

Abstract
Human diseases are caused by alleles that encompass the full range of variant types, from single-nucleotide changes to copy-number variants, and these variations span a broad frequency spectrum, from the very rare to the common. The picture emerging from analysis of whole-genome sequences, the 1000 Genomes Project pilot studies, and targeted genomic sequencing derived from very large sample sizes reveals an abundance of rare and private variants. One implication of this realization is that recent mutation may have a greater influence on disease susceptibility or protection than is conferred by variations that arose in distant ancestors.
Genetic Contributions to Disease
Common chronic diseases such as diabetes, coronary heart disease, stroke, neuropsychiatric illness (including schizophrenia, autism, and developmental disabilities), chronic respiratory disease, and cancer account for an overwhelmingly large fraction of mortality, morbidity, and health care expenditure (http://www.cdc.gov/nchs/). These diseases disproportionately affect aging populations and burden the health care systems and economies of industrialized nations throughout the world. Understanding the underlying causes of such disorders is a key step toward enabling earlier and more precise diagnosis, prognosis, interventional therapy, and potentially prevention.
Most common diseases are complex or multifactorial with both environmental and genetic contributions along with their nearly intractable interaction effects. In general, the environmental components are challenging to identify and quantitate. In contrast, as a result of the emergence of powerful genomic technologies, the analysis of the genetic components is becoming increasingly tractable and relatively inexpensive to investigate. These technical improvements have fueled a pipeline of discovery of the genes and variants that predispose to human maladies. The technical improvements have also impacted genetic diagnostics, as it is now practical to sequence an entire individual’s genome for less than the cost of a comprehensive set of whole-body imaging scans. Furthermore, the cost of whole-genome sequencing is rapidly becoming less expensive than current clinically implemented “multigene panel testing” for molecular diagnosis of disease traits with even modest genetic heterogeneity.
Allele Frequency Distributions and Human Disease
A “common disease/common variant” (CDCV) hypothesis has been popularized as an explanation for common disorders and has garnered much support (Reich and Lander, 2001). This model presupposes that different combinations of common alleles aggregate in specific individuals to increase disease risk. The CDCV hypothesis was a major intellectual impetus for the International Haplotype Mapping (HapMap) project and ultimately led to a proliferation of genome-wide association studies (GWAS) identifying regions influencing disease status or risk factor levels (http://www.genome.gov/gwastudies). As a consequence, insights into potential new pathways underlying common disease have emerged.
Manolio et al. (2009) suggested that the genetic variance explained per se is of interest for what it might suggest about effective research paradigms. Accounting for the genetic variance is not the same as achieving utility and impact. If the goal of our shared research program is to achieve mechanistic understanding and lessen the impact of human disease, then the magnitude of the genetic effect is less important than the possible insight provided by the newly identified loci. Genes identified through their weakly acting common alleles may give important clues about pathways and/or be excellent targets for therapeutic and preventive strategies.
However impressive the information gleaned from GWAS has been, the results have explained only a few percent of the apparent genetic variance contributing to common diseases. Furthermore, these studies have not yet delivered medically actionable variants that inform medical decision making by helping to establish an etiological diagnosis and lead to a more efficacious treatment or prevention plan. Both diagnostic utility and classification of pathogenicity are closely associated with the magnitude of each variant-specific effect. For most complex diseases, unless a variant clearly partitions the affected into distinct biological subgroups or can be incorporated into risk-prediction models, there is likely to be limited diagnostic usefulness. Morrison et al. (2007) proposed the use of a composite “genetic risk score” for risk assessment. However, it remains to be determined whether variants identified by GWAS have a role as biomarkers in risk assessment and clinical decision making. Overall, these data do not support a simple additive version of the CDCV model as an explanation for the majority of the genetic component underlying risk for common disease.
Research efforts have shifted to exploration of less frequent variants in common disorders. It has been noted for decades that the mutational changes that underlie rare and highly penetrant Mendelian disease may share features with genetic factors that underlie more common forms of the disease (Boerwinkle and Utermann, 1988; Goldstein and Brown, 2001). Clearly, the relationship between rare and common disease is not a simple one, but there are emerging examples wherein specific loci that cause “Mendelian disease” are contributing to the background risk to a parallel common disorder. Although a common disease/rare variant (CDRV) hypothesis is attractive, it demands tenable and complete explanations as to how the functional roles of individual alleles can work to produce the ultimate phenotypic effects. The models must consider the range of variant types, including single-base or simple-nucleotide variants (SNV), short insertions or deletions (indels), structural variants, and copy-number variants (CNV), the penetrance of individual alleles, and allelic and locus interactions (dominance and epistasis, respectively) and show how these all combine to produce the population frequency and the phenotypic complexity of different disorders.
Fortunately, the current state of knowledge of key examples supports models that close the gap between complex and Mendelian traits. These examples show how mutations in single genes can fulfill the definition of Mendelian disease—but in different context are parts of the menu of causal contributors to complex disorders (Greeley et al., 2010; Voight et al., 2010). As we begin to observe instances wherein variation at more than one locus contributes to perturbations of networks and ultimate phenotype, the relevance of assessing genome-wide variation becomes more apparent. Thus, inferences about individual mutation burden by geneticists in the last century are now open to direct observation (Muller, 1950).
Variation and Disease Susceptibility—We Are All Truly Unique
The interplay between different types of variation and their contribution to disease are highly dependent on our understanding of the normal patterns of genetic variation. For rare variants, this has been a particular challenge, as highly accurate data need to be generated from many samples in order to properly determine the frequency and population distribution of the genetic variants. To illustrate this: the successful HapMap project (International HapMap 3 Consortium, 2010; International HapMap Consortium, 2005; Frazer et al., 2007) that provided an early survey of single-base variation across major human populations cataloged only a fraction of the genetic variation above a frequency of 5%. Even the 1000 Genomes Project pilot studies comprehensively captured only variation at greater than 1% frequency (1000 Genomes Project Consortium, 2010).
Our view of the site frequency spectrum of these rare variants (<1%) has been more influenced recently as a result of the generation of personal genome data using whole-exome sequencing and whole-genome sequencing (Gonzaga-Jauregui et al., 2011). The number of diploid human genome sequences available for analyses is growing rapidly. Remarkably, from the small number determined and publicly available to date, it is apparent that even more genetic variation exists between individuals than was previously expected (Ahn et al., 2009; Bentley et al., 2008; Kim et al., 2009; Levy et al., 2007; Lupski et al., 2010; Schuster et al., 2010; Wang et al., 2008; Wheeler et al., 2008). When compared with the haploid reference, each individual human genome on average contains some three and a half million SNV and about 1,000 CNV (>450 bp) (Conrad et al., 2010), many of which appear to be rare in the population from which the individual was sampled. In addition, each individual personal genome sequence still reveals 200,000–500,000 SNV that have not been observed in other publicly available personal genomes, many of which may be unique to that individual’s family or clan. In parallel, recent studies that deeply sequence relatively large samples (hundreds to thousands of individuals) show that the rate of identification of variants that have not been seen (private variants) continues unabated with every new individual sampled (Coventry et al., 2010; Turner et al., 2008). The extent of some of this nucleotide variation may have been anticipated from human genetic studies during the previous three decades that established that an SNV occurs about every 1 Kb; however, the extent of rare and “private” SNV was not anticipated, and the extent of CNV was unexpected. There are technical limitations to some of these studies—including a background of variation introduced in cultured cells—as well as in the mutation detection methods themselves, particularly for CNV in the 100 bp to 500 bp range, low-copy repeat sequences, and simple repeats. Nevertheless, the enormous extent of private variation has been clearly established.
Rare Variants and New Mutation
A number of factors may have led to the observed skewing of the allele frequency spectrum toward rare and private variants. The explosion of human populations in the current historical epoch could, by itself, account for the short branch lengths and low frequencies of the most distal segments of human variant genealogies (Boyko et al., 2008; Coventry et al., 2010; Turner et al., 2008). In addition, secular factors that have enabled the explosion of the population, such as abundant food supplies, improved sanitation, and routine vaccinations, may directly participate in the relaxation of the most important selective pressures that have constrained the population in the past. Even the widespread availability of minimal routine health care may be artificially slowing negative selection. Dramatic reductions in maternal death and infant mortality, properly celebrated in the last 100 years, may be influencing the distribution of genetic variation and contribute to relaxed selection. Finally, mutation rate, perhaps partially driven by increased paternal age (Crow, 2008) and undiscovered environmental factors, may contribute to the observed rare variant spectra.
The conceptual shift to emphasizing studies of abundant, rare, and heterogeneous variants profoundly impacts our approaches to studying the genetic architecture of human disease, leading to a genome-wide, versus a locus- or gene-specific, emphasis. Genetic architecture here refers to the types of variation (SNV, CNV, etc., both coding and noncoding), their allele frequency distribution (common, rare, intermediate), the size of an allele’s effects, and new mutation rates. For a given individual, what is important to know is not only the number and location of pathogenic variants taken one at a time but also the unique composition of his or her genome-wide mutational burden. If this is the case, then the risk conferred by any particular allele estimated from the population risk would be much less relevant than the personal risk emerging from the total mutational burden in each individual. The shift in emphasis to a whole-genome view changes how we should consider the way in which harmful combinations of mutant alleles assemble or accumulate in each genome. Each personal genome has a collection or “ecology” of deleterious and protective variations, which in combination (not necessarily in sum) dictate the health of the individual. Understanding this genome ecology will be a substantial challenge in human genetics and has ramifications for the extent to which genetic information can be maximized for medical utility.
Each personal genome combines inherited alleles and new variation introduced by de novo mutation. Interestingly, CNV may contribute in a significant way, from both the novel combinations inherited from each parent and the new mutations. This is the very type of variation not fully taken into account when previous mutation models were being considered. Locus-specific mutation rates for SNV are 2.0–2.5 × 10−8 and have recently been shown to potentially differ in male versus female germ cells (Conrad et al., 2011); for CNV, new mutation rates can be substantially higher: between 10−6 and 10−4, 100 to 10,000 times more frequent than in SNV (Lupski, 2007a). The latter figures implicate CNV in sporadic traits (Lupski, 2007a) including birth defects (Lu et al., 2008) and highlight the contribution of new mutation to individual mutational burden (Potocki et al., 1999). Either new or recent (i.e., arising in close relatives or “clan members”) de novo mutations could substantially contribute to phenotypic extremes, such as birth defects and disease.
Although de novo CNV have been detectable now for some time with microarray technologies, identifying smaller de novo events (e.g., SNV) has become feasible only recently with the advent of large-scale DNA sequencing technologies. Recent exome sequencing studies of family trios with patients manifesting sporadic intellectual disability (previously more frequently referred to as mental retardation [MR]) identified a high frequency of de novo mutations in “MR genes” (Vissers et al., 2010). Such studies support established theory that if the mutational target is large (and hence the observed gene mutation rate is high), de novo mutations may account for a high incidence of disease even when the selection coefficient is close to 1.0. These early studies suggest a resolution to the question of why the frequency of neurodevelopmental disabilities is high despite near genetic lethality for such traits. Relatedly, sequencing studies of multiple ion channel genes in patients with epilepsy (Klassen et al., 2011) and of known autism susceptibility genes in subjects with high-functioning autism (Schaaf et al., 2011) reveal many rare variants and also de novo mutations that may be contributing to disease.
The concept of new mutation in X-linked lethal disorders was well established by Haldane (Haldane, 1935). However, the new mutation contribution to many human disease traits may be greater than anticipated (Hoischen et al., 2010), particularly for genetically heterogeneous conditions in which hundreds of genes could be involved but only one or a few loci are responsible in an individual patient. The developmental timing of new somatic mutations is perhaps underappreciated (Lupski, 2010) as previous studies have emphasized germline events. New mutations may occur in the germline, during any stage of development of the organism, in stem cells, or in differentiated somatic cells.
Chromothripsis in cancer (Stephens et al., 2011) and complex genomic rearrangements (CGR) associated with selected genomic disorders (Liu et al., 2011) both illustrate the potential gene(s) alteration—complexities that can be brought about by new mutation CNV events. In each case, a single mutational event can result in a cataclysmic chromosomal catastrophe and alter the copy number or structure of several different genes.
“Clan Genomics”
Most sites of variation have low minor allele frequencies (that is, are rare) and are of recent origin, and therefore the major contributors to inherited disease susceptibility are likely to be those alleles that arose recently in an extended pedigree. Purifying natural selection is expected to eliminate highly deleterious variants before they reach a high frequency, such that disease risk alleles with large effects should be enriched at the lower frequencies (Marth et al., 2011). The idea that there are unique combinations of rare variants characteristic of a recent family lineage and that these combinations can have a causative role in disease is encapsulated by what we refer to as “clan genomics” (Figure 1). The population from which one comes and its collection of older common variants may have less influence on an individual’s disease susceptibility than the collection of recently arisen rare variants and de novo mutations (Figures 2A and 2B). The most important thing that an individual needs to consider in terms of their genetic variation with relation to disease susceptibility is therefore recent “genetic history” of their extended pedigree or clan. From the standpoint of delivering personalized genomic medicine, the medically actionable alleles are the ones of most interest; and these may be highly weighted toward recent rare variants.
Figure 1. Clan Genomics.
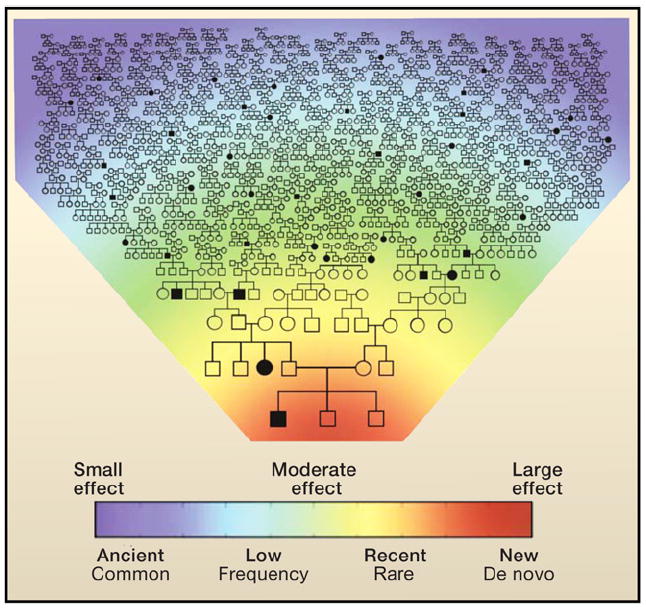
Heat map and extended pedigree showing the conceptual relationship among de novo mutations leading to disease (red), recent mutations with moderate effects arising within a clan (yellow and green), and older common variants with small effects segregating in the population (blue). An individual’s genetic disease risk emerges from the collection of variants he or she has inherited from both parental lineages of distant ancestors (typically common and of individually small effect), more recent ancestors (rare, but potentially larger effect), and de novo mutations.
Figure 2. Phenotypic Consequences of Allele Combinations.

This figure demonstrates “clan genomics,” wherein the combinations of alleles one inherits from his or her nearest relatives profoundly affect clinical outcome. In these illustrative pedigrees, different combinations of ABCA4 alleles can affect age-of-onset of Stargardt macular dystrophy (STGD; MIM 601691) (A), Mendelian versus multifactorial trait (i.e., Stargardt disease versus age-related macular degeneration [AMD]) (B), or retinal disease type (i.e., Stargardt disease versus retinitis pigmentosa [RP; MIM 601718]) (C).
(D) Differing SH3TC2 alleles result in recessive Charcot-Marie-Tooth disease (CMT; MIM 601596), dominant axonal neuropathy, or the complex trait of carpal tunnel syndrome.
Nevertheless, the most important thing is not to focus disproportionately on specific variants, but rather to integrate across all classes of risk-associated variants. In some individuals, risk may be caused by an unusual combination of common variants, whereas in others it will be due to a smaller number of large effect rare variants.
Mendelian Disease and Complex Traits
Resequencing studies of genes that can cause rare Mendelian forms of common complex traits reveal that rare variants can contribute to hypertension (Ji et al., 2008; Wagner, 2008), hypercholesterolemia (Kotowski et al., 2006), hypertriglyceridemia (Romeo et al., 2009), and nonalcoholic fatty liver disease (Romeo et al., 2008) in the population at large. These examples inform models where individual alleles with high penetrance contribute to common complex traits. In addition, when GWAS signals have identified variants for common traits, their molecular mechanistic underpinnings often support those already established by Mendelian forms of the condition (Sankaran et al., 2008, 2009; Vernimmen et al., 2009).
The idea that genes responsible for Mendelian disease can also have a role in the common form of the same or a similar condition is not new. For example, the pioneering studies of Michael Brown and Joseph Goldstein showed that individuals with compound heterozygous mutations in the low-density lipoprotein receptor (LDLR) gene manifest the Mendelian disorder familial hypercholesterolemia (FH) (Brown and Goldstein, 1986; Goldstein and Brown, 1987). FH patients have extremely high cholesterol levels and can have coronary atherosclerotic heart disease and myocardial infarctions in their teenage years. Interestingly, the type of LDLR gene mutation predicts cardiovascular risk in children with familial hypercholesterolemia (Guardamagna et al., 2009). Heterozygous rare variant mutations at the LDLR locus can also cause the complex traits of early onset hypercholesterolemia, coronary atherosclerotic heart disease, and myocardial infarctions in carriers with disease manifesting in the fourth or fifth decades of life.
Recessive Mendelian Mutations Can Increase Complex Disease Risk in Carriers
Heterozygous carriers for recessive disease genes do not manifest the recessive disease but may be susceptible to a milder or related malady, which may consist of a complex trait with a similar phenotype. For example, heterozygote carriers of mutations in the ataxia telangiestasia locus are susceptible to breast cancer (Athma et al., 1996), and similar heterozygous carrier susceptibilities are also manifest for other recessive human cancer predisposition syndromes (Heim et al., 1991). Carriers for mutations in the Gaucher disease causative gene, GBA encoding glucocerebrosidase, are at increased risk for Parkinson disease (Goker-Alpan et al., 2004; Sidransky et al., 2009). Heterozygous carriers of mutations in the cystic fibrosis transmembrane regulator gene, CFTR, can be susceptible to idiopathic pancreatitis (Cohn et al., 1998; Sharer et al., 1998; Weiss et al., 2005), chronic obstructive pulmonary disease (COPD) (Divac et al., 2004), and even chronic rhinosinusitis (Wang et al., 2000, 2005). Carriers of α-1-antitrypsin (AAT) deficiency can also be susceptible to COPD (Hersh et al., 2004; Poller et al., 1990). Interestingly, even such common traits as age-related macular degeneration (AMD) and carpal tunnel syndrome are associated with heterozygous carrier status for mutations in ABCA4, the gene responsible for Stargardt macular dystrophy (Bacq et al., 2009), and Charcot-Marie-Tooth neuropathy genes (Lupski et al., 2010), respectively (Figures 2C and 2D). In the latter case, haploinsufficiency due to either heterozygous SNV (Lupski et al., 2010) or heterozygous CNV (Del Colle et al., 2003) can convey the trait. Whereas most carrier states may have rare allele frequencies, others will actually have a significant carrier frequency in selected populations (e.g., CFTR ~4% in European descendants).
Genes and Single Loci Implicated in Mendelian Disease and in Complex Disease Risk
In addition to variants that cause Mendelian disease-informing complex traits, there is a striking reciprocity of genes implicated by GWAS that are also known to underlie rare Mendelian diseases. For example, 11 of 30 genes associated with serum lipid levels are implicated in single-gene disorders of lipid metabolism (Kathiresan et al., 2009). We reviewed the current listing of annotated genes with significant associations in 891 GWAS studies (http://www.genome.gov/gwastudies/). We found that at least 268 genes implicated by GWAS are also known to bear mutations in rare single-gene disorders. Some of these associations are intuitive, such as those associated with biochemical traits and related inborn errors of metabolism. There are also a significant number of genes that underlie developmental disorders that harbor common variants affecting risk of cancer, body growth, and cardiovascular traits (Table S1 available online). This raises the testable hypothesis that genetic influences on human diseases can largely be accounted for by a subset of genes that play roles in a restricted set of pathways. Immune and inflammatory pathways provide a robust example as do those genes involved in lipid metabolism. It is important to note that in our survey, for most cases of GWAS the causal gene underlying a given GWAS signal is unknown.
Whereas GWAS can indirectly implicate “Mendelian genes” in complex disease risk, different mutations of a single gene, or a CNV at a single locus, are directly implicated in complex disease risk. A poignant example of the different phenotypic consequences of distinct allelic variants at a locus is provided by the fragile X mental retardation 1 (FMR1) locus. Triplet repeat expansion of the CGG repeat element in the 5′ untranslated region (UTR) of the FMR1 gene—an especially unstable form of indel mutation—causes severe X-linked mental retardation in both males and females. Alleles with lower numbers of CGG repeats (55–200 repeats; called premutation alleles), however, cause adult onset tremor/ataxia syndrome (FXTAS) in approximately 33% of males and 10% of females (Hagerman et al., 2004; Jacquemont et al., 2004). Thus, premutation variants that have been considered nonpathogenic can have phenotypic consequences for common complex traits such as tremor and ataxia.
Rare CNV at different loci have also recently been associated with complex traits including Alzheimer disease (Rovelet-Lecrux et al., 2006), Parkinson disease (Farrer et al., 2004; Singleton et al., 2003), lupus glomerulonephritis (Aitman et al., 2006), Crohn disease (Fellermann et al., 2003, 2006; McCarroll et al., 2008), psoriasis (Hollox et al., 2008), pancreatitis (Le Maréchal et al., 2006), and obesity (Bochukova et al., 2010). Many rare CNV have also been associated with intellectual disability (Stankiewicz and Beaudet, 2007) and with some forms of neuropsychiatric illness, including schizophrenia (Consortium, 2008; Lupski, 2008; McCarthy et al., 2009; Stefansson et al., 2008) and autism (Kumar et al., 2008; Shinawi et al., 2010; Weiss et al., 2008).
Same Gene, Different Mutations, Diseases, and Modes of Inheritance
A further illustrative example of the connection between Mendelian and complex traits is provided by variants at the MECP2 locus and their contribution to disease. Heterozygous loss-of-function SNV in MECP2 result in the X-linked dominant trait of Rett Syndrome in girls (Amir et al., 1999); however, hemizygous loss-of-function mutations are thought to be lethal in males. Recently, duplication of CNV including MECP2 has been associated with an intellectual disability plus seizure disorder in males (Carvalho et al., 2009; del Gaudio et al., 2006; Friez et al., 2006; Meins et al., 2005; Van Esch et al., 2005) and autism spectrum disorder (Ramocki et al., 2009; Schaaf et al., 2011). Male patients with triplication of MECP2 have a more severe phenotype (del Gaudio et al., 2006). Of note, maternal carriers of the MECP2 duplication (CNV) appear more susceptible to psychiatric symptoms unrelated to having a child with a disability (Ramocki et al., 2009). Thus, at a single locus, the genetic variation can cause an X-linked dominant disorder in females and an X-linked recessive trait in males and can be associated with susceptibility to a common complex trait in carrier mothers.
For mutations at a single locus, allelic interactions can profoundly affect clinical phenotype. At the ABCA4 locus, the disease severity is related to the residual activity of encoded transporter protein (Figures 2A–2C). Recessive Stargardt disease is caused by compound heterozygous mutations at this locus (Allikmets et al., 1997b). Homozygous or compound heterozygous mutations, if both null, result in retinitis pigmentosa. Within a single pedigree or clan, different combinations of alleles can result in differing ages of onset (Lewis et al., 1999), completely different diseases (Shroyer et al., 2001a), or both a recessive Stargardt disease and susceptibility to a complex trait, age-related macular degeneration, due to a heterozygous carrier state (Figure 2) (Shroyer et al., 1999, 2001b).
Multiple Mutated Genes Underlying Clinical Phenotypes
Rare point mutations (either functional noncoding SNV [Kurotaki et al., 2005] or coding SNV with incomplete penetrance [Shy et al., 2006]) in combination with a deletion CNV have been shown to contribute together to particular phenotypes. A combination of a rare deletion CNV with a de novo duplication CNV can also result in a phenotype that appears to be a complex trait (Potocki et al., 1999). Sometimes SNV mutations at two different loci, i.e., digenic inheritance, are required to manifest a trait segregating as a recessive disease, and the mutational load required may have a single mutant allele at each of the two loci (Kajiwara et al., 1994) (double heterozygous) or two mutant alleles at one locus and one at the other (triallelic inheritance) (Figure 3) (Katsanis et al., 2001). With respect to models for Mendelian transmission, a deletion CNV renders a locus monoallelic, whereas a duplication CNV results in a triallelic locus (Figure 3).
Figure 3. Models of Disease Allele Transmission.
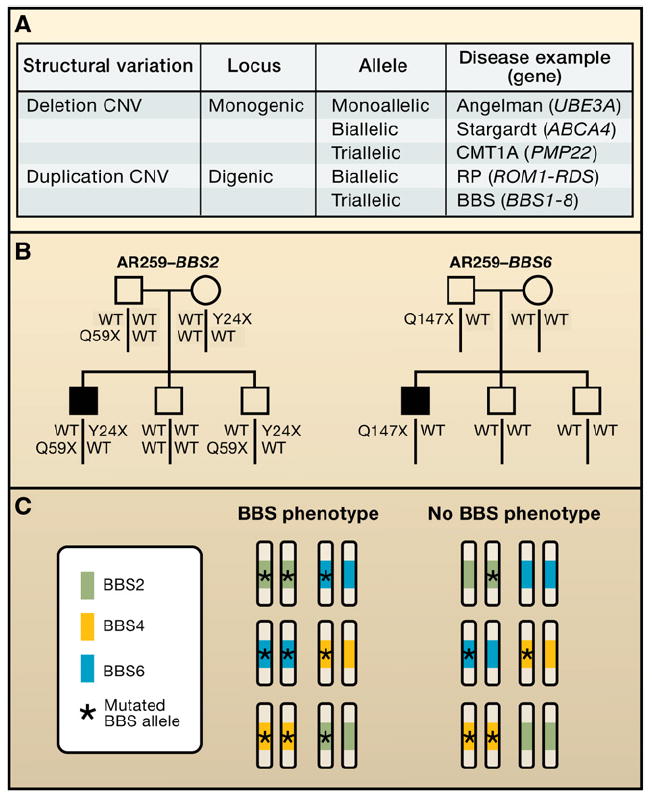
(A) In classical Mendelian disease, for a recessive, monogenic disease, at that single locus there is biallelic inheritance (highlighted in box). Examples could be either Stargardt macular dystrophy or cystic fibrosis, which are both due to point mutations in ATP-binding cassette (ABC) transporter genes. However, at some loci in the human genome, imprinting results in monoallelic expression, and the disease phenotype will occur in a manner dependent on the parent of origin of the specific mutation, either by deletion copy number variants (CNV) or uniparental disomy (UPD). The example given is the Angelman syndrome with point mutations in the UBE3A gene. The CMT1A locus (17p12) represents a triallelic locus whereby because of the duplication, there are three copies of the PMP22 gene. None of the copies have point mutations in them, but it takes three copies to convey the clinical phenotype. Other examples of disease allele transmission include interactions between two or potentially more genes. In the classic model of digenic inheritance, the phenotype of retinitis pigmentosa has been shown to be due to heterozygous point mutations in the ROM1 gene in combination with heterozygous point mutations at the RDS locus. Thus there is biallelic digenic inheritance. Note that a genomic deletion CNV renders a locus monoallelic, whereas a duplication CNV results in a triallelic locus.
(B) Bardet-Biedl syndrome (BBS), traditionally thought of as a recessive trait, can sometimes result from three mutant alleles, two of which come from one locus, and one from another locus. This is an example of digenic triallelic inheritance.
(C) A single pedigree illustrates triallelic inheritance for BBS. Standard pedigree symbols are used; filled squares, affected with BBS. Alleles segregating at two distinct loci (BBS2 and BBS6) are shown, one in each pedigree. WT, wild-type or normal allele.
It is now well established that even simple Mendelian traits can have modifier loci (Badano and Katsanis, 2002; Dipple and McCabe, 2000), demonstrating the potential importance of nonhomologous allelic interaction and epistasis. For example, severity of disease for CMT can be due to a combination of mutations at more than one CMT locus (Chung et al., 2005; Hodapp et al., 2006; Meggouh et al., 2005) (Figures 4A–4C).
Figure 4. Totality of Pathogenic Variants, Disease Severity, and Clan Genomics.
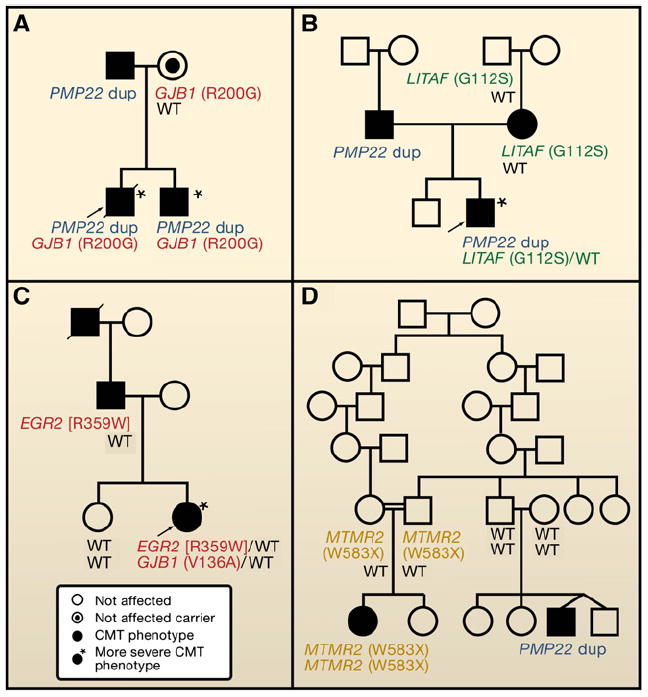
Pedigrees of families segregating Charcot-Marie-Tooth (CMT) neuropathy, illustrating that disease severity is directly related to pathogenic mutational burden.
(A–C) Mutations at two different CMT loci result in a more severe phenotype. These double heterozygotes may be due to either a single-nucleotide variant (SNV) + copy-number variant (CNV) (A and B) or two SNV (C) (Chung et al., 2005; Hodapp et al., 2006; Meggouh et al., 2005).
(D) In a single family, disease results from homozygous MTMR2 mutation (likely related to consanguinity) or de novo CNV—the CMT1A duplication (PMP22) (Verny et al., 2004); an example of clan genomics.
Large CNV or Aneuploidy Can Simultaneously Affect Multiple Genes
In contrast to the trans-genetics of Mendelism (Figures 2-4), genetic interactions occurring on the same chromosome or in cis (Figures 5A–5B) can also have profound consequences as exemplified at the alpha globin locus. For structural variants, the genomic mutational load can reflect the size of the CNV and inclusion of additional dosage-sensitive genes or genomic segments in cis (Bi et al., 2009; Lupski et al., 1991, 1992; Roa et al., 1996). Two extreme examples of this “cis-genetics” effect are segmental aneuploidy (Figure 5) and complete aneuploidy (e.g., trisomy 21) that convey complex phenotypes related to the size of the CNV and number of dosage-sensitive genes and/or genomic segments involved. For Down syndrome associated with trisomy 21, this includes an endophenotype of early onset Alzheimer disease; the amyloid precursor protein (APP) gene maps to chromosome 21, and duplications involving this gene have indeed been associated with Alzheimer disease (Rovelet-Lecrux et al., 2006).
Figure 5. Bridging the Gap between Chromosomal Syndromes and Mendelian Disorders.
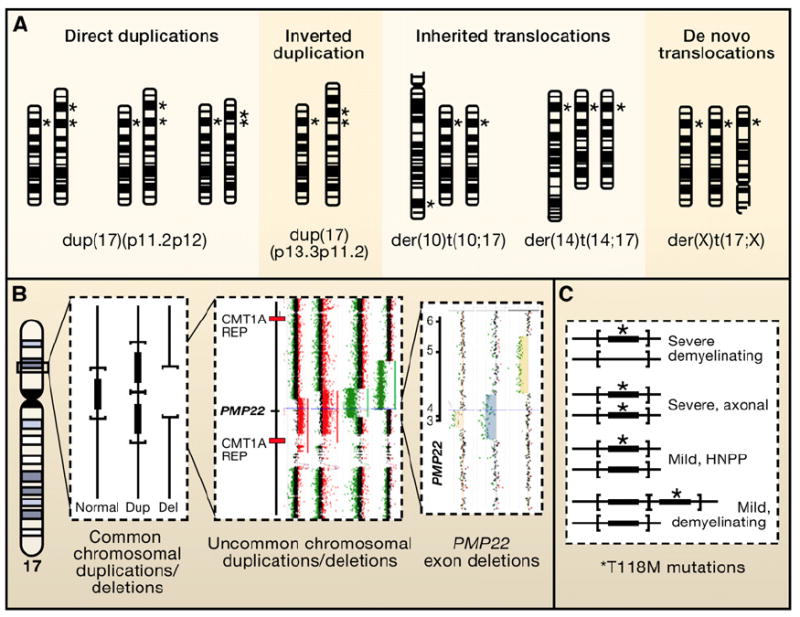
(A) Chromosomal duplication mapping wherein chromosomally visible duplication abnormalities, as evidenced by altered G-banding patterns, are used to delineate the portion of the genome responsible for the reduced motor nerve conduction velocities that accompany the demyelinating form of Charcot-Marie-Tooth disease (CMT1A; MIM 118220). Several different chromosomal abnormalities have been reported in association with a CMT1 phenotype. Note, different chromosome 17 abnormalities including direct duplications, inverted duplications, and inherited as well as de novo translocations have been reported with complex phenotypes that include CMT. If the duplicated genomic interval encompasses the 17p12 dark G-band where the PMP22 gene maps (*), then the patient will have a demyelinating neuropathy, as evidenced by decreased motor nerve conduction velocities, as part of their clinical phenotype.
(B) Submicroscopic genomic rearrangements associated with neuropathy. Vertical lines represent a “blow-up” of the genomic interval within 17p12 containing the PMP22 gene (filled rectangle). The horizontal parentheses delimit the rearranged interval for the common deletion (depicted by absence of vertical line) and duplication (two copies of gene and interval). To the right are rare-sized copy-number variants (CNV) depicting genomic deletion (green dots on array CGH) versus duplication (red dots on array).
(C) shows genotype/phenotype correlations between PMP22 point mutations associated with neuropathy. The T118M missense amino acid substitution in PMP22 appears to be a reduced penetrance loss-of-function allele. As a heterozygous mutation it can result in a mild hereditary neuropathy with liability to pressure palsies (HNPP) phenotype in some individuals; as a homozygous allele it can convey a severe axonal neuropathy. Interestingly, when the T118M allele occurs in combination with the HNPP deletion, a severe demyelinating phenotype results. Of further interest, when the T118M allele occurs in combination with the CMT1A duplication, the loss-of-function missense amino substitution appears to mitigate some of the consequences of the gain-of-function duplication CNV.
For intellectual disability, recent studies suggest the possibility that two independent CNV (El-Hattab et al., 2010; Girirajan et al., 2010; Potocki et al., 1999) can contribute to the ultimate phenotype, as shown in individual patients and as predicted by previous models (Lupski, 2007b).
In aggregate these data show that rare variants and the genome-wide totality of pathogenic alleles contribute to complex traits (Allikmets, 2000; Allikmets et al., 1997a; Douros et al., 2008; Hersh et al., 2004; Lupski, 2007b; Poller et al., 1990; Wittrup et al., 1997, 2006). Unfortunately, such rare variants are not being accounted for in many current GWAS, and CNV and noncoding SNV are not detected by typical whole-exome sequencing approaches.
A Unified Genetic Model for Human Disease
In the past, focused, locus-specific, single-gene analyses have elucidated genetic etiologies for disease, but it is now emerging that whole-genome sequencing will produce a more complete assessment of genetic variation contributing to personal health. The genome of each individual contains the inherited contribution of common variants that segregate within the population, the inherited contributions of rare variants that emerged in recent history in the clan, the new combinations of such recently arising variants from both parents, and the new mutation contributions yielding the total mutational burden (Figure 1). Highly penetrant rare variants, and often de novo mutations, contribute medically actionable alleles to Mendelian disease and perhaps extremes of phenotypes in common disease. Common variants can contribute to medically actionable variants for pharmacogenomics traits.
What emerges is a unified picture whereby previously distinct entities or categories of human diseases, chromosomal syndromes, genomic disorders, Mendelian traits, and common diseases or complex traits, can now be considered as part of one continuum (Figure 6), whereby common and rare variants including de novo mutations in the context of environmental influences result in perturbation of the biological balance of a restricted set of networks activating final common pathways that ultimately cause disease. Even though there may be many loci that contribute to interindividual inherited susceptibility of a phenotype in a population, in any one individual rare or common variants from just a few may be responsible for the trait (i.e., oligogenic inheritance). Extreme genetic heterogeneity and the contributions of new mutation may underlie some of the apparent complexity of complex traits.
Figure 6. A Continuum for the Genetics of Human Disease.
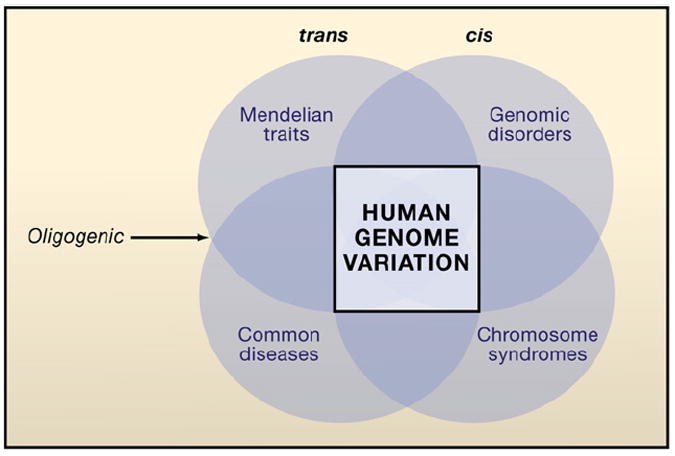
The square (center) represents genomic variation that can influence the different categories of genetic disease. The circles represent the overlapping categories of human disease with darker regions depicting intersection with greater overlap in the underlying genetic influences on these given disease categories. A unified model for human genetic disease proposes that all major categories of disease with genetic influence—Mendelian disease, common disease or complex traits, genomic disorders, and chromosomal syndromes—can be explained by variation in DNA sequence (SNV) or copy number (CNV) from a “wild-type” diploid state. Whereas trans-genetic interactions at a single locus (alleles) or between loci may contribute to Mendelian disease and complex traits, cis-genetic interactions can be important to phenotypic manifestations in genomic disorders (CNV) and chromosomal syndromes (segmental aneuploidy). Digenic and triallelic inheritance bridge Mendelian traits and complex disease; each represents an oligogenic inheritance model.
A unified genetic model for human disease breaks down the artificial boundaries between categories of human disease (Figure 6). It views all human disease categories including complex traits, Mendelian disease, genomic disorders, and even chromosomal syndromes as representing a spectrum of phenotypic manifestations reflecting the totality of pathogenic variants: ancestral alleles, those arising in recent ancestors (clan), unique combinations inherited from parents, and de novo variants (Figure 1). A full accounting of individual mutational load genome-wide and expansion of the current genocentric, locus-specific model opens the door to reinvestigation of classic problems in human genetics. These challenges include understanding the molecular basis of incomplete penetrance and variable expressivity of monogenic traits, clinical manifestations of “recessive alleles” (i.e., weak semidominance), homologous allelic interaction and nonhomologous allelic interaction, and their effects on disease and health. This new synthesis is required to interpret the ecology of individual genomes in the context of complete individual genetic variation data, population genetics, and evolution.
Genome-wide assays including whole-genome sequencing, copy-number arrays, and transcriptional profiling are among the current technologies that can be used to further explore and test the “genome-wide totality of pathogenic variants” hypothesis. These genome analysis methods can now generate a massive data flow, opening up to experimental exploration fundamental questions that have occupied the minds of generations of scientists and philosophers. Yet, such genome-wide experimental assays alone will be insufficient. Other challenges include: How many types of variants (repeat expansions, CNV between 100 and 500 bp, etc.) are we missing with current techniques? How will we validate the phenotypic effects of variants observed in a single individual or family? What analytical approaches should clinical genome sequencing projects adopt given the sheer complexity of some of the gene-disease associations described herein? How can we integrate disease risk emerging from common and rare variants in an individual genome? Can disease phenotypes be refined and redefined by molecular correlates such as gene expression, chromatin conformation, DNA methylation, and all of the other ‘omics? Can individual serial observation of molecular phenotypes, much as we currently do for routine lab measures such as glucose and lipids, show us stronger effects of underlying genetic variation that are otherwise poorly captured by cross-sectional studies and lead us to yet new models?
Supplementary Material
Acknowledgments
This work was supported in part by the National Human Genome Research Institute (5 U54 HG003273) to R.A.G. and the National Institute of Neurological Disorders and Stroke (R01NS058529) to J.R.L. J.R.L. is a consultant for Athena Diagnostics, has stock ownership in 23andMe and Ion Torrent Systems, and is a coinventor on multiple United States and European patents for DNA diagnostics. R.A.G. and J.W.B. are founding shareholders in Seq-Wright, Inc. The Department of Molecular and Human Genetics derives revenue from clinical testing by high-resolution human genome analyses.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes one table and can be found with this article online at doi:10.1016/j.cell.2011.09.008.
Contributor Information
James R. Lupski, Email: jlupski@bcm.edu.
Richard A. Gibbs, Email: agibbs@bcm.edu.
References
- 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn SM, Kim TH, Lee S, Kim D, Ghang H, Kim DS, Kim BC, Kim SY, Kim WY, Kim C, et al. The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome Res. 2009;19:1622–1629. doi: 10.1101/gr.092197.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, Mangion J, Roberton-Lowe C, Marshall AJ, Petretto E, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- Allikmets R The International ABCR Screening Consortium. Further evidence for an association of ABCR alleles with age-related macular degeneration. Am J Hum Genet. 2000;67:487–491. doi: 10.1086/303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, Peiffer A, Zabriskie NA, Li Y, Hutchinson A, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997a;277:1805–1807. doi: 10.1126/science.277.5333.1805. [DOI] [PubMed] [Google Scholar]
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997b;15:236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Athma P, Rappaport R, Swift M. Molecular genotyping shows that ataxia-telangiectasia heterozygotes are predisposed to breast cancer. Cancer Genet Cytogenet. 1996;92:130–134. doi: 10.1016/s0165-4608(96)00328-7. [DOI] [PubMed] [Google Scholar]
- Bacq Y, Gendrot C, Perrotin F, Lefrou L, Chrétien S, Vie-Buret V, Brechot MC, Andres CR. ABCB4 gene mutations and single-nucleotide polymorphisms in women with intrahepatic cholestasis of pregnancy. J Med Genet. 2009;46:711–715. doi: 10.1136/jmg.2009.067397. [DOI] [PubMed] [Google Scholar]
- Badano JL, Katsanis N. Beyond Mendel: an evolving view of human genetic disease transmission. Nat Rev Genet. 2002;3:779–789. doi: 10.1038/nrg910. [DOI] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W, Sapir T, Shchelochkov OA, Zhang F, Withers MA, Hunter JV, Levy T, Shinder V, Peiffer DA, Gunderson KL, et al. Increased LIS1 expression affects human and mouse brain development. Nat Genet. 2009;41:168–177. doi: 10.1038/ng.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochukova EG, Huang N, Keogh J, Henning E, Purmann C, Blaszczyk K, Saeed S, Hamilton-Shield J, Clayton-Smith J, O’Rahilly S, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature. 2010;463:666–670. doi: 10.1038/nature08689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerwinkle E, Utermann G. Simultaneous effects of the apolipoprotein E polymorphism on apolipoprotein E, apolipoprotein B, and cholesterol metabolism. Am J Hum Genet. 1988;42:104–112. [PMC free article] [PubMed] [Google Scholar]
- Boyko AR, Williamson SH, Indap AR, Degenhardt JD, Hernandez RD, Lohmueller KE, Adams MD, Schmidt S, Sninsky JJ, Sunyaev SR, et al. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- Carvalho CM, Zhang F, Liu P, Patel A, Sahoo T, Bacino CA, Shaw C, Peacock S, Pursley A, Tavyev YJ, et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum Mol Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KW, Sunwoo IN, Kim SM, Park KD, Kim WK, Kim TS, Koo H, Cho M, Lee J, Choi BO. Two missense mutations of EGR2 R359W and GJB1 V136A in a Charcot-Marie-Tooth disease family. Neurogenetics. 2005;6:159–163. doi: 10.1007/s10048-005-0217-4. [DOI] [PubMed] [Google Scholar]
- Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998;339:653–658. doi: 10.1056/NEJM199809033391002. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. Wellcome Trust Case Control Consortium. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 1000 Genomes Project. Variation in genome-wide mutation rates within and between human families. Nat Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium IS International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coventry A, Bull-Otterson LM, Liu X, Clark AG, Maxwell TJ, Crosby J, Hixson JE, Rea TJ, Muzny DM, Lewis LR, et al. Deep resequencing reveals excess rare recent variants consistent with explosive population growth. Nat Commun. 2010;1:131. doi: 10.1038/ncomms1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF. Maintaining evolvability. J Genet. 2008;87:349–353. doi: 10.1007/s12041-008-0057-8. [DOI] [PubMed] [Google Scholar]
- Del Colle R, Fabrizi GM, Turazzini M, Cavallaro T, Silvestri M, Rizzuto N. Hereditary neuropathy with liability to pressure palsies: electrophysiological and genetic study of a family with carpal tunnel syndrome as only clinical manifestation. Neurol Sci. 2003;24:57–60. doi: 10.1007/s100720300072. [DOI] [PubMed] [Google Scholar]
- del Gaudio D, Fang P, Scaglia F, Ward PA, Craigen WJ, Glaze DG, Neul JL, Patel A, Lee JA, Irons M, et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med. 2006;8:784–792. doi: 10.1097/01.gim.0000250502.28516.3c. [DOI] [PubMed] [Google Scholar]
- Dipple KM, McCabe ER. Phenotypes of patients with “simple” Mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet. 2000;66:1729–1735. doi: 10.1086/302938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divac A, Nikolic A, Mitic-Milikic M, Nagorni-Obradovic L, Petrovic-Stanojevic N, Dopudja-Pantic V, Nadaskic R, Savic A, Radojkovic D. High frequency of the R75Q CFTR variation in patients with chronic obstructive pulmonary disease. J Cyst Fibros. 2004;3:189–191. doi: 10.1016/j.jcf.2004.05.049. [DOI] [PubMed] [Google Scholar]
- Douros K, Loukou I, Doudounakis S, Tzetis M, Priftis KN, Kanavakis E. Asthma and pulmonary function abnormalities in heterozygotes for cystic fibrosis transmembrane regulator gene mutations. Int J Clin Exp Med. 2008;1:345–349. [PMC free article] [PubMed] [Google Scholar]
- El-Hattab A, Zhang F, Maxim R, Christensen KM, Ward JC, Scaglia F, Lupski JR, Cheung SW. Deletion and duplication of 15q24: molecular mechanisms and potential modification by additional copy number variants. Genet Med. 2010;12:573–586. doi: 10.1097/GIM.0b013e3181eb9b4a. [DOI] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- Fellermann K, Wehkamp J, Herrlinger KR, Stange EF. Crohn’s disease: a defensin deficiency syndrome? Eur J Gastroenterol Hepatol. 2003;15:627–634. doi: 10.1097/00042737-200306000-00008. [DOI] [PubMed] [Google Scholar]
- Fellermann K, Stange DE, Schaeffeler E, Schmalzl H, Wehkamp J, Bevins CL, Reinisch W, Teml A, Schwab M, Lichter P, et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am J Hum Genet. 2006;79:439–448. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, et al. International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friez MJ, Jones JR, Clarkson K, Lubs H, Abuelo D, Bier JA, Pai S, Simensen R, Williams C, Giampietro PF, et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118:e1687–e1695. doi: 10.1542/peds.2006-0395. [DOI] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet. 2004;41:937–940. doi: 10.1136/jmg.2004.024455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of low-density lipoprotein receptors: implications for pathogenesis and therapy of hypercholesterolemia and atherosclerosis. Circulation. 1987;76:504–507. doi: 10.1161/01.cir.76.3.504. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Molecular medicine. The cholesterol quartet. Science. 2001;292:1310–1312. doi: 10.1126/science.1061815. [DOI] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C, Lupski JR, Gibbs R. Human genome sequencing in health and disease. Ann Rev Med. 2011 doi: 10.1146/annurev-med-051010-162644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greeley SA, Tucker SE, Worrell HI, Skowron KB, Bell GI, Philipson LH. Update in neonatal diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17:13–19. doi: 10.1097/MED.0b013e328334f158. [DOI] [PubMed] [Google Scholar]
- Guardamagna O, Restagno G, Rolfo E, Pederiva C, Martini S, Abello F, Baracco V, Pisciotta L, Pino E, Calandra S, et al. The type of LDLR gene mutation predicts cardiovascular risk in children with familial hypercholesterolemia. J Pediatr. 2009;155:199–204. e192. doi: 10.1016/j.jpeds.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ, Leavitt BR, Farzin F, Jacquemont S, Greco CM, Brunberg JA, Tassone F, Hessl D, Harris SW, Zhang L, et al. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am J Hum Genet. 2004;74:1051–1056. doi: 10.1086/420700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS. The rate of spontaneous mutation of a human gene. J Genet. 1935;31:317–326. doi: 10.1007/BF02717892. [DOI] [PubMed] [Google Scholar]
- Heim RA, Lench NJ, Swift M. Heterozygous manifestations in four autosomal recessive human cancer-prone syndromes: ataxia telangiectasia, xeroderma pigmentosum, Fanoni anemia, and Bloom syndrome. Mutat Res. 1991;284:25–36. doi: 10.1016/0027-5107(92)90022-t. [DOI] [PubMed] [Google Scholar]
- Hersh CP, Dahl M, Ly NP, Berkey CS, Nordestgaard BG, Silverman EK. Chronic obstructive pulmonary disease in alpha1-antitrypsin PI MZ heterozygotes: a meta-analysis. Thorax. 2004;59:843–849. doi: 10.1136/thx.2004.022541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodapp JA, Carter GT, Lipe HP, Michelson SJ, Kraft GH, Bird TD. Double trouble in hereditary neuropathy: concomitant mutations in the PMP-22 gene and another gene produce novel phenotypes. Arch Neurol. 2006;63:112–117. doi: 10.1001/archneur.63.1.112. [DOI] [PubMed] [Google Scholar]
- Hoischen A, van Bon BW, Gilissen C, Arts P, van Lier B, Steehouwer M, de Vries P, de Reuver R, Wieskamp N, Mortier G, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- Hollox EJ, Huffmeier U, Zeeuwen PL, Palla R, Lascorz J, Rodijk-Olthuis D, van de Kerkhof PC, Traupe H, de Jongh G, den Heijer M, et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet. 2008;40:23–25. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap 3 Consortium. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Hagerman RJ, Leehey MA, Hall DA, Levine RA, Brunberg JA, Zhang L, Jardini T, Gane LW, Harris SW, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291:460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- Ji W, Foo JN, O’Roak BJ, Zhao H, Larson MG, Simon DB, Newton-Cheh C, State MW, Levy D, Lifton RP. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994;264:1604–1608. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, Hoskins BE, Scambler PJ, Davidson WS, Beales PL, Lupski JR. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–2259. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- Kim JI, Ju YS, Park H, Kim S, Lee S, Yi JH, Mudge J, Miller NA, Hong D, Bell CJ, et al. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–1015. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, McPherson J, Bourquin T, Lewis L, Villasana D, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotowski IK, Pertsemlidis A, Luke A, Cooper RS, Vega GL, Cohen JC, Hobbs HH. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet. 2006;78:410–422. doi: 10.1086/500615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH, Jr, Dobyns WB, Christian SL. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- Kurotaki N, Shen JJ, Touyama M, Kondoh T, Visser R, Ozaki T, Nishimoto J, Shiihara T, Uetake K, Makita Y, et al. Phenotypic consequences of genetic variation at hemizygous alleles: Sotos syndrome is a contiguous gene syndrome incorporating coagulation factor twelve (FXII) deficiency. Genet Med. 2005;7:479–483. doi: 10.1097/01.gim.0000177419.43309.37. [DOI] [PubMed] [Google Scholar]
- Le Maréchal C, Masson E, Chen JM, Morel F, Ruszniewski P, Levy P, Férec C. Hereditary pancreatitis caused by triplication of the trypsinogen locus. Nat Genet. 2006;38:1372–1374. doi: 10.1038/ng1904. [DOI] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RA, Shroyer NF, Singh N, Allikmets R, Hutchinson A, Li Y, Lupski JR, Leppert M, Dean M. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet. 1999;64:422–434. doi: 10.1086/302251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SCS, Dhar SU, Kolodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, Bacino CA, et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;146:889–903. doi: 10.1016/j.cell.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XY, Phung MT, Shaw CA, Pham K, Neil SE, Patel A, Sahoo T, Bacino CA, Stankiewicz P, Kang SH, et al. Genomic imbalances in neonates with birth defects: high detection rates by using chromosomal microarray analysis. Pediatrics. 2008;122:1310–1318. doi: 10.1542/peds.2008-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet Suppl. 2007a;39:S43–S47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Structural variation in the human genome. N Engl J Med. 2007b;356:1169–1171. doi: 10.1056/NEJMcibr067658. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Schizophrenia: Incriminating genomic evidence. Nature. 2008;455:178–179. doi: 10.1038/455178a. [DOI] [PubMed] [Google Scholar]
- Lupski JR. New mutations and intellectual function. Nat Genet. 2010;42:1036–1038. doi: 10.1038/ng1210-1036. [DOI] [PubMed] [Google Scholar]
- Lupski JR, Wise CA, Kuwano A, Pentao L, Parke JT, Glaze DG, Ledbetter DH, Greenberg F, Patel PI. Gene dosage is a mechanism for Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;1:29–33. doi: 10.1038/ng0492-29. [DOI] [PubMed] [Google Scholar]
- Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362:1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth GT, Yu F, Indap AR, Garimella K, Gravel S, Leong WF, Tyler-Smith C, Bainbridge M, Blackwell T, Zheng-Bradley X, et al. The functional spectrum of low-frequency coding variation. Genome Biol. 2011 doi: 10.1186/gb-2011-12-9-r84. Published online September 14 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy SE, Makarov V, Kirov G, Addington AM, McClellan J, Yoon S, Perkins DO, Dickel DE, Kusenda M, Krastoshevsky O, et al. Wellcome Trust Case Control Consortium. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–1227. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggouh F, de Visser M, Arts WF, De Coo RI, van Schaik IN, Baas F. Early onset neuropathy in a compound form of Charcot-Marie-Tooth disease. Ann Neurol. 2005;57:589–591. doi: 10.1002/ana.20434. [DOI] [PubMed] [Google Scholar]
- Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, Epplen JT. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet. 2005;42:e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AC, Bare LA, Chambless LE, Ellis SG, Malloy M, Kane JP, Pankow JS, Devlin JJ, Willerson JT, Boerwinkle E. Prediction of coronary heart disease risk using a genetic risk score: the Atherosclerosis Risk in Communities Study. Am J Epidemiol. 2007;166:28–35. doi: 10.1093/aje/kwm060. [DOI] [PubMed] [Google Scholar]
- Muller HJ. Our load of mutations. Am J Hum Genet. 1950;2:111–176. [PMC free article] [PubMed] [Google Scholar]
- Poller W, Meisen C, Olek K. DNA polymorphisms of the alpha 1-antitrypsin gene region in patients with chronic obstructive pulmonary disease. Eur J Clin Invest. 1990;20:1–7. doi: 10.1111/j.1365-2362.1990.tb01769.x. [DOI] [PubMed] [Google Scholar]
- Potocki L, Chen KS, Koeuth T, Killian J, Iannaccone ST, Shapira SK, Kashork CD, Spikes AS, Shaffer LG, Lupski JR. DNA rearrangements on both homologues of chromosome 17 in a mildly delayed individual with a family history of autosomal dominant carpal tunnel syndrome. Am J Hum Genet. 1999;64:471–478. doi: 10.1086/302240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Peters SU, Tavyev YJ, Zhang F, Carvalho CM, Schaaf CP, Richman R, Fang P, Glaze DG, Lupski JR. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol. 2009;66:771–782. doi: 10.1002/ana.21715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- Roa BB, Greenberg F, Gunaratne P, Sauer CM, Lubinsky MS, Kozma C, Meck JM, Magenis RE, Shaffer LG, Lupski JR. Duplication of the PMP22 gene in 17p partial trisomy patients with Charcot-Marie-Tooth type-1 neuropathy. Hum Genet. 1996;97:642–649. [PubMed] [Google Scholar]
- Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo S, Yin W, Kozlitina J, Pennacchio LA, Boerwinkle E, Hobbs HH, Cohen JC. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119:70–79. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrière A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, Mikkola HK, Hirschhorn JN, Cantor AB, Orkin SH. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Xu J, Ragoczy T, Ippolito GC, Walkley CR, Maika SD, Fujiwara Y, Ito M, Groudine M, Bender MA, et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460:1093–1097. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Sabo A, Sakai Y, Crosby J, Muzny D, Hawes A, Lewis L, Akbar H, Varghese R, Boerwinkle E, et al. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum Mol Genet. 2011;20:3366–3375. doi: 10.1093/hmg/ddr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SC, Miller W, Ratan A, Tomsho LP, Giardine B, Kasson LR, Harris RS, Petersen DC, Zhao F, Qi J, et al. Complete Khoisan and Bantu genomes from southern Africa. Nature. 2010;463:943–947. doi: 10.1038/nature08795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharer N, Schwarz M, Malone G, Howarth A, Painter J, Super M, Braganza J. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med. 1998;339:645–652. doi: 10.1056/NEJM199809033391001. [DOI] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang S-HL, Shen JJ, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010;47:332–341. doi: 10.1136/jmg.2009.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroyer NF, Lewis RA, Allikmets R, Singh N, Dean M, Leppert M, Lupski JR. The rod photoreceptor ATP-binding cassette transporter gene, ABCR, and retinal disease: from monogenic to multifactorial. Vision Res. 1999;39:2537–2544. doi: 10.1016/s0042-6989(99)00037-1. [DOI] [PubMed] [Google Scholar]
- Shroyer NF, Lewis RA, Yatsenko AN, Lupski JR. Null missense ABCR (ABCA4) mutations in a family with stargardt disease and retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2001a;42:2757–2761. [PubMed] [Google Scholar]
- Shroyer NF, Lewis RA, Yatsenko AN, Wensel TG, Lupski JR. Cosegregation and functional analysis of mutant ABCR (ABCA4) alleles in families that manifest both Stargardt disease and age-related macular degeneration. Hum Mol Genet. 2001b;10:2671–2678. doi: 10.1093/hmg/10.23.2671. [DOI] [PubMed] [Google Scholar]
- Shy ME, Scavina MT, Clark A, Krajewski KM, Li J, Kamholz J, Kolodny E, Szigeti K, Fischer RA, Saifi GM, et al. T118M PMP22 mutation causes partial loss of function and HNPP-like neuropathy. Ann Neurol. 2006;59:358–364. doi: 10.1002/ana.20777. [DOI] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Beaudet AL. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev. 2007;17:182–192. doi: 10.1016/j.gde.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietiläinen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, et al. GROUP. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, Blayney ML, Beck S, Hurles ME. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernimmen D, Marques-Kranc F, Sharpe JA, Sloane-Stanley JA, Wood WG, Wallace HA, Smith AJ, Higgs DR. Chromosome looping at the human alpha-globin locus is mediated via the major upstream regulatory element (HS -40) Blood. 2009;114:4253–4260. doi: 10.1182/blood-2009-03-213439. [DOI] [PubMed] [Google Scholar]
- Verny C, Ravisé N, Leutenegger AL, Pouplard F, Dubourg O, Tardieu S, Dubas F, Brice A, Genin E, LeGuern E. Coincidence of two genetic forms of Charcot-Marie-Tooth disease in a single family. Neurology. 2004;63:1527–1529. doi: 10.1212/01.wnl.0000142082.65144.ee. [DOI] [PubMed] [Google Scholar]
- Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, et al. MAGIC investigators; GIANT Consortium. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner CA. How much is blood pressure in the general population determined by rare mutations in renal salt-transporting proteins? J Nephrol. 2008;21:632–634. [PubMed] [Google Scholar]
- Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Moylan B, Leopold DA, Kim J, Rubenstein RC, Togias A, Proud D, Zeitlin PL, Cutting GR. Mutation in the gene responsible for cystic fibrosis and predisposition to chronic rhinosinusitis in the general population. JAMA. 2000;284:1814–1819. doi: 10.1001/jama.284.14.1814. [DOI] [PubMed] [Google Scholar]
- Wang X, Kim J, McWilliams R, Cutting GR. Increased prevalence of chronic rhinosinusitis in carriers of a cystic fibrosis mutation. Arch Otolaryngol Head Neck Surg. 2005;131:237–240. doi: 10.1001/archotol.131.3.237. [DOI] [PubMed] [Google Scholar]
- Weiss FU, Simon P, Bogdanova N, Mayerle J, Dworniczak B, Horst J, Lerch MM. Complete cystic fibrosis transmembrane conductance regulator gene sequencing in patients with idiopathic chronic pancreatitis and controls. Gut. 2005;54:1456–1460. doi: 10.1136/gut.2005.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, et al. Autism Consortium. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- Wittrup HH, Andersen RV, Tybjaerg-Hansen A, Jensen GB, Nordestgaard BG. Combined analysis of six lipoprotein lipase genetic variants on triglycerides, high-density lipoprotein, and ischemic heart disease: cross-sectional, prospective, and case-control studies from the Copenhagen City Heart Study. J Clin Endocrinol Metab. 2006;91:1438–1445. doi: 10.1210/jc.2005-1725. [DOI] [PubMed] [Google Scholar]
- Wittrup HH, Tybjaerg-Hansen A, Abildgaard S, Steffensen R, Schnohr P, Nordestgaard BG. A common substitution (Asn291Ser) in lipoprotein lipase is associated with increased risk of ischemic heart disease. J Clin Invest. 1997;99:1606–1613. doi: 10.1172/JCI119323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.