

Abstract
Fast scan cyclic voltammetry in brain slices (slice voltammetry) has been used over the last several decades to increase substantially our understanding of the complex local regulation of dopamine release and uptake in the striatum. This technique is routinely used for the study of changes that occur in the dopamine system associated with various disease states and pharmacological treatments, and to study mechanisms of local circuitry regulation of dopamine terminal function. In the context of this Review, we compare the relative advantages of voltammetry using striatal slice preparations versus in vivo preparations, and highlight recent advances in our understanding of dopamine release and uptake in the striatum specifically from studies that use slice voltammetry in drug-naïve animals and animals with a history of psychostimulant self-administration.
Keywords: Dopamine, voltammetry, ex vivo, in vivo, striatum, dopamine transporter, release, uptake, phasic, tonic, brain slices
1. Introduction
Voltammetric detection methods for oxidizable chemical species are widely used to study the kinetics of monoamine neurotransmitters.1,2 Fast scan cyclic voltammetry (FSCV) is a powerful electrochemical technique for studying neurotransmitter signaling, including release and uptake kinetics.3,4 FSCV can be performed both in vivo and ex vivo, where it can be used to investigate dopamine (DA) signaling, as well as changes that occur after various neurochemical, genetic, pharmacological, and behavioral manipulations.5,6 The purpose of this review is to (1) compare strengths and limitations of striatal slice preparations to strengths and limitations of in vivo anesthetized, freely moving, and freely behaving preparations, (2) review both direct and indirect modulators of DA release from studies using varying stimulation frequencies to model tonic and phasic firing of DA neurons, and (3) review recent findings from investigations on the modulation of striatal DA uptake and DA transporter (DAT) function following rodent self-administration of stimulants.
1.1. General Methodology, Modeling, and Analysis
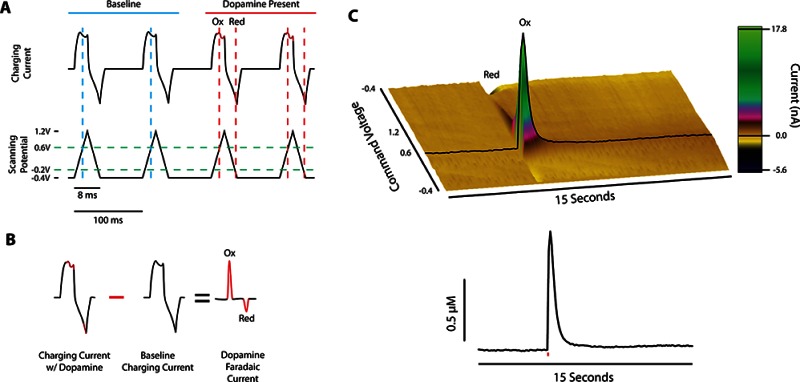
In relation to other monoamine detection methods, such as microdialysis, voltammetry has a high temporal and spatial resolution, making it a suitable method for examining rapid neurochemical signaling similar to that associated with phasic burst firing observed in dopaminergic neurons.7−9 For this technique, a triangular voltage ramp with parameters that can vary across laboratories and experiments (e.g., range of −0.4 to +1.2 V vs Ag/AgCl at 400 V/s) is generated by a computer (Figure 1A) and sent to a potentiostat which applies the voltage ramp to the carbon fiber electrode, producing a primarily capacitive charging current. During a voltage scan, DA is oxidized into dopamine-o-quinone during the rising phase (at 0.6 V) and reduced back to DA during the declining phase (at 0.2 V). The current produced by oxidation and reduction of a species is referred to as a faradaic current, and is the basis for voltammetric detection of DA. Since this faradaic current is additive to the charging current waveform, a simple background subtraction can be performed to remove the background current, while still maintaining the faradaic current, resulting in a voltammogram that aids in the identification of DA (Figure 1B). A series of background subtracted faradaic currents are plotted across time to produce a three-dimensional array of data, as seen in the pseudocolor plot in Figure 1C. The amplitude of the current (z-axis) at the oxidation potential (y-axis) over time (x-axis) is linearly proportional to the concentration of DA present at the electrode tip, so that electrodes can be exposed to known concentrations following an experiment to determine each electrode’s calibration constant.
Figure 1.

Fast scan cyclic voltammetry methodology for subsecond detection of dopamine. (A) Black traces show the charging current (top) resulting from a voltage sweep (bottom) in the absence (blue) and presence (red) of dopamine. Dopamine oxidation and reduction occur at 0.6 and −0.2 V (intercept of green and red dashed lines), respectively. (B) Charging currents containing dopamine are subtracted from baseline currents, resulting in a faradaic current proportional to the concentration of dopamine present. (C) Color plot of background subtracted data after an electrical stimulation shows dopaminergic changes in faradaic current across time (top panel), which can be represented two-dimensionally as change in DA concentration across time (bottom panel). Red line in bottom panel represents single-pulse electrical stimulation. The oxidation current traces from a color plot are compared with electrode calibration factors to determine release concentrations, and dopamine clearance is calculated to determine dopamine transporter function.
Since voltammetric measurement of DA relies on the presence of its signature voltammogram, it is of note that other neurochemicals such as norepinephrine and epinephrine have redox potentials similar to DA. To avoid confusion from signal contamination, researchers typically examine DA release in brain areas where DA is the predominant catecholamine, or use pharmacological techniques to demonstrate that the measured catecholamine is DA.10,11 For example, much work focuses on striatal recordings of DA, in relation to drug effects and learning mechanisms.12−17
There are multiple software packages designed for collecting and analyzing voltammetric data which are currently being used in ex vivo, in vivo anesthetized, freely moving, and freely behaving preparations.18−21 For example, we have recently developed a software program for voltammetric data collection and analysis called Demon Voltammetry and Analysis.21 This program contains a number of automatic signal analyses including Michaelis-Mentin kinetics (e.g., Vmax, apparent Km), parameters describing time constants and exponential decay functions for uptake (e.g., tau, half-life), principle component analysis for in vivo measurements, as well as ongoing development of collection and analysis features.
1.2. Comparing the Strengths and Limitations of Ex Vivo and In Vivo Voltammetry
Voltammetric recordings are commonly performed in in vivo and ex vivo preparations12−17 with each paradigm presenting its own set of strengths and limitations. Here we compare each preparation, and the suitability of these preparations in assessing DA activity.
1.2.1. Voltammetry Performed In Vivo
Voltammetry has been performed in vivo using a wide range of species including (but not limited to) mice, rats, monkeys, and humans.5,22−24 In vivo voltammetry can be further categorized by the specific preparation that is used, which includes anesthetized, freely moving, and freely behaving preparations. There is considerable overlap in their strengths when compared to ex vivo voltammetry; nevertheless, the variety of options stems from researchers’ need to answer a specific questions that may not easily be addressed by a single preparation. Compared to ex vivo voltammetry, the most notable strengths of in vivo voltammetry are that (1) neural circuitry remains intact and (2) DA neurochemistry can be explored in situ. These are common advantages for all in vivo preparations, and they allow experimenters to investigate the systems that modulate and/or elicit DA release in addition to studying effects of drugs following systemic administration on DA activity.
1.2.1.1. Voltammetry Performed in Anesthetized Preparations
For the anesthetized preparation, electrical, optical, or pharmacological-induced stimulation of DA cell bodies (or excitatory efferent projections to DA cell bodies) is required to elicit DA release.22,26,27 An advantage of electrical or optical stimulations is that DA release typically reaches levels that are sufficient for saturating the DAT, a prerequisite for accurately estimating the maximal rate of DA uptake.28 This saturation is often not present for in vivo preparations that do not artificially elicit DA release, which makes investigating the role of the DAT in DA signaling difficult. The ability to model DA uptake makes preparations that stimulate DA cell bodies inherently appealing. For example, voltammetry in anesthetized rodents is ideal for examining the pharmacokinetics of drug action, such as rate of drug onset.29,30 These studies require systemic drug delivery in combination with the ability to model DA uptake accurately. Understanding the rate of drug onset is critical for understanding drug abuse liability, and several previous studies have explored drug onset by measuring electrically evoked DA release and uptake at several time points after an intravenous injection.29−32 One inherent disadvantage for electrical stimulation, however, is the lack of specificity for activating DA. Indeed, electrical stimulation of the VTA, for example, will also activate GABA and glutamatergic cell bodies, which could modulate the release of DA. This lack of specificity has been reduced with the advent of optogenetic stimulation of DA cell bodies.26 Nevertheless, both electrical and optogenetic stimulation remains artificial, and may not fully recapitulate natural firing of DA neurons in response to stimuli in the environment, even under optimized conditions. An additional caveat to anesthetized preparations is that anesthetics are powerful modulators of brain neurochemistry, and may alter DA signaling.33 For these reasons, many researchers prefer performing voltammetry in freely moving, nonanesthetized preparations.
1.2.1.2. Voltammetry Performed in Freely Moving Preparations
Voltammetry in a freely moving preparation is similar to voltammetry in an anesthetized preparation, except that animals are allowed to recover from the anesthesia administered for probe implantation surgery, in order to measure DA signals in awake, freely moving subjects. Electrical, optical, or pharmacologically induced stimulation of DA is sometimes used, but is not required, to elicit DA release in freely moving preparations. Therefore, all of the strengths and caveats of electrical and optogenetic stimulation described for anesthetized preparations are the same for freely moving preparations. One additional advantage to freely moving preparations is that DA release is not influenced by anesthetic drugs, which typically prevent spontaneous, nonexperimenter evoked DA release events; termed DA transients. Therefore, voltammetry in freely moving animals is required to investigate the neural circuitry that is involved in modulating and eliciting DA transients. Freely moving studies have demonstrated that the main excitatory effects of ethanol and cannabinoids on DA release are through increased frequency of DA transients.34,35 In fact, all psychostimulant drugs of abuse (that have been investigated) have demonstrated increased frequency and/or magnitude of DA transients following systemic administration,36−38 an effect not typically observed following systemic administration of psychostimulants in anesthetized preparations .25,27 However, not all DA transients are spontaneous. Using voltammetry in freely moving animals, researchers have demonstrated that passive, non-contingent receipt of reward or food can elicit a DA burst that is time-locked to the receipt of the reward.39 There are a few practical considerations when deciding to perform voltammetry in freely moving animals. One is increased attrition rate given the tendency for animals to displace recording electrodes, particularly when administering psychostimulant drugs that increase locomotion and stereoptyped grooming behavior. Second, DA transients are smaller in magnitude compared to DA release from direct stimulation of DA cell bodies. Third, baseline signals tend to be less stable given greater shifts in pH and other transmitters in the brain of awake, freely moving animals. Points two and three combined means smaller signal-to-noise ratios, and therefore, voltammetry in freely moving animals requires sophisticated chemometric and principle component analyses to isolate and quantify DA signals.20,40
1.2.1.3. Voltammetry Performed in Freely Behaving Preparations
Voltammetry in freely behaving animals expands the freely moving preparation to monitor DA release events that are time-locked to behavior and encounters with meaningful environmental stimuli. This preparation is necessary for demonstrating behavioral correlates of spontaneous and environment-evoked DA release.6 This preparation has shed considerable light on our understanding of how DA signaling relates to learning, motivation, and reinforcement. For example, seminal work using this technique demonstrated neurochemical correlates to Pavlovian/associative learning as well as operant conditioning within the nucleus accumbens (NAc).12,39,41,42 Namely, voltammetry paired with either contingent or noncontingent receipt of rewards has documented increases in DA release in response to environmental cues that are paired with or predict the reward, as well as to the rewards themselves.39,41 Furthermore, cues that predict footshock, aversive stimuli, or even extended delays in receipt of a cocaine infusion engenders a decrease in DA signals.42−44 This preparation has been advanced in recent years by the development of chronic electrodes that can monitor rapid DA signals for weeks to months45,46 as well as the use of electrode arrays which can be used to monitor DA activity in multiple brain areas simultaneously during behavior.46
1.2.2. Voltammetry Performed Ex Vivo
The strengths and limitations of voltammetry performed in brain slices are interwoven throughout the remainder of the Review. However, this section is included specifically to highlight a number of strengths that set the background for studies reviewed herein. Slice preparations as a whole are a more attractive paradigm for measuring kinetics. For instance, release signals are more robust in slices, allowing a more accurate measurement of maximal uptake rates. It is generally accepted that in order to get an accurate measure of uptake, DA levels must be approximately 10× the affinity of DA for the DAT (0.16 μM28,47). Anesthetized preparations do reach levels that meet these criteria when DA cell bodies are stimulated; however, the ease and precision of electrode placement for slice preparation is unmatched. This spatial resolution is particularly attractive given the established heterogeneity in both uptake and release measures within microdomains in the striatum. An additional strength of slice voltammetry is the ability to take measurements from multiple regions within the same slice, as well as multiple slices from the same animal.48 This can provide more powerful experimental designs, in that multiple drug concentration–response curves can be tested within a single animal, allowing for correlational analyses, and reducing subject numbers. Also, ex vivo voltammetry is a more powerful technique for measuring receptor potency and efficacy shifts of hetero and autoreceptor activity. The power of this technique comes from the ability to measure the effects of a drug across a number of increasing concentrations. The ability to run full concentration–response curves within a single animal allows for the determination of pharmacological measures such as efficacy, IC50, Ki, and Kd values, and other measures of shifts in potency. This is often difficult, if not impossible, within a single animal using in vivo preparations. This difficulty arises because pharmacological measures cannot be determined with a single dose, since it is impossible to determine whether the shift in the dose–response is an upward/downward efficacy shift or a leftward/rightward potency shift, both of which may result from different pharmacological changes. For example, the D2-type receptor agonist quinpirole reduces evoked DA release in a concentration dependent manner, which can be used to assess shifts in quinpirole potency after a treatment such as cocaine self-administration. This attenuation of DA release is shifted after cocaine self-administration, so that it requires greater concentrations of quinpirole to have the same effect on stimulated DA release (decreased potency), indicative of overall decreased endogenous autoreceptor activity.49
Another strength of voltammetry in slices is the ability to generate stimulations that mimic both tonic and phasic DA firing that occur in awake, behaving animals. It is typically more difficult to detect DA release from low pulse, low frequency stimulations that model tonic firing of DA neurons in in vivo anesthetized and freely moving preparations. As discussed in section 2.1, modeling of tonic and phasic firing patterns ex vivo yields similar patterns of DA release compared to cue-evoke DA detected using voltammetry in freely behaving animals.
While one limitation of voltammetry in brain slices is that much of brain circuitry is removed, a corresponding strength is the ability to isolate brain regions and explore local circuitry and regulation of DA release and uptake more readily than in vivo preparations. Moreover, it is often difficult to infuse drugs and manipulate local circuitry in vivo while not interfering with the recording electrode.50 As reviewed more below, voltammetry in brain slices allows for direct examination of local modulators of DA release and uptake. This includes, but is not limited to, acetylcholine (ACh) interneuron modulation of DA release,51 retrograde signaling and modulation from medium spiny neurons (MSN),52 and modulation from receptors located directly on DA nerve terminals, such kappa opioid53 and nicotinic ACh receptors (nAChR).54,55
1.2.2.1. Assessing DA Kinetics Ex Vivo Following In Vivo Drug Administration: A Two-Pronged Approach
One approach commonly used in our laboratory is to measure shifts in drug potency and efficacy using slice voltammetry following in vivo administration of illicit and therapeutic drugs. This two-pronged approach allows for correlation of drug-induced shifts in behavior with shifts in local DA circuitry and DA nerve terminal function. Specific findings on DA release and uptake that have emerged from this two pronged approach is almost solely reviewed in sections 2.3. and 3.4, respectively. While voltammetry in freely behaving animals also allows for correlating neurochemistry and behavior, slice voltammetry bypasses potential drug-induced changes in pharmacokinetics that could contaminate DA signals following systemic administration. The concentration–response curves that are generated from voltammetry in slices can correspond directly to multiple doses of drugs that are administered systemically. Additionally, voltammetry in slices bypasses nonspecific and spontaneous release of DA and other transmitters as well as shifts in pH that occur in freely moving animals.23 Depending on the specific research question, these signals could occlude the investigation of changes in the ability of specific receptors and neurotransmitters to modulate DA release and uptake.
2. Using Slice Voltammetry to Study Local Regulation of DA Release in the Striatum
This section reviews some of the primary findings from slice voltammetry studies that use both tonic and phasic stimulations to explore mechanisms and modulators of DA release. DA cell bodies in the substantia nigra pars compacta (SNc) and ventral tegmental area (VTA) project to dorsal and ventral striatum, respectively.56 Although some degree of overlap exists both topographically and functionally, SNc projections to dorsal caudate (CPU) are generally thought to be involved in motor behaviors and habit formation/learning, while VTA projections to ventral striatum (i.e., NAc) are involved in reward, motivation, salience and associative learning.57,58 DA release from nerve terminals in the striatum is linked to the excitability of DA cell bodies in the SNc and VTA, while local striatal circuits can both directly and indirectly modulate the magnitude54 and onset59,60 of DA release from axons and terminals.
2.1. Characteristics of DA Release in the Striatum
Since the mid-1980s, researchers have used voltammetry in rodent brain slices to understand local DA release and uptake dynamics.61,62 While some work exploring stimulated DA release was performed by pioneers in the field in the 1980s and 1990s,61−67 the vast majority of published work using ex vivo voltammetry was dedicated to understanding uptake kinetics via the DAT.68−71 Indeed, voltammetry in a brain slice preparation is arguably among the most powerful approaches for exploring the functional implications of genetic and drug-induced alterations in DAT function. Nevertheless, there has been a substantial increase in the number of publications using slice voltammetry to explore DA release dynamics throughout the striatum. Given the lack of spontaneous DA signals in striatal brain slices as well as the post-mortem nature of the experiments, the general approach for ex vivo studies of DA release has been to use electrical stimulations to model endogenous DA firing that occurs in response to environmental stimuli. Early work using electrically evoked DA showed substantial heterogeneity in the magnitude of DA release elicited by a single electrical pulse across subregions of the striatum. A gradient in the magnitude of DA release from relatively small signals in the ventromedial striatum (NAc) to larger signals in the dorsolateral striatum48,71,72 has been shown to be present in primate (marmoset and rhesus macaque), guinea pig, rat, and mouse striatal tissue; albeit with a significant heterogeneity in the range of DA release magnitudes across species.48,72 Interestingly, this heterogeneity across regions extends to paired-pulse stimulations. Relative to the first pulse, a second pulse occurring within 100 ms elicits greater DA signals in the NAc/ventromedial striatum and elicits the same or smaller signals in the dorsolateral striatum.73 The gradient in the relative magnitude of the second pulse has been shown to be dependent on calcium availability and not regional variation in DA uptake, D2 autoreceptors, or GABAA activation.73
Single-pulse stimulations and multiple-pulse stimulations less than 20 Hz are thought to best model endogenous tonic firing of DA neurons, although the characteristics of endogenous firing vary to some degree from what can be modeled with electrical stimulations (Figure 2). Namely, electrical stimulations that model tonic firing will invariably elicit synchronized DA release from populations of DA nerve terminals, whereas endogenous tonic DA release stems from random firing from single neurons.74,75 This population firing from electrical stimulation leads to greater magnitude DA release when compared to endogenous firing. Despite differences in absolute magnitude, however, the utility of tonic stimulations mainly reside in their relation to multiple pulse stimulations that are equal to or exceed the 20 Hz frequency, which model phasic bursting of DA neurons (Figure 2). Indeed, Dani and colleagues have recently substantiated the face validity of slice voltammetry for measuring phasic burst firing of DA neurons by comparing ex vivo phasic/tonic ratios to phasic bursts in awake, freely moving animals receiving unexpected rewards.76 In this study, tonic firing was modeled ex vivo using multiple pulses at 0.2 Hz followed by a switch to a phasic firing stimulation (5 pulses @ 20 Hz). They demonstrated increased phasic/tonic ratios in the NAc compared with little to no phasic facilitation of release in the dorsolateral striatum, which is consistent with work reviewed earlier.73 They extended this work to demonstrate the same relationship in awake, freely moving animals receiving unexpected rewards, whereby ventromedial phasic stimulations are much greater in magnitude compared to dorsolateral striatum.76
Figure 2.

Representative dopamine traces highlighting the frequency response in the magnitude of dopamine in the nucleus accumbens shell. Electrical stimulations during voltammetric recording of evoked DA in brain slices may vary in pulse number, current amplitude, and frequency to model both tonic and phasic firing of DA neurons that occur in vivo. One common approach is to evaluate evoked DA release to single pulse and multiple pulses (e.g., 5) across frequencies that range from 5 to 100 Hz, with 20 Hz as a general tipping point in the shift from tonic to phasic signaling of DA neurons. Frequency response is more robust in the ventromedial striatum (i.e., nucleus accumbens shell) as shown here compared to the dorsolateral striatum, although the phasic frequency that elicits the highest peak amplitude can vary. Ratios can be calculated that compare the peak-height of the phasic signals to either the peak height of single pulse signal (dotted line), or the peak-height of a multiple pulse tonic signal.
2.2. Modulation of DA Release in the Striatum
Voltammetry in brain slices has proven to be a useful technique to quickly and efficiently explore striatal circuitry and the regulation of rapid DA signals, which have been implicated in motivation, reward, and reinforcement. There are a number of neurotransmitters and other chemical messengers that have been shown with slice voltammetry to modulate DA release by both single/tonic pulses as well as multiple pulses at phasic frequencies.
There is extensive research over the last several decades investigating facilitation of electrically evoked DA release in brain slices from DAT blockade and from inhibition of DA release via activation of D2 autoreceptors. Given a number of excellent reviews on these topics,77,78 these direct pathways will not be covered in the current Review. In addition to the effects of DAT and D2 autoreceptors, a number of other receptors have been shown to directly modulate electrically stimulated DA release via their localization of DA terminals. For example, kappa opioid receptors are located on DA nerve terminals in the striatum and decrease DA release to both single and multiple pulses.53,79 Additionally, the GABAB agonist, R(+)-Raclopride, has been shown to decrease DA release to single-pulse stimulations,80 while the GABAA antagonist, Baclofen, does not modify either single- or multiple-pulse DA release.73 The most extensively studied modulator of DA release in recent years, outside of the direct effects of DAT inhibition and D2 activation, is the nicotinic acetylcholine receptor (nAChR). This is attributed to the localization of beta2-containing nicotinic receptors on DA terminals, which when desensitized or blocked decrease DA release in response to single pulse and tonic frequency stimulations while having no effect or even facilitating DA release in response to phasic stimulations.54,55,81
In fact, this early work on nAChR modulation of DA release highlights one of at least two predominant indirect pathways by which many drugs and neurotransmitters modulate striatal release. The first indirect pathway is through tonically active acetylcholine (ACh) interneurons (TAN). ACh itself, nitric oxide,82 and opioids can modulate DA nerve terminals both directly and indirectly through modulation of ACh interneurons or nAChRs. Indeed, activation of the muscarinic M2 and M4 receptors,83 the mu and delta opioid receptors,79 or deletion of TANs themselves84 mimics the effects shown with blockade of nAChRs. Not only does this body of work have implications for nicotine administration via cigarette smoking, which has been shown to desensitize nAChRs, but also it demonstrates that pauses in TAN firing and subsequent decreases in ACh tone can amplify phasic/tonic ratios which modulate learning and motivation signals in the striatum.51,77 In addition to demonstrating that pauses in TAN signaling modulates DA release, recent voltammetry studies in brain slices demonstrated that activation of TANs directly elicits DA release at terminals in the striatum. Indeed, photostimulation of TANs expressing channelrhodhopsin resulted in DA release directly from nerve terminals similar to that observed from electrical stimulation.59,60 Therefore, it appears that activation of TANs can elicit DA release directly from nerve terminal while inhibition of TANs can modulate DA release in a frequency dependent manner.
Rice and colleagues have outlined another indirect pathway, which has been shown to mediate many (but possibly not all) of the effects of glutamate, GABA, and cannabinoids in the CPU.52,85 DA modulation in this pathway is through generation of hydrogen peroxide (H2O2). This modulatory pathway only affects multiple pulse stimulations given the time required for diffusion of H2O2. AMPA receptor activation (likely on medium spiny neurons (MSN)) is necessary for H2O2 release, and H2O2 suppresses DA release directly via activation of ATP sensitive potassium channels on DA terminals.52,86,87 In fact, modulation of H2O2 has been hypothesized to be an intermediary step for cannabinoid and GABA receptor modulation of DA release.52,85,88 Specifically, it has been proposed or shown directly that CB1 receptor activation facilitates H2O2 which suppresses DA signals, while GABA release opposes MSN activation and release of H2O2, thereby facilitating DA signals.50,52,85 The recently developed ability to detect and isolate H2O2 using voltammetry89 makes slice voltammetry a powerful tool for quantifying both DA and H2O2 levels simultaneously. Another modulator of electrically stimulated DA release in the striatum is hypocretin, which has recently been shown to facilitate multiple-pulse stimulations in the NAc shell through an indirect pathway. Since AMPA receptor antagonists block hypocretin’s effect, H2O2 may be involved but more work is needed to outline the pathway.
2.3. Plasticity of DA Release in the Striatum by Drugs of Abuse
Using slice voltammetry to explore changes in DA release magnitude in response to both single and multiple pulses following chronic administration of drugs of abuse is significantly underutilized compared to investigations of acute modulators of DA release. In naïve brain slices, DAT blockers dose-dependently modulate DA release in response to single pulses in an inverted U-shaped manner, while reverse-transport DA releasers linearly decrease stimulated release.16 The blocker-induced facilitation is due to uptake inhibition which decreases at higher concentrations due to some combination of sodium channel blockade by some blockers (i.e., cocaine) and D2 activation by released DA. For example, D2 autoreceptor antagonists fail to fully reverse the descending limb of the cocaine dose response curve (Ferris et al., unpublished), suggesting sodium channel blockade as a possible mechanism. Releasers linearly decrease stimulated release possibly through both stimulated and nonstimulated DA efflux and through subsequent D2 receptor activation.16
Our laboratory has demonstrated that high (1.5 mg/kg/inf, i.v.) and moderate (0.75 mg/kg/inf. i.v.) dose cocaine self-administration decreases single pulse DA release anywhere between 25% and 50%, and decreases the facilitation of DA release caused by perfusion of cocaine and methylphenidate (MPH) at concentrations less than or equal to 3 μM.15,16 This latter effect is likely due to a reduced ability of these compounds and other blockers to inhibit DA uptake (reviewed in section 3.4). The mechanism for decreased DA release at baseline is unclear, but D2 autoreceptors have been ruled out since cocaine self-administration decreases the sensitivity of D2 receptors as measured by a quinpirole dose response curve.49 Decreased release could be due to redistribution of DA pools; however, no experiment has investigated DA release in response to multiple pulses following cocaine self-administration, with or without compounds that selectively target releasable versus storage pools of DA. Interestingly, however, cocaine self-administration had no effect on the ability of amphetamine (AMPH) and non-AMPH based releasers to linearly decrease stimulated DA release.16
In contrast to cocaine self-administration, we recently showed that MPH self-administration actually facilitates DA release to single pulse stimulation.17 We chose a MPH dose (0.56 mg/kg/inf, i.v.) that matched the behavioral profile (intake and breakpoints) of the cocaine dose used in previous studies.17 Furthermore, AMPH self-administration has no effect on single-pulse DA release parameters in our studies.15 The substantial variability in plasticity of DA release (or lack thereof) caused by self-administration of blockers when compared to releasers, and even cocaine when compared MPH, suggests a complex system that may rely on variations in terminal expression of proteins such as the DAT (reviewed below), tyrosine hydroxlase, or differential redistribution of intracellular pools of DA.
Unlike other psychostimulants, the plasticity of DA release in response to both tonic and phasic stimulations has been investigated following nicotine treatment. A recent study demonstrated that three to six month oral intake of nicotine in monkeys reduces both single and multiple pulse stimulation of DA in the NAc shell, and even completely blocks frequency-dependent increases in DA release in this area.90 Perfusion of the alpha-6 antagonist, α-Ctx-MII, or mecamylamine, was able to reduce single pulse release, but failed to enhance stimulations that model phasic bursts. Likewise, a different study demonstrated that oral intake of nicotine for 12 weeks following by a 1, 5, or 10 day withdrawal period reduced stimulated DA release in response to both single and multiple-pulse frequencies.91 In contrast with the study performed in monkeys, however, the decrease in tonic stimulations was greater in magnitude than the decrease in phasic stimulations, thereby causing enhanced phasic/tonic ratios in animals receiving nicotine followed by 1 and 5 days of withdrawal (with a trend toward recovery after 10 days of withdrawal). It is unclear whether the differential effects on the phasic/tonic ratio between these studies represents species, dose, time administering, or withdrawal differences.
2.4. Conclusions
Slice voltammetry has been at the forefront of techniques used to understand local modulators of DA release in the striatum. Seminal research using slice voltammetry has outlined the role of TANs (i.e., ACh) in modulating and eliciting DA release, as well as the role of retrograde signaling and direct receptor/terminal modulation of tonic and phasic DA release. We have gained unprecedented insight into drug-induced modulation of release and plasticity of release following a history of drug abuse. This work has highlighted therapeutic avenues for human diseases that alter DA release such as Parkinson’s disease and drug addiction.15,92,93
3. Using Slice Voltammetry to Study DA Uptake in the Striatum
3.1. Modeling DA Signals to Determine Kinetic Parameters
In addition to assessing presynaptic DA release, it is possible to measure subsecond uptake kinetics, which allows for assessment of DAT function after drug treatments or genetic manipulations.11 In general, a Michaelis–Menten based kinetic model is used to determine uptake kinetics, which has been reviewed extensively in the past,4,5,94 although some groups report clearance using decay constants such as tau.21 With Michaelis–Menten modeling, in order to determine the maximal rate of uptake (Vmax) two assumptions are made: First, stimulated release concentrations are high enough to saturate the DAT. The second assumption is that uptake through the DAT is the primary mechanism responsible for clearing DA from the synapse, thus the clearance portion of the curve (the downward slope) is due to DAT-mediated uptake. Determination of Vmax using Michaelis–Menten kinetic analysis is best evaluated using single pulse stimulation parameters, but these analyses can also be done using multiple pulse stimulations in brain regions where there are lower DA release values in order to get DA release that is at a saturating level. In order to estimate Vmax using a Michaelis–Menten model of DA uptake, the apparent affinity of DA for the DAT (app. Km) is fixed to a range between 160 and 200 nM.28,47 While fixing app. Km is appropriate under conditions where no drug is present in the perfusion medium, drugs that have been shown to alter the affinity of DA for the DAT require app. Km to vary, as discussed in section 3.3.
3.2. Factors Influencing Maximal Rate of Uptake
Vmax is a rate term (measured as [DA] per second), which is inversely related to the time it takes released DA to be cleared, that can be affected by two main factors: surface DAT expression and allosteric modifications to the DAT protein. First, Vmax is most often a functional measure of the DATs present at the cell surface. A number of studies have demonstrated that when Vmax is increased, DAT expression is increased to a similar extent.17 In addition, genetic mouse strains support this, where DAT knockout mice have no measurable Vmax as the DA is cleared purely by diffusion,69,95 DAT heterozygous knockout mice show a reduced Vmax, and DAT transgenic overexpressing mice, which show an increase in DAT surface expression have a significantly increased Vmax.96 Transgenic animals allow for variation in DAT levels; however, it has also been demonstrated that brain regions with higher DAT levels, such as the dorsal striatum, have higher Vmax as compared to regions with lower DAT levels, such as the NAc.72 Because the change in DAT surface expression often results in similar changes in uptake rate, Vmax measurements may be able to be used to estimate surface DAT expression after a treatment or genetic manipulation.
One caveat of using voltammetric measures to estimate DAT levels is that Vmax can be changed independent of cell surface expression levels. Functional regulation of the DAT protein occurs by a number of processes including phosphorylation, gycosylation, palmitoylation, or oligomerization of the DAT protein itself, as well as interactions with other presynaptic receptor subtypes such as D2 autoreceptors.97 For example, activation of protein kinase C reduces DAT recycling and results in reduced uptake capacity, which results in decreased Vmax.98−100 Also, mechanisms involved in DAT coupling with the vesicular monoamine transporter (VMAT2) can lead to uptake rate changes independent of DAT levels via interactions with various proteins such as synaptogyrin-3 and Hsc70, which lead to increased uptake into vesicles and concomitant uptake via the DAT.101,102 The complex of VMAT2 and DAT can lead to regulation of extracellular tone via their modification in uptake rates.103 In addition to indirect allosteric modulators of DAT function, there is some evidence that sites on the DAT protein lead to direct allosteric modification of DAT function. The allosteric modulator N-(2,2-diphenylethyl)-2-phenyl-4-quinazolinamine (SoRI-20040) results in decreased Vmax and increased apparent Km and Kd values of DA for the DAT.104 Further, it has been demonstrated that the DAT forms oligomer complexes with itself and other proteins such as D2 autoreceptors.105 It has been suggested that the oligomerization of DAT proteins could lead to changes in uptake rates, where by the complexes have reduced uptake function than the same number of monomer DAT. Because of the ability of DA uptake to change independent of DAT trafficking to and from the membrane, assaying DAT levels by Western blot hybridization or other quantitative methods is not sufficient to make inferences about uptake rates. Voltammetric methods, although they are not necessarily quantitative measures of DAT protein levels, are sensitive to both protein expression and allosteric alterations in DAT function, and provide information that has relevance for synaptic DA levels.
3.3. Assessing Drug Induced Inhibition of the DAT
In slice preparations, the potency of drugs can be assessed before and after a treatment or genetic manipulation by running cumulative concentration–response curves. As reviewed earlier, this is a particularly powerful characteristic of voltammetry in slices because the half-life of many drugs can be hours to days, making such dose response curves difficult when using in vivo preparations. In regards to slices, at each concentration, the signal can be modeled to determine the amount of uptake inhibition elicited by each drug across groups or treatments. Once the baseline Vmax measurement is determined (as reviewed in section 3.1), drugs can be added to slice preparations. The Vmax is typically held constant and the app. Km value is allowed to change in order to determine uptake inhibition as a function of the affinity of DA for the DAT. App. Km is inversely related to the affinity of DA for the DAT, thus as app. Km increases, DA affinity decreases, which leads to slowed uptake of DA and prolonged DA in the extracellular space, which is measured by voltammetry as a increased time for the DA to be cleared and return to baseline.
3.4. Assessing Potency Shifts in Psychostimulant Effects Following Drug Self-Administration
Our group has focused on assessing the effects of psychostimulant self-administration on subsequent drug potency using voltammetry in brain slices. It has been demonstrated repeatedly that ligand affinity for receptors can vary after pharmacological treatments.106 This concept is not as widely studied for transporters such as the DAT. However, we have shown that the affinity of psychostimulant ligands at the DAT, such as cocaine, can be changed. While tolerance to the psychological effects of cocaine had been demonstrated previously, Mateo et al.49 showed that cocaine self-administration results in pharmacodynamic tolerance at the DAT, whereby cocaine was less effective at inhibiting DA uptake in the NAc core and shell (Figure 3A). Subsequently, cocaine administration resulting in cocaine tolerance at the DAT has been observed in a number of studies using different self-administration paradigms.15−17 Furthermore, we have demonstrated that this reduced ability of cocaine to inhibit DA uptake is accompanied by a reduction in cocaine-induced DA overflow as measured by microdialysis.15,49 Altered Vmax is an unlikely mechanism for reduced effects of cocaine at the DAT. This is primarily attributed to the fact that a number of studies have consistently demonstrated reduced effects of cocaine at the DAT following a history cocaine self-administration while observing increases,49 decreases,15,17 and no change16 in Vmax. Variation in Vmax across these studies seems to rely on the schedule of cocaine self-administration or the amount of time between the final self-administration session and voltammetry experiments. Nevertheless, these studies collectively demonstrate that changes in Vmax are not a mechanism for the changes in cocaine potency. This is further supported by the fact that cocaine self-administration does not reduce the ability of other substrates, such as AMPH, to inhibit DA uptake and increase extracellular DA as measured by voltammetry and microdialysis, respectively.15 Because the effects cannot be due to changes in Vmax/DAT levels, it is likely that some allosteric modification to the DAT protein itself, such as phosphorylation, glycosylation, or the formation of DAT complexes, is responsible for the change in cocaine potency following cocaine self-administration.
Figure 3.
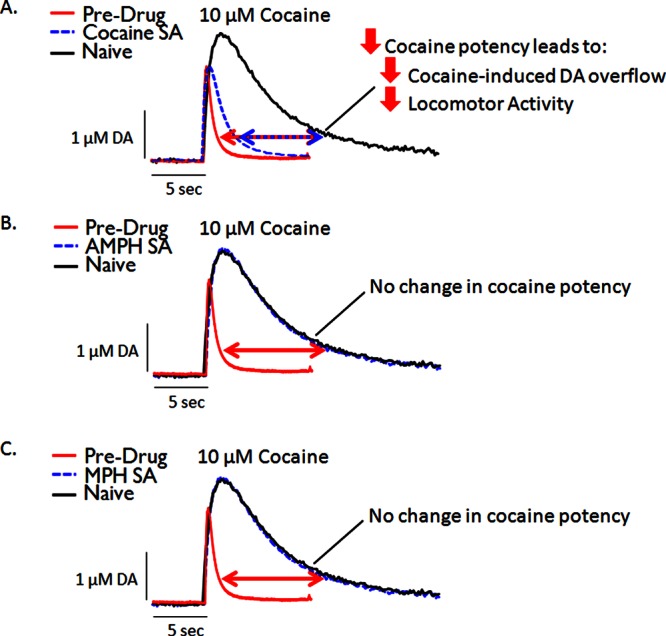
Representative voltammetric dopamine traces highlighting the effects of cocaine, amphetamine (AMPH), and methylphenidate (MPH) self-administration (SA) on cocaine potency. For all panels (not real data), red traces represent typical baseline (predrug) release and uptake of dopamine in both naïve and cocaine SA animals. Black traces represent typical dopamine release and uptake following application of cocaine in naïve animals. Uptake is inhibited when cocaine is applied, leading to greater maximal peak-height and slower return to baseline. Blue traces represent inhibition of dopamine uptake following application of cocaine in animals with a history of cocaine (A), AMPH (B), or MPH (C) SA. Note that the blue trace in Panel A (cocaine SA) exhibits a reduced maximal peak-height and reduced rightward-shift in the descending limb of the curve relative to the black trace (naïve). Therefore, cocaine SA produces tolerance to the neurochemical effects of cocaine (A), while AMPH (B) or MPH (C) self-administration does not affect cocaine potency. (See refs (15−17 and 49) for supporting data.)
In addition to assessing cocaine potency, slice voltammetry allows researchers to assess easily and effectively potency shifts for a wide range of compounds following a treatment or paradigm. Ferris et al.16 assessed the potency of a number of psychostimulants following a history of cocaine self-administration. We determined that the reduced ability of drugs to inhibit DA uptake was afforded to a number of structurally dissimilar DA blockers, but not DA releasers or methylphenidate (MPH). MPH is categorized as a psychostimulant blocker that is structurally similar to AMPH, prompting us to hypothesize that the lack of effect for MPH was due to the structure of the compound as an AMPH-like compound. However, the structurally dissimilar, non-AMPH releaser benzylpiperidine, was also unaffected by cocaine self-administration. Therefore, our working model is that a compound’s reduced ability to inhibit DA uptake after a history of cocaine self-administration is primarily based on the functional classification as a DA blocker, rather than a structural similarity to cocaine or AMPH per se. Although MPH is categorized as a blocker, it has been demonstrated to have release capabilities at high concentrations and binds to the DAT in a way that is dissimilar from other blockers. Because of this, it is possible that MPH’s unique effects are due to either (1) its ability to release DA or (2) the way that it binds to the transporter.
Given the divergent effects following a history of cocaine self-administration, we aimed to determine the effects of both AMPH and MPH self-administration on psychostimulant potency as it compared to cocaine self-administration (Figure 3). AMPH self-administration resulted in no change in cocaine or AMPH potency, indicating that the tolerance effects were specific to cocaine self-administration15 (Figure 3B). Surprisingly, MPH self-administration also resulted in no change in cocaine potency17 (Figure 3C), but increased the potency of MPH on a slice. This demonstrates that self-administration does not always result in tolerance to the self-administered drug and related compounds, but rather both tolerance and sensitization can be observed, depending on the drug.
3.5. Conclusions
Utilizing slice voltammetry in combination with stimulant self-administration is a tool for assessing DA uptake changes that occur following drug abuse paradigms. The ability to assess potency shifts for a wide range of psychostimulant compounds that are structurally and functionally distinct allows for the determination of how self-administration-induced DAT changes affect specific classes of psychostimulants. Future research will focus on behavioral consequences of drug-specific shifts in potency by correlating these changes with measures of reward and reinforcement. For example, ongoing research in our lab has shown that plasticity in the ability of cocaine to inhibit the DAT is associated with acquisition rates of cocaine self-administration.107 This work, then, not only has implications for long-term effects of psychostimulants in human addicts, but correlations of changes in DA uptake with behaviors that model drug abuse vulnerability may inform individual differences in susceptibility to addiction.
4. General Conclusions
Because of the ability to isolate release and uptake parameters, as well as the high temporal and spatial resolution, slice voltammetry is a powerful technique for assessing sub second DA kinetics. Even though slice voltammetry has been used to assess DA release and uptake for some time, the utility of slice voltammetry has not diminished and is still growing. Researchers are incorporating new ways to study dopaminergic function in slices by using optogenetic stimulation techniques, changing stimulation parameters to mimic tonic and phasic bursting, and studying the effects of transgenic and chronic drug treatments on DA function. These approaches not only allow for understanding mechanisms of local circuitry regulation, but also understanding changes that occur in the DA system following psychiatric disorders, including drug abuse and addiction.
Glossary
Abbreviations
- DA
dopamine
- DAT
dopamine transporter
- NAc
nucleus accumbens
- FSCV
fast scan cyclic voltammetry
- nAChR
nicotinic acetylcholine receptor
- ACh
acetylcholine
- SNc
substantia nigra pars compacta
- VTA
ventral tegmental area
- CPU
caudate putamen
- Vmax
maximal rate of uptake
- MPH
methylphenidate
- AMPH
amphetamine
- GABA
gamma-aminobutyric acid
- TAN
tonically active neuron
- H2O2
hydrogen peroxide
- VMAT2
vesicular monoamine transporter 2
This work was funded by NIH Grants R01 DA024095, R01 DA03016 (S.R.J.), P50DA006634 (S.R.J.), T32 DA007246, F31 DA031533 (E.S.C.), and K99 DA031791 (M.J.F.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Wightman R. M. (2006) Probing cellular chemistry in biological systems with microelectrodes. Science 311(5767), 1570–1574. [DOI] [PubMed] [Google Scholar]
- Michael D. J.; Wightman R. M. (1999) Electrochemical monitoring of biogenic amine neurotransmission in real time. J. Pharm. Biomed. Anal. 19(1–2), 33–46. [DOI] [PubMed] [Google Scholar]
- John C. E.; Jones S. R. (2007) Voltammetric characterization of the effect of monoamine uptake inhibitors and releasers on dopamine and serotonin uptake in mouse caudate-putamen and substantia nigra slices. Neuropharmacology 52(8), 1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D. L.; Hermans A.; Seipel A. T.; Wightman R. M. (2008) Monitoring rapid chemical communication in the brain. Chem. Rev. 108(7), 2554–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D. L.; Venton B. J.; Heien M. L. A. V.; Wightman R. M. (2003) Detecting subsecond dopamine release with fast-scan cyclic voltammetry in vivo. Clin. Chem. 49(10), 1763–1773. [DOI] [PubMed] [Google Scholar]
- Carelli R. M.; Wightman R. M. (2004) Functional microcircuitry in the accumbens underlying drug addiction: insights from real-time signaling during behavior. Curr. Opin. Neurobiol. 14(6), 763–768. [DOI] [PubMed] [Google Scholar]
- Grace A. A.; Bunney B. S. (1984) The Control of Firing Pattern in Nigral Dopamine Neurons - Burst Firing. J. Neurosci. 4(11), 2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland B. I.; Reynolds J. N. J.; Hay J.; Perk C. G.; Miller R. (2002) Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience 114(2), 475–492. [DOI] [PubMed] [Google Scholar]
- Robinson D. L.; Wightman R. M. (2004) Nomifensine amplifies subsecond dopamine signals in the ventral striatum of freely-moving rats. J. Neurochem. 90(4), 894–903. [DOI] [PubMed] [Google Scholar]
- Park J.; Kile B. M.; Wightman R. M. (2009) In vivo voltammetric monitoring of norepinephrine release in the rat ventral bed nucleus of the stria terminalis and anteroventral thalamic nucleus. Eur. J. Neurosci. 30(11), 2121–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo B.; Muller T.; Koch H. (1999) Effects of cannabinoids on dopamine release in the corpus striatum and the nucleus accumbens in vitro. J. Neurochem. 73(3), 1084–1089. [DOI] [PubMed] [Google Scholar]
- Roitman M. F.; Stuber G. D.; Phillips P. E. M.; Wightman R. M.; Carelli R. M. (2004) Dopamine operates as a subsecond modulator of food seeking. J. Neurosci. 24(6), 1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan J. O.; Walton M. E.; Phillips P. E. M. (2010) Dissociable cost and benefit encoding of future rewards by mesolimbic dopamine. Nat. Neurosci. 13(1), 25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat M. J.; Kuhnen C. M.; Phillips P. E. M. (2010) Delays Conferred by Escalating Costs Modulate Dopamine Release to Rewards But Not Their Predictors. J. Neurosci. 30(36), 12020–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris M. J.; Mateo Y.; Roberts D. C. S.; Jones S. R. (2011) Cocaine-Insensitive Dopamine Transporters with Intact Substrate Transport Produced by Self-Administration. Biol. Psychiatry 69(3), 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris M. J.; Calipari E. S.; Mateo Y.; Melchior J. R.; Roberts D. C. S.; Jones S. R. (2012) Cocaine Self-Administration Produces Pharmacodynamic Tolerance: Differential Effects on the Potency of Dopamine Transporter Blockers, Releasers, and Methylphenidate. Neuropsychopharmacology 37(7), 1708–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari E. S.; Ferris M. J.; Melchior J. R.; Bermejo K.; Salahpour A.; Roberts D. C. S.; Jones S. R. (2012) Methylphenidate and cocaine self-administration produce distinct dopamine terminal alterations. Addict. Biol. 10.1111/j.1369-1600.2012.00456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heien M. L. A. V.; Johnson M. A.; Wightman R. M. (2004) Resolving neurotransmitters detected by fast-scan cyclic voltammetry. Anal. Chem. 76(19), 5697–5704. [DOI] [PubMed] [Google Scholar]
- Keithley R. B.; Heien M. L.; Wightman R. M. (2009) Multivariate concentration determination using principal component regression with residual analysis. TrAC, Trends Anal. Chem. 28(9), 1127–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keithley R. B.; Wightman R. M. (2011) Assessing Principal Component Regression Prediction of Neurochemicals Detected with Fast-Scan Cyclic Voltammetry. ACS Chem. Neurosci. 2(9), 514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason J. T.; Espana R. A.; Jones S. R. (2011) Demon Voltammetry and Analysis software: Analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J. Neurosci. Methods 202(2), 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleson E. B.; Salek J.; Bonin K. D.; Jones S. R.; Budygin E. A. (2009) Real-time voltammetric detection of cocaine-induced dopamine changes in the striatum of freely moving mice. Neurosci. Lett. 467(2), 144–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariansen J. L.; Heien M. L. A. V.; Hermans A.; Phillips P. E. M.; Hernadi I.; Bermudez M. A.; Schultz W.; Wightman M. (2012) Monitoring extracellular pH, oxygen, and dopamine during reward delivery in the striatum of primates. Front. Behav. Neurosci. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida K. T.; Sandberg S. G.; Lohrenz T.; Comair Y. G.; Saez I.; Phillips P. E. M.; Montague P. R. (2011) Sub-Second Dopamine Detection in Human Striatum. PLoS One 6(8), e23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Aragona B. J.; Kile B. M.; Carelli R. M.; Wightman R. M. (2010) In Vivo Voltammetric Monitoring of Catecholamine Release in Subterritories of the Nucleus Accumbens Shell. Neuroscience 169(1), 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. M.; Stuber G. D. (2012) Optogenetic Strategies to Dissect the Neural Circuits that Underlie Reward and Addiction. Cold Spring Harb. Perspect. Med. 10.1101/cshperspect.a011924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venton B. J.; Wightman R. M. (2007) Pharmacologically induced, subsecond dopamine transients in the caudate-putamen of the anesthetized rat. Synapse 61(1), 37–39. [DOI] [PubMed] [Google Scholar]
- Wightman R. M.; Amatore C.; Engstrom R. C.; Hale P. D.; Kristensen E. W.; Kuhr W. G.; May L. J. (1988) Real-Time Characterization of Dopamine Overflow and Uptake in the Rat Striatum. Neuroscience 25(2), 513–523. [DOI] [PubMed] [Google Scholar]
- Mateo Y.; Budygin E. A.; Morgan D.; Roberts D. C. S.; Jones S. R. (2004) Fast onset of dopamine uptake inhibition by intravenous cocaine. Eur. J. Neurosci. 20(10), 2838–2842. [DOI] [PubMed] [Google Scholar]
- Espana R. A.; Roberts D. C. S.; Jones S. R. (2008) Short-acting cocaine and long-acting GBR-12909 both elicit rapid dopamine uptake inhibition following intravenous delivery. Neuroscience 155(1), 250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heien M. L. A. V.; Khan A. S.; Ariansen J. L.; Cheer J. F.; Phillips P. E. M.; Wassum K. M.; Wightman R. M. (2005) Real-time measurement of dopamine fluctuations after cocaine in the brain of behaving rats. Proc. Natl. Acad. Sci. U.S.A. 102(29), 10023–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason J. T.; Jones S. R.; Espana R. A. (2011) Low and High Affinity Dopamine Transporter Inhibitors Block Dopamine Uptake Within 5 Sec of Intravenous Injection. Neuroscience 182, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphalen R. I.; Desai K. M.; Hemmings H. C. (2013) Presynaptic inhibition of the release of multiple major central nervous system neurotransmitter types by the inhaled anaesthetic isoflurane. Br. J. Anaesth. 110(4), 592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D. L.; Howard E. C.; McConnell S.; Gonzales R. A.; Wightman R. M. (2009) Disparity Between Tonic and Phasic Ethanol-Induced Dopamine Increases in the Nucleus Accumbens of Rats. Alcohol.: Clin. Exp. Res. 33(7), 1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheer J. F.; Wassum K. M.; Heien M. L. A. V.; Phillips P. E. M.; Wightman R. M. (2004) Cannabinoids enhance subsecond dopamine release in the nucleus accumbens of awake rats. J. Neurosci. 24(18), 4393–4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber G. D.; Roitman M. F.; Phillips P. E. M.; Carelli R. M.; Wightman R. M. (2005) Rapid dopamine signaling in the nucleus accumbens during contingent and noncontingent cocaine administration. Neuropsychopharmacology 30(5), 853–863. [DOI] [PubMed] [Google Scholar]
- Aragona B. J.; Cleaveland N. A.; Stuber G. D.; Day J. J.; Carelli R. M.; Wightman R. M. (2008) Preferential enhancement of dopamine transmission within the nucleus accumbens shell by cocaine is attributable to a direct increase in phasic dopamine release events. J. Neurosci. 28(35), 8821–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daberkow D. P.; Brown H. D.; Bunner K. D.; Kraniotis S. A.; Doellman M. A.; Ragozzino M. E.; Garris P. A.; Roitman M. F. (2013) Amphetamine Paradoxically Augments Exocytotic Dopamine Release and Phasic Dopamine Signals. J. Neurosci. 33(2), 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips P. E. M.; Stuber G. D.; Heien M. L. A. V.; Wightman R. M.; Carelli R. M. (2003) Subsecond dopamine release promotes cocaine seeking. Nature 422(6932), 614–618. [DOI] [PubMed] [Google Scholar]
- Keithley R. B.; Carelli R. M.; Wightman R. M. (2010) Rank Estimation and the Multivariate Analysis of in Vivo Fast-Scan Cyclic Voltammetric Data. Anal. Chem. 82(13), 5541–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day J. J.; Roitman M. F.; Wightman R. M.; Carelli R. M. (2007) Associative learning mediates dynamic shifts in dopamine signaling in the nucleus accumbens. Nat. Neurosci. 10(8), 1020–1028. [DOI] [PubMed] [Google Scholar]
- Oleson E. B.; Gentry R. N.; Chioma V. C.; Cheer J. F. (2012) Subsecond Dopamine Release in the Nucleus Accumbens Predicts Conditioned Punishment and Its Successful Avoidance. J. Neurosci. 32(42), 14804–14808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitman M. F.; Wheeler R. A.; Wightman R. M.; Carelli R. M. (2008) Real-time chemical responses in the nucleus accumbens differentiate rewarding and aversive stimuli. Nat. Neurosci. 11(12), 1376–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler R. A.; Aragona B. J.; Fuhrmann K. A.; Jones J. L.; Day J. J.; Cacciapaglia F.; Wightman R. M.; Carelli R. M. (2011) Cocaine Cues Drive Opposing Context-Dependent Shifts in Reward Processing and Emotional State. Biol. Psychiatry 69(11), 1067–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J. J.; Sandberg S. G.; Wanat M. J.; Gan J. O.; Horne E. A.; Hart A. S.; Akers C. A.; Parker J. G.; Willuhn I.; Martinez V.; Evans S. B.; Stella N.; Phillips P. E. M. (2010) Chronic microsensors for longitudinal, subsecond dopamine detection in behaving animals. Nat. Methods 7(2), 126–U58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willuhn I.; Burgeno L. M.; Everitt B. J.; Phillips P. E. M. (2012) Hierarchical recruitment of phasic dopamine signaling in the striatum during the progression of cocaine use. Proc. Natl. Acad. Sci. U.S.A. 109(50), 20703–20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q.; Reith M. E. A.; Wightman R. M.; Kawagoe K. T.; Garris P. A. (2001) Determination of release and uptake parameters from electrically evoked dopamine dynamics measured by real-time voltammetry. J. Neurosci. Methods 112(2), 119–133. [DOI] [PubMed] [Google Scholar]
- Calipari E. S.; Huggins K. N.; Mathews T. A.; Jones S. R. (2012) Conserved dorsal-ventral gradient of dopamine release and uptake rate in mice, rats and rhesus macaques. Neurochem. Int. 61(7), 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo Y.; Lack C. M.; Morgan D.; Roberts D. C. S.; Jones S. R. (2005) Reduced dopamine terminal function and insensitivity to cocaine following cocaine binge self-administration and deprivation. Neuropsychopharmacology 30(8), 1455–1463. [DOI] [PubMed] [Google Scholar]
- Herr N. R.; Belle A. M.; Daniel K. B.; Carelli R. M.; Wightman R. M. (2010) Probing Presynaptic Regulation of Extracellular Dopamine with Iontophoresis. ACS Chem. Neurosci. 1(9), 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg S. J. (2006) Meaningful silences: how dopamine listens to the ACh pause. Trends Neurosci 29(3), 125–131. [DOI] [PubMed] [Google Scholar]
- Avshalumov M. V.; Chen B. T.; Marshall S. P.; Pena D. M.; Rice M. E. (2003) Glutamate-dependent inhibition of dopamine release in striatum is mediated by a new diffusible messenger, H2O2. J. Neurosci. 23(7), 2744–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosser B.; Kudernatsch M. B.; Sutor B.; Tenbruggencate G. (1995) Delta,Mu and Kappa-Opioid Receptor Agonists Inhibit Dopamine Overflow in Rat Neostriatal Slices. Neurosci. Lett. 191(1–2), 126–130. [DOI] [PubMed] [Google Scholar]
- Rice M. E.; Cragg S. J. (2004) Nicotine amplifies reward-related dopamine signals in striatum. Nat. Neurosci. 7(6), 583–584. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Sulzer D. (2004) Frequency-dependent modulation of dopamine release by nicotine. Nat. Neurosci. 7(6), 581–582. [DOI] [PubMed] [Google Scholar]
- Zahm D. S.; Cheng A. Y.; Lee T. J.; Ghobadi C. W.; Schwartz Z. M.; Geisler S.; Parsely K. P.; Gruber C.; Veh R. W. (2011) Inputs to the Midbrain Dopaminergic Complex in the Rat, with Emphasis on Extended Amygdala-Recipient Sectors. J. Comp. Neurol. 519(16), 3159–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reep R. L.; Cheatwood J. L.; Corwin J. V. (2003) The associative striatum: Organization of cortical projections to the dorsocentral striatum in rats. J. Comp. Neurol. 467(3), 271–292. [DOI] [PubMed] [Google Scholar]
- Haber S. N.; Fudge J. L. (1997) The primate substantia nigra and VTA: Integrative circuitry and function. Crit. Rev. Neurobiol. 11(4), 323–342. [DOI] [PubMed] [Google Scholar]
- Threlfell S.; Lalic T.; Platt N. J.; Jennings K. A.; Deisseroth K.; Cragg S. J. (2012) Striatal Dopamine Release Is Triggered by Synchronized Activity in Cholinergic Interneurons. Neuron 75(1), 58–64. [DOI] [PubMed] [Google Scholar]
- Cachope R.; Mateo Y.; Mathur B. N.; Irving J.; Wang H. L.; Morales M.; Lovinger D. M.; Cheer J. F. (2012) Selective Activation of Cholinergic Interneurons Enhances Accumbal Phasic Dopamine Release: Setting the Tone for Reward Processing. Cell Rep. 2(1), 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly R. S.; Wightman R. M. (1987) Detection of Dopamine Overflow and Diffusion with Voltammetry in Slices of Rat-Brain. Brain Res. 423(1–2), 79–87. [DOI] [PubMed] [Google Scholar]
- Palij P.; Bull D. R.; Sheehan M. J.; Humphrey P. P. A.; Millar J.; Stamford J. A.; Kruk Z. L. (1988) A New Technique for Quantitative Measurement of Stimulated Endogenous Dopamine Release in Brain-Slices Using Voltammetry. Br. J. Pharmacol. 94, 347.3395782 [Google Scholar]
- Stamford J. A.; Kruk Z. L.; Millar J. (1986) Sub-2Nd Striatal Dopamine Release Measured by Invivo Voltammetry. Brain Res. 381(2), 351–355. [DOI] [PubMed] [Google Scholar]
- Stamford J. A.; Kruk Z. L.; Millar J. (1988) Stimulated Limbic and Striatal Dopamine Release Measured by Fast Cyclic Voltammetry - Anatomical, Electrochemical and Pharmacological Characterization. Brain Res. 454(1–2), 282–288. [DOI] [PubMed] [Google Scholar]
- Toner C. C.; Stamford J. A. (1997) Involvement of N- and P/Q- but not L- or T-type voltage-gated calcium channels in ischaemia-induced striatal dopamine release in vitro. Brain Res. 748(1–2), 85–92. [DOI] [PubMed] [Google Scholar]
- Davidson C.; Stamford J. A. (1993) Neurochemical Evidence of Functional A10 Dopamine Terminals Innervating the Ventromedial Axis of the Neostriatum - In-Vitro Voltammetric Data in Rat-Brain Slices. Brain Res. 615(2), 229–239. [DOI] [PubMed] [Google Scholar]
- Phillips P. E. M.; Stamford J. A. (2000) Differential recruitment of N-, P- and Q-type voltage-operated calcium channels in striatal dopamine release evoked by “regular” and “burst” firing. Brain Res. 884(1–2), 139–146. [DOI] [PubMed] [Google Scholar]
- Jones S. R.; Gainetdinov R. R.; Wightman R. M.; Caron M. G. (1998) Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J. Neurosci. 18(6), 1979–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S. R.; Gainetdinov R. R.; Jaber M.; Giros B.; Wightman R. M.; Caron M. G. (1998) Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc. Natl. Acad. Sci. U.S.A. 95(7), 4029–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo Y.; Budygin E. A.; John C. E.; Banks M. L.; Jones S. R. (2004) Voltammetric assessment of dopamine clearance in the absence of the dopamine transporter: no contribution of other transporters in core or shell of nucleus accumbens. J. Neurosci. Methods 140(1–2), 183–187. [DOI] [PubMed] [Google Scholar]
- Jones S. R.; Odell S. J.; Marshall J. F.; Wightman R. M. (1996) Functional and anatomical evidence for different dopamine dynamics in the core and shell of the nucleus accumbens in slices of rat brain. Synapse 23(3), 224–231. [DOI] [PubMed] [Google Scholar]
- Cragg S. J.; Hille C. J.; Greenfield S. A. (2000) Dopamine release and uptake dynamics within nonhuman primate striatum in vitro. J. Neurosci. 20(21), 8209–8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg S. J. (2003) Variable dopamine release probability and short-term plasticity between functional domains of the primate striatum. J. Neurosci. 23(10), 4378–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace A. A.; Bunney B. S. (1984) The Control of Firing Pattern in Nigral Dopamine Neurons - Burst Firing. J. Neurosci. 4(11), 2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace A. A.; Bunney B. S. (1984) The Control of Firing Pattern in Nigral Dopamine Neurons - Single Spike Firing. J. Neurosci. 4(11), 2866–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. F.; Doyon W. M.; Clark J. J.; Phillips P. E. M.; Dani J. A. (2009) Controls of Tonic and Phasic Dopamine Transmission in the Dorsal and Ventral Striatum. Mol. Pharmacol. 76(2), 396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice M. E.; Patel J. C.; Cragg S. J. (2011) Dopamine Release in the Basal Ganglia. Neuroscience 198, 112–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Sulzer D. (2012) Regulation of striatal dopamine release by presynaptic auto- and heteroreceptors. Basal Ganglia 2(1), 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt J. P.; McGehee D. S. (2008) Presynaptic opioid and nicotinic receptor modulation of dopamine overflow in the nucleus accumbens. J. Neurosci. 28(7), 1672–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz Y.; Schmauss C.; Sulzer D. (2002) Altered dopamine release and uptake kinetics in mice lacking D-2 receptors. J. Neurosci. 22(18), 8002–8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R.; McIntosh J. M.; Marks M. J.; Maskos U.; Cragg S. J. (2012) Striatal alpha 5 Nicotinic Receptor Subunit Regulates Dopamine Transmission in Dorsal Striatum. J. Neurosci. 32(7), 2352–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung H.; Threlfell S.; Cragg S. J. (2011) Nitric Oxide Donors Enhance the Frequency Dependence of Dopamine Release in Nucleus Accumbens. Neuropsychopharmacology 36(9), 1811–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S.; Clements M. A.; Khodai T.; Pienaar I. S.; Exley R.; Wess J.; Cragg S. J. (2010) Striatal Muscarinic Receptors Promote Activity Dependence of Dopamine Transmission via Distinct Receptor Subtypes on Cholinergic Interneurons in Ventral versus Dorsal Striatum. J. Neurosci. 30(9), 3398–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J. C.; Rossignol E.; Rice M. E.; Machold R. P. (2012) Opposing regulation of dopaminergic activity and exploratory motor behavior by forebrain and brainstem cholinergic circuits. Nat. Commun. 3, 1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avshalumov M. V.; Patel J. C.; Rice M. E. (2008) AMPA receptor-dependent H2O2 generation in striatal medium spiny neurons but not dopamine axons: One source of a retrograde signal that can inhibit dopamine release. J. Neurophys. 100(3), 1590–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J. C.; Witkovsky P.; Coetzee W. A.; Rice M. E. (2011) Subsecond regulation of striatal dopamine release by pre-synaptic K-ATP channels. J. Neurochem. 118(5), 721–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J. C.; Rice M. E. (2012) Classification of H2O2 as a Neuromodulator that Regulates Striatal Dopamine Release on a Subsecond Time Scale. ACS Chem. Neurosci 3(12), 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo B.; Muller T.; Koch H. (1999) Effects of cannabinoids on dopamine release in the corpus striatum and the nucleus accumbens in vitro. J. Neurochem. 73(3), 1084–1089. [DOI] [PubMed] [Google Scholar]
- Roberts J. G.; Hamilton K. L.; Sombers L. A. (2011) Comparison of electrode materials for the detection of rapid hydrogen peroxide fluctuations using background-subtracted fast scan cyclic voltammetry. Analyst 136(17), 3550–3556. [DOI] [PubMed] [Google Scholar]
- Perez X. A.; Ly J.; McIntosh J. M.; Quik M. (2012) Long-Term Nicotine Exposure Depresses Dopamine Release in Nonhuman Primate Nucleus Accumbens. J. Pharmacol. Exp. Ther. 342(2), 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. F.; Dong Y.; Doyon W. M.; Dani J. A. (2012) Withdrawal from Chronic Nicotine Exposure Alters Dopamine Signaling Dynamics in the Nucleus Accumbens. Biol. Psychiatry 71(3), 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez X. A.; Bordia T.; McIntosh J. M.; Quik M. (2010) alpha 6 beta 2*and alpha 4 beta 2*Nicotinic Receptors Both Regulate Dopamine Signaling with Increased Nigrostriatal Damage: Relevance to Parkinson’s Diseas. Mol. Pharmacol. 78(5), 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venda L. L.; Cragg S. J.; Buchman V. L.; Wade-Martins R. (2010) alpha-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 33(12), 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagoe K. T.; Zimmerman J. B.; Wightman R. M. (1993) Principles of Voltammetry and Microelectrode Surface-States. J. Neurosci. Methods 48(3), 225–240. [DOI] [PubMed] [Google Scholar]
- Gainetdinov R. R.; Jones S. R.; Fumagalli F.; Wightman R. M.; Caron M. G. (1998) Re-evaluation of the role of the dopamine transporter in dopamine system homeostasis. Brain Res. Rev. 26(2–3), 148–153. [DOI] [PubMed] [Google Scholar]
- Salahpour A.; Ramsey A. J.; Medvedev I. O.; Kile B.; Sotnikova T. D.; Holmstrand E.; Ghisi V.; Nicholls P. J.; Wong L.; Murphy K.; Sesack S. R.; Wightman R. M.; Gainetdinov R. R.; Caron M. G. (2008) Increased amphetamine-induced hyperactivity and reward in mice overexpressing the dopamine transporter. Proc. Natl. Acad. Sci. U.S.A. 105(11), 4405–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Reith M. E. A. (2008) Role of the Dopamine Transporter in the Action of Psychostimulants, Nicotine, and Other Drugs of Abuse. CNS Neurol. Disord.: Drug Targets 7(5), 393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels G. M.; Amara S. G. (1999) Regulated trafficking of the human dopamine transporter - Clathrin-mediated internalization and lysosomal degradation in response to phorbol esters. J. Biol. Chem. 274(50), 35794–35801. [DOI] [PubMed] [Google Scholar]
- Melikian H. E. (2004) Neurotransmitter transporter trafficking: endocytosis, recycling, and regulation. Pharmacol. Ther. 104(1), 17–27. [DOI] [PubMed] [Google Scholar]
- Zahniser N. R.; Sorkin A. (2004) Rapid regulation of the dopamine transporter: role in stimulant addiction?. Neuropharmacology 47, 80–91. [DOI] [PubMed] [Google Scholar]
- Requena D. F.; Parra L. A.; Baust T. B.; Quiroz M.; Leak R. K.; Garcia-Olivares J.; Torres G. E. (2009) The molecular chaperone Hsc70 interacts with the vesicular monoamine transporter-2. J. Neurochem. 110(2), 581–594. [DOI] [PubMed] [Google Scholar]
- Egana L. A.; Cuevas R. A.; Baust T. B.; Parra L. A.; Leak R. K.; Hochendoner S.; Pena K.; Quiroz M.; Hong W. C.; Dorostkar M. M.; Janz R.; Sitte H. H.; Torres G. E. (2009) Physical and Functional Interaction between the Dopamine Transporter and the Synaptic Vesicle Protein Synaptogyrin-3. J. Neurosci. 29(14), 4592–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergo S.; Johansen J. L.; Leist M.; Lotharius J. (2007) Vesicular monoamine transporter 2 regulates the sensitivity of rat dopaminergic neurons to disturbed cytosolic dopamine levels. Brain Res. 1185, 18–32. [DOI] [PubMed] [Google Scholar]
- Pariser J. J.; Partilla J. S.; Dersch C. M.; Ananthan S.; Rothman R. B. (2008) Studies of the biogenic amine transporters. 12. Identification of novel partial inhibitors of amphetamine-induced dopamine release. J. Pharmacol. Exp. Ther. 326(1), 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N. H.; Reith M. E. A. (2008) Substrates dissociate dopamine transporter oligomers. J. Neurochem. 105(3), 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callier S.; Snapyan M.; Le Crom S.; Prou D.; Vincent J. D.; Vernier P. (2008) Evolution and cell biology of dopamine receptors in vertebrates. Biol. Cell. 95(7), 489–502. [DOI] [PubMed] [Google Scholar]
- Ferris M. J.; Calipari E. S.; Melchior J. R.; Roberts D. C.; Espana R. A.; Jones S. R.. Paradoxical tolerance to cocaine after initial supersensitivity in drug-use prone animals. Eur. J. Neurosci. 2013, in press. [DOI] [PMC free article] [PubMed]