

Abstract
Following the “finished,” euchromatic, haploid human reference genome sequence, the rapid development of novel, faster, and cheaper sequencing technologies is making possible the era of personalized human genomics. Personal diploid human genome sequences have been generated, and each has contributed to our better understanding of variation in the human genome. We have consequently begun to appreciate the vastness of individual genetic variation from single nucleotide to structural variants. Translation of genome-scale variation into medically useful information is, however, in its infancy. This review summarizes the initial steps undertaken in clinical implementation of personal genome information, and describes the application of whole-genome and exome sequencing to identify the cause of genetic diseases and to suggest adjuvant therapies. Better analysis tools and a deeper understanding of the biology of our genome are necessary in order to decipher, interpret, and optimize clinical utility of what the variation in the human genome can teach us. Personal genome sequencing may eventually become an instrument of common medical practice, providing information that assists in the formulation of a differential diagnosis. We outline herein some of the remaining challenges.
Keywords: whole-genome sequencing (WGS), exome sequencing, simple nucleotide variation (SNV), structural variation, personal genomics
THE HUMAN REFERENCE GENOME
The 2001 draft sequence of the human genome, by the Human Genome Project (HGP) (1), was unquestionably a great scientific achievement, a turning point for human genetics, and the starting point for human genomics. Three years later, international efforts delivered a high-quality finished human genome assembly representing 99% of the euchromatic sequence (2). Although by far the highest-quality genome for any organism, it was still incomplete. In addition, the human reference genome is a haploid consensus mosaic sequence derived from multiple individuals. The assembly and refine-ment of the reference genome were able to provide a snapshot of genetic variation, mainly in the form of single nucleotide polymorphisms (SNPs), and also a glimpse into the complex architecture of segmental duplications (3) and low-copy repeats (4). Simple nucleotide variation (SNV), which includes SNPs and small indels, has been further surveyed in many individuals. The HGP combined with the HapMap Project populated dbSNP, a database of SNPs (http://www.ncbi.nlm.nih.gov/snp/), with ~10 million well-characterized common variants in different world populations; the HapMap also provided a backbone of common haplotypes in human genomes (5–8).
Some of the most challenging regions to be cloned, sequenced, and assembled for the reference sequence were those enriched in highly repetitive (e.g., Alus, LINEs) and near-identical low-copy repeat sequences. Later, with the availability of genome-wide assays, it became apparent that the complex architecture of the human genome can result in a broader spectrum of variation known as structural variation, which includes inversions and copy-number variants (CNVs). Remarkably, CNVs may account for more variable base pairs between individuals than SNPs alone. Such architectural complexity could also result in genome instability and susceptibility to rearrangements that can cause disease—a group of conditions referred to as genomic disorders (4, 9).
Humans are diploid, and to understand the genetics of Mendelian and complex diseases, as well as to grasp the extent of human variation, we must consider the interplay between the pairs of alleles of genes and of all the genes within the genome, as well as the nongenic sequences. Only then can we build complete models of genetic interactive networks.
During the decade after the HGP, technical development enabled massively parallel next-generation DNA sequencing (NGS), ushering in a new era for human genomics. The period began with a series of key examples of individual genomes that established the basis for the analysis of subsequent genomes (Table 1). This increased availability of an individual’s genetic information may provide a useful tool for the practicing physician, eventually assisting in differential diagnosis and potentially enabling anticipatory guidance and possibly preventive genomic medicine.
Table 1.
Comparison of sequenced personal human genomes
| Individual | Ploidy | Technology | Av Depth | Total SNPs [M] | Known SNPs [M] (%) | Novel SNPs [M] (%) | Heterozygous SNPs [M] (%) | Homozygous SNPs [M] (%) | cSNPs | nsSNPs | InDels | CNVs (≥100 bp) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Venter | 2n | Sanger | 7.5× | 3.21 | 2.80 (87.22%) | 0.41 (12.77%) | 1.76 (54.85%) | 1.45 (45.15%) | 21,152 | 6,114 | 214,691 | 6,485 |
| Watson | 2n | Roche 454 | 7.4× | 3.32 | 2.71 (81.73%) | 0.61 (18.27%) | 1.67 (50.53%) | 1.64 (49.47%) | 22,041 | 10,659 | 222,718 | 1,674 |
| Chinese (YH) | 2n | Illumina | 36.0× | 3.07 | 2.65 (87.13%) | 0.41 (12.87%) | 1.72 (56.03%) | 1.35 (43.97%) | 15,759 | 7,062 | 135,262 | 2,682 |
| African (NA18507)* | 2n | Illumina | 40.6× | 3.61 | 2.72 (75.50%) | 0.88 (24.50%) | 2.28 (63.21%) | 1.32 (36.79%) | 26,140 | 5,361 | 404,416 | 8,470 |
| African (NA18507)* | 2n | AB SOLiD | 17.9× | 3.86 | 3.13 (81.00%) | 0.73 (19.00%) | 2.33 (60.30%) | 1.53 (39.70%) | 68,624 | 9,902 | 226,529 | 6,714 |
| Korean (SJK) | 2n | Illumina | 28.9× | 3.43 | 3.01 (87.79%) | 0.42 (12.21%) | 2.00 (58.21%) | 1.43 (41.79%) | 27,118 | 9,334 | 342,965 | 3,303 |
| Korean (AK1) | 2n | Illumina | 27.8× | 3.45 | 2.86 (83.30%) | 0.59 (16.70%) | 2.11 (61.11%) | 1.34 (38.89%) | 21,606 | 10,162 | 170,202 | 414 |
| Khoisan (KB1) | 2n | Roche 454 | 10.2× | 4.05 | 3.31 (81.65%) | 0.74 (18.35%) | 2.39 (59.00%) | 1.66 (41.00%) | 22,119 | na | 463,788 | na |
| D. Tutu (ABT) | 2n | AB SOLiD | 30.0× | 3.62 | 3.21 (88.61%) | 0.41 (11.39%) | 2.17 (60.00%) | 1.44 (40.00%) | 17,342 | na | 3,395 | na |
| Lupski | 2n | AB SOLiD | 29.6× | 3.42 | 2.85 (83.58%) | 0.56 (16.42%) | 2.00 (58.72%) | 1.41 (41.28%) | 18,406 | 9,069 | na | 530 |
Same HapMap sample was independently sequenced and reported using two different technologies.
Abbreviations: cSNPs, coding SNPs; nsSNPs, nonsynonymous SNPs; CNVs, copy-number variants; na, data not available.
PERSONAL HUMAN GENOMES
The Venter Genome
In 2007, the personal genome sequence of J. Craig Venter, developed using whole-genome shotgun and traditional Sanger dideoxy sequencing and consisting of 2.8 Gb of reference-matched genome sequence, was published (10). The analysis of the Venter genome sequence identified 1.2 million novel variants when compared to the human reference assembly; non-SNP variants ranging from small indels to large CNVs and inversions accounted for 74% of the total number of variant bases. Some of the CNVs identified overlapped with 95 genes, including seven OMIM (Online Mendelian Inheritance in Man; http://www.ncbi.nlm.nih.gov/omim) genes for traits such as blood-group determination and diseases such as Zellweger syndrome (MIM #214100). Nonsynonymous variants in 4,107 genes were identified; 10,208 genes were found to harbor at least one heterozygous variant.
For J. Craig Venter, having his genome sequenced revealed that he is heterozygous for several variants in genes associated with coronary artery disease, hypertension, and myocardial infarction. There is a family history of cardiovascular disease; nevertheless, JCV is also heterozygous for some cardiac-protective variants. Thus, it remains unknown whether or how the different variants might account for cardiovascular disease risk in this particular individual, so clinical utility could not be documented. He is also heterozygous for a null allele in the GSTM1 gene, which is important for detoxification and the metabolism of xenobiotics. Carriers of null alleles for this gene have increased susceptibility to environmental toxins and possibly increased risk of developing a variety of cancers (11). This variant might be relevant to this individual’s history of skin cancer.
The Watson Genome
The genomic sequence of the codiscoverer of the structure of DNA, James D. Watson, was published in 2008 (12). This first complete human genome sequenced by NGS technology marked the beginning of a revolution in human genome resequencing and personal genomics. Watson’s genome was sequenced in just two months; however, the analysis required a significantly greater amount of time. The Watson genome was the first diploid genome to be publicly released (in May 2007).
The comparison of Watson’s genome with the human reference sequence led to the identification of SNPs, plus small indels and CNVs. More deletion events than insertions were identified at a 2.3:1 ratio. Most of the coding indels identified were heterozygous and multiples of three in length, and therefore unlikely to inactivate genes. There was a significant enrichment of indels in the size range of 300–350 bp, consistent with the size of Alu sequences. The Watson genome had 23 large CNVs that ranged in size from 26 kb to 1.6 Mb, which were thought to represent benign variation, including CNVs of olfactory receptor gene clusters. Thirty-four genes are located within these CNVs; whether their expression or function is altered owing to the CNVs remains to be determined.
In a comparison of nonsynonymous variants to the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk), 32 variants that matched previously reported disease-causing mutations were found. Twelve of these were linked to autosomal recessive diseases or traits, such as retinitis pigmentosa or congenital nephrotic syndrome; the other 20 were associated with variable risk of developing common diseases. Interestingly, three SNPs that were homozygous in the subject and annotated to be highly penetrant disease-causing mutations were identified. Subsequent confirmation of these SNPs demonstrated that one of them was heterozygous and that the population frequencies of the other two were >0.15, indicating that although present at low frequencies, the homozygous genotypes are present in the general population, and these variants are not likely disease causing.
African Genomes
Later in 2008, the genomic sequence of a Yoruban individual, HapMap sample NA18507, was determined (13). This same genome was sequenced again in 2009 using a different NGS technology (14); a comparison demonstrates the importance of the NGS method used and the annotation approach applied for analysis. An enrichment of heterozygous (versus homozygous) SNPs was observed in this genome. Some homozygous SNPs were found to be associated with pharmacogenetic traits or susceptibility to cancer or some other complex disease. Indels affected exons of 2,241 genes, with an enrichment of indels in the first and last exons. Some of the deletions found were also observed in other published personal genomes, suggesting that they are actually insertions in the reference human genome assembly.
More recently, the complete genomes of two Khoisan and Bantu individuals from southern Africa were sequenced in addition to the exomes of three other individuals originating from different indigenous groups across the Kalahari Desert (KB1, ABT, NB1, TK1, and MD8 respectively) (15). The ABT genome was derived from Archbishop Desmond Tutu. The KB1 genome was sequenced using long reads in order to create a de novo assembly because of the diversity expected from African genomes. The scaffold for this assembly covers ~3.09 Gb. It was found that on average two Bushmen (KB1, ABT, NB1, TK1, and MD8) differ from each other at 1.2 nucleotides per kilobase, which is more than the average interindividual variation of 1.0 nucleotide per kilobase observed from studies performed mostly in subjects of European descent. It is important to note that this latter difference (1 nt/kb) is derived from exonic sequences; the variation between two Bushmen may well be greater for noncoding regions. An enrichment of SNPs in promoter regions was observed, which might cause differences in expression and phenotypes in these individuals. There was an aggregate of 27,641 nonsynonymous substitutions among all the sequenced individuals; of these 47.55% were novel, affecting 7,720 genes.
Several of the 621 previously known SNPs that were found in the southern African genomes have functional associations. One SNP in the promoter region of LCT is associated with lactase persistence in European populations. The non-European allele was found homozygous in all the Bushmen analyzed, consistent with lactose intolerance expected in foraging (rather than farming) populations. Variants in the SLC24A5 gene associated with skin color and increased production of melanin were also identified. Interestingly, some associations with enhanced physiological traits were observed in the majority of these individuals, such as homozygosity for a VDR allele associated with increased bone mineral density, and homozygosity for an allele in ACTN3 associated with increased muscle power performance and sprint. SNPs associated with metabolism of xenobiotics, chloride reabsorption, and enhanced hearing were also identified. Alleles for common traits such as phenylthiocarbamide (PTC) tasting were found as fixed variants in the Bushmen, suggesting that they might still be relevant for plant tasting and toxic-compound discrimination in these foraging populations. In addition, over-representation of nonsynonymous changes were seen in gene ontology categories related to sensory perception, muscular and skeletal development, and inflammatory response and wound healing.
CNVs were found to alter the copy number of 193 genes in the KB1 genome compared to the NA18507 Yoruban genome. These included increased CNVs at the well-known variable amylase (16) and alpha defensins loci (17), probably reflecting differences in the dietary habits of populations across Africa and previously discussed environmental adaptation variation.
Mitochondrial sequences for these southern African individuals revealed approximately five times more variation than is usually observed between the reference mitochondrial genome and a Caucasian genome, and approximately four times more variation between any two of the sequenced individuals.
Asian Genomes
The first Asian genome sequence to be published was the YH genome, derived from a Han Chinese individual (18). Complete or partial deletions of 33 genes were detected in this genome. A heterozygous mutation in the GJB2 gene responsible for autosomal recessive deafness was identified among the nonsynonymous SNPs. In addition, several alleles associated with tobacco addiction and increased risk for Alzheimer disease were found in this self-reported heavy smoker. A familial history of Alzheimer disease could not be assessed.
Following this first Asian genome, the genomes of two Korean individuals were published, SJK (19) and AK1 (20). Nonsynonymous SNPs in the SJK genome were distributed throughout 5,365 genes, and 1,348 of these nonsynonymous SNPs were novel. The majority of indels detected were in introns, while only 49 were found in coding regions; however, these alter the reading frames of 27 different genes, probably affecting their function. Some mitochondrial genome variants were found, including 44 novel SNPs, of which 6 were non-synonymous, plus 3 insertions and 1 deletion.
In the AK1 genome, 70 of the indels detected were homozygous and 26 were in OMIM genes, of which 13 have been associated with some disease. Seven hundred seventy-three SNPs were predicted to be potentially associated with a clinical phenotype, including 269 known SNPs having associations with variable risk of developing certain types of cancer, diabetes, or Alzheimer disease. There were 504 nonsynonymous SNPs identified in genes associated with Mendelian diseases or traits, of which 22 were nonsense mutations and 5 were homozygous in the AK1 individual. Among these traits were dry earwax, apparently common in the Korean population, and drug metabolism variants that are of pharmacogenetic relevance. The CNVs detected might affect the function or expression of the 106 genes they encompass in this individual.
Insights from these personal genomes led to recognition of the tremendous variation that the human genome harbors and the importance of sequencing more genomes in order to get a more comprehensive catalogue of that variation, especially low-frequency and rare variants (Figure 1). Collaborative projects that aimed to catalogue human variation in different populations and include variants with minor allele frequency (MAF) ≥ 1% in addition to common SNP variants (MAF ≥ 5%) were therefore initiated, including The 1000 Genomes Project (TGP).
Figure 1.

Comparison of single nucleotide polymorphisms (SNPs) in 10 personal genomes. All SNPs in any of 10 sequenced personal genomes were compared with the other 9 genomes. Altogether, the 10 genomes contribute 14,608,404 nonredundant SNPs (first bar). The second bar pictures all SNPs that are unique to each of the personal genomes; the third bar represents all the SNPs that are unique in a given personal genome but also novel; the fourth bar shows the SNPs shared by individuals of the same ethnic group. Abbreviations: AF1, NA18507(1) Illumina; AF2, NA18507(2) SOLiD; KB1, Khoisan genome; ABT, Archbishop Desmond Tutu; YH, Chinese genome; SJK, Korean genome 1; AK1, Korean genome 2; JCV, J. Craig Venter; JDW, James D. Watson; JRL, James R. Lupski.
The 1000 Genomes Project
Similar to the initial goal of the International HapMap Project (5), TGP aims to characterize human variation of all types by high-throughput and unbiased sequencing of 1000+ human genomes from diverse populations. To test different general approaches, the pilot included three elements:
Low-coverage sequencing (2–4× average depth) of 179 samples of the three main HapMap population groups in order to identify all the variants with a MAF ≥ 1%. From this, 14.89 million SNPs and 1.33 million indels were identified, of which 54% and 57% were novel, respectively.
Deep sequencing of a Caucasian and a Yoruban trio at ~40× coverage.
Deep resequencing of 8,140 exons in 697 samples in order to capture most of the “normal” coding variation.
This third part of the pilot identified 12,758 SNPs but only 96 indels; interestingly 70% of the SNPs identified were novel. Included in these were 68,300 nonsynonymous SNPs, of which ~50% were novel; some of these were validated to be polymorphic across several samples. Interestingly, nonsense mutations, splice-site variants, and frameshifting variants were biased toward lower allele frequencies and even private to some populations or individuals. Six hundred seventy-one apparent disease-causing mutations included in the Human Gene Mutation Database were identified in this resequencing project.
De novo assembly was performed using the data for the low-coverage pilot, and pooling all samples together, this process identified 3.7 Mb of sequence not present in the reference assembly. Of this sequence, 87% matched other known human or primate sequences, while 79% matched sequence present in the Venter genome. It was observed that variation across the genome was not evenly distributed; some highly polymorphic regions such as the HLA locus showed more variation, whereas other gene-rich, highly conserved regions showed less variation.
Overall, the initial pilot phase of TGP identified 16.78 million variants in 742 samples from different populations. The final goal of the project is to sequence 2,500 additional individuals of diverse populations from five geographical areas at ~4× average depth of coverage in an attempt to identify most of the variation that occurs at ≥0.1% frequency in the population. The most recent data report that TGP has identified 38.9 million SNP sites (G. Marth, personal communication). Interestingly, many of these variants are private, i.e., present in very few individuals or just one individual.
STRUCTURAL VARIATION IN HUMAN GENOMES
Resequencing of personal human genomes has provided further insight into the extent of an important, but until recently unappreciated, form of variation; i.e., structural variation (SV).
In 2004, two articles (21, 22) marked a turning point in our appreciation and understanding of human genetic variation. Until then it was well recognized that rearrangements in the human genome could occur, but these were regarded as rare events that could be the cause of syndromes known as genomic disorders (4, 9). It was understood that genomic rearrangements could be incited by the local architecture of the genome and that they were often produced by nonallelic homologous recombination between highly identical segments of the genome named low-copy repeats (23). In 2001 it had been appreciated that the human genome was rich in segmental duplications (3), but it was in 2004 that two groups, using genome-wide assays enabled by the HGP, systematically assessed and visualized genome-wide differences in the copy number of regions in normal, healthy, unrelated individuals. These CNVs were observed as gains or losses of genomic segments spanning a few kilobases to several hundred kilobases and even megabases in size. Approximately 34%–40% of the CNVs detected in these initial studies were observed in more than one individual, and some of them in more than 10% of the individuals. Many of these CNV regions were observed to overlap segmental duplications. Together these studies reported the identification of 20 CNV regions in the human genome. Further genome-wide studies of CNVs in multiple genomes discovered and added many more regions to the increasing list of known polymorphic CNVs (24). Higher-resolution genome-wide arrays led to the estimate that on average every individual possesses ~1,000 polymorphic CNVs (MAF ≥ 5%) that range in size from 500 bp to 1.2 Mb (median = 2.9 kb) (25).
As of August 2011, the Database of Genomic Variants (http://projects.tcag.ca/variation/) reports 15,963 structurally variable loci in the genome. These loci are dispersed throughout the genome, although clustered in certain regions such as pericentromeric and sub-telomeric regions. In aggregate, structural variation encompasses large indel polymorphisms (100 bp–1 kb), CNVs (>1 kb), and inversions. In addition to accounting for more total variable base pairs across the genome than SNPs, CNVs are probably an important source of biochemical, metabolic, and phenotypic variations among individuals in the population. Approximately 35% of the genes in the human genome are encompassed either totally or partially by a CNV that can alter their expression or even their structure, possibly giving rise to novel fusion transcripts.
Although array comparative genomic hybridization (aCGH) is efficient at detecting CNVs, the technique is limited by the resolution of the arrays and the spacing between probes that tile the reference genome. Furthermore, arrays only assay for the sequence found in the reference assembly. In addition, aCGH is always performed using a control DNA, which may confound the overall interpretation of CNVs. (Is the observation a gain in the test sample or a loss in the reference sample utilized for aCGH?) Whole-genome sequencing (WGS) combined with high-resolution aCGH in personal genomes has revealed a higher number of CNVs in the low size range (<5 kb) than expected, usually averaging ~2 kb. Thus, the allele frequency spectrum for CNVs in a human genome reveals a much higher frequency for smaller sized (<1 kb) CNVs—the very size range beyond the capability of most conventional aCGH.
Less is known about inversions; like deletions and duplications, inversions can result from nonallelic homologous recombination between highly identical repeated sequences, but in inverted orientation. A study aimed to identify all the potential recombinogenic inverted sequences in the human reference genome showed that some of these predicted sequences can recombine mitotically and produce somatic inversions, suggesting that the human genome is a structural mosaic at the somatic level (26). These data also suggest that the size of the recombining sequences and the distance between them can influence the rate at which they recombine and produce rearrangements. Interestingly, one of the first pathogenic inversion rearrangements identified disrupts the factor VIII gene and is responsible for ~20% of all hemophilia A (MIM + 306700) and about half of the severe cases (27). One of the most common polymorphic inversions is a 900-kb region in chromosome 17q21.31 that was found to be under selection in the European population (28), but this inversion predisposes to a deletion rearrangement that causes the 17q21.31 microdeletion syndrome (MIM #613533) (29).
Genomic rearrangements are known to cause “rare” genomic disorders such as Charcot-Marie-Tooth disease (CMT1A; MIM #118220), hereditary neuropathy with liability to pressure palsies (HNPP; MIM #162500), Smith-Magenis syndrome (SMS; MIM #182290), Potocki-Lupski syndrome (PTLS; MIM #610883), Williams-Beuren syndrome (WBS; MIM #194050), and Pelizaeus-Merzbacher disease (PMD; MIM #312080). However, our better understanding and detection of CNVs has enabled the identification of rare CNVs as a cause of other, not-so-rare diseases and complex disorders such as Parkinson disease, Alzheimer disease, psoriasis, autism, schizophrenia, and HIV susceptibility (30, 31). It is probable that further studies of CNVs in other complex diseases will unveil new CNVs implicated in susceptibility to disease. As a consequence, detection of structural variation is imperative in any WGS study.
Recent genome-wide studies to search for CNVs using data from WGS approaches have greatly increased the catalogue of known structural variants. These studies have also started to define the boundaries of some of these variants at the sequence level, enabling the elucidation of the breakpoint or junctional sequence from which to infer the mechanisms involved in CNV generation in the human genome (32). Data generated by TGP identified and characterized ~28,000 structural variants of >50 bp in 185 individuals; ~55% of these variants were novel. TGP found many structural variants that had not been previously identified, with a bias toward smaller CNVs.
Furthermore, we have just begun to appreciate that the structure of each individual genome varies tremendously with regard to the location of individual retrotransposed inserted elements such as long interspersed elements (LINEs) and short interspersed elements (SINEs), especially Alu sequences (33). Several of the personal genomes sequenced to date revealed that the distribution of indel polymorphisms and CNVs consistently has two peaks (Figure 2): one at 300–350 bp, consistent with insertion/deletion polymorphisms of Alu sequences, and a second at ~6 kb, consistent with the size of L1 elements, representing repetitive sequence dimorphisms in diploid genomes. In the Venter genome, a total of 1,316 Alu indels were identified, 53% of which were inserted in this genome as compared to the reference genome and were not present in any of the databases for SINE or retrotransposon insertion polymorphisms. Likewise, ~900 Alu indel differences were identified in the Watson genome when compared to the reference assembly.
Figure 2.

Size distribution of large indels (100 bp–1 kb) and copy-number variants (CNVs) (>1 kb) in sequenced personal human genomes. Distribution of large indels and CNVs in 8 personal genomes is shown by size. We can observe peaks between 300 and 400 bp, consistent with Alu indel polymorphisms, and at ~1–2 kb. Few polymorphic CNVs are larger than 200 kb. Abbreviations: AF1, NA18507(1) Illumina; AF2, NA18507(2) SOLiD; KB1, Khoisan genome; ABT, Archbishop Desmond Tutu; YH, Chinese genome; SJK, Korean genome 1; AK1, Korean genome 2; JCV, J. Craig Venter; JDW, James D. Watson; JRL, James R. Lupski.
More recently, several studies have interrogated retrotransposed insertional polymorphisms in the human genome. Surprisingly, retrotransposed elements in the human genome are far from dormant. These elements show considerable unanticipated activity; thus, each personal human genome is far more diverse structurally than we had appreciated initially. Alu and L1 elements are the most common retrotransposed sequences in the human genome, believed to have been the most recent to be active. One study shows that L1 elements can transpose at frequencies higher than expected both somatically and in the germline (34). Another study (35) surveyed 68 L1s that were not present in the reference assembly in different individuals and found that 54% of them are actually complete and active L1 elements that can transpose, as documented by a functional assay in vitro. This is consistent with observations made to determine the insertion sites of most of the human-specific L1 family of retrotransposons in several individuals (36). It was observed that L1 elements are dimorphic in the human genome and that any two individuals differ on average at 285 insertion sites. It is estimated that the total number of dimorphic L1 elements in the population ranges between 3,000 and 10,000.
Although no current sequencing technology can accurately detect and specifically assay all the structural variants in a given genome, increase in the length of reads produced by NGS technologies and improvement in the algorithms for CNV calling and de novo assembly of personal genomes may eventually allow the unbiased detection of structural variants of all sizes and types and with sequence-level resolution of breakpoints.
EXOME SEQUENCING IN MEDICAL GENETICS
The exome comprises the coding sequences of all annotated protein-coding genes (~23,000) and is equivalent to ~1% of the total haploid genomic sequence (~30 Mb). To sequence this subset of the genome through massively parallel sequencing, targeted-capture methodologies were developed using arrays (37), and later beads in solution (38), that hybridize to the exonic sequences to be captured.
The first targeted attempt to sequence only this “whole-exome” fraction of the genome, as validation for a disease-gene identification approach, was reported in 2009 (39). The exomes of eight HapMap individuals were examined and the accuracy of the approach validated using HapMap data from these individuals. In addition, the exomes of four other individuals affected by Freeman-Sheldon syndrome (MIM #193700), which is known to be caused by mutations in the MYH3 gene, were also sequenced. A total of 56,240 nonredundant coding SNPs were identified across the 12 exomes, most of which were already present in dbSNP; 23% were novel variants. Combining the variants in the Freeman-Sheldon syndrome patients to search for mutations in a common gene among them, and subsequent filtering of these variants for those known in dbSNP or found in the HapMap samples, narrowed the list of candidate genes to just one: MYH3. This experiment demonstrated that it was possible to capture most of the variation contained in the exome and that by applying bioinformatic filtering steps it was possible to identify the pathogenic variants for a genetic disease.
Choi et al. showed that it was possible to reach a more accurate diagnosis of a patient after exome sequencing (40). They applied this approach to diagnose a patient referred for possible Bartter syndrome, a disease of impairment in salt reabsorption. The patient was born from healthy parents who were first cousins. Thus, absence of heterozygosity was an additional filter to narrow the list of candidate genes in which to search for homozygous mutations. A novel homozygous mutation in SLC26A3, a gene in which homozygous or compound heterozygous loss-of-function mutations are known to cause congenital chloride-losing diarrhea (CLD; MIM #214700), was identified. After the identification of this mutation, clinical follow-up revealed that the patient indeed had CLD, not considered in the initial differential diagnosis, and definitely did not have Bartter syndrome.
The utility of exome sequencing for gene discovery in a recessive Mendelian disorder with unknown genetic cause was also realized for Miller syndrome (MIM #263750) (41). The approach consisted of sequencing the exomes of four affected individuals, including a pair of siblings. Because Miller syndrome was thought to be a recessive disorder, special attention was given to genes that contained at least two variants. Comparison of genes shared among the affected individuals narrowed the list to just one gene, DHODH, which encodes dihydroorotate dehydrogenase and is involved in the biosynthesis of pyrimidines. All of the sequenced individuals harbored compound heterozygous mutations in this gene, and all sets of parents were shown to be carriers for the mutations, fulfilling Mendelian expectations.
Next, exome sequencing was performed to identify the gene responsible for an autosomal dominant disorder, Schinzel-Giedion syndrome (MIM #269150), another syndrome with an unknown genetic cause (42). The exomes of four affected unrelated individuals were sequenced at ~43× coverage. The variants were filtered for known variants and then compared to identify common candidate genes in which all the affected individuals carried at least one novel variant. Heterozygous novel mutations were found and further confirmed in the SETBP1 gene. Testing the presence of the identified variants in the parents of the affected individuals showed that all mutations arose de novo, consistent with dominant mutations in this sporadic syndrome.
This approach for gene discovery was also applied to Kabuki syndrome (MIM #147920) (43). The exomes of ten unrelated individuals with this rare syndrome were sequenced to ~40× coverage. When attempting to identify common genes with novel variants shared by all the cases, the authors failed to identify a suitable candidate gene. However, ranking of the affected individuals based on the canonical phenotype for Kabuki syndrome and subsequent analysis of variants in shared genes within subsets or ranked individuals uncovered MLL2 as the gene most probably responsible for this syndrome. Exome sequencing was able to identify nonsense and frameshifting mutations in seven out of ten cases resequenced. Sanger sequencing of MLL2 exons in the remaining cases identified small frameshifting indels in two additional cases. Further validation and Sanger sequencing of the identified gene in additional cases of Kabuki syndrome showed a success rate of 66% for identification of mutations in MLL2. This suggests that Kabuki syndrome might be a genetically heterogeneous disease, with other genes responsible for the phenotype in selected patients. This study underlines the importance of an adequate phenotypic characterization of patients in order to reduce genetic heterogeneity that may confound the analysis.
A recent success story for the application of exome sequencing for genetic diagnosis and patient management is reported by Worthey et al. (44). Whole-exome sequencing was performed in a male child referred for inflammatory bowel disease (IBD) phenotypically similar to Crohn disease. Because in some instances congenital immune deficiencies can present with IBD-like illness, the child was immunologically and genetically tested for several possible autoimmune disorders, all of which were nonproductive for an etiological diagnosis. Exome sequencing was performed in an attempt to identify potential genetic susceptibility variants. The sequencing approach revealed 15,272 coding variants, of which 6,799 were nonsynonymous SNPs, including 706 novel variants and 13 nonsense changes. Assuming a recessive model for this patient’s disease, Worthey et al. examined genes with homozygous, hemizygous, or compound heterozygous variants. Remarkably, a hemizygous change of a highly conserved residue in the X-linked inhibitor of apoptosis gene (XIAP) was identified. The mutation was confirmed by Sanger sequencing in the patient and his mother; the mother was a heterozygous carrier showing skewed X-linked inactivation in lymphocytes. Based on this result, and considering that XIAP deficiency is a life-threatening condition, hematopoietic stem cell progenitor transplantation was implemented. After some post-transplant complications, the patient was reported to be improving and thriving.
Most recently, exome sequencing and analyses of healthy compared to affected tissues from patients diagnosed with Proteus syndrome (MIM #176920) (45) were able to identify an activating mutation that occurs somatically in the AKT1 gene of the abnormal tissues. This activating mutation apparently results in overgrowth of the mutant cells Whole-exome resequencing (WES) has become more widely used for genetic diagnosis and gene discovery because it is less costly than WGS. However, despite the recent explosion of successful and useful applications of WES (Table 2), one must realize that it assesses nucleotide variation in only ~2% of the genome, the part that we believe we know how to interpret; 98% of the human genome is not assayed (Figure 3). This unappreciated variation might be particularly important when we investigate genetic and genomic variants associated with complex, heterogeneous, or more subtle phenotypes than the fully penetrant Mendelian diseases studied to date.
Table 2.
Exome sequencing in human diseases
| Disease | MIM # | Inheritance | Capture platform | Sequencing Technology | Samples | Identified gene | Reference |
|---|---|---|---|---|---|---|---|
| Congenital chloride diarrhea | #214700 | AR | Roche NimbleGen | Illumina | 1 affected | SLC26A3 | 40 |
| Miller syndrome | #263750 | AR | Agilent SureSelect | Illumina | 4 affecteds (1 sib-pair) | DHODH* | 41 |
| Schinzel-Giedion syndrome | #269150 | AD | Agilent SureSelect | SOLiD | 4 unrelated affecteds | SETBP1* | 42 |
| Nonsyndromic hearing loss DFNB82 | #613557 | AR | Agilent SureSelect | Illumina | 1 affected in family | GPSM2* | 46 |
| Perrault syndrome | #233400 | AR | Agilent SureSelect | Illumina | 1 affected in family | HSD17B4* | 47 |
| Kabuki syndrome | #147920 | AD | Agilent SureSelect | Illumina | 10 unrelated affecteds | MLL2* | 43 |
| Severe brain malformations | #600176 | AR | Roche NimbleGen | Illumina | 1 affected in family | WDR62* | 48 |
| Sensenbrenner syndrome/cranioectodermal dysplasia (CED) | #613610 | AR | Agilent SureSelect | SOLiD | 2 unrelated affecteds | WDR35* | 49 |
| Mabry syndrome/hyperphosphatasia with mental retardation | #239300 | AR | Agilent SureSelect | SOLiD | 3 affected siblings | PIGV* | 50 |
| Autosomal-dominant spinocerebellar ataxia | AD | Roche NimbleGen | Illumina | 4 related affecteds | TGM6* | 51 | |
| Mental retardation | AD | Agilent SureSelect | SOLiD | 10 parent-case trios | DYNC1H1, ZNF599*, RAB39B, YY1, BPIL3*, PGA5*, DEAF1, CIC, SYNGAP1, JARID1C | 52 | |
| Mitochondrial complex I deficiency | #611126 | AR | Agilent SureSelect | SOLiD | 1 affected | ACAD9* | 53 |
| Familial combined hypolipidemia | #605019 | AR | Agilent SureSelect | Illumina | 2 related affecteds | ANGPTL3* | 54 |
| Amyotrophic lateral sclerosis | AD | Agilent SureSelect | Illumina | 2 related affecteds | VCP* | 55 | |
| Autoimmune lymphoproliferative syndrome (ALPS) | #601859 | AR | Agilent SureSelect | Illumina | 1 affected | FADD* | 56 |
| Seckel syndrome | #210600 | AD | Agilent SureSelect | Illumina | 1 affected | CEP152* | 57 |
| CMT1X | #302800 | XL | Agilent SureSelect | Illumina | 2 related affecteds | GJB1 | 58 |
| Inflammatory bowel disease/X-linked inhibitor of apoptosis deficiency | XL | Roche NimbleGen | Roche 454 | 1 affected | XIAP | 44 | |
| Severe skeletal dysplasia | AR | Roche NimbleGen | Illumina | 2 affecteds and parents | POP1* | 59 | |
| Hajdu-Cheney syndrome (HCS) | #102500 | AD | Agilent SureSelect | Illumina | 3 unrelated affecteds | NOTCH2* | 60 |
| Osteogenesis imperfecta (OI) | AR | Agilent SureSelect | SOLiD | 1 affected in family | SERPINF1* | 61 | |
| Hereditary hypotrichosis simplex (HHS) | AD | Roche NimbleGen | Illumina | 1 affected in family | RPL21* | 62 | |
| Acne inversa/hidradenitis suppurativa | #142690 | AD | Agilent SureSelect | Illumina | 2 affecteds in family | NCSTN | 63 |
| Primary lymphoedema | AD | Agilent SureSelect | Illumina | 1 affected in family | GJC2* | 64 | |
| Hereditary sensory neuropathy with dementia and hearing loss (HSAN1) | #162400 | AR | Agilent SureSelect/Nimblegen | Illumina/Roche 454 | 4 kindreds | DNMT1* | 65 |
| Hereditary spastic paraparesis (HSP) | AR | Agilent SureSelect | Illumina | 1 parent-case trio | KIF1A* | 66 | |
| Hereditary progeroid syndrome | AR | Agilent SureSelect | Illumina | 2 affecteds | BANF1* | 67 | |
| Chondrodysplasia and abnormal joint development | AR | Agilent SureSelect | SOLiD | 3 affecteds | IMPAD1* | 68 | |
| Amelogenesis imperfecta and gingival hyperplasia syndrome | AR | Agilent SureSelect | SOLiD | 1 affected | FAM20A* | 69 | |
| Hypertrophic mitochondrial cardiomyopathy | AR | Agilent SureSelect | Illumina | 1 affected | AARS2* | 70 | |
| Mosaic variegated aneuploidy syndrome (MVA) | #257300 | AR | Agilent SureSelect | Illumina | 2 affected siblings | CEP57* | 71 |
| Autism spectrum disorder (ASD) | AD | Roche NimbleGen | Illumina | 20 parent-case trios | Potential genes identified | 72 | |
| Immunodeficiency–centromeric instability–facial anomalies syndrome type 2 (ICF2) | #614069 | AR | Roche NimbleGen | Illumina | 1 affected | ZBTB24* | 73 |
| High myopia | AD | Roche NimbleGen | Illumina | 2 affecteds | ZNF644* | 74 | |
| 3-M syndrome | AR | Agilent SureSelect | SOLiD | 3 affecteds | CCDC8* | 75 | |
| Late-onset Parkinson disease | #168600 | AD | Agilent SureSelect & Roche Nimblegen | Illumina | 2 affecteds & 2 affecteds | VPS35* | 76, 77 |
| Leber congenital amaurosis (LCA) | #204000 | AR | Agilent SureSelect | Illumina | 1 affected | KCNJ13* | 78 |
| Gray platelet syndrome (GPS) | #139090 | AR | Agilent SureSelect | Illumina | 4 affecteds | NBEAL2* | 79 |
| KBG syndrome | #148050 | AD | Roche NimbleGen | Illumina | 3 affecteds | ANKRD11* | 80 |
| Hereditary pheochromocytoma (PCC) | #171300 | AD | Agilent SureSelect | Illumina | 3 affecteds | MAX* | 81 |
| Bohring-Opitz syndrome | #605039 | AD | Agilent SureSelect | SOLiD | 3 affecteds | ASXL1* | 82 |
| Acromicric and geleophysic dysplasias | #231050 | AR | Agilent SureSelect | SOLiD | 2 affecteds | FBN1 | 83 |
| Hajdu-Cheney syndrome (HCS) | #102500 | AD | Agilent SureSelect | Illumina | 6 affecteds | NOTCH2* | 84 |
| Mitochondrial cardiomyopathy | AR | Agilent SureSelect | Illumina | 1 affected | MRPL3* | 85 | |
| Proteus syndrome | #176920 | somatic | Agilent SureSelect | Illumina | 17 samples from 12 affecteds | AKT1* | 45 |
Novel disease gene.
Figure 3.
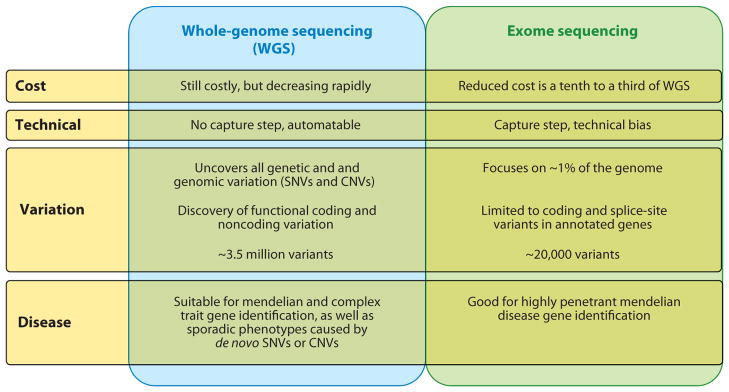
A comparison of the weaknesses and strengths of whole-genome sequencing (WGS) and exome sequencing approaches for disease-gene identification. Abbreviations: CNVs, copy-number variants; SNVs, simple nucleotide variants.
WHOLE-GENOME SEQUENCING FOR GENETIC DIAGNOSIS AND PATIENT MANAGEMENT
Initial personal genome projects delivered a number of individual diploid human genomes, but all of them were from individuals with no explicit clinical phenotype.
Although exome sequencing has successfully identified the causative mutations of selected highly penetrant Mendelian diseases, it interrogates SNVs for only the coding fraction of the genome that we have annotated as functional. Many other variants, including SNVs as well as CNVs, in noncoding, conserved, or regulatory regions can confer disease. These cannot be analyzed by sequencing only the exome (Figure 3).
The true challenge for personalized genomics is to identify disease-causing mutations among the approximately 3.0–3.5 million SNVs (on average) and ~1,000 CNVs in a given human diploid genome.
The Lupski Genome
In 2010, Lupski et al. reported the complete genome sequencing of an individual with Charcot-Marie-Tooth neuropathy (CMT1) and the identification of the disease-causing mutations in this individual and his family (86). This personal genome was obtained by NGS − at ~30× average depth coverage to ensure the identification of most of the variants. In addition to NGS, multiple aCGH platforms were utilized for independent detection, validation, and analysis of CNV.
Comparison of this individual’s genome sequence to the human genome reference assembly and filtering of the SNP variants identified 1.16 million SNPs in intragenic regions, of which 9,069 were nonsynonymous coding substitutions, including 121 nonsense substitutions.
A candidate gene analysis of functional SNPs in 40 known neuropathy-associated genes revealed compound heterozygosity for mutations in the SH3 domain and tetratricopeptide repeats 2 (SH3TC2) gene. The first variant was identified at ~7× coverage; additional sequencing revealed a second variant in the same gene. The first variant was a novel missense mutation (p.Y169H) and the second was a previously identified disease-causing nonsense mutation (p.R954X). Further tests showed that the two identified variants segregated with the disease and that only those individuals who had inherited both pathogenic variant alleles at this locus presented the CMT1 phenotype. Interestingly, the authors noted cosegregation of each of the heterozygous variants in other family members with one of three distinguishable electrophysiological phenotypes from studies performed on the entire family. These analyses suggested that individuals heterozygous for the missense mutation presented with an axonal neuropathy phenotype, whereas carriers for the nonsense mutation exhibited median nerve findings consistent with susceptibility to carpal tunnel syndrome.
In addition, other variants were identified in the proband’s genome that might be of clinical signficance. Some were associated with pharmacogenetic traits, including a homozygous variant associated with drug-induced cholestasis or warfarin sensitivity. Carrier status for other Mendelian diseases, such as Cockayne syndrome (MIM #133540), erythropoietic protoporphyria (MIM #177000), and Refsum disease (MIM #266500) was identified, as well as variants associated with risk and protection from different types of cancer. Of note, a presumed pathogenic variant was found in the ABCD1 gene, responsible for X-linked adrenoleukodystrophy (MIM #300100); the proband does not present the disease.
Whole-genome resequencing was applied to determine the cause of hypercholesterolemia in an 11-month-old girl with a family history negative for hypercholesterolemia, who was born to unrelated healthy parents (87). After Sanger sequencing all the genes suspected to be responsible for hypercholesterolemia without finding any disease-causing mutations, the genome of this patient was sequenced at ~49× average coverage in order to identify the genetic cause of her disease. Comparison to the human reference genome identified 3.29 million SNPs and 502,000 indels and other variants. Initial analysis focused on coding variants, mainly non-synonymous and splice-site variants, of which there were 9,726. Filtering for novel variants reduced the number of variants to 699 in 604 genes. The authors adopted a recessive model for this child’s disease and consequently looked for genes that contained at least two nonsynonymous variants.
Functional classification of the variants in the candidate genes identified compound heterozygous nonsense mutations in ABCG5. Confirmation of the mutations in the proband by Sanger sequencing showed that these were true positive variants and that the mother was a carrier for the p.Q16X mutation, whereas, consistent with Mendelian expectations, the father was heterozygous for the p.R446X mutation. The latter mutation had been previously reported as causative for sitosterolemia (MIM #210250), while the p.Q16X mutation was novel. It is interesting to note that the original diagnosis for this patient was hypercholesterolemia and not sitosterolemia because the initial plasma levels of plant sterols were deemed nondiagnostic for sitosterolemia. Therefore, the known genes for sitosterolemia, ABCG5 and ABCG8, were not tested. However, this was because the patient was breast-fed at the time of testing and therefore her dietary consumption of plant sterols was minimal and not accumulating in plasma, although this imbalance caused increased levels of cholesterol. Later testing, at two years of age, for plasma sterols and cholesterol levels showed values consistent with the sitosterolemia diagnosis. Treatment with a sterol absorption inhibitor and a low-cholesterol, low-plant-sterol diet helped to lower this patient’s plasma cholesterol levels.
Most recently, WGS proved useful in the molecular diagnosis and therapeutic management of a pair of twins with dopa-responsive dystonia (DRD; MIM #605407) of unknown genetic cause (88). The genomes of a pair of fraternal twins with childhood-onset dystonia were sequenced at ~30× coverage. In total 2.50 million and 2.42 million SNPs were found, of which both twins shared 1.63 million. Analysis of the variants identified 9,531 shared coding SNPs of which 4,605 were shared nonsynonymous. Assuming a recessive model for DRD in these twins, the authors searched for genes that had two or more variants, which narrowed the list of candidates to three genes. Interestingly, one of the candidate genes was SPR (sepiapterin reductase), a gene in which mutations have been previously associated with DRD. However, SPR is thought responsible for <3% of all cases of this rare disorder and therefore there was initially no clinically available specific gene test for it. A missense (p.R150G) and a nonsense (p.K251X) mutation were found as compound heterozygous in SPR; these were further confirmed by Sanger sequencing in both twins, and both parents were shown to be heterozygous.
The enzyme sepiapterin reductase is involved in the biosynthesis of tetrahydrobiopterin (BH4), an important cofactor for the enzymes involved in the metabolism of aromatic amino acids, including tyrosine hydroxylase (involved in the biosynthesis of dopamine) and tryptophan hydroxylase (involved in the biosynthesis of serotonin). An interesting unanticipated aspect of this study was drawn from the variant information provided. Not only did WGS allow elucidation of the genetic cause of DRD in these patients, but identification of the mutated gene suggested therapeutic management changes to further optimize treatment. Although being treated with L-Dopa greatly improved the condition of these patients, residual clinical signs and symptoms remained. With SPR mutations, the serotonin pathway is also unbalanced because of insufficient BH4. Supplemental therapy with adjuvant 5-hydroxy-tryptophan (5HTP) was shown to compensate the serotonin-production pathway, resulting in documented clinical improvement in these patients. Adjuvant therapy for this form of DRD can also include selective serotonin reuptake inhibitors (SSRIs). Interestingly, the heterozygous nonsense variant was, as expected, identified in the obligate carrier mother, but furthermore found in the maternal grandmother, both of whom had been diagnosed previously with fibromyalgia, a condition that can respond to SSRIs. WGS led to unanticipated insights that provided new therapeutic avenues based on the medically actionable variants identified, and the applied medical treatment resulted in amelioration of symptoms, marking a true landmark in personal medical genomics.
Probably the most immediate applicability of genomic sequencing in clinical practice, in addition to reaching an accurate genetic diagnosis of a given disease, is in the field of pharma-cogenomics (89). It is now possible to identify the genome-wide totality of potentially clinically relevant pharmacogenomic variants and ascertain if an individual is a fast or slow metabolizer of a certain drug, allowing individualized dosage adjustment to maximize therapeutic effect and minimize side effects.
HUMAN GENOME VARIATION: OUR CURRENT VIEW
Sequencing and analysis of personal human genomes has revealed that each individual differs from the human genome reference sequence at 3.5 million SNPs on average (Table 1). Some variants identified in personal genomes have in fact represented the common alleles in the population, suggesting that “rare SNPs” may be overrepresented in the reference. However, the number of novel variants found in each genome does not seem to decrease as more genomes are sequenced. A given personal genome has on average 400,000–600,000 novel SNPs when compared to the dbSNP only; remarkably, additional comparison with other personal genomes of unrelated individuals reveals on average ~200,000 novel unique variants per individual. The sequencing of the first personal genomes yielded ~14.6 million nonredundant SNPs that differed from the reference assembly (Figure 1). As predicted, we have observed that the genomes of older world populations (e.g., Africans) contain more SNPs, and some SNPs have become fixed in certain populations and not others.
Considering all the different types of variation, we have come to realize that on average a pair of homologous chromosomes in a given individual is ~99.5% identical in total number of base pairs, in contrast to the assumption that any two human individuals are 99.99% identical at the DNA level.
The aggregate of human genomic information and the catalogue of human variation from the human genomic projects—including the HGP, HapMap, and more recently the sequencing of personal genomes and TGP, as well as population studies of genome-wide CNVs, have taught us lessons regarding our species’ genome architecture, variation, evolution, and function (see sidebar, “Lessons Learned from Personal Genomes”). The foundation provided by the HGP with the human genome reference sequence enabled these gains in knowledge.
CHALLENGES
As more personal genomes are sequenced and made publicly available, we will uncover more genomic variation. This will likely further illuminate how the totality of genomic variation accounts for polymorphic traits and both complex and Mendelian diseases.
LESSONS LEARNED FROM PERSONAL GENOMES.
The human genome is highly variable. Each personal genome differs from the reference human assembly in ~ 3.5 million SNPs and 1000 large (>500 bp) CNVs.
SNPs are more frequent in autosomes than in the sex chromosomes.
The human genome is under purifying selection. There is a bias against SNP and indel ocurrence in internal exons; their occurrence is enriched in the first and last exons of genes. There is a bias favoring indels of multiples of three in order not to disrupt the reading frame.
The capability to call SNPs accurately from whole-genome sequencing (WGS) data increases with the average depth of coverage. Homozygous positions require 10–15× average depth of coverage, and sensitivity to detect >99% of the heterozygous positions starts at ~30×.
A predominance of heterozygous SNPs is observed among the novel variants. These probably represent rare variants that have arisen recently and are private to families or “clans.” However, these may add to the mutation load of the individual and should be considered when analyzing for disease associations and carrier status.
On average, the genome of any individual will contain 20,000–25,000 coding variants, of which 9,000–11,000 are nonsynonymous and a slightly higher number are synonymous.
It has been estimated that a “normal, healthy” individual is a heterozygous carrier of 40–100 highly penetrant deleterious variants that can potentially cause a Mendelian disease; many of these represent recessive carrier states (90). However, this estimate is based only on the coding regions and the approximately 5%–10% of genes and diseases that we currently understand; it might be that we all carry many more deleterious changes or potentially pathogenic variants than we now predict.
Comparison of the nonsynonymous SNPs in personal genomes provides a glimpse of variation patterns. Some genes are prone to accumulating changes, either because they are less essential for the survival and fitness of the individual or because they might tolerate more genetic diversity. Thus, new mutation may play a much greater role in evolutionary adaptation to a particular environment than anticipated. This could be particularly relevant to disease states.
Genes with functions associated with environmental adaptation, such as those involved in sensory functions (e.g., olfactory and taste receptors) or immunological functions and signal transduction (e.g., GPCRs) seem to be enriched for nonsynonymous SNPs. For example, it is well recognized that some of the genes that vary the most in humans are those for olfactory receptors (91, 92).
In several of the personal genomes published, supposedly highly penetrant mutations causative of Mendelian disease were identified in homozygous or hemizygous states even though the subjects were healthy. One explanation is that owing to an uneven distribution of reads throughout the genome, there might be fewer reads to accurately call the variants in these positions and these variants may actually be heterozygous. Another possibility is that these are rare polymorphisms that were identified in a disease-affected patient and mistakenly reported to be the disease-causing mutations. Alternatively, penetrance of Mendelian disease-associated variants may be lower than anticipated, as they have often not been studied in unaffected individuals.
In each genome sequenced, there have been megabases of DNA sequence that cannot be mapped to the haploid reference genome assembly or to any other genome. This sequence is enriched for repeated elements but also contains functional elements including genes, many of which are known to be relevant to environmental perception and adaptation.
Structural variation in the human genome is unexpectedly high. Clear patterns are observed, such as the peaks of retro-transposable element dimorphisms. Furthermore, the CNV allele frequency spectrum reveals a much higher frequency of smaller CNVs (<1 kb) and indels (<100 bp) (Figure 2).
Our current understanding of the human genome has led us to prioritize coding non-synonymous variants over the other >3 million SNVs and ~1,000 CNVs that we currently do not understand, in our quest to identify disease-causing mutations. Although this approach will most probably uncover highly penetrant and functionally relevant variation that is disease causing, it does not address potential consequences of a tremendous number of variants, not only coding but also noncoding, which may also be functionally relevant.
Caution is necessary with analysis approaches that filter out variants by using computational predictions of the functional impact of new variants. Many times the algorithms to predict if a given nonsynonymous variant is pathogenic have been useful, but many other known disease-causing mutations are predicted to be benign or polymorphic. Consequently, although computational predictions can aid the prioritization of identified variants, they do not provide sufficient adequate criteria to eliminate candidates (93).
Despite the availability of several diploid individual human genomes, we still know little about haplotypes in most of these genomes. It is important to consider haplotypes if we are to fully understand the potential effects of variants and their interplay with other genes. The cis interactions between variants on the same chromosome and the trans interactions between those on homologous or different nonhomologous chromosomes should also be considered when evaluating expression of genes. The importance of knowing the haplotypes might be more evident when considering variants in imprinted genes and their functional consequences. More subtle perhaps is their impact as expression-quantitative trait loci and the variability some loci exhibit due to either allele-specific or parent-of-origin-specific expression. Expression levels differ among individuals, largely because of inherited variation in the genome (94). Gene expression studies have identified DNA variation and several noncoding regulatory regions that influence the differential expression of genes (95). Genome-wide approaches to sequence the total transcriptome (RNA-seq) of a specific cell type or particular physiological state are beginning to show how and which genes are expressed differentially, in addition to linking these changes in expression with specific genetic variants (96). This type of variation in expression is also important to consider because in some cases disease might be the result of altered expression (97), as also revealed by CNV-associated pathogenic gene-dosage effects. The contribution of quantitative trait loci to specific traits is still not well determined even in well-studied and common traits such as height and skin color.
Understanding not only the functional but more importantly the medical significance of variants is a challenging and still evolving task. For fully penetrant mutations in known disease genes, the functional impact of variants can be readily determined. However, the challenge remains for the ~20,000 genes for which function has not been assigned and phenotypes or associated traits have not been elucidated. Other key questions are how the variants in different genes modify a given phenotype, how genes interact, and how the alleles within a pair interact. The recognition of what constitutes a medically actionable variant is currently an imperfect science. Guidelines for clinical interpretation of WGS are beginning to appear (98).
Because the main research objective of exome sequencing and WGS is to discover the genetic causes of rare and complex diseases, we must consider other factors that may confound our analysis and filtering criteria as we analyze candidate variants (Figure 4). For example, in attempts to identify recessive traits and diseases, “normal” control individuals might be carriers, and some recessive alleles may be low-penetrance alleles that exist in the general population but do not confer any phenotype unless combined with a null or other mutant allele for that gene. Filtering candidate variants against population databases might be counterproductive in these cases. Allele frequency spectra may become an important parameter for determining if a variant is likely benign.
Figure 4.

Schematic workflow of whole-genome/exome sequencing data analysis. After sequencing, the sequence reads are mapped and aligned against the human reference genome assembly in order to obtain a list of variants at every position that does not match the reference. Quality filters are applied to obtain high-quality variant calls. Various filtering criteria are applied to prioritize the candidate variants. Most variants will be excluded because they are known, meaning that they are already in variation databases, such as the database of single nucleotide polymorphisms (dbSNP), The 1000 Genomes Project database, etc. The focus is mainly on novel variants, which can be tiered in functional classes according to their annotation. For coding variants, priority is given to nonsense, frameshifting, splice-site, and then missense mutations. Computational prediction of the functional impact of these variants can also help prioritize candidate mutations. Based on the characteristics of the trait or disease of interest, variants can be examined under a dominant or recessive model. Additional confirmation through other resources can strengthen the hypotheses of the functional significance of identified variants. Genetic and functional confirmation of the candidate disease-causing variants is the final, most important step.
A substantial and not yet entirely appreciated problem for personal genome sequence analysis, especially for medical diagnosis and applications, is the veracity of the current mutation databases. If we are to use WGS in clinical practice, it is of the utmost importance that mutation databases recognize potentially pathogenic variants of clinical significance, i.e., distinguish medically actionable variants from benign variation.
FUTURE ISSUES
Large-scale human genome sequencing projects and other disease-focused sequencing projects will add more variants to the databases. The challenge that remains is the analysis of this information and the knowledge to be gained concerning the biology of our own genome. Structural variation is still challenging to assess using only NGS platforms. Comparison and standardization of sequencing technologies and improvement in mapping and de novo assembly algorithms will eventually allow the accurate prediction of indels, CNVs, and inversions at the nucleotide level of resolution.
Other technical challenges are storing and accessing the vast amounts of data that genomic projects produce. Should we store the data files produced by the sequencing machines or just the processed and analyzed data? Will access be public or restricted for research purposes only? In addition, the bioinformatic analysis still remains a bottleneck; even with automated pipelines, processing these vast amounts of information still requires extensive computational power and time. Furthermore, current algorithms are suboptimal for some purposes; improvement of current and development of novel algorithms are necessary. A goal for WGS should be the ability to provide all of the variants in an individual’s genome in a highly reliable manner. Nevertheless, clinical utility can be achieved for many patients long before that laudable goal.
Next-generation resequencing of personal genomes will in the very near future become common practice. An initial clinical application may be to assay for genetic susceptibilities and factors that may contribute to a disease state for a genetically heterogeneous condition if current panel testing is prohibitively expensive. Complete genome sequencing is leading the way toward making personalized genomic medicine possible in the near future. Legal and ethical issues may arise—some anticipated, others not (99).
Eventually, decreasing costs may allow personal genome sequencing to be available for everyone. However, analysis, annotation, and interpretation of variant information are essential to provide clinicians and patients with information that can be used to better manage an individual’s health or disease (100).
Acknowledgments
J.R.L. receives support from the National Institute of Neurological Disorders and Stroke (R01 NS058529) and the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center (HD024064). R.A.G. receives support from the National Human Genome Research Institute (5 U54 HG003273).
Glossary
- HGP
Human Genome Project
- SNV
simple nucleotide variation
- Inversion
a genomic segment that differs in orientation compared with the human reference genome
- Copy-number variant (CNV)
a genomic segment that deviates from the expected locus-specific diploid state, either through deletions or amplifications
- NGS
next-generation DNA sequencing
- MAF
minor allele frequency
- TGP
The 1000 Genomes Project
- Segmental duplication
a region of the human genome >1 kb in size that is duplicated, sharing at least 90% identity with its other copy located elsewhere in the genome
- WGS
whole-genome sequencing
Footnotes
DISCLOSURE STATEMENT
J.R.L. is a consultant for Athena Diagnostics and Ion Torrent Systems Inc., and holds multiple U.S. and European patents for DNA diagnostics. R.A.G. has co-investments with Life Technologies. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular diagnostic testing (Medical Genetics Laboratories: http://www.bcm.edu/geneticlabs/).
Contributor Information
Claudia Gonzaga-Jauregui, Email: gonzagaj@bcm.edu.
James R. Lupski, Email: jlupski@bcm.edu.
Richard A. Gibbs, Email: agibbs@bcm.edu.
LITERATURE CITED
- 1.International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 3.Bailey JA, Yavor AM, Massa HF, et al. Segmental duplications: organization and impact within the current Human Genome Project assembly. Genome Res. 2001;11:1005–17. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–22. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 5.The International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 6.The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The International HapMap Consortium. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lupski J. Genomic disorders ten years on. Genome Med. 2009;1:42.2–42.11. doi: 10.1186/gm42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy S, Sutton G, Ng PC, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ginsberg G, Smolenski S, Neafsey P, et al. The influence of genetic polymorphisms on population variability in six xenobiotic-metabolizing enzymes. J Toxicol Environ Health Part B: Crit Rev. 2009;12:307–33. doi: 10.1080/10937400903158318. [DOI] [PubMed] [Google Scholar]
- 12.Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–76. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 13.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKernan KJ, Peckham HE, Costa GL, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–41. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuster SC, Miller W, Ratan A, et al. Complete Khoisan and Bantu genomes from southern Africa. Nature. 2010;463:943–47. doi: 10.1038/nature08795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perry GH, Dominy NJ, Claw KG, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet. 2007;39:1256–60. doi: 10.1038/ng2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aldred PMR, Hollox EJ, Armour JAL. Copy number polymorphism and expression level variation of the human alpha-defensin genes DEFA1 and DEFA3. Hum Mol Genet. 2005;14:2045–52. doi: 10.1093/hmg/ddi209. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Wang W, Li R, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahn S-M, Kim T-H, Lee S, et al. The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome Res. 2009;19:1622–29. doi: 10.1101/gr.092197.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J-I, Ju YS, Park H, et al. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–15. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–28. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 22.Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–51. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 23.Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 24.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conrad DF, Pinto D, Redon R, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2009;464:704–12. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flores M, Morales L, Gonzaga-Jauregui C, et al. Recurrent DNA inversion rearrangements in the human genome. Proc Natl Acad Sci. 2007;104:6099–106. doi: 10.1073/pnas.0701631104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lakich D, Kazazian HH, Antonarakis SE, et al. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet. 1993;5:236–41. doi: 10.1038/ng1193-236. [DOI] [PubMed] [Google Scholar]
- 28.Stefansson H, Helgason A, Thorleifsson G, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37:129–37. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- 29.Koolen DA, Sharp AJ, Hurst JA, et al. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet. 2008;45:710–20. doi: 10.1136/jmg.2008.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–81. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–55. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 32.Conrad DF, Bird C, Blackburne B, et al. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet. 2010;42:385–91. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lupski JR. Retrotransposition and structural variation in the human genome. Cell. 2010;141:1110–12. doi: 10.1016/j.cell.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 34.Iskow RC, McCabe MT, Mills RE, et al. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141:1253–61. doi: 10.1016/j.cell.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beck CR, Collier P, Macfarlane C, et al. LINE-1 retrotransposition activity in human genomes. Cell. 2010;141:1159–70. doi: 10.1016/j.cell.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ewing AD, Kazazian HH., Jr Whole-genome resequencing allows detection of many rare LINE-1 insertion alleles in humans. Genome Res. 2011;21(6):985–90. doi: 10.1101/gr.114777.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albert TJ, Molla MN, Muzny DM, et al. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4:903–5. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- 38.Bainbridge M, Wang M, Burgess D, et al. Whole exome capture in solution with 3 Gbp of data. Genome Biol. 2010;11:R62. doi: 10.1186/gb-2010-11-6-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–76. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci. 2009;106:19096–101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoischen A, van Bon BWM, Gilissen C, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42:483–85. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 43.Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–93. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–62. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 45.Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611–19. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walsh T, Shahin H, Elkan-Miller T, et al. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am J Hum Genet. 2010;87:90–94. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pierce SB, Walsh T, Chisholm KM, et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault syndrome. Am J Hum Genet. 2010;87:282–88. doi: 10.1016/j.ajhg.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bilguvar K, Öztürk AK, Louvi A, et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature. 2010;467:207–10. doi: 10.1038/nature09327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilissen C, Arts HH, Hoischen A, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010;87:418–23. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krawitz PM, Schweiger MR, Rödelsperger C, et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010;42:827–29. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 51.Wang JL, Yang X, Xia K, et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain. 2010;133:3510–18. doi: 10.1093/brain/awq323. [DOI] [PubMed] [Google Scholar]
- 52.Vissers LELM, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–12. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 53.Haack TB, Danhauser K, Haberberger B, et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;42:1131–34. doi: 10.1038/ng.706. [DOI] [PubMed] [Google Scholar]
- 54.Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363:2220–27. doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–64. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolze A, Byun M, McDonald D, et al. Whole-exome-sequencing-based discovery of human FADD deficiency. Am J Hum Genet. 2010;87:873–81. doi: 10.1016/j.ajhg.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalay E, Yigit G, Aslan Y, et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet. 2010;43:23–26. doi: 10.1038/ng.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Montenegro G, Powell E, Huang J, et al. Exome sequencing allows for rapid gene identification in a Charcot-Marie-Tooth family. Ann Neurol. 2011;69:464–70. doi: 10.1002/ana.22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Glazov EA, Zankl A, Donskoi M, et al. Whole-exome re-sequencing in a family quartet identifies POP1 mutations as the cause of a novel skeletal dysplasia. PLoS Genet. 2011;7:e1002027. doi: 10.1371/journal.pgen.1002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simpson MA, Irving MD, Asilmaz E, et al. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat Genet. 2011;43:303–5. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- 61.Becker J, Semler O, Gilissen C, et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2011;88:362–71. doi: 10.1016/j.ajhg.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou C, Zang D, Jin Y, et al. Mutation in ribosomal protein L21 underlies hereditary hypotrichosis simplex. Hum Mutat. 2011;32:710–14. doi: 10.1002/humu.21503. [DOI] [PubMed] [Google Scholar]
- 63.Liu Y, Gao M, Lv Y-m, et al. Confirmation by exome sequencing of the pathogenic role of NCSTN mutations in acne inversa (hidradenitis suppurativa) J Invest Dermatol. 2011;131:1570–72. doi: 10.1038/jid.2011.62. [DOI] [PubMed] [Google Scholar]
- 64.Ostergaard P, Simpson MA, Brice G, et al. Rapid identification of mutations in GJC2 in primary lymphoedema using whole exome sequencing combined with linkage analysis with delineation of the phenotype. J Med Genet. 2010;48:251–55. doi: 10.1136/jmg.2010.085563. [DOI] [PubMed] [Google Scholar]
- 65.Klein CJ, Botuyan M-V, Wu Y, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet. 2011;43:595–600. doi: 10.1038/ng.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Erlich Y, Edvardson S, Hodges E, et al. Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res. 2011;21:658–64. doi: 10.1101/gr.117143.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puente Xose S, Quesada V, Osorio Fernando G, et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet. 2011;88:650–56. doi: 10.1016/j.ajhg.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vissers Lisenka ELM, Lausch E, Unger S, et al. Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP. Am J Hum Genet. 2011;88:608–15. doi: 10.1016/j.ajhg.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O’Sullivan J, Bitu CC, Daly SB, et al. Whole-exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome. Am J Hum Genet. 2011;88:616–20. doi: 10.1016/j.ajhg.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Götz A, Tyynismaa H, Euro L, et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:635–42. doi: 10.1016/j.ajhg.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Snape K, Hanks S, Ruark E, et al. Mutations in CEP57 cause mosaic variegated aneuploidy syndrome. Nat Genet. 2011;43:527–29. doi: 10.1038/ng.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.O’Roak BJ, Deriziotis P, Lee C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–89. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Greef JC, Wang J, Balog J, et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am J Hum Genet. 2011;88:796–804. doi: 10.1016/j.ajhg.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shi Y, Li Y, Zhang D, Zhang H, et al. Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet. 2011;7:e1002084. doi: 10.1371/journal.pgen.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hanson D, Murray PG, O’Sullivan J, et al. Exome sequencing identifies CCDC8 mutations in 3-M syndrome, suggesting that CCDC8 contributes in a pathway with CUL7 and OBSL1 to control human growth. Am J Hum Genet. 2011;89:148–53. doi: 10.1016/j.ajhg.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zimprich A, Benet-Pagès A, Struhal W, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–75. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vilariño-Güell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–67. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sergouniotis PI, Davidson AE, Mackay DS, et al. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause Leber congenital amaurosis. Am J Hum Genet. 2011;89:183–90. doi: 10.1016/j.ajhg.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Albers CA, Cvejic A, Favier R, et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat Genet. 2011;43:735–37. doi: 10.1038/ng.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sirmaci A, Spiliopoulos M, Brancati F, et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011;89:289–94. doi: 10.1016/j.ajhg.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43:663–67. doi: 10.1038/ng.861. [DOI] [PubMed] [Google Scholar]
- 82.Hoischen A, van Bon BW, Rodríguez-Santiago B, et al. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet. 2011;43:729–31. doi: 10.1038/ng.868. [DOI] [PubMed] [Google Scholar]
- 83.Le Goff C, Mahaut C, Wang LW, et al. Mutations in the TGFβ binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am J Hum Genet. 2011;89:7–14. doi: 10.1016/j.ajhg.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Majewski J, Schwartzentruber JA, Caqueret A, et al. Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum Mutat. 2011;32:1114–17. doi: 10.1002/humu.21546. [DOI] [PubMed] [Google Scholar]
- 85.Galmiche L, Serre V, Beinat M, et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum Mutat. 2011;32:1225–31. doi: 10.1002/humu.21562. [DOI] [PubMed] [Google Scholar]
- 86.Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362:1181–91. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rios J, Stein E, Shendure J, et al. Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia. Hum Mol Genet. 2010;19:4313–18. doi: 10.1093/hmg/ddq352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bainbridge MN, Wiszniewski W, Murdock DR, et al. Whole genome sequencing enables optimized patient management. Sci Translational Med. 2011:87re3. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med. 2011;364:1144–53. doi: 10.1056/NEJMra1010600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.The 1000Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hasin-Brumshtein Y, Lancet D, Olender T. Human olfaction: from genomic variation to phenotypic diversity. Trends Genet. 2009;25:178–84. doi: 10.1016/j.tig.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 92.Hasin Y, Olender T, Khen M, et al. High-resolution copy-number variation map reflects human olfactory receptor diversity and evolution. PLoS Genet. 2008;4:e1000249. doi: 10.1371/journal.pgen.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hicks S, Wheeler DA, Plon S, et al. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum Mut. 2011;32:661–68. doi: 10.1002/humu.21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yan H, Yuan W, Velculescu VE, et al. Allelic variation in human gene expression. Science. 2002;297:1143. doi: 10.1126/science.1072545. [DOI] [PubMed] [Google Scholar]
- 95.Cheung VG, Spielman RS. Genetics of human gene expression: mapping DNA variants that influence gene expression. Nat Rev Genet. 2009;10:595–604. doi: 10.1038/nrg2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–77. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Emilsson V, Thorleifsson G, Zhang B, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–28. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 98.Berg JS, Khoury MJ, Evans JP. Deploying whole genome sequencing in clinical practice and public health: meeting the challenge one bin at a time. Genet Med. 2011;13:499–504. doi: 10.1097/GIM.0b013e318220aaba. [DOI] [PubMed] [Google Scholar]
- 99.McGuire AL, Lupski JR. Personal genome research: what should the participant be told? Trends Genet. 2010;26:199–201. doi: 10.1016/j.tig.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mardis E. The $1,000 genome, the $100,000 analysis? Genome Med. 2010;2:84. doi: 10.1186/gm205. [DOI] [PMC free article] [PubMed] [Google Scholar]