceptor beads and streptavidin donor beads (120 μg/mL) in 2 μL assay buffer (final concentration 20 μg/mL each). After incubation in the dark at room temperature for 60 min, the AlphaScreen signal was measured on an Envision plate reader. Background signals in the presence of nonbiotinylated AF4 27-mer were subtracted at each concentration of biotin-AF4 27-mer to obtain the corrected value. The equilibrium dissociation constant (Kd) for biotin-AF4 27-mer binding to AF9-FLAG was determined using a nonlinear regression fit of the corrected data to a one-site binding model in GraphPad Prism.
Competition with Nonbiotinylated AF4 Peptide
Nonbiotinylated AF4-derived peptides of varying lengths (Table 1) were used as control inhibitors to verify that competitive inhibition of the interaction between biotin-AF4 27-mer and AF9-FLAG could be detected. A 14-point concentration–response curve was prepared for each nonbiotinylated AF4 peptide using semilog dilution in water/0.01% Tween-20. Each concentration of peptide (in 4 μL of water/0.01% Tween-20) was added to a low-volume 384-well AlphaPlate. The remainder of the assay was performed as described in Steps 3–8 in Table 2. The final concentrations of a nonbiotinylated AF4 peptide in the 10 μL AF4-AF9 binding assay were 3.6 pM–3.6 μM. In the peptide competition assay, the control (equivalent to highest signal) refers to biotin-AF4 27-mer and AF9 in the absence of the nonbiotinylated peptide. The background (equivalent to lowest signal) refers to biotin-AF4 27-mer in the absence of AF9.
Table 2.
AF4-AF9 Binding Assay Protocol
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Water/0.01% Tween-20 | 4 μL | Add to 384-well plate |
| 2 | Compound in DMSO | 50 nL | Pintool transfer compound into water |
| 3 | Biotin-AF4 27-mer peptide | 3 μL | Tip transfer on JANUS |
| 4 | AF9-FLAG | 3 μL | Tip transfer on JANUS |
| 5 | Incubation time | 90 min | At room temperature |
| 6 | Anti-FLAG beadsStreptavidin beads | 2 μL | BioTek reagent dispenser or tip transfer on Janus |
| 7 | Incubation time | 60 min | Plates maintained in low light at room temperature |
| 8 | AlphaScreen readout | cps | Perkin Elmer EnVision® Multilabel Reader with standard AlphaScreen option (Em=570 nm) |
Step Notes
1. Water with 0.01% Tween-20 (or 750 nM unlabeled AF4 27-mer for low-control wells) added by BioTek reagent dispenser during the MicroSource collection screen and added by tip transfer on the Janus during the TimTec screen. The switch from BioTek to Janus reagent dispensing resulted in improved assay statistics (Table 3).
3. Added 8 nM biotin-AF4 27-mer peptide in 2.3×PBS buffer. Final concentration 2.4 nM in 10 μL assay volume before bead addition.
4. Added 8 nM FLAG-AF9 in 1× PBS buffer. Final concentration 2.4 nM in 10 μL assay volume before bead addition. AF9 is a highly disorganized protein prone to aggregation. It is added last because it is stabilized by binding to AF4.
5. Plates were sealed during the incubation period.
6. Beads were premixed and added simultaneously in 1× buffer under subdued lighting. For the MicroSource collection screen, beads (120 μg/mL each, final concentration 20 μg/mL in 12 μL) were added by reagent dispenser. For the TimTec screen, beads (48 μg/mL each, final concentration 8 μg/mL in 12 μL) were added by tip transfer on the Janus.
7. Plates were sealed during the incubation period.
DMSO, dimethyl sulfoxide; PBS, phosphate-buffered saline.
MicroSource Spectrum Collection Screen
Table 2 describes the step-by-step screening protocol. The MicroSource Spectrum collection was screened for inhibition of the AF4-AF9 binding interaction at a single concentration (20 μM); no replicates were performed. Compounds were diluted to 4 mM in DMSO, and 50 nL was transferred using a Janus MDT Automated workstation (Perkin Elmer) fitted with a hydrophobic pintool (V & P Scientific, San Diego, CA) into assay plates containing 4 μL of water/0.01% Tween-20. Biotin-AF4 27-mer (3 μL in 2.3× assay buffer) and AF9-FLAG (3 μL in 1× assay buffer) were added using a Janus MDT Automated workstation. Final concentrations for the AF4-AF9 binding step were 20 μM test compound, 2.4 nM AF9-FLAG, and 2.4 nM biotin-AF4 27-mer in 10 μL of assay buffer. The plates were incubated at room temperature for 90 min, followed by the addition of a mixture of anti-FLAG-coated acceptor and streptavidin-conjugated donor beads (120 μg/mL each in 2 μL) to give a final concentration of 20 μg/mL in 12 μL total volume. Assay plates were incubated at room temperature for an additional 60 min under subdued lighting; high levels of ambient light can result in a significant decrease in signal. AlphaScreen signals were measured on the Envision plate reader.
Each plate contained 32 replicates of controls (columns 1 and 23) and background (columns 2 and 24). The control wells (100% signal, 0% inhibition) consisted of 2.4 nM biotin-AF4 27-mer and 2.4 nM AF9-FLAG; the background wells (0% signal, 100% inhibition) also contained 300 nM inhibitor (nonbiotinylated AF4 27-mer peptide). This inhibitor concentration was chosen from a dose–response curve (shown in Fig. 5) as the concentration that completely abolishes signal. Concentrations are based on a volume of 10 μL in the AF4-AF9 binding assay before the addition of beads. Assay plates (containing compounds) were sandwiched between quality control (QC) plates that consisted of 16-point serial dilutions of the nonbiotinylated AF4 27-mer (5 μM–3 pM) in the presence of biotin-AF4 27-mer and AF9-FLAG.
Fig. 5.

Competition of nonbiotinylated AF4 peptides with biotin–AF4 27-mer for binding to the AF9-FLAG protein. Nonbiotinylated AF4 peptides of varying lengths were tested for inhibition of binding between biotin-AF4 27-mer peptide and AF9-FLAG. The unlabeled peptides in the order of potency were AF4 27-mer (●), 12-mer (■), and 10-mer (▲). The 8-mer (♦) and inactive 12-mer (□) showed no inhibition of binding. Values=mean±SEM; n=3.
Compounds selected as actives in the initial screen at 20 μM were re-evaluated in dose–response in triplicate. A range of concentrations (10 mM–305 nM) of each compound of interest was prepared by 16-point 2-fold serial dilution in DMSO. Each compound dilution was added by 50 nL pintool, and the assay was performed under identical conditions to the initial pilot screen. The final concentration range for compounds in the assay was 50 μM–1.5 nM. Serially diluted nonbiotinylated AF4 27-mer was added by pintool to columns 3 and 22 of each plate as a QC (final concentration: 3 fM–500 nM in the 10 μL binding assay).
Biotin–FLAG Counterscreen
Actives from the initial screen were also counterscreened in parallel with the binding assay, as dose–response (in triplicate) using a biotin–FLAG peptide (Perkin Elmer) in place of biotin-AF4 27-mer and AF9-FLAG as the linker between anti-FLAG acceptor beads and streptavidin-coated donor beads. Any compounds that caused a decrease in counterscreen signal were considered artifacts that interfere with either AlphaScreen signal generation or bead capture. The same set of compounds that were serially diluted for dose–response confirmation in the AF4-AF9 binding assay were delivered by pintool and assayed under identical conditions to the binding assay. In place of biotin-AF4 27-mer peptide and AF9, 6 μL of biotin–FLAG peptide in 1.67× assay buffer was added to give a final concentration of 7.5 nM in 10 μL. This concentration provided a signal equivalent to the highest signal observed for the AF4-AF9 assay. Control (100% signal) comprised 7.5 nM biotin–FLAG in the absence of compounds; for the background (0% signal), the buffer was substituted for biotin–FLAG.
TimTec NPL and NDL Collection Screen
Compounds were screened, and actives were confirmed at 10 μM under identical conditions to the MicroSource pilot screen. QC plates containing 320 wells of nonbiotinylated AF4 27-mer (14 nM, final concentration before bead addition) in the presence of biotin-AF4 27-mer and AF9-FLAG were included. Active compounds were retested in triplicate at a single concentration (10 μM in a 10-μL assay volume) in the AF4-AF9 binding assay and the counterscreen assay using biotin-FLAG from Perkin Elmer. Compounds confirmed as active in the AF4-AF9 binding assay and inactive in the counterscreen were further evaluated in dose–response in both assays as described for the MicroSource Spectrum collection screen. The counterscreen in dose–response was performed using biotin–FLAG from Biomatik.
Data Analysis
AlphaScreen signals were converted to percent activity (Eq. 1) for dose–response data analysis, and further to percent inhibition (Eq. 2) for analysis of primary screening data by normalizing to plate controls.
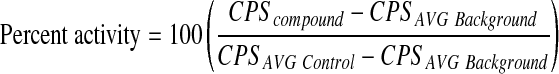 |
(1) |
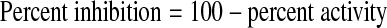 |
(2) |
S/B (Eq. 3) and Z′-factors (Eq. 4), measures of assay performance, were determined using equations previously described.19
 |
(3) |
 |
(4) |
where μ and σ represent the mean and standard deviation, respectively. Dose–response curves and IC50s were generated using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA).
Results
Development of 384-Well–Format AlphaScreen Assay
This study aimed to establish a high-throughput assay to screen for inhibitors of the interaction between the transcription factors AF4 and AF9. To this end, we designed an assay that measures binding between a biotinylated peptide derived from the AF9-binding site of AF4 (biotin-AF4 27-mer; Table 1, key binding residues in bold type) and FLAG-tagged AF9 protein (full-length). The protein–peptide interaction is detected by AlphaScreen upon the addition of streptavidin-coated donor beads and anti-FLAG-coated acceptor beads (Fig. 1B).
AF9-FLAG and biotin-AF4 27-mer were cotitrated to establish the optimal concentrations and determine assay sensitivity (Fig. 2). Maximal signal (known as the hook point20,21) was reached at 36 nM biotin-AF4 peptide or 12 nM AF9-FLAG, respectively, in the presence of a fixed concentration of the other binding partner, above which bead saturation results in decreased signal. Background remained constant in both biotin-AF4 peptide and AF9 titrations, while S/B increased up to the hook point. For subsequent assays, 2.4 nM each of biotin-AF4 27-mer and AF9-FLAG was used, because these concentrations provided a robust signal and are well below the binding capacity of the beads. (The theoretical binding capacity for 20 μg/mL beads is reported by Perkin Elmer to be 100 nM and 30 nM, for anti-FLAG acceptor and streptavidin donor beads, respectively. However, because the size and affinity of the binding partners influence the bead-binding capacity, it is more appropriate to rely upon the experimental determination of the hook point as a measure of bead saturation for a particular assay.22)
Fig. 2.

AF9 protein and biotin-AF4 27-mer titrations. (A) Titration of biotin-AF4 27-mer against a fixed concentration of AF9-FLAG (3.6 nM) gave a maximal signal at 36 nM biotin-AF4 27-mer. Above 36 nM, the so-called hook point, a decrease in signal and S/B was observed consistent with bead saturation. (B) Titration of AF9-FLAG against a fixed concentration of biotin-AF4 27-mer (3.6 nM) gave a maximal signal and S/B at 12 nM AF9-FLAG. Values=mean±SEM; n=3. S/B, signal-to-background ratio.
The kinetics of the interaction between AF9 and biotin-AF4 27-mer were also assessed. Timecourse experiments demonstrated that equilibrium binding between biotin-AF4 27-mer and AF9 was reached after 90 min (Fig. 3A). Since the length of bead incubation and order of reagent addition can have a significant impact on assay sensitivity, these parameters were also investigated (Supplementary Fig. S1 summarizes the different order of addition protocols). Maximal signal was achieved when biotin-AF4 27-mer and AF9-FLAG were allowed to preincubate for 90 min, followed by a 60-min incubation with a mixture of the anti-FLAG and streptavidin beads (Fig. 3B). A drop in signal was observed when the beads were added immediately to AF9-FLAG before addition of biotin-AF9 27-mer, or when biotin-AF4 27-mer and AF9-FLAG were combined simultaneously with anti-FLAG and streptavidin beads (Fig. 3C). After optimization of incubation periods and order of addition, a saturation binding experiment was performed. Binding of biotin-AF4 27-mer to AF9-FLAG was saturable with an apparent Kd of 0.52 nM (95% confidence interval: 0.41–0.63 nM; Fig. 3D). A similar high affinity (Kd=0.17 nM) was reported by Leach et al.,23 using fluorescence anisotropy to measure binding between a slightly shorter AF4 peptide (residues 758–778) and the ANC1 homology domain of AF9 (residues 490–568) fused to a maltose-binding protein.
Fig. 3.

Assay development and optimization. (A) Time course for biotin-AF4 27-mer peptide binding to AF9-FLAG protein was determined by preincubating the peptide and protein for the times shown, followed by incubation with the acceptor and donor beads for 1 h. (B) Time course for bead capture was determined by preincubating biotin-AF4 27-mer and AF9-FLAG for 90 min, and varying the incubation time after bead addition. (C) The order of addition was tested in 4 sequences: (A) biotin-AF4 27-mer preincubated for 90 min with AF9-FLAG, followed by the addition of a mixture of both beads and additional 60 min incubation, (B) biotin-AF4 27-mer preincubated with AF9-FLAG for 90 min, followed by addition of acceptor beads and 60-min incubation, then addition of donor beads, and a final 60-min incubation before reading, (C) AF9-FLAG preincubated with beads for 60 min, followed by addition of biotin-AF4 27-mer, and a second incubation of 90 min; and (D) biotin-AF4 27-mer, AF9-FLAG, and beads added simultaneously and incubated for 90 min before read. (D) The dissociation constant was determined for the peptide–protein interaction using 0.12 nM AF9 and 0.06–12 nM biotin-AF4 27-mer in 10μL prior to bead addition. Background wells also included 300 nM nonbiotinylated AF4 27-mer. Values from background ranged from 320 to 650 cps and were subtracted from the totals to get the corrected value. Measurements were made in triplicate, and the mean±SD (n=3) are plotted. Plots show values after background subtraction. The apparent Kd was calculated as 0.52 nM. (E) DMSO compatibility was evaluated by incubating biotin-AF4 27-mer and AF9-FLAG in the presence of varying concentrations of DMSO for 90 min, followed by 60-min bead incubation. The AF4-AF9 binding signal in the presence of DMSO was expressed as a percentage compared to the signal generated in the absence of DMSO. For panels A–E, the final concentrations in the 12-μL assay were 2 nM Biotin-AF4 27-mer, 2 nM AF9-FLAG, and 20 μg/mL each of donor and acceptor beads in the assay buffer. In all graphs (unless otherwise noted), values=mean±SD; n=6; and arrows indicate final assay conditions.
The assay buffer was optimized to increase the S/B. AF4-AF9 binding was evaluated in Tris, phosphate, and HEPES buffers from pH 7–9. Buffers with high- and low-salt conditions were also investigated (data not shown). PBS (pH 7.4) was ultimately determined to provide the most robust conditions. BSA (0.1%) and 0.01% Tween-20 were included in the buffer to minimize nonspecific binding and reagent or compound aggregation, respectively. Additionally, since compound libraries are stored in DMSO, we determined the compatibility of DMSO with the assay. AF4-AF9 binding signals were unaffected by up to 0.5% DMSO (Fig. 3E).
To explore ways to decrease reagent costs, we evaluated the effect of reducing the final concentration of AlphaScreen beads below the 20 μg/mL recommended by Perkin Elmer. Figure 4A shows that the magnitude of both control and background signals was reduced at lower bead concentrations, resulting in no overall change in S/B. Calculation of Z′-factors at bead concentrations of 8 and 20 μg/mL showed an improvement in assay robustness at the lower bead concentration (Fig. 4B). These two different bead concentrations were further evaluated during pilot screening; the MicroSource library was screened using 20 μg/mL beads, whereas the TimTec library was screened using 8 μg/mL (see Screen of the TimTec NPL and NDL Collections).
Fig. 4.

Optimal bead concentrations for AF4-AF9 binding assays. (A) To determine optimal bead concentrations, biotin-AF4 27-mer and AF9-FLAG (2.4 nM each) were preincubated for 90 min, followed by 60-min incubation with varying concentrations of AlphaScreen beads. Controls (●) consisted of biotin-AF4 27-mer and AF9-FLAG; background (■) also included 300 nM nonbiotinylated AF4 27-mer. Values=mean±SD; n=3. (B) AF4-AF9 AlphaScreen control signals for 8 and 20 μg/mL bead concentrations in a total 12-μL assay volume. Biotin-AF4 27-mer and AF9-FLAG (2.4 nM each) were preincubated for 90 min, followed by 60-min incubation with AlphaScreen beads. Controls are shown in gray and background shown in black. Values=mean±SD; n=24.
Finally, stability studies were undertaken to ensure that beads, biotin-AF4 27-mer, and AF9-FLAG would be stable for the extended periods of time necessary for liquid-handling protocols. Each of the reagents in turn was allowed to stand at room temperature for 0–160 min after which the AF4-AF9 binding assay was run under standard conditions. There was no observed loss in activity for any of the reagents up to 160 min (data not shown).
Competition Experiments with Nonbiotinylated AF4 Peptides
To demonstrate specificity and reversibility of AF4 peptide binding to AF9, we performed a competition experiment with nonbiotinylated peptides of varying lengths (Fig. 5 and Table 1). Nonbiotinylated AF4 27-mer effectively competed with biotin-AF4 27-mer. Applying a sigmoidal dose–response model gave an IC50 of 2.7 nM. As the length of the nonbiotinylated AF4 peptide decreased, the IC50 increased: a 12-mer peptide gave an IC50 of 5.8 nM, and a 10-mer peptide gave an IC50 of 13.2 nM, whereas truncating the sequence to 8 amino acids abolished activity entirely. Changing only two residues of the 12-amino-acid peptide also completely abolished activity. This potency ranking correlates well with the data reported by Srinivasan et al.13
To evaluate the reproducibility of the assay response to inhibition between plates and from one screening run to the next, QC plates were placed at the beginning and end of each screening run. During pilot screening of the MicroSource library, QC plates were set up containing 16-point serial dilutions of nonbiotinylated-AF4 27-mer in the presence of biotin-AF4 27-mer and AF9-FLAG. Serially diluted nonbiotinylated AF4 27-mer (500 nM–3 fM) was also included in columns 3 and 22 of each MicroSource dose–response plate as an internal QC. Figure 6 shows high concordance between the IC50 values observed in a series of 9 MicroSource dose–response plates. The control titration of the nonbiotinylated AF4 inhibitor on these plates yielded an interplate average IC50 of 3.23±1.5 nM and a minimum significant ratio (MSR) of 1.62, indicating a highly stable run according to the definition given by Eastwood and coworkers.24 (One is the best possible ratio. An MSR of <4 suggests an assay with high test-to-test dose–response reproducibility). During TimTec screening, assay plates were sandwiched between QC plates containing 320 wells of 27-mer peptide at a single concentration expected to give 70–80% inhibition based on the calculated IC50 (Supplementary Fig. S2).
Fig. 6.

Plate-to-plate variability. Graph compares results for a 16-point dose–response quality control that was included in columns 3 and 22 of all 9 MicroSource dose–response plates (n=18). These wells contained 2.4 nM biotin-AF4 27-mer, 2.4 nM AF9-FLAG, and 500 nM–3 pM nonbiotinylated AF4 27-mer. Data points represent a single IC50 determination. Solid line represents mean IC50, and dashed lines represent 1 SD from the mean.
Screen of the MicroSource Spectrum Collection
To validate that the AF4-AF9 binding assay was suitable for HTS, a pilot screen was undertaken using an assay protocol adapted for automated liquid handling (Table 2). The MicroSource Spectrum library of 2,000 drugs and natural products was screened for inhibitors of the AF4-AF9 interaction at 20 μM. The assay performed adequately over the 8-plate screen giving a median Z′-factor of 0.53 (Table 3). The median percent coefficient of variation (CV) for the controls within each of the eight plates was 13.8% with an S/B of 19. Based on the CV for the controls (0% inhibition) averaged across all eight plates (14.5%), the threshold for hit selection was set at 45% inhibition. Inhibition values >45% were considered to be statistically significant, since they differ from the mean control by >3 standard deviations. Forty-five compounds met this threshold (2.25% hit rate) and were retested for AF4-AF9 inhibition in dose–response using 2-fold serial dilutions from 50 μM down to 1.5 nM. These hits were also tested in a parallel counterscreen in which biotin-AF4 27-mer and AF9-FLAG were replaced by a biotin–FLAG peptide. Compounds active against both the AF4-AF9 screen and the biotin–FLAG counterscreen were considered artifacts that interfered with bead capture or AlphaScreen signal generation. Thirty-six compounds gave an IC50<50 μM against AF4-AF9. Of those compounds, 13 were inactive (<20% inhibition at 50 μM) in the biotin–FLAG counterscreen, yielding a confirmed hit rate of 0.65% (Fig. 7).
Table 3.
Summary Statistics for Pilot Screen with MicroSource and TimTec Libraries
| MicroSource | TimTec | |
|---|---|---|
| Number of plates | 8 | 15 |
| High controls | ||
| Median | 22,058 | 19,005 |
| Range | 19,836–23,439 | 16,673–22,346 |
| Median CV | 13.8% | 8.4% |
| CV range | 10.1–19.5% | 6.4–12.6% |
| Low controls | ||
| Median | 1,125 | 892 |
| Range | 1,049–1,306 | 775–1,111 |
| Median CV | 16.9% | 11.5% |
| CV range | 12.1–22.6% | 7.5–19.1% |
| Z′-factor | ||
| Median | 0.53 | 0.71 |
| Range | 0.36–0.68 | 0.59–0.78 |
| S/B | ||
| Median | 19.0 | 21.2 |
| Range | 17.5–20.8 | 16.0–22.0 |
The table above represents the median and range for the average plate control cps for a total of n=8 (MicroSource) or 15 plates (TimTec).
CV, coefficient of variation; S/B, signal-to-background ratio.
Fig. 7.

Summary schematic for hit identification process. MicroSource and TimTec libraries were screened using the AlphaScreen AF4-AF9 binding assay. After counterscreen and evaluation by dose–response, we identified 18 confirmed hits (one of which was independently identified in both libraries).
Screen of the TimTec NPL and NDL Collections
Based on our experience screening the MicroSource collection, several parameters were changed for screening of the TimTec collection of 3,680 natural product and natural product-derived synthetic compounds in the AF4-AF9 binding assay. To improve CVs and Z′-factors, we changed the instrument used for reagent addition and further optimized the Envision plate reader for AlphaScreen detection. In the MicroSource collection screen, a BioTek reagent dispenser was used to add water/Tween before compound delivery and to dispense the AlphaScreen beads after peptide–protein binding. For lower-volume dispenses (0–5 μL), we consistently found that CVs were better with the Janus compared to the BioTek (Table 3). Consequently, for the TimTec screen, all reagents were added using disposable tips on a Janus MDT Automated workstation. We also reduced the final concentration of AlphaScreen beads in the assay from 20 to 8 μg/mL. This change was primarily intended to save reagent costs, but our TimTec screening results appeared to confirm our earlier finding that lowering the bead concentration improved the Z′-factor (Fig. 4B). Overall assay statistics for the TimTec screen were considerably improved over the MicroSource collection screen (Table 3 and Fig. 8). We confirmed that changing the bead concentration did not affect the assay response by comparing all dose–response curves for nonbiotinylated AF4 27-mer obtained during the MicroSource and TimTec screens. We observed no significant difference in IC50s between the two different bead concentrations (Supplementary Fig. S3).
Fig. 8.

TimTec screening plate controls. Controls (●) consisted of 2.4 nM biotin-AF4 27-mer and 2.4 nM AF9-FLAG. Background (■) also contained 300 nM nonbiotinylated AF4 27-mer. Values=mean±SD for each plate in the screening run. Z′-factors are depicted by open circles (○).
After encountering a high initial hit rate of 2.25% in the MicroSource screen and attrition of >60% of these hits after counter-screening to eliminate artifacts, we modified our strategy during the TimTec screen to exclude the hits that interfere with assay detection. Rather than retesting all initial hits in dose–response, we immediately followed the single-point screen at 10 μM with retesting of hits at 10 μM in triplicate in both the AF4-AF9 screen and the Biotin-FLAG counterscreen. Following the same strategy used for the MicroSource library and screening the 131 hits from the TimTec library in dose–response for both the AF4-AF9 screen and Biotin-FLAG counterscreen would have required 48 plates. With the modified screening strategy (Fig. 7), the number of plates for confirmation, counterscreen, and dose–response in the AF4-AF9 assay and counterscreen was reduced to 12.
Screening of the TimTec library at 10 μM resulted in 131 hits with <36% inhibition. The hit threshold was derived from the percent CV for the mean of the controls on each plate. These CVs ranged from 6.4% to 12.6% (Table 3), so a hit threshold of 36% was selected to be ∼3 times the maximum percent CV. QC plates containing 14 nM AF4 27-mer (final concentration before bead addition) had an average of 81% inhibition (SD=3.5%). Confirmation and counterscreen of hits at 10 μM in triplicate resulted in 46 compounds confirmed for inhibition >36%; of these, 14 appeared to be selective for inhibition of AF4-AF9 as compared to the biotin–FLAG counterscreen (selectivity was defined as follows: inhibition of AF4-AF9 assay – inhibition of counterscreen >20%. The threshold was set at >20% difference in inhibition, since this represents 3 times the maximum CV [6.7%] for the biotin–FLAG counterscreen controls.). Dose–response testing of these 14 compounds revealed 10 compounds with IC50<50 μM. These 10 compounds were also tested in dose–response in the counterscreen, and 6 were found to be inactive (<20% inhibition at 50 μM), giving a confirmed hit rate of 0.16%. These results showed that initial counterscreening at a single concentration was effective in identifying 6 hits confirmed by dose–response to be selective for AF4-AF9 binding; although an additional 4 compounds appeared selective when tested at a single concentration, these were subsequently shown by dose–response testing to be artifacts inhibiting both the AF4-AF9 binding assay and the counterscreen (Fig. 7).
Confirmed Hits from the Two Pilot Screens
Overall, the MicroSource and the TimTec library screens revealed 13 and 6 confirmed hits, respectively. Compound 6 was present in both libraries and identified as active in both screens. Compound 10, the salt form of compound 6, was also present and active in the MicroSource screen. Thus, the total number of structurally unique hits with IC50<50 μM was 17 after screening of 5,680 compounds and eliminating artifacts that interfered with the assay readout. Representative IC50 curves for the most potent hits that were selective for the AF4-AF9 binding assay over the biotin–FLAG counterscreen are shown in Figure 9. The mean compound IC50 values are listed in Table 4, and structures are shown in Figure 10. Structure searching in PubChem revealed that screening data have been deposited for 13 out of the 18 compounds listed in Table 4. The PubChem activity profile indicates the frequency with which these compounds have been found to be active against other targets. The promiscuity index as defined by Schurer et al.25 ranks cross-screen activity on a scale from 0 to 1; the lower the value, the greater the selectivity of the compound for a small number of targets. Using a promiscuity index threshold of 0.05, five compounds listed in Table 4 are selective, while eight are promiscuous. The eight promiscuous compounds are active in >5% of the screens in which they have been tested, and will be excluded from further study.
Fig. 9.

Dose–response curves for MicroSource and TimTec hits. Data points represent the percent activity in the AF4-AF9 binding assay (■) and biotin–FLAG counterscreen (○). Dose–response curves were fitted using one data point per concentration per plate; plates were run in triplicate to obtain 3 independent IC50 determinations. The mean value of IC50s from these curves are reported in Table 4.
Table 4.
Summary of Hits from AF4-AF9 Binding Pilot Screens
| Compound numbera | PubChem CIDb | IC50 (μM)c | PubChem activity profiled | Promiscuity indexe |
|---|---|---|---|---|
| 1 | 26041 | 3.32±1.14 | 66/171 | 0.39 |
| 2 | 3983972 | 3.51±2.32 | Not tested | N/A |
| 3 | 2767 | 13.7±0.61 | 342/1108 | 0.31 |
| 4 | 2874122 | 15.3±0.92 | Not tested | N/A |
| 5 | 102678 | 16.9±1.14 | 12/141 | 0.085 |
| 6f | 6674 | 18.9±2.83 | 4/758 | 0.005 |
| 7 | 16667745 | 19.0±1.75 | 2/65 | 0.031 |
| 8 | 3699557 | 22.8±3.59 | 7/719 | 0.01 |
| 9 | 3598 | 26.9±2.66 | 258/1129 | 0.23 |
| 10 | 23682217 | 27.2±5.41 | 1/44 | 0.023 |
| 11 | 31239 | 27.5±2.04 | 34/299 | 0.11 |
| 12 | 309343 | 32.6±3.66 | 0/20 | 0 |
| 13 | 3928673 | 34.6±3.55 | Not tested | N/A |
| 14 | 392616 | 35.8±4.23 | 34/279 | 0.12 |
| 15 | 45114149 | 37.2±5.15 | Not tested | N/A |
| 16 | 426756 | 45.4±2.65 | 61/438 | 0.14 |
| 17 | 6842 | 46.6±6.35 | 74/406 | 0.18 |
| 18 | 3049 | 48.6±5.15 | Not tested | N/A |
aStructures are shown in Figure 10.
bPubChem compound ID that may be used to access test data deposited at pubchem.ncbi.nlm.nih.gov
cData are mean values±SD (n=3).
dNumber of PubChem Bioassays in which the compound is flagged as active/total number of PubChem bioassays in which compound has been tested.
ePubChem activity profile expressed as a decimal.
fThis compound was identified in the screens of both the MicroSource and TimTec libraries.
N/A, not available.
Fig. 10.

Structures of confirmed hits from MicroSource and TimTec libraries. Compound numbers refer to those in Table 4.
Confirmed AlphaScreen Artifacts
As described above, compounds active against both the AF4-AF9 screen and the biotin–FLAG counterscreen were considered artifacts that interfered with bead capture or AlphaScreen signal generation. To analyze further the nature of these artifacts and their prevalence in AlphaScreen-based HTS, we examined their structures and searched PubChem for activity in similar assays. This analysis was focused on hits from the MicroSource library, as most of these compounds are well-represented in PubChem. Screening data have not been reported for the majority of the TimTec compounds, so these were not analyzed further.
Table 5 lists 19 compounds that were active in both the HTS assay and the counterscreen, and gave IC50<50 μM in the latter. Structures are shown in Figure 11, grouped by five compound types as indicated in the figure and the table. In the case of four of the compounds, the mechanism by which they interfere with AlphaScreen detection is clear: compound 19, biotin, disrupts capture of the biotinylated peptide by the streptavidin beads; and compounds 20, 21, and 23 are all highly colored and suppress luminescence by the inner filter effect. These types of AlphaScreen artifact have been well documented in the academic and vendor literature.20,22,26 The remaining compounds do not stand out so obviously as likely artifacts, but several possess structural features that may be problematic. A total of seven compounds (including compounds 20, 21, and 23 mentioned above) contain amino- or azo-benzene moieties, and a further three are quinone derivatives. These may undergo redox chemistry that can interfere with AlphaScreen signal generation.20,22 Tertiary aminobenzene derivatives have been reported as AlphaScreen artifacts due to quenching of singlet oxygen, which is essential for energy transfer between donor and acceptor beads.26 These types of compounds are notorious for interfering with other types of assays as well, as indicated by their promiscuity index values listed in Table 5; all but one of the amino- and azo-benzenes and quinones were reported active in >15% of PubChem screens. The remaining compounds in Table 5 are more heterogeneous, and may be broadly classified as conjugated aromatics or heterocycles. One of these compounds (compound 30) has not previously been reported as active, while the remaining compounds were less promiscuous than the amino- and azo-benzenes and quinones, but nevertheless were active in between 6% and 16% of PubChem screens.
Table 5.
Compounds Identified in Counterscreen as AlphaScreen Artifacts
| |
|
|
|
PubChem AlphaScreen bioassay IC50 (μM)d |
|
|
|
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound numbera | Compound typea | PubChem CIDb | Counterscreen IC50 (μM)c | AID 595 | AID 485360 | AID 504332 | AID 504333 | AID 504339 | AID 540317 | Median PubChem IC50e | Median PubChem IC50/counterscreen IC50 | PubChem activity profilef | Promiscuity indexg |
| 19h | Biotin | 253 | 0.1±0 | ND | 0.35 | 0.8 | 0.8 | 11.2 | 10 | 0.8 | 8.0 | 20/715 | 0.028 |
| 20i | Amino- and azo-benzene derivates | 11057 | 0.2±0 | 12.6 | ND | 0.4 | ND | ND | ND | 6.5 | 32.5 | 209/513 | 0.41 |
| 21j | Amino- and azo-benzene derivates | 5702227 | 0.2±0 | ND | ND | ND | ND | ND | ND | N/A | N/A | 18/102 | 0.18 |
| 27 | Quinone derivates | 4212 | 0.3±0 | 12.6 | ND | 4 | ND | 6.3 | 25.1 | 9.45 | 31.5 | 385/1105 | 0.35 |
| 22 | Amino- and azo-benzene derivates | 2794 | 1.2±0.1 | >57 | ND | 2.5 | ND | 7.1 | 4.5 | 5.8 | 4.8 | 97/648 | 0.15 |
| 23k | Amino- and azo-benzene derivates | 9566046 | 1.7±0.1 | ND | ND | ND | ND | ND | ND | N/A | N/A | N/A | N/A |
| 24 | Amino- and azo-benzene derivates | 8691 | 8±1.2 | 39.8 | >57 | 22.9 | 44.5 | >114 | >114 | >50.8 | >6.4 | 32/813 | 0.039 |
| 30 | Other heterocycles | 16758142 | 12.2±3.2 | ND | ND | ND | ND | ND | ND | N/A | N/A | 0/58 | 0 |
| 25 | Amino and azo benzene derivates | 54736625 | 12.4±0.5 | ND | 31.6 | 12.6 | 12.6 | 50.1 | 35.5 | 31.6 | 2.5 | 87/349 | 0.25 |
| 31 | Other heterocycles | 66062 | 14.2±2.9 | ND | >57 | 25.1 | 31.6 | 50.1 | 39.8 | 39.8 | 2.8 | 83/864 | 0.096 |
| 32 | Other heterocycles | 4499 | 17.3±0.4 | ND | ND | 11.2 | ND | 20 | 15.8 | 15.8 | 0.9 | 58/580 | 0.1 |
| 26 | Amino- and azo-benzene derivates | 14710 | 20.6±4.4 | >57 | ND | 2.8 | ND | 50.1 | 70.8 | 60.5 | 2.9 | 168/905 | 0.19 |
| 35 | Other conjugated aromatics | 2733525 | 22.8±2.1 | >57 | 39.8 | 22 | 44.7 | 56.2 | >114 | 50.5 | 2.2 | 133/828 | 0.16 |
| 28 | Quinone derivates | 5967872 | 28.1±8.6 | ND | ND | ND | ND | ND | ND | N/A | N/A | 18/73 | 0.25 |
| 36 | Other conjugated aromatics | 439664 | 28.7±3.1 | ND | ND | ND | ND | ND | ND | N/A | N/A | N/A | N/A |
| 37 | Other conjugated aromatics | 3005572 | 29.4±2.6 | ND | ND | ND | ND | 25.1 | 57 | 41.05 | 1.4 | 26/416 | 0.062 |
| 33 | Other heterocycles | 16362 | 32.3±4 | >57 | >57 | 22.6 | 22.4 | 39.8 | 44.7 | 42.3 | 1.3 | 124/1166 | 0.11 |
| 34 | Other heterocycles | 4748 | 40.5±5.4 | >57 | >57 | 23.4 | 25.1 | 56.2 | >114 | >56.6 | >1.4 | 114/1201 | 0.095 |
| 29 | Quinone derivates | 443939 | 48±11.9 | ND | >57 | 22.4 | 35.5 | >114 | >114 | >57 | >1.2 | 440/1210 | 0.36 |
aSee Figure 11.
bPubChem compound ID that may be used to access test data deposited at pubchem.ncbi.nlm.nih.gov
cData are mean values±SD (n=3) determined in biotin-FLAG counterscreen as described in the Materials and Methods section.
dData are IC50 values reported from quantitative HTS performed at the National Institutes of Health Chemical Genomics Center. AID refers to assay ID in PubChem.
eMedian of data in preceding 6 columns.
fNumber of PubChem Bioassays in which the compound is flagged as active/total number of PubChem bioassays in which the compound has been tested.
gPubChem activity profile expressed as a decimal.
hBiotin—competes with the biotin-labeled reagent.
iGentian Violet—inner filter effect.
jGentian Violet analog—inner filter effect.
kChicago Sky Blue—inner filter effect.
ND, not determined.
Fig. 11.

Structures of AlphaScreen artifacts from MicroSource library. Compound numbers refer to those in Table 5.
To determine whether the counterscreen hits are likely to interfere routinely with AlphaScreen, we identified 6 assays in PubChem that used AlphaScreen to monitor the protein–peptide interactions. In all 6 cases, quantitative HTS (qHTS) was run at the National Institutes of Health Chemical Genomics Center, generating IC50 values for the entire screening library. Fourteen out of the 19 compounds listed in Table 5 were tested in at least 2 out of the 6 assays. In almost all cases, measurable IC50 values were obtained. The data contain some outliers, so for each compound, a median IC50 was calculated rather than the arithmetic mean. Eleven compounds gave median IC50 values ranging from 0.8 to 60.5 μM. The remaining 3 compounds were inactive in several of the assays, so a lower limit (>50.8, >56.6, and >57 μM, for compounds 24, 34, and 29, respectively) may be placed on the median, but not an exact value. The ratio of median PubChem IC50/counterscreen IC50 was calculated to determine the extent of correlation between the data sets. In all but one case, the PubChem IC50 values were higher than the counterscreen IC50s. In the majority of cases, the ratio was <3, although five of the most potent compounds in the counterscreen were between 4.8-fold and 32.5-fold less active in the PubChem screens. Some discrepancy is not unexpected, because the compounds in the PubChem screens were obtained from different sources than the MicroSource compounds reported here. Overall, given the differences in the assay design and compound source, the correlation between the counterscreen and PubChem data is quite strong (at least qualitatively), supporting the conclusion that the 19 compounds in Table 5 are commonly encountered AlphaScreen artifacts and not target-directed inhibitors.
Discussion
We have established an HTS-compatible assay for identification of small-molecule compounds that modulate the interaction between MLL-AF4 and the transcription factor AF9 using the AlphaScreen® technology. The assay is robust and sensitive with Z′-factor values >0.5. HTS assay readiness was demonstrated by screening this assay against 5,680 pharmacologically active compounds, natural products, and natural product-derived synthetic compounds from MicroSource and TimTec libraries. Hits were confirmed by dose–response, and artifacts were identified and eliminated through counterscreening against a biotin–FLAG control peptide.
AlphaScreen is a versatile and sensitive technology for the detection of the protein–protein and protein–peptide interactions. Ultimately, assays can be sufficiently robust for large HTS campaigns. However, extensive and careful optimization is required. The optimal sequence length of the biotinylated peptide must be determined empirically. While some published methods use biotinylated peptides as short as 13 amino acids,27 others require longer peptides to see a robust signal. The linker, or nonbinding, sequence length must accommodate opposing factors such as steric hindrance by bound beads and proximity required for singlet oxygen transfer. In our case, initial tests with a 12-amino-acid biotinylated AF4 peptide were unsuccessful; hence, we subsequently selected a 27-amino-acid peptide derived from the AF9-binding region of AF4. AlphaScreen is also affected by ambient lighting conditions and changes in temperature. The beads are sensitive to intense light or long exposure to ambient light, a problem that is resolved by handling the beads in the dark or under subdued or green filtered lighting conditions. Temperature can influence the equilibrium binding of antibodies and analytes and the chemical generation and transfer of singlet oxygen. Perkin Elmer notes that changes in signal can be as high as 10% per degree Celsius,20 and consistent with this, we found that it is important for the temperature in the plate reader to be very close to the temperature at which the plates are incubated before reading.
Even after optimization, the assay is prone to compound effects unrelated to the targeted protein–peptide binding interaction. The susceptibility of AlphaScreen to artifacts interfering with bead capture or energy transfer was evident from the high primary hit rate obtained in both pilot screens. However, with a simple counterscreen using a reagent that contains both bead capture tags in a single molecule, it is possible to readily identify these artifacts and exclude them from further study. Using a biotin–FLAG peptide, we were thus able to eliminate 60% of the hits. A search for compound activity in related assays in PubChem provided further evidence that the counterscreen hits are indeed artifacts that repeatedly disrupt HTS using AlphaScreen. Activity attributed to these compounds and structurally related analogs in future HTS should therefore be treated with caution.
Counterscreening confirmed that 18 hits are not AlphaScreen artifacts and therefore disrupt the binding of AF4-derived peptide to AF9 protein. However, it cannot be assumed that the inhibition of the AF4-AF9 interaction shown by these compounds is mediated through binding to the intact AF9 protein. Although we included BSA and the detergent Tween-20 in the assay buffer to minimize the likelihood of compound aggregation, protein denaturation, or other nonspecific effects, these cannot be ruled out without further characterization of the interaction of the hits with AF9. Important evidence for selective inhibition of AF4-AF9 binding will come from direct measurement of compound binding to the AF9 protein. This will serve as a useful alternative assay to confirm inhibition results observed in the AlphaScreen assay. We are currently working with the Broad Institute (Cambridge, MA) to develop a thermal-shift assay,28 and we are also evaluating other label-free methods capable of detecting small-molecule binding to AF9.
For hits to be useful as chemical probes to explore the biology of MLL-R leukemia and to be advanced as potential therapeutic leads, it is essential that they selectively disrupt MLL-AF4 binding to AF9 in MLL-R leukemia cells. Hemenway and coworkers have shown that cell-penetrating peptides derived from AF4 disrupt the AF4-AF9 interaction and cause cell death preferentially in MLL-R leukemia cell lines.13,14 Thus, AF9 binding compounds will be studied for their effect on cell viability in leukemic cell lines. Based on the peptide work in the Hemenway group, it is our expectation that AF9-binding compounds will selectively kill leukemic cells that harbor the MLL-AF4 fusion over those that do not bear the fusion. Such compounds should hold significant promise as potential therapeutic leads, and will be further advanced to animal testing in a mouse model of engrafted leukemia.29
Successful assay development and pilot screening in our laboratory formed the basis for transfer of the primary HTS assay and secondary assays to the Broad Institute for screening against the 350,000-compound MLSMR. Hits identified from HTS at the Broad Institute and at Nemours will be characterized in a battery of biochemical and cell-based secondary assays, and the most promising chemical series will be optimized by medicinal chemistry. Ultimately, compounds identified as potent and selective hits through these endeavors may serve as leads in the development of targeted therapies for MLL-R leukemias.
Supplementary Material
Abbreviations
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- CV
coefficient of variation
- DMSO
dimethyl sulfoxide
- HEPES
4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid
- HOX
homeobox
- HTS
high-throughput screening
- MLL
mixed-lineage leukemia
- MLL-R
MLL-rearranged
- MLSMR
Molecular Libraries Small Molecule Repository
- MSR
minimum significant ratio
- NaCl
sodium chloride
- NDL
Natural Derivates Library
- NPL
Natural Products Library
- QC
quality control
- qHTS
quantitative HTS
- S/B
signal-to-background ratio
Acknowledgments
The authors acknowledge support to A.D.N. from the National Institutes of Health (R21 NS073046), Alex Lemonade Stand Foundation, I Care I Cure Foundation, and the Nemours Foundation, and funding provided by the B+ Foundation to purchase the assay development and screening instrumentation used in this study. V.G.W. is supported by a UNCF-Merck Postdoctoral Fellowship. The authors would also like to give special thanks to Carl Apgar and Matthew Reuter of Perkin Elmer, Charles Hemenway of Loyola University, and Nicky Tolliday and colleagues of the Broad Institute for their many helpful discussions and suggestions.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Mackall CL. In search of targeted therapies for childhood cancer. Front Oncol. 2011;1:18. doi: 10.3389/fonc.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Slany RK. The molecular biology of mixed lineage leukemia. Haematologica. 2009;94:984–993. doi: 10.3324/haematol.2008.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson BW. Devidas M. Carroll AJ, et al. Specific MLL partner genes in infant acute lymphoblastic leukemia (ALL) associated with outcome are linked to age, white blood cell count (WBC) at diagnosis: a report on the Children's Oncology Group (COG) P9407 Trial. American Society of Hematology Annual Meeting and Exposition; New Orleans, LA. 2009. [Google Scholar]
- 4.Meyer C. Kowarz E. Hofmann J, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23:1490–1499. doi: 10.1038/leu.2009.33. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong SA. Staunton JE. Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong SA. Golub TR. Korsmeyer SJ. MLL-rearranged leukemias: insights from gene expression profiling. Semin Hematol. 2003;40:268–273. doi: 10.1016/s0037-1963(03)00196-3. [DOI] [PubMed] [Google Scholar]
- 7.Faber J. Krivtsov AV. Stubbs MC, et al. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood. 2009;113:2375–2385. doi: 10.1182/blood-2007-09-113597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bitoun E. Oliver PL. Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- 9.Monroe SC. Jo SY. Sanders DS, et al. MLL-AF9, MLL-ENL alter the dynamic association of transcriptional regulators with genes critical for leukemia. Exp Hematol. 2011;39:77–86. doi: 10.1016/j.exphem.2010.09.003. e71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huret JL. Dessen P. Bernheim A. An atlas of chromosomes in hematological malignancies. Example: 11q23 and MLL partners. Leukemia. 2001;15:987–989. doi: 10.1038/sj.leu.2402135. [DOI] [PubMed] [Google Scholar]
- 11.Meyer C. Schneider B. Jakob S, et al. The MLL recombinome of acute leukemias. Leukemia. 2006;20:777–784. doi: 10.1038/sj.leu.2404150. [DOI] [PubMed] [Google Scholar]
- 12.Erfurth F. Hemenway CS. de Erkenez AC. Domer PH. MLL fusion partners AF4 and AF9 interact at subnuclear foci. Leukemia. 2004;18:92–102. doi: 10.1038/sj.leu.2403200. [DOI] [PubMed] [Google Scholar]
- 13.Srinivasan RS. Nesbit JB. Marrero L. Erfurth F. LaRussa VF. Hemenway CS. The synthetic peptide PFWT disrupts AF4-AF9 protein complexes and induces apoptosis in t(4;11) leukemia cells. Leukemia. 2004;18:1364–1372. doi: 10.1038/sj.leu.2403415. [DOI] [PubMed] [Google Scholar]
- 14.Palermo CM. Bennett CA. Winters AC. Hemenway CS. The AF4-mimetic peptide, PFWT, induces necrotic cell death in MV4-11 leukemia cells. Leuk Res. 2008;32:633–642. doi: 10.1016/j.leukres.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett CA. Winters AC. Barretto NN. Hemenway CS. Molecular targeting of MLL-rearranged leukemia cell lines with the synthetic peptide PFWT synergistically enhances the cytotoxic effect of established chemotherapeutic agents. Leuk Res. 2009;33:937–947. doi: 10.1016/j.leukres.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kazmierski WM. Kenakin TP. Gudmundsson KS. Peptide, peptidomimetic and small-molecule drug discovery targeting HIV-1 host-cell attachment and entry through gp120, gp41, CCR5 and CXCR4†. Chem Biol Drug Des. 2006;67:13–26. doi: 10.1111/j.1747-0285.2005.00319.x. [DOI] [PubMed] [Google Scholar]
- 17.MicroSource Spectrum Collection. www.msdiscovery.com/spectrum.html. [Oct 22;2012 ]. www.msdiscovery.com/spectrum.html
- 18.TimTec Nature Inspired Screening Libraries. www.TimTec.net/Natural-Products.html. [Oct 22;2012 ]. www.TimTec.net/Natural-Products.html
- 19.Zhang JH. Chung TD. Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 20.PerkinElmer LAS literature 007011_01. Boston: PerkinElmer; 2004. PerkinElmer: A Practical Guide to Working with Alpha Screen™. [Google Scholar]
- 21.Arkin MR. Glicksman MA. Fu H. Havel JJ. Du Y. Inhibition of protein-protein interactions: non-cellular assay formats. In: Sittampalam GS, editor; Gal-Edd N, editor; Arkin MR, editor. Assay Guidance Manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences; Bethesda, MD: 2012. [Nov 2;2012 ]. [PubMed] [Google Scholar]
- 22.PerkinElmer LAS literature 009625_01. Boston: PerkinElmer; 2011. PerkinElmer: User's Guide to Alpha Assays Protein:Protein Interactions™. [Google Scholar]
- 23.Leach BI. Kuntimaddi A. Schmidt CR. Cierpicki T. Johnson SA. Bushweller JH. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure. 8(21):176–183. doi: 10.1016/j.str.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eastwood BJ. Farmen MW. Iversen PW, et al. The minimum significant ratio: a statistical parameter to characterize the reproducibility of potency estimates from concentration-response assays and estimation by replicate-experiment studies. J Biomol Screen. 2006;11:253–261. doi: 10.1177/1087057105285611. [DOI] [PubMed] [Google Scholar]
- 25.Schurer SC. Vempati U. Smith R. Southern M. Lemmon V. BioAssay ontology annotations facilitate cross-analysis of diverse high-throughput screening data sets. J Biomol Screen. 2011;16:415–426. doi: 10.1177/1087057111400191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baell JB. Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 27.Mayasundari A. Ferreira AM. He L. Mahindroo N. Bashford D. Fujii N. Rational design of the first small-molecule antagonists of NHERF1/EBP50 PDZ domains. Bioorg Med Chem Lett. 2008;18:942–945. doi: 10.1016/j.bmcl.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 28.Niesen FH. Berglund H. Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 29.Lee EM. Bachmann PS. Lock RB. Xenograft models for the preclinical evaluation of new therapies in acute leukemia. Leuk Lymphoma. 2007;48:659–668. doi: 10.1080/10428190601113584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.