

Abstract
The mineralocorticoid aldosterone plays an important role in regulating blood pressure, with excess causing hypertension and exacerbating cardiovascular disease. Previous studies have indicated a role for both phospholipase D (PLD) and protein kinase D (PKD) in angiotensin II (AngII)-regulated aldosterone production in adrenal glomerulosa cells. Therefore, the relationship between AngII-activated PLD and PKD was determined in two glomerulosa cell models, primary bovine zona glomerulosa (ZG) and the HAC15 human adrenocortical carcinoma cells, using two inhibitors, 1-butanol and the reported PLD inhibitor, fluoro-2-indolyl des-chlorohalopemide (FIPI). FIPI was first confirmed to decrease PLD activation in response to AngII in the two glomerulosa cell models. Subsequently, it was shown that both 1-butanol and FIPI inhibited AngII-elicited PKD activation and aldosterone production. These results indicate that PKD is downstream of PLD and suggest that PKD is one of the mechanisms through which PLD promotes aldosterone production in response to AngII in adrenal glomerulosa cells.
Keywords: adrenal glomerulosa, protein kinase D (PKD), phospholipase D (PLD), aldosterone, angiotensin II
1. INTRODUCTION
The mineralocorticoid aldosterone is important for normal sodium homeostasis and blood pressure control, with excess aldosterone resulting in hypertension and exacerbates congestive heart failure and cardiovascular disease. The production of this hormone is regulated by angiotensin II (AngII), which binds to the AngII type I receptor to activate phospholipase C. Phospholipase C-catalyzed phosphoinositide hydrolysis generates two second messengers: inositol 1,4,5-trisphosphate, which triggers calcium release from the endoplasmic reticulum, and diacylglycerol (DAG), which can activate DAG-responsive enzymes, such as members of the protein kinase C family. DAG can also be produced by the action of phospholipase D (PLD), which hydrolyzes phosphatidylcholine to yield phosphatidic acid (and choline). Phosphatidic acid is a lipid signal, activating such enzymes as Raf-1, mTOR and phosphatidylinositol 4-phosphate 5-kinase (reviewed in (Cummings et al., 2002; Shapiro and Bollag, 2008)). Phosphatidic acid, in turn, is dephosphorylated by lipid phosphate phosphatases and phosphatidic acid phosphatases (or lipins) [reviewed in (Brindley et al., 2009)] to generate the signal DAG.
Another DAG-responsive enzyme is the serine/threonine protein kinase, protein kinase D (PKD). PKD possesses two cysteine-rich C1 regions that bind DAG (or phorbol ester), and the enzyme can be activated by this binding. In addition, we have recently shown that in primary cultures of bovine zona glomerulosa (ZG) cells PKD activity can be stimulated by protein kinase C-mediated phosphorylation (manuscript in preparation), as has been demonstrated in other cell types [reviewed in (Rozengurt, 2011)]. Previous reports have shown that 1-butanol inhibited PKD activation stimulated by lysophosphatidic acid and platelet-derived growth factor in fibroblasts (Kam and Exton, 2004) and antibody ligation in a lymphocyte cell line (Caloca et al., 2008). Because 1-butanol can be used by PLD in place of water in a transphosphatidylation reaction to generate phosphatidylbutanol, thereby inhibiting the production of phosphatidic acid, this result suggests that PLD-derived lipid signals are important in PKD activation. However, a recent report indicates that 1-butanol can have effects that are not related to PLD inhibition in certain cell responses and in some cell types (Su et al., 2009), [although it should be noted that Caloca et al. (2008) did not rely solely on 1-butanol effects and also supported their conclusions with genetic manipulation of PLD and PKD.]. Other investigators have also obtained evidence of PKD activation mediated by PLD in response to the pharmacologic agents ilimaquinone (Sonoda et al., 2007) and reactive oxygen species (Cowell et al., 2009). Therefore, we investigated the effect of inhibiting PLD on AngII-induced PKD activation in primary cultures of bovine ZG cells and the human adrenocortical carcinoma cell line, HAC15, a clone of the H295R cell line (Parmar et al., 2008; Wang and Rainey, 2012; Wang et al., 2012). Since PLD activation has not been characterized in the HAC15 cells, we also determined the dose and time dependence of AngII-stimulated PLD activity.
2. MATERIALS AND METHODS
2.1. Materials
DMEM/F12, was purchased from Gibco (Grand Island, NY). penicillin-streptomycin-fungizone and penicillin-streptomycin were purchased from Biologos, Inc. (Naperville, IL). Cosmic calf serum, was obtained from Thermo Scientific (South Logan, UT). Horse serum and fetal bovine serum (FBS) were obtained from Atlanta Biologicals (Norcross, GA). ITS+ (insulin-transferrin-selenous acid with linoleic acid and bovine serum albumin) was from Becton-Dickinson Labware (Franklin Lakes, NJ). Phosphatidylethanol (PEt) and phosphatidic acid were purchased from Avanti Polar Lipids (Alabaster, AL). Butylated hydroxyanisole (BHA), α-tocopherol, ascorbic acid, gentamycin and 22(R)-hydroxycholesterol were obtained from Sigma (St. Louis, MO). The solid-phase radioimmunoassay kit was from Siemens (Tarrytown, NY). [3H]Oleic acid and En3Hance were purchased from Dupont NEN (Boston, MA). The rabbit anti-phosphoserine916PKD and anti-PKD (total) antibodies were obtained from Cell Signaling Technology (Danvers, MA). The goat anti-rabbit (phosphoserine916PKD and total PKD secondary antibody), and rabbit anti-goat (actin secondary antibody) were from Sigma. Immobilon P-FL was obtained from Millipore (Billerica, MA).
2.2. Cell Culture
Bovine adrenal glomerulosa cells were prepared from near-term fetal calf adrenal glands obtained from a local meat-packing plant. Zona glomerulosa-enriched tissure slices were dissected from the adrenal glands. Cells were released from collagenase-digested slices using mechanical agitation, collected by centrifugation and cultured in a serum-containing DMEM/F12 medium as previously described (Shapiro et al., 2010).
Human adrenocortical carcinoma HAC15 cells (Parmar et al., 2008)), which is actually a clone of the H295R cell line (Wang and Rainey, 2012; Wang et al., 2012), were grown in DMEM/F12 medium containing 1% ITS, 1% penicillin-streptomycin and 10% Cosmic calf serum to approximately 70–75% confluence. The cells were then incubated for 20–24 hours in low serum medium containing 1% Cosmic calf serum prior to experimentation.
2.3. Measurement of Aldosterone Production
Primary cultures of bovine ZG cells were cultured overnight in serum-containing medium and then refed with serum-free medium as in (Shapiro et al., 2010). After 20–24 hours the cells were placed into bicarbonate-buffered Kreb’s Ringer containing 2.5 mM sodium acetate (KRB+) equilibrated with 5% CO2 at 37°C. Cells were then incubated with KRB+ alone or KRB+ containing 10 μM 22(R)-hydroxycholesterol in the presence and absence of 750 nM FIPI (or the DMSO vehicle control) for 60 minutes at 37°C in an atmosphere of 5% CO2. Supernatants were collected and stored frozen until assayed.
Cultured HAC15 cells were incubated with equilibrated low-serum DMEM/F12 medium containing the appropriate agents for the indicated time. The supernatants were collected and stored frozen −20°C until aldosterone was assayed using a solid phase radioimmunoassay kit (Diagnostic Products, Los Angeles, CA)
2.4. Phopholipase D Activity Assay
PLD activity was monitored as the production of radiolabeled phosphatidylethanol (PEt) in [3H]oleate-prelabelled cells. Cells were incubated for 20–24hrs in low-serum medium containing 5μCi/mL [3H]oleate, equilibrated in KRB+ and stimulated with appropriate agents in the presence of 0.5% ethanol. Cells were solubilized in 0.2% SDS containing 5 mM EDTA, and lipids were extracted into chloroform/methanol. Phospholipids were separated by thin-layer chromatography, visualized with autoflourography using En3Hance, identified by co-migration with authentic standards and cut out and quantified by liquid scintillation spectrometry.
2.5. Western Analysis
Cells incubated for 20–24 hours in low-serum medium were treated with the appropriate agents as indicated. Cells were lysed, protein levels in the lysates determined and equal amounts of protein analyzed by western blotting using an anti-phosphoserine916PKD-specific or anti-actin primary antibodies and alkaline phosphatase- or IRDye-conjugated secondary antibodies. Immunoreactive proteins were visualized using an ECF system (Amersham, Piscataway, NJ) with imaging on a Typhoon imager (Molecular Dynamics, Sunnyvale, CA). Alternatively, an Odyssey imaging system ((Licor Biosciences, Lincoln, NE) was utilized and immunoreactivity quantified with Odyssey application software (version 2.1). Proteins were normalized to actin as indicated. Actin was used for normalization because both the total PKD and phosphoserine910PKD antibodies were raised in rabbit, necessitating stripping and reprobing of the blot, with possibly nonhomogeneous loss of immunoreactivity, if total PKD were to be used for normalization. Nevertheless, in several instances we compared values using actin versus total PKD as the normalization control and observed essentially no differences.
2.6. Viability Assay
HAC15 cells were incubated with or without FIPI (750 nM) in the presence or absence of AngII (10 nM) for 24 hours. The pretreated cells were trypsinized and incubated with 0.4% trypan blue. The number of trypan blue-excluding cells, expressed as a percentage relative to the total cell number, was determined using a T10™ Automated Cell Counter (Biorad, Hercules, CA).
2.7. Statistical Analysis
Experiments were performed a minimum of three times and the values obtained expressed as the means ± SEM. Significant differences were statistically determined with ANOVA followed by a Newman-Keuls post-hoc test using Graphpad Prism or Instat (La Jolla, CA).
3. RESULTS
3.1. 1-Butanol inhibits AngII-induced PKD activation and aldosterone production in primary cultures of bovine adrenal glomerulosa cells
PLD hydrolyzes phosphatidylcholine to yield phosphatidic acid; however, in the presence of small amounts of a primary alcohol, such as 1-butanol. PLD catalyzes a transphosphatidylation reaction to generate a phosphatidylalcohol (i.e., phosphatidylbutanol). Thus, the primary alcohol diverts production away from phosphatidic acid, decreasing the levels of this lipid messenger. Therefore, a primary alcohol such as 1-butanol can be used to inhibit PLD-mediated lipid signal generation, as we have previously shown in primary cultures of bovine adrenal glomerulosa cells (Bollag et al., 2002). We have also previously shown that AngII induces the activation of PKD in these cells (Shapiro et al., 2010), as determined by monitoring autophosphorylation at serine910, a reported marker of PKD activation status (Matthews et al., 1999). We found that 0.3% 1-butanol significantly (p<0.05, n = 3) inhibited AngII-elicited PKD activation, as monitored by the levels of phosphoserine910 PKD immunoreactivity (Figure 1A and B). In these same samples 1-butanol also inhibited AngII-stimulated aldosterone production (Figure 1C).
Figure 1. Inhibition of PLD-mediated signal production with 1-butanol inhibits AngII-induced PKD activation in primary cultures of bovine adrenal glomerulosa cells.

Primary cultures of bovine adrenal glomerulosa cells were incubated for 30 minutes with or without 10 nM AngII in the presence or absence of 0.3% 1-butanol. Cells were harvested from duplicate wells and analyzed by western blotting for PKD phosphorylation at serine 910. Panel A shows a representative immunoblot, whereas Panel B illustrates the combined results of 3 separate experiments performed in duplicate. Immunoreactivity of pSer910 was normalized using actin and then expressed relative to the normalized control value. Panel C demonstrates the inhibition by 1-butanol of AngII-elicited aldosterone secretion in the collected supernatants. *p<0.05, **p<0.01, ***p<0.001 versus the control, †p<0.05, ††p<0.01 versus AngII alone by ANOVA followed by a Student-Newmann-Keuls post-hoc test. (Note that the lower band seen in some lanes of the representative western blot was not consistently observed and is likely non-specific.)
3.2. The reported PLD inhibitor FIPI inhibits AngII-induced PLD and PKD activation in primary cultures of bovine adrenal glomerulosa cells
Recently, a novel PLD inhibitor FIPI has been reported to inhibit dose-dependently both PLD1 and PLD2 in vitro and in situ. Indeed, Frohman and colleagues showed that in intact cells, a dose of 750 nM is sufficient to block (essentially completely) PLD1 activation in response to phorbol 12-myristate 13-acetate (PMA) or the constitutive activity of overexpressed PLD2 (Su et al., 2009). Therefore, using 750 nM FIPI we first sought to verify the ability of FIPI to inhibit AngII-induced PLD activation in primary cultures of bovine zona glomerulosa cells. [3H]Oleate-prelabeled bovine glomerulosa cells were pretreated with 750 nM FIPI, stimulated with 10 nM AngII for 30 minutes in the presence of 0.5% ethanol and radiolabeled PEt levels monitored. As observed previously (Bollag et al., 1990; Bollag et al., 2002), AngII induced PLD activation, increasing PEt levels by about 5-fold over the control value (Figure 2A). Whereas FIPI by itself had no significant effect on PLD activation, the inhibitor decreased AngII-stimulated PEt levels (Figure 2A). We then investigated whether FIPI could inhibit AngII-elicited PKD activation in bovine ZG cells. Again cells were pretreated with 750 nM FIPI prior to stimulation with 10 nM AngII. As shown in Figure 2B, AngII induced PKD activation in bovine ZG cells, as previously demonstrated (Shapiro et al., 2010), and FIPI significantly (p<0.05, n = 4) inhibited this activation (Figure 2B).
Figure 2. FIPI inhibits both AngII-induced PLD and PKD activation in primary cultures of bovine adrenal glomerulosa cells.

(A) [3H]Oleate-prelabeled glomerulosa cells were pretreated for 20–30 minutes with or without 750 nM FIPI (or the DMSO vehicle) prior to incubation in the presence or absence of 10nM AngII for 30 minutes. Cell lipids were extracted using chloroform-methanol and analyzed by thin-layer chromatography. Results represent the means ± SEM of 6 samples from 3 separate experiments; ***p<0.001 versus the control, †††p<0.001 versus AngII alone. (B) Glomerulosa cells were pretreated for 30 minutes with or without 750 nM FIPI (or the 0.1% DMSO vehicle) prior to incubation in the presence or absence of 10nM AngII for 30 minutes. Autophosphorylation of PKD at serine910 was determined by western analysis of duplicate samples, with a representative blot shown in the upper panel. In the lower panel phosphoserine910 levels were normalized to actin levels and expressed as the -fold over the control. Values represent the means ± SEM of 4 separate experiments performed in duplicate; †p<0.05 versus AngII alone. (D) Medium from the experiments shown was collected and aldosterone levels measured. Values represent the means ± SEM of 4 separate experiments performed in duplicate; *p<0.05, **p<0.01, ***p<0.001 versus the control, †p<0.05, †††p<0.001 versus AngII alone by ANOVA followed by a Student-Newmann-Keuls post-hoc test.
3.3 FIPI inhibits AngII-induced, but not 22(R)-hydroxycholesterol-mediated aldosterone production in primary cultures of bovine adrenal glomerulosa cells
To examine the effect of FIPI on AngII-elicited steroidogenesis, medium from the experiments performed above was collected and aldosterone production measured. As shown in Figure 2D, FIPI (at a dose of 750 nM) had no significant effect on basal aldosterone production but significantly inhibited AngII-induced steroid hormone synthesis (p<0.001, n = 4). We also examined the effect of FIPI on aldosterone production in response to a one-hour incubation with 22(R)-hydroxycholesterol. Water-soluble 22(R)-hydroxycholesterol can bypass normal signaling mechanisms and the rate-limiting step of cholesterol transport from the outer to the inner mitochondrial membrane, where the enzyme initiating steroidogenesis is located. Therefore, steroidogenesis in response to incubation with 22(R)-hydroxycholesterol is a measure of glomerulosa cell health and aldosterone biosynthetic ability. Cells were incubated with 22(R)-hydroxycholesterol in the presence and absence of 750 nM FIPI and medium collected after one hour. FIPI had no effect FIPI on 22(R)-hydroxycholesterol-mediated aldosterone production, as aldosterone amoints of 1468 ± 727 versus 1390 ± 504 pg/mL/hr, or 72- ± 26- versus 71 ± 21-fold over the control, for 22(R)-hydroxycholesterol without and with FIPI, respectively, were not statistically different (p>0.9, n = 4). Thus, these results indicate that FIPI is not simply exhibiting non-specific cytotoxicity.
3.4. AngII activates PLD in a concentration-dependent and sustained manner in HAC15 cells
We have previously shown that AngII activates PLD in a dose-dependent manner in both primary cultures of bovine adrenal glomerulosa cells (Bollag et al., 1990; Jung et al., 1998; Bollag et al., 2002) and in the human adrenocortical carcinoma cell line, NCI H295R (Zheng and Bollag, 2003). However, while AngII induces sustained PLD activation in the bovine ZG cells (Jung et al., 1998), the hormone produces only a transient response in H295R cells (Zheng and Bollag, 2003). The HAC15 cell line (Parmar et al., 2008) is a newly identified clone of H295R cells (Wang and Rainey, 2012), which demonstrates good aldosterone responses to AngII(Wang et al., 2012). However, to date there have been no reports concerning the ability of AngII to stimulate PLD activity in these HAC15 cells. Therefore, we determined whether AngII induced a dose-dependent activation of PLD in [3H]oleate-prelabeled HAC15 cells. As shown in Figure 3A, AngII significantly stimulated PLD activity in a dose-dependent manner with a maximal increase at a 10 nM concentration (p<0.05, n = 3). We next determined the time course of AngII-induced PLD activation in these cells. In contrast to H295R cells (Zheng and Bollag, 2003) but similarly to primary bovine ZG cells (Jung et al., 1998), HAC15 cells exhibited sustained PLD activation in the HAC15 cells in response to AngII (Figure 3B), with significantly increased radiolabeled PEt levels between 5 and 60 minutes of stimulation (p<0.01, n = 5).
Figure 3. AngII activates PLD in a dose-dependent and sustained manner in HAC15 cells.
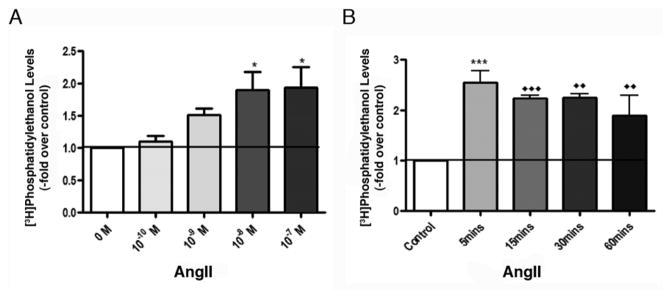
(A) Dose dependence of AngII-elicited PLD activation in HAC15 cells. [3H]Oleate-prelabeled HAC15 cells were treated for 60 minutes with various doses of AngII in the presence of 0.5% ethanol. Data represent the means ± SEM of 4 experiments performed in duplicate; *p<0.05 versus the control value. (B) Sustained AngII induced-PLD activation in HAC15 cells. [3H]Oleate-prelabeled cells were exposed to 0.5% ethanol and 10 nM AngII was added in a staggered fashion 0, 30 and 45 mins later, with all samples terminated at the same time. For the 5 min time point, ethanol was added for 5 min before stimulation with or without 10 nM AngII for 5 min. Data represent the means ± SEM of 4 experiments performed in duplicate; ***p< 0.001 versus the 5-min control; ◆◆p < 0.01, ◆◆◆p < 0.001 versus the 60-min control.
3.5. FIPI inhibits AngII-induced PLD and PKD activation iand aldosterone production in HAC15 cells
The ability of FIPI to inhibit AngII-elicited PLD activation in the HAC15 cells was determined by monitoring AngII-increased radiolabeled PEt levels in the presence and absence of 750 nM FIPI in [3H]oleate-prelabeled cells. As in the primary bovine ZG cells (Figure 2), FIPI had no effect on basal PLD activity but inhibited AngII-induced PLD activation (Figure 4A). When cells were again stimulated with (or without) AngII and the presence or absence of FIPI and PKD activation monitored by western analysis of autophosporylation of serine910, FIPI was observed to significantly (p<0,05, n = 3) inhibit AngII-elicited PKD activation (Figure 4B and C). Likewise, FIPI also inhibited AngII-increased aldosterone production in HAC15 cells, by approximately 50%, without significantly altering basal or 22(R)-hydroxycholesterol-mediated steroidogenesis (Figure 5).
Figure 4. FIPI inhibits AngII-induced PLD and PKD activation in the human adrenocortical carcinoma cell line, HAC15.

(A) [3H]Oleate-prelabeled HAC15 cells were pretreated for 20–30 minutes with or without 750 nM FIPI (or the DMSO vehicle) prior to incubation in the presence or absence of 10nM AngII for 30 minutes. Cell lipids were extracted using chloroform-methanol and analyzed by thin-layer chromatography. Results represent the means ± SEM of 3 separate experiments performed in duplicate; *p<0.05 versus the control, †p<0.05 versus AngII alone. (B) HAC15 cells were pretreated for 30 minutes with or without 750 nM FIPI (or the DMSO vehicle) prior to incubation in the presence or absence of 10nM AngII for 30 minutes. Autophosphorylation of PKD at serine910 was determined by western analysis, with a representative blot of duplicate samples shown in panel B. (C) Phosphoserine910 levels were normalized to total PKD levels and expressed as fold relative to control. Values represent the means ± SEM of 5 separate experiments performed in duplicate; *p<0.05 versus the control, †p<0.05 versus AngII alone.
Figure 5. FIPI inhibits AngII-induced, but not 22(R)hydroxycholesterol-mediated aldosterone production in HAC15 human adrenocortical carcinoma cells.
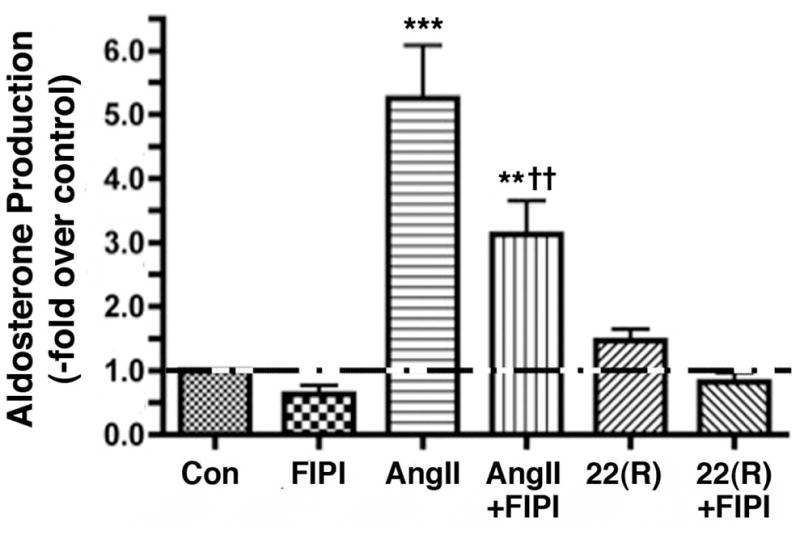
HAC15 cells incubated for 20–24 hours in low-serum medium were treated with low-serum medium in the presence of 750 nM FIPI or the vehicle control (0.1% DMSO) with or without 10 nM AngII for 18 hours. In other wells 22(R)-Hydroxycholesterol (2 μM) was added, and experimental medium was collected 6 hours later (for a total incubation time of 24 hours) and stored frozen until assayed. Aldosterone levels were then measured using a solid-phase radioimmunoassay and expressed as the –fold relative to the control (basal) levels. Values represent the means ± SEM of 3 separate experiments performed in triplicate; *p<0.05 versus the control; †p<0.05 versus AngII alone.
3.6. Incubation with FIPI for 24 hours does not alter HAC15 cell viability
Whereas in the primary bovine ZG cells, addition of 22(R)-hydroxycholesterol results in the production of large amounts of aldosterone, because the HAC15 cells show low expression of aldosterone synthase basally (Parmar et al., 2008), provision of 22(R)-hydroxycholesterol in these cells mediates minimal aldosterone production (Figure 5). Thus, as an additional measure of potential cytotoxicity in stimulated cells, we also examined the ability of FIPI to affect HAC15 cell viability in the prsence and absence of AngII exposure using a trypan blue exclusion assay and a Countess Cell Counter. As shown in Figure 6, there was no significant effect of 750 nM FIPI on cell viability, either in the presence or absence of 10 nM AngII (n = 3), suggesting that the inhibitory effect of FIPI on aldosterone production in these cells is unlikely to be the result on nonspecific toxicity.
Figure 6. FIPI, in the absence of presence of AngII, has no significant cytotoxic effect on HAC15 human adrenocortical carcinoma cells.
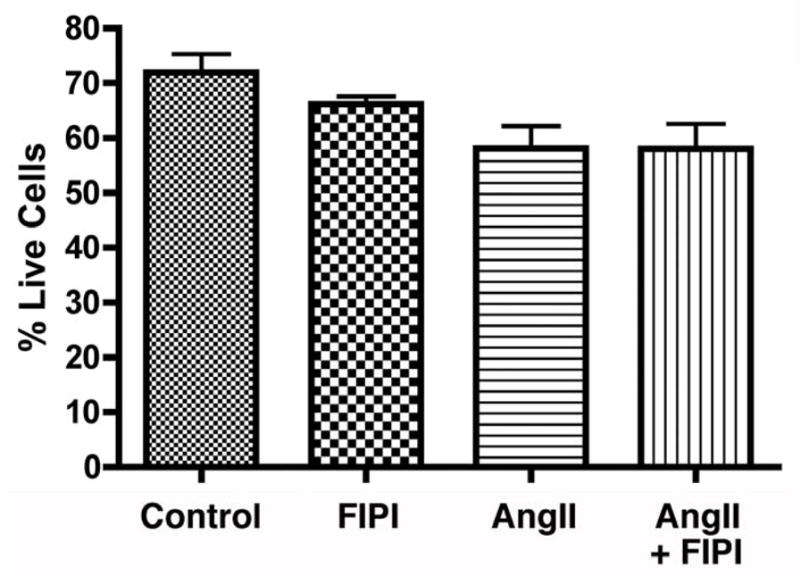
HAC15 cells were incubated for 24 hours with or without 750 nM FIPI (or the 0.1% DMSO vehicle control) in the presence or absence of 10 nM AngII in low-serum medium. Cells were then incubated with 0.4% trypan blue and the number of cells excluding trypan blue were counted using a T10™ Automated Cell Counter and expressed as the percentage of total cells. Values represent the means ± SEM of 3 separate experiments performed in triplicate; there were no significant differences between conditions.
4. DISCUSSION
In this report using two different methods of inhibiting PLD-mediated lipid signal generation, we have shown that AngII-stimulated PLD and its products are important for the activation of PKD by AngII in glomerulosa cell models. Thus, the primary alcohol 1-butanol diverts production away from physiological lipid signals to non-physiological phosphatidylbutanol thereby decreasing levels of phosphatidic acid and diacylglycerol. We have previously shown that 1-butanol inhibits the AngII-induced increase in phosphatidic acid and diacylglycerol levels, as well as aldosterone production (Bollag et al., 2002). Similarly, in H295R cells 1-butanol also inhibits aldosterone production in response to AngII (Zheng and Bollag, 2003). 1-Butanol also inhibited AngII-elicited PKD activation and aldosterone production in primary cultures of bovine ZG cells (Figure 1). Similarly, inhibition of AngII-stimulated PLD activity using the PLD inhibitor FIPI also decreased AngII-induced PKD activation and aldosterone production in these cells (Figures 2 and 3). This inhibition was not the result of non-specific cytotoxicity, as aldosterone production mediated by 22(R)-hydroxycholesterol was not affected. We also investigated the effect of FIPI on AngII-elicited PKD activation in another glomerulosa cell model, the HAC15 human adrenocortical carcinoma cell line. As in the primary bovine ZG cells, FIPI also inhibited PKD activation and aldosterone production in response to AngII in the HAC15 cells. Taken together, these results indicate that PKD is downstream of PLD-mediated lipid signal generation in AngII-stimulated adrenal glomerulosa cell models.
PLD activation in response to AngII has heretofore not been characterized in the HAC15 cell line, a derivative of the H295R cell line (Qin et al., 2010). We found that as in primary bovine ZG and H295R cells (Jung et al., 1998; Zheng and Bollag, 2003), AngII activated PLD in a dose-dependent manner with maximal stimulation at a concentration of 10 nM (Figure 4A). In contrast to the H295R cell line from which they were derived, however, HAC15 cells exhibited sustained PLD activation when stimulated with AngII (Figure 4B). Based on our data indicating the importance of PLD to aldosterone production (Bollag et al., 2002; Zheng and Bollag, 2003; Qin et al., 2010), it is perhaps then not surprising that these cells demonstrate such a robust aldosterone steroidogenic response to AngII (Wang and Rainey, 2012; Wang et al., 2012). Also based on previous reports of the involvement of PLD2 in AngII-induced steroidogenesis (Qin et al., 2010), it is interesting to speculate that the sustained PLD activation in HAC15 cells results perhaps from enhanced PLD2 expression and/or coupling to AngII-activated signal transduction mechanisms.
It should be noted that even the essentially complete inhibition of PLD activity by FIPI, as evidenced by the return of radiolabeled PEt levels to a near-basal value (Figures 2 and 5), only partially inhibited AngII-induced PKD activation. This result is consistent with the finding in other cell types (Waldron and Rozengurt, 2000; Storz et al., 2003; Qin et al., 2006; Arun et al., 2011) that PKD can be activated by multiple mechanisms, including Src tyrosine kinase family- and protein kinase C-mediated transphosphorylation [reviewed in (Rozengurt, 2011)]. Data from our laboratory indicate similar mechanisms of PKD activation in primary cultures of bovine ZG cells (manuscript in preparation). The existence of multiple and somewhat redundant mechanisms of PKD activation suggest the importance of this enzyme to the AngII-elicited aldosterone synthetic response. PKD also possesses two cysteine-rich C1 domains capable of binding DAG (or phorbol ester). Thus, PLD-generated DAG could potentially contribute to AngII-induced PKD activation in two ways: (1) by binding to the C1 domains in PKD to promote activation directly [reviewed in (Brose and Rosenmund, 2002)] and (2) by activating protein kinase C and triggering transphosphorylation [reviewed in (Rozengurt, 2011)], and thereby activation, of PKD indirectly. Whether or not the phosphatidic acid produced directly by PLD’s hydrolysis of phospholipids also plays a role in AngII-elicited PKD activation is at present unknown.
On the other hand, in other cell types phosphatidic acid is known to recruit and activate Raf-1 kinase upstream of the mitogen-activated protein kinase, extracellular signal-regulated kinase 1 and 2 (ERK-1/2) (Rizzo et al., 1999; Rizzo et al., 2000). In bovine ZG cells ERK-1/2 has also been shown to phosphorylate and activate cholesterol ester hydrolase (Cherradi et al., 2003), the enzyme which releases the aldosterone precursor cholesterol from its cholesterol ester storage form. In this way, PLD could potentially contribute to AngII-induced aldosterone production independently from its ability to activate PKD. Nevertheless, PLD inhibition by FIPI also induced only a small inhibition of aldosterone secretion acutely (Figure 3), although the inhibitor had a more pronounced effect on chronic production (Figure 6). This result indicates that AngII utilizes multiple non-overlapping signaling pathways to trigger its effects on aldosterone synthesis, as has been reviewed by several authors (Foster, 2004; Spät and Hunyady, 2004; Hattangady et al., 2012).
In summary, our results demonstrate that in adrenal glomerulosa cells, AngII-induced PLD activity and the lipid signals thus generated lie upstream of PKD and the cellular response, aldosterone production. These effects were demonstrated in two models of adrenal glomerulosa cells, primary cultures of bovine ZG and HAC15 human adrenocortical carcinoma cells, and using two inhibitors, 1-butanol and FIPI. Thus, our data argue for a mechanism by which PLD mediates aldosterone production in part through PKD activation in adrenal glomerulosa cells stimulated with AngII.
Highlights.
AngII induces sustained phospholipase D (PLD) activity in HAC15 adrenocortical cells
Two inhibitors of PLD signaling decrease AngII-induced protein kinase D activation
These inhibitors reduce AngII-induced aldosterone synthesis in glomerulosa cells
AngII-activated PLD is upstream of protein kinase D activation in glomerulosa cells
Acknowledgments
The authors gratefully acknowledge the generosity of Dr. William Rainey for providing the HAC15 cells as well as for his helpful suggestions and discussions. The authors also appreciate the expert technical assistance of Mr. Peter Parker and Ms. Mariya V. George. This research was supported by a VA Merit Award, a Georgia Health Sciences University Diabetes and Obesity Discovery Institute Synergy Award and National Institutes of Health # HL70046.
Footnotes
Abbreviations: AngII, angiotensin II; DAG, diacylglycerol; FIPI, fluoro-2-indolyl des-chlorohalopemide; KRB+, Bicarbonate-buffered Kreb’s Ringer containing 2.5 mM sodium acetate; PKD, protein kinase D; PLD, phospholipase D; ZG, zona glomerulosa
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arun SN, Kaddour-Djebbar I, Shapiro BA, Bollag WB. Ultraviolet B irradiation and activation of protein kinase D in primary mouse epidermal keratinocytes. Oncogene. 2011;30:1586–1596. doi: 10.1038/onc.2010.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag WB, Barrett PQ, Isales CM, Liscovitch M, Rasmussen H. A potential role for phospholipase-D in the angiotensin-II-induced stimulation of aldosterone secretion from bovine adrenal glomerulosa cells. Endocrinology. 1990;127:1436–1443. doi: 10.1210/endo-127-3-1436. [DOI] [PubMed] [Google Scholar]
- Bollag WB, Jung EM, Calle RA. Mechanism of angiotensin II-induced phospholipase D activation in adrenal glomerulosa cells. Mol Cell Endocrinol. 2002;192:7–16. doi: 10.1016/s0303-7207(02)00134-x. [DOI] [PubMed] [Google Scholar]
- Brindley DN, Pilquil C, Sariahmetoglu M, Reue K. Phosphatidate degradation: phosphatidate phosphatases (lipins) and lipid phosphate phosphatases. Biochim Biophys Acta. 2009;1791:956–961. doi: 10.1016/j.bbalip.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Rosenmund C. Move over protein kinase C, you’ve got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci. 2002;115:4399–4411. doi: 10.1242/jcs.00122. [DOI] [PubMed] [Google Scholar]
- Caloca MJ, Zugaza JL, Bustelo XR. Mechanistic analysis of the amplification and diversification events induced by Vav proteins in B-lymphocytes. J Biol Chem. 2008;283:36454–36464. doi: 10.1074/jbc.M803814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherradi N, Pardo B, Greenberg AS, Kraemer FB, Capponi AM. Angiotensin II activates cholesterol ester hydrolase in bovine adrenal glomerulosa cells through phosphorylation mediated by p42/p44 mitogen-activated protein kinase. Endocrinology. 2003;144:4905–4915. doi: 10.1210/en.2003-0325. [DOI] [PubMed] [Google Scholar]
- Cowell CF, Doppler H, Yan IK, Hausser A, Umezawa Y, Storz P. Mitochondrial diacylglycerol initiates protein-kinase D1-mediated ROS signaling. J Cell Sci. 2009;122:919–928. doi: 10.1242/jcs.041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings R, Parinandi N, Wang L, Usatyuk P, Natarajan V. Phospholipase D/phosphatidic acid signal transduction: role and physiological significance in lung. Mol Cell Biochem. 2002:234–235. 99–109. [PubMed] [Google Scholar]
- Foster RH. Reciprocal influences between the signalling pathways regulating proliferation and steroidogenesis in adrenal glomerulosa cells. J Mol Endocrinol. 2004;32:893–902. doi: 10.1677/jme.0.0320893. [DOI] [PubMed] [Google Scholar]
- Hattangady NG, Olala LO, Bollag WB, Rainey WE. Acute and chronic regulation of aldosterone production. Mol Cell Endocrinol. 2012;350:151–162. doi: 10.1016/j.mce.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung EM, Betancourt-Calle S, Mann-Blakeney R, Foushee T, Isales CM, Bollag WB. Sustained phospholipase D activation in response to angiotensin II but not carbachol in bovine adrenal glomerulosa cells. Biochem J. 1998;330:445–451. doi: 10.1042/bj3300445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam Y, Exton JH. Role of phospholipase D in the activation of protein kinase D by lysophosphatidic acid. Biochem Biophys Res Commun. 2004;315:139–143. doi: 10.1016/j.bbrc.2004.01.034. [DOI] [PubMed] [Google Scholar]
- Matthews SA, Rozengurt E, Cantrell D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/protein kinase Cμ. J Biol Chem. 1999;274:26543–26549. doi: 10.1074/jbc.274.37.26543. [DOI] [PubMed] [Google Scholar]
- Parmar J, Key RE, Rainey WE. Development of an adrenocorticotropin-responsive human adrenocortical carcinoma cell line. J Clin Endocrinol Metab. 2008;93:4542–4546. doi: 10.1210/jc.2008-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Frohman MA, Bollag WB. Phospholipase D2 mediates acute aldosterone secretion in response to angiotensin II in adrenal glomerulosa cells. Endocrinology. 2010;151:2162–2170. doi: 10.1210/en.2009-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Zeng H, Zhao D. Requirement of protein kinase D tyrosine phosphorylation for VEGF-A165-induced angiogenesis through its interaction and regulation of phospholipase Cγ phosphorylation. J Biol Chem. 2006;281:32550–32558. doi: 10.1074/jbc.M604853200. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Shome K, Vasudevan C, Stolz DB, Sung T-C, Frohman MA, Watkins SC, Romero G. Phospholipase D and its product, phosphatidic acid, mediate agonist-dependent Raf-1 translocation to the plasma membrane and the activation of the mitogen-activated protein kinase pathway. J Biol Chem. 1999;274:1131–1139. doi: 10.1074/jbc.274.2.1131. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Shome K, Watkins SC, Romero G. The recruitment of raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with ras. J Biol Chem. 2000;275:23911–23918. doi: 10.1074/jbc.M001553200. [DOI] [PubMed] [Google Scholar]
- Rozengurt E. Protein kinase D signaling: multiple biological functions in health and disease. Physiology (Bethesda) 2011;26:23–33. doi: 10.1152/physiol.00037.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro BA, Bollag WB. Angiotensin II signaling in the adrenal cortex: a role for protein kinases C and D in acute aldosterone secretion? In: Miura H, Sasaki Y, editors. Angiotensin Research Progress. NovaScience Publishers, Inc; Hauppauge, New York: 2008. pp. 1–33. [Google Scholar]
- Shapiro BA, Olala L, Arun SN, Parker PM, George MV, Bollag WB. Angiotensin II-activated protein kinase D mediates acute aldosterone secretion. Mol Cell Endocrinol. 2010;317:99–105. doi: 10.1016/j.mce.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda H, Okada T, Jahangeer S, Nakamura S. Requirement of phospholipase D for ilimaquinone-induced Golgi membrane fragmentation. J Biol Chem. 2007;282:34085–34092. doi: 10.1074/jbc.M705593200. [DOI] [PubMed] [Google Scholar]
- Spät A, Hunyady L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev. 2004;84:489–539. doi: 10.1152/physrev.00030.2003. [DOI] [PubMed] [Google Scholar]
- Storz P, Doppler H, Johannes F-J, Toker A. Tyrosine phosphorylation of protein kinase D in the pleckstrin homology domain leads to activation. J Biol Chem. 2003;278:17969–17976. doi: 10.1074/jbc.M213224200. [DOI] [PubMed] [Google Scholar]
- Su W, Yeku O, Olepu S, Genna A, Park JS, Ren H, Du G, Gelb MH, Morris AJ, Frohman MA. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol Pharmacol. 2009;75:437–446. doi: 10.1124/mol.108.053298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron RT, Rozengurt E. Oxidative stress induces protein kinase D activation in intact cells. J Biol Chem. 2000;275:17114–17121. doi: 10.1074/jbc.M908959199. [DOI] [PubMed] [Google Scholar]
- Wang T, Rainey WE. Human adrenocortical carcinoma cell lines. Mol Cell Endocrinol. 2012;351:58–65. doi: 10.1016/j.mce.2011.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Rowland JG, Parmar J, Nesterova M, Seki T, Rainey WE. Comparison of aldosterone production among human adrenocortical cell lines. Horm Metab Res. 2012;44:245–250. doi: 10.1055/s-0031-1298019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Bollag WB. AngII induces transient phospholipase D activity in the H295R glomerulosa cell model. Mol Cell Endocrinol. 2003;206:113–122. doi: 10.1016/s0303-7207(03)00211-9. [DOI] [PubMed] [Google Scholar]