

Abstract
Accumulating evidence supports the value of 5-HT1A receptor (5-HT1AR) agonists for dyskinesias that arise with long-term L-DOPA therapy in Parkinson’s disease (PD). Yet, how 5-HT1AR stimulation directly influences the dyskinetogenic D1 receptor (D1R)-expressing striatonigral pathway remains largely unknown. To directly examine this, one cohort of hemiparkinsonian rats received systemic injections of Vehicle + Vehicle, Vehicle + the D1R agonist SKF81297 (0.8 mg/kg), or the 5-HT1AR agonist ±8-OH-DPAT (1.0 mg/kg) + SKF81297. Rats were examined for changes in abnormal involuntary movements (AIMs), rotations, striatal preprodynorphin (PPD), and glutamic acid decarboxylase (GAD; 65 and 67) mRNA via RT-PCR. In the second experiment, hemiparkinsonian rats received intrastriatal pretreatments of Vehicle (aCSF), ±8-OH-DPAT (7.5 mM), or ±8-OH-DPAT + the 5-HT1AR antagonist WAY100635 (4.6 mM), followed by systemic Vehicle or SKF81297 after which AIMs, rotations, and extracellular striatal glutamate and nigral GABA efflux were measured by in vivo microdialysis. Results revealed D1R agonist-induced AIMs were reduced by systemic and intrastriatal 5-HT1AR stimulation while rotations were enhanced. Although ±8-OH-DPAT did not modify D1R agonist-induced increases in striatal PPD mRNA, the D1R/5-HT1AR agonist combination enhanced GAD65 and GAD67 mRNA. When applied locally, ±8-OH-DPAT alone diminished striatal glutamate levels while the agonist combination increased nigral GABA efflux. Thus, presynaptic 5-HT1AR stimulation may attenuate striatal glutamate levels, resulting in diminished D1R-mediated dyskinetic behaviors, but maintain or enhance striatal postsynaptic factors ultimately increasing nigral GABA levels and rotational activity. The current findings offer a novel mechanistic explanation for previous results concerning 5-HT1AR agonists for the treatment of dyskinesia.
Keywords: 5-HT1A receptor, D1 receptor, GABA, Glutamate, Dyskinesia, Parkinson’s disease
Chronic dopamine (DA) replacement therapy with l-3,4-dihydroxyphenylalanine (L-DOPA) for Parkinson’s disease (PD) patients often results in debilitating, abnormal involuntary movements known as L-DOPA-induced dyskinesia (LID). In recent years, interest in the role of the serotonin (5-HT) system in LID has increased. While a variety of 5-HT receptors have been examined, the 5-HT1A receptor (5-HT1AR) has received the most attention. Several 5-HT1AR agonists have displayed acute and chronic antidyskinetic effects in both animal models1−4 and in the clinic.5,6 Indeed, these compounds can maintain/improve L-DOPA’s efficacy2,7 and exert antiparkinsonian effects.8,9 Unfortunately, a number of studies have also shown that 5-HT1AR agonists may, in some cases, worsen PD symptoms. For example, the more potent enantiomer +8-OH-DPAT reduced LID but worsened motor disability,10 inducing a 5-HT-like syndrome.11 Clinically, high doses of the 5-HT1AR agonists tandospirone and sarizotan have been reported to exacerbate parkinsonian features.12 Thus, understanding the neural mechanisms of action of 5-HT1AR agonists in the parkinsonian brain should improve and hasten their potential use for PD patients.
Several key mechanisms appear to be involved in the development and expression of LID. First, substantial evidence indicates that LID results from supraphysiological increases in L-DOPA-derived DA within the striatum.13,14 The cause for this aberrant and excessive release of striatal DA likely involves the 5-HT system. Following DA depletion, serotonergic neurons of the raphe nuclei are thought to actively convert exogenously administered L-DOPA into DA and release it into the striatum in an unregulated fashion.1 Interestingly, 5-HT1A/1B receptor agonism has been shown to attenuate both LID and this enhanced effect on striatal DA levels, suggesting that one mechanism of action for 5-HT1AR agonists in the treatment of LID includes tempering raphe-striatal DA release.15,16
Another mechanism of LID may involve the corticostriatal glutamate pathway.17 Various studies have determined that increased levels/expression of striatal extracellular glutamate, glutamate transporters, and glutamate receptors are positively associated with dyskinetic motor behaviors in animal models of PD18−20 and in PD patients.21,22 Conversely, a variety of glutamate receptor antagonists are known to reduce dyskinesia,23 including amantadine, which is the only compound recommended by the American Academy of Neurology (Level C evidence) for the treatment of dyskinesia. Recently, our laboratory has demonstrated that stimulation of striatal 5-HT1AR reduced local glutamate and the concurrent expression of LID.20 These results suggest an additional mechanism of action for the antidyskinetic effects of 5-HT1AR agonists against L-DOPA involves the reduction of corticostriatal glutamate release.
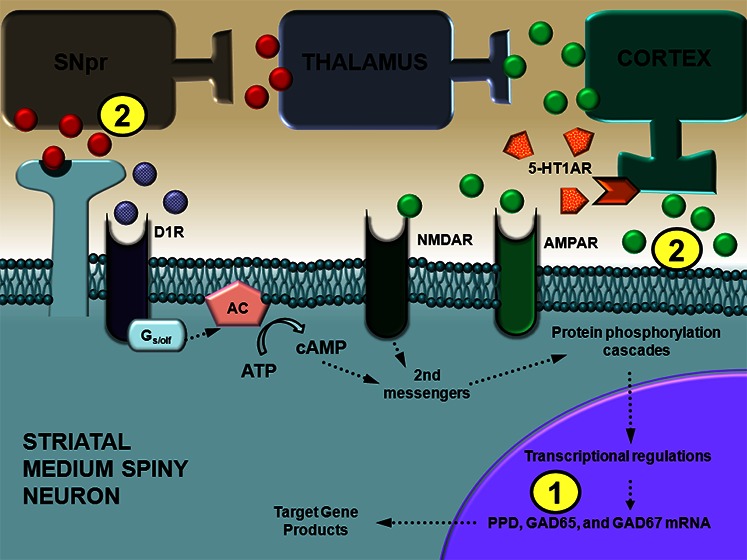
Lastly, LID is also due in part to supersensitive striatal DA receptors, especially D1 receptors (D1R), and increased striatal D1R direct pathway activity.24,25 Approximately 95% of all striatal neurons are γ-aminobutyric-acid (GABA)ergic medium spiny neurons that, in general, give rise to two major output pathways commonly referred to as the “direct” and “indirect” pathway. Striatal output neurons of the direct pathway express chiefly D1R and produce substance P and dynorphin,26 whereas striatal output neurons of the indirect pathway express mainly D2 receptors (D2R) and contain enkephalin.27 While L-DOPA treatment increases striatal preprodynorphin (PPD) mRNA in the DA-lesioned striatum, its effects on striatal preproenkephalin (PPE) mRNA are less consistent.28,29 Medium spiny neurons also express two isoforms of the GABA-synthesizing enzyme, glutamic acid decarboxylase (GAD) 65 and 67.30 Although the relationship between D2R and GAD65/67 is somewhat unclear, L-DOPA or D1R agonism increases GAD65 and GAD67 mRNA in the DA-depleted striatum, particularly in PPE-unlabeled neurons.28,31,32 As such, increases in PPD and GAD mRNA in the DA-depleted striatum in dyskinetic states are often associated with activation D1R and the direct pathway. Interestingly, 5-HT1AR agonism has been shown to attenuate L-DOPA-induced increases in striatal dynorphin/PPD and GAD mRNA,7,32 suggesting 5-HT1AR stimulation may reduce striatal D1R activity in LID.
In addition to the enhancement of striatal PPD and GAD mRNA, the occurrence of LID is paralleled by surges of GABA release in the substantia nigra pars reticulata (SNpr),33 which is the major output of the D1R direct pathway, but not the globus pallidus external segment,29 which is the major output of the striatal D2R indirect pathway. Likewise, striatal D1R but not D2R blockade attenuated LID and coincident L-DOPA-induced increases in nigral GABA.33 Collectively, the data provide significant evidence for the involvement of D1R and the direct striatonigral pathway in the generation of LID. Furthermore, D1R agonists can independently induce dyskinesia in experimental and clinical models of PD.34−36 In a series of studies, we have found that 5-HT1AR agonist administration diminishes D1R agonist-induced dyskinesia while increasing contralateral rotations and improving forelimb akinesia in hemiparkinsonian rats.20,36,37 While previous work indicates that these effects may occur at the level of the striatum,37 the specific impact of 5-HT1AR agonism on the D1R direct striatonigral pathway has not yet been investigated.
The goal of the current research, therefore, was to determine the effects of 5-HT1AR stimulation on D1R-mediated striatonigral function in hemiparkinsonian rats. The D1R agonist SKF81297 was employed to circumvent raphe-striatal DA release mechanisms and more effectively isolate dyskinetogenic features of the striatonigral pathway. In the first experiment, the effects of systemic administration of the full 5-HT1AR agonist ±8-OH-DPAT on striatal PPD, GAD65, and GAD67 mRNA were examined. In the second experiment, ±8-OH-DPAT was striatally perfused using in vivo microdialysis to examine changes in extracellular striatal glutamate and nigral GABA levels. The present results indicate that reductions in striatal glutamate may be related to antidyskinetic effects of local 5-HT1AR stimulation, while increases in striatal GAD mRNA and nigral GABA levels may explain the enhancement of D1R-mediated rotational activity. These findings implicate a novel striatonigral mechanism of action for 5-HT1AR agonists that may be important for understanding their potential therapeutic actions in the treatment of LID.
Results and Discussion
Experiment 1: Effects of 5-HT1AR Stimulation on D1R Agonist-Mediated Striatal PPD and GAD65/67 mRNA
Systemic 5-HT1AR Agonist Administration Attenuates D1R-Induced Dyskinesia while Enhancing Rotations
Two weeks following surgery, hemiparkinsonian rats were injected with Vehicle (20% DMSO in 0.9% NaCl, sc) or the D1R agonist SKF81297 (0.8 mg/kg, sc) every 2–3 days for a total of 3 days and abnormal involuntary movements (AIMs; see description in Methods) were rated for those that received SKF81297 (see Figure 1A). Two days following this priming regimen, Vehicle-primed rats received Vehicle treatment and SKF81297-primed rats received 1 of the following: Vehicle + Vehicle, Vehicle + SKF81297, or the full 5-HT1AR agonist ±8-OH-DPAT (1.0 mg/kg, sc) + SKF81297. Immediately following these treatments, rats were rated for axial, limb, and orolingual (ALO) AIMs and rotations every 10 min for 2 h. Vehicle-treated rats in both the Vehicle- and SKF81297-primed groups displayed neither ALO AIMs nor rotational activity. Pretreatment with ±8-OH-DPAT significantly reduced moderate to severe SKF81297-induced ALO AIMs at all time points (Figure 2A), while significantly enhancing SKF81297-induced rotations at 50–120 min postinjection (Figure 2B).
Figure 1.

Experimental Design. Schematic outlines of the timecourses for experiments 1 and 2. 1 In Experiment 1, all rats (N = 43) received unilateral 6-OHDA lesions of the MFB. Two weeks later, animals were tested for lesion using the forepaw adjusting steps (FAS) test. Priming commenced 1 week later, in which rats were injected every 2–3 days for a total of 3 days with either Vehicle (VEH; 20% DMSO in 0.9% NaCl, sc) or the D1 receptor agonist SKF81297 (SKF; 0.8 mg/kg, sc; n = 33) and rated for abnormal involuntary movements (AIMs). Median scores (±MAD) for axial, limb, and orolingual (ALO) AIMs are displayed. At the end of priming, dyskinetic rats (n = 32) were split into three equally dyskinetic groups based on ALO AIMs median scores. Two days later on test day, VEH-primed rats (n = 10) received Vehicle (VEH; 0.9% NaCl, sc) + VEH, while SKF-primed rats received one of the following: VEH + VEH (n = 10); VEH + SKF (n = 12); or the full 5-HT1AR agonist ±8-OH-DPAT (DPAT; 1.0 mg/kg, sc) + SKF (n = 10). Priming is denoted in parentheses. ALO AIMs were rated and rotations were counted every 10 min for 2 h immediately following injections. Following 2 h of behavioral ratings, rats were killed and striata immediately dissected and placed in RNALater for subsequent analyses of PPD, GAD65, and GAD67 mRNA using real time RT-PCR. (B) In Experiment 2, all rats (N = 26) received unilateral 6-OHDA lesioned of the MFB and were fitted unilaterally with microdialysis guide cannulae targeting the striatum and SNpr ipsilateral to the lesion. Three weeks later, all rats were primed with SKF81297 (SKF; 0.8 mg/kg, sc) and split into equally dyskinetic groups, as described in Experiment 1. At 2 to 7 days later, microdialysis testing began, which included 60 min habituation, 40 min baseline, 120 min vehicle treatment, 120 min drug treatment, and 60 min postdrug treatment sampling (dialysate collected every 20 min). Intrastriatal drug infusion included: Vehicle (VEH; aCSF), the full 5-HT1AR agonist ±8-OH-DPAT (DPAT; 7.5 mM), or combined DPAT (7.5 mM) + the 5-HT1AR antagonist WAY100635 (WAY; 4.6 mM), followed 10 min later by systemic treatment injections of Vehicle (VEH; 20% DMSO, 0.9% NaCl) or SKF. ALO AIMs and rotations were observed every 10 min during the 120 min drug treatment and 60 min postdrug treatment times. Approximately 3 days following microdialysis testing for all rats, rats were killed and whole brains were taken for histological probe placements or striatal tissue was dissected for HPLC-ED analyses of DOPAC and DA.
Figure 2.

Effects of 5-HT1AR stimulation on striatal factors related to D1R agonist-induced dyskinesia. Rats (N = 43) in Experiment 1 received unilateral 6-OHDA lesions of the MFB and 3 weeks later were primed with Vehicle (VEH; 20% DMSO in 0.9% NaCl, sc) or the D1R agonist SKF81297 (SKF; 0.8 mg/kg, sc) every 2–3 days for a total of 3 days (priming is denoted in parentheses). Two days following priming on test day, VEH-primed rats (n = 10) received Vehicle (VEH; 0.9% NaCl, sc) + VEH. Dyskinetic SKF-primed rats (n = 32) received one of the following: VEH + VEH (n = 10); VEH + SKF (n = 12); or the full 5-HT1AR agonist ±8-OH-DPAT (DPAT; 1.0 mg/kg, sc) + SKF (n = 10). ALO AIMs were rated and rotations were counted every 10 min for 2 h immediately following injections. Treatment effects for (A) ALO AIMs (expressed as medians ± MAD) and (B) rotations (expressed as means ± SEM) were analyzed by employing nonparametric Kruskal–Wallis tests and two-way ANOVA, respectively. Following 2 h of behavioral ratings, rats were killed and striata immediately dissected and placed in RNALater for subsequent analyses of PPD, GAD65, and GAD67 mRNA using real time RT-PCR. Bars depict the effects of treatment on striatal (C) PPD, (D) GAD65, and (E) GAD67 mRNA in the intact and lesioned striata, graphed as percent change from ultimate control [intact striata from VEH + VEH (VEH)] ± SEM. Main effects of treatment and lesion and treatment-by-lesion interactions were determined by two-way ANOVAs. Significant differences between treatments were determined by Mann–Whitney post hoc comparisons for ALO AIMs and LSD post hoc tests for rotations and mRNA. *p < 0.05 vs VEH + SKF (SKF); ×p < 0.05 vs VEH + VEH (VEH). #p < 0.05 vs VEH + VEH (SKF); +p < 0.05 vs Intact side.
The observed reduction of ALO AIMs and increase in contralateral rotations is in agreement with our previous reports.36,37 While contralateral rotations have historically been considered a reflection of a drug’s antiparkinsonian properties, they have also been linked to dyskinesia and more generally are thought to illustrate DA receptor sensitization. It is possible that the present behavioral results may be a result of competing motor behaviors, whereby the reduction in ALO AIMs leads to an increase in rotations. However, the time course does not entirely support this (Figure 2A,B), given that ALO AIMs were reduced at every time point and rotations did not increase until 50 min postinjection. Furthermore, recent evidence suggests that AIMs and rotations represent distinct motor behaviors that should be analyzed separately.38 Indeed, it was found that amantadine, the only recommended adjunct for LID,39 reduced AIMs while maintaining or increasing rotations. These collective findings suggest that AIMs and rotations may be somewhat independent and may have distinct mechanistic substrates. In order to help understand the neural underpinnings of our behavioral outcomes, changes in striatal PPD, GAD65, and GAD67 mRNA were examined.
Systemic 5-HT1AR Agonist Administration Further Enhances D1R-Induced Increases in Striatal GAD65/67 mRNA but Does Not Modify PPD mRNA
Immediately following 2 h post-treatment, rats were killed by decapitation and striatal tissue was dissected for real time RT-PCR analyses (see Figure 1A and Methods). No main effects or an interaction were found for the housekeeper gene, GAPDH (p > 0.05), thereby allowing for comparison of lesion- and treatment-induced effects.
The direct striatonigral pathway in the basal ganglia expresses chiefly DA D1R, produces substance P and dynorphin, and expresses two isoforms of the GABA-synthesizing enzyme, GAD65 and GAD67.26,30 Thus, PPD and GAD mRNA are often used as markers of D1R direct striatonigral pathway activity. In the intact striatum, D1R agonists have been shown to increase extracellular striatal dynorphin B levels and striatal GAD65 mRNA but not GAD67 mRNA.40−43 Likewise, in the current experiment, administration of the D1R agonist SKF81297 to D1R agonist-primed rats significantly enhanced PPD and GAD65 mRNA, but not GAD67, in the intact striatum compared to both Vehicle-treated groups (Figure 2C–E). Pretreatment with ±8-OH-DPAT did not modify SKF81297-induced increases in PPD or GAD65 but significantly increased GAD67 mRNA in the intact striatum compared to Vehicle-primed, Vehicle-treated rats (Figure 2E).
In DA-depleted striatonigral neurons, L-DOPA and D1R agonists increase dynorphin/PPD, GAD65, and GAD67 gene expression7,28,32,43,44 and these changes are positively correlated with dyskinesia.7,28,32 Corroborating previous reports, treatment with SKF81297 on test day induced pronounced dyskinesia (Figure 2A) and augmented PPD, GAD65, and GAD67 mRNA in the DA-depleted striatum compared to vehicle treatment (Figure 2C–E). Moreover, the enhancements of PPD and GAD67 were greater in the DA-lesioned hemisphere, suggesting that DA loss sensitized these responses and further implicates their involvement in dyskinetic behaviors.28
Despite a significant reduction in D1R-induced ALO AIMs (Figure 2A), pretreatment with ±8-OH-DPAT did not modify SKF81297-induced enhancement of striatal PPD mRNA and further potentiated GAD65 and GAD67 in the lesioned hemisphere (Figure 2C–E). These novel findings are in contrast to those demonstrating that 5-HT1AR stimulation attenuated LID while blocking L-DOPA-induced increases in striatal dynorphin/PPD and GAD mRNA.7,32 However, in those studies, ±8-OH-DPAT attenuated L-DOPA-induced rotations, whereas previously36,37 and here (Figure 2B) D1R agonist-mediated rotations were increased. Therefore, the increase in striatal GAD mRNA may reflect an enhancement of the striatonigral pathway, manifesting behaviorally in an increase in rotational activity. Unfortunately, the current experiment did not include a group receiving only ±8-OH-DPAT, and thus, it is not known whether the current results are attributable to stimulation of 5-HT1AR alone or whether they are due to a synergistic effect of concurrent 5-HT1AR and D1R stimulation. Indeed, studies have revealed that 5-HT1AR agonist treatment alone can increase rotational activity.8,9 Moreover, it is unknown whether these changes in striatal gene expression were due to a local population of 5-HT1AR or the result of indirect effects via distal 5-HT1AR stimulation. Thus, for the second experiment using in vivo microdialysis, striatal 5-HT1AR were directly targeted and ±8-OH-DPAT treatment alone was included. The goal was to determine whether combined 5-HT1AR/D1R stimulation enhances the striatonigral pathway as indicated by extracellular nigral GABA release.
Experiment 2: Effects of Striatal 5-HT1AR Stimulation on D1R Agonist-Mediated Extracellular Striatal Glutamate and Nigral GABA Levels
Striatal ±8-OH-DPAT Reduces D1R-Mediated Dyskinesia while Increasing Contralateral Rotations
In Experiment 2, hemiparkinsonian rats with microdialysis guide cannulae targeting the striatum and SNpr ipsilateral to the lesion were primed with SKF81297 (0.8 mg/kg, sc; see Figure 1B for design and Figure 3 for placements). Following this, microdialysis testing began, which included intrastriatal treatments of Vehicle (aCSF), ±8-OH-DPAT (7.5 mM), or ±8-OH-DPAT (7.5 mM) + WAY100635 (4.6 mM), followed by systemic injections of Vehicle (20% DMSO in 0.9% NaCl, sc) or SKF81297 (0.8 mg/kg, sc). As shown in Figure 4, intrastriatal ±8-OH-DPAT reduced ALO AIMs compared to SKF81297 alone at every time point and significantly increased SKF81297-induced rotations at 50–70 min and 130–140 min post-SKF81297 injection. The 5-HT1AR antagonist WAY100635 blocked ±8-OH-DPAT’s effects on ALO AIMs from 30 to 140 min post-SKF81297 (Figure 4A) and at every time point for rotations (Figure 4B). Intrastriatal ±8-OH-DPAT infusion alone did not induce ALO AIMs but produced mild contralateral rotational activity (Figure 4B). Importantly, the effects of striatal 5-HT1AR stimulation alone or in combination with D1R stimulation reflect the systemic effects displayed in Experiment 1 and are commensurate with previous findings.8,9,20,36,37
Figure 3.

Microdialysis Probe Placements. Schematic representations of coronal rat brain sections taken from Paxinos and Watson (1998). Shaded cylinders depict the distribution of (A) striatal (bregma 1.20 mm) and (B) nigral (bregma −5.30 mm) probe sites in all rats used in Experiment 2 (N = 26). Relevant anatomical structures: Aq, aqueduct (Sylvius); Cc, corpus callosum; Cpu, caudate putamen; LV, lateral ventricle; SNpr, substantia nigra pars reticulata.
Figure 4.

Effects of striatal 5-HT1AR stimulation on D1R-mediated ALO AIMs and rotations in 6-OHDA-lesioned, SKF81297-primed rats. Rats in Experiment 2 received unilateral 6-OHDA of the MFB and 3 weeks later were primed with the D1R agonist SKF81297 (SKF; 0.8 mg/kg, sc) every 2–3 days for a total of 3 days. Dyskinetic rats (N = 26) underwent a microdialysis procedure including 40 min baseline, 120 min vehicle treatment, 120 min drug treatment, and 60 min postdrug treatment sampling (dialysate collected every 20 min). Intrastriatal drug infusion included: Vehicle (VEH; aCSF), the full 5-HT1AR agonist ±8-OH-DPAT (DPAT; 7.5 mM), or combined DPAT (7.5 mM) + the 5-HT1AR antagonist WAY100635 (WAY; 4.6 mM), followed 10 min later by systemic treatment injections of Vehicle (VEH; 20% DMSO, 0.9% NaCl) or SKF. ALO AIMs and rotations were observed during this time. Treatment groups consist of n = 8–12/treatment group. Treatment effects for (A) ALO AIMs (expressed as medians ± MAD) and (B) rotations (expressed as means ± SEM) were analyzed by employing nonparametric Kruskal–Wallis tests and two-way ANOVAs, respectively. Significant differences between treatments were determined by Mann–Whitney post hoc comparisons for ALO AIMs and LSD post hoc tests for rotations. *p < 0.05 for DPAT (7.5 mM) + SKF vs VEH + SKF. +p < 0.05 for DPAT (7.5 mm)+ SKF vs DPAT (7.5 mM) + WAY (4.6 mM) + SKF.
Striatal ±8-OH-DPAT Alone and with SKF81297 Reduces Local Glutamate Levels
Enhanced activity of the glutamatergic corticostriatal pathway is believed to be a contributing mechanism of LID.17 However, whether D1R agonists directly induce glutamate release is less studied. In agreement with our previous work,20 despite significant dyskinesia, treatment with the D1R agonist SKF81297 in hemiparkinsonian rats did not modify striatal glutamate levels (Figure 5). However, striatal 5-HT1AR stimulation alone diminished local glutamate (by ∼25%) and reduced it further (by ∼40%) when given with the D1R agonist. Notably, like its behavioral effects, the ±8-OH-DPAT-induced reduction of striatal glutamate was reversed with the 5-HT1AR antagonist WAY100635 (Figure 5B). The current findings support previous work demonstrating that systemic 5-HT1AR agonists reduce striatal glutamate levels in intact45 and 6-OHDA-lesioned46 rats, as well as our recent results showing that the same concentration of striatal ±8-OH-DPAT simultaneously diminished local glutamate and LID.20
Figure 5.

Effects of striatal 5-HT1AR stimulation on local glutamate in a D1R-mediated model of dyskinesia. Rats in Experiment 2 received unilateral 6-OHDA of the MFB and 3 weeks later were primed with the D1R agonist SKF81297 (SKF; 0.8 mg/kg, sc) every 2–3 days for a total of 3 days. Dyskinetic rats (N = 26) underwent a microdialysis procedure including 40 min baseline, 120 min vehicle treatment, 120 min drug treatment, and 60 min postdrug treatment sampling (dialysate collected every 20 min). Intrastriatal drug infusion included: Vehicle (VEH; aCSF), the full 5-HT1AR agonist ±8-OH-DPAT (DPAT; 7.5 mM), or combined DPAT (7.5 mM) + the 5-HT1AR antagonist WAY100635 (WAY; 4.6 mM), followed 10 min later by systemic treatment injections of Vehicle (VEH; 20% DMSO, 0.9% NaCl) or SKF. ALO AIMs and rotations were observed during this time. Treatment groups consist of n = 8–12/treatment group. (A) Lines depict the means (expressed as % baseline ± SEM) of striatal glutamate. (B) Bars depict the means (expressed as % baseline ± SEM) of striatal glutamate during the 120 min of vehicle (VEH (aCSF) + VEH) and 120 min of drug treatments. Effects over time were determined by two-way mixed design ANOVA, main effects for treatment collapsed across time were analyzed with one-way ANOVAs, and LSD post hoc tests were utilized. *p < 0.05 vs VEH + SKF; +p < 0.05 vs DPAT (7.5 mM) + WAY (4.6 mM) + SKF. #p < 0.05 vs VEH + VEH; ×p < 0.05 vs DPAT (7.5 mM) + VEH.
Since previous work demonstrated that systemic ±8-OH-DPAT reduced SKF81297-induced ALO AIMs without modifying extracellular striatal glutamate levels,20 the current findings are more likely attributable to striatal 5-HT1AR stimulation. However, striatal 5-HT1AR mRNA levels are very low47 or even absent48 in the intact striatum. Moreover, unilateral 6-OHDA-lesions in rats have not been shown to modify 5-HT1AR mRNA levels.49 Unfortunately, those results stem from a relatively small number of animals that incorporated several variables, including both male and female rats, two different placements for 6-OHDA-lesions, and extracting tissue anywhere from 1 to 5 weeks postlesion. In contrast, 5-HT1AR in the DA-depleted striata have been shown to increase (∼50%) in MPTP-treated monkeys50,51 and further enhanced (200–300%) in MPTP-treated, dyskinetic macaques and PD patients who received L-DOPA.51,52 While future studies are necessary in the PD rodent model, it is possible that 5-HT1AR expression in our D1R agonist-treated, dyskinetic rats is also enhanced.
Interestingly, 5-HT neurons express vesicular glutamate transporter 3 mRNA and protein, which is a marker of glutamate transmission.53 Because striatal 5-HT1AR expression is low, they are believed to be located presynaptically within the striatum and stimulation would decrease the release of neurotransmitters.54 Thus, striatal 5-HT1AR may be acting presynaptically on corticostriatal glutamatergic neurons or possibly, but less likely, via raphestriatal neurons to diminish local glutamate. In support of this, recent evidence indicates that L-DOPA treatment increases 5-HT1AR expression within the middle layers of the motor cortex and parts of the striatum.51,52 The authors have suggested that the observed increase in striatal 5-HT1AR is the result of an enhancement of 5-HT1AR located on the terminals of corticostriatal neurons. Indeed, by reducing corticostriatal glutamate, 5-HT1AR stimulation may disrupt the dysfunctional glutamatergic/dopaminergic activity occurring at corticostriatal synapses55,56 and alleviate associated dyskinetic behaviors.
Striatal ±8-OH-DPAT Alone Reduces Nigral GABA Levels while Combined with SKF81297 Leads to an Increase in Nigral GABA
Acute and chronic L-DOPA has been shown to increase GABA release in the SNpr in animal models of PD,29,33,57 while in vitro D1R stimulation increases GABA neurotransmission in DA-depleted striatonigral neurons.58 Although L-DOPA-induced nigral GABA efflux appears to be D1R dependent,33 direct D1R agonist treatment had not been previously investigated in vivo in DA-depleted animals. In the present experiment, systemic SKF81297 induced robust ALO AIMs (Figure 4A) but did not enhance extracellular nigral GABA levels (Figure 6).
Figure 6.

Effects of striatal 5-HT1AR stimulation on extracellular nigral GABA in a D1R-mediated model of dyskinesia. Rats in Experiment 2 received unilateral 6-OHDA of the MFB and 3 weeks later were primed with the D1R agonist SKF81297 (SKF; 0.8 mg/kg, sc) every 2–3 days for a total of 3 days. Dyskinetic rats (N = 26) underwent a microdialysis procedure including 40 min baseline, 120 min vehicle treatment, 120 min drug treatment, and 60 min postdrug treatment sampling (dialysate collected every 20 min). Intrastriatal drug infusion included: Vehicle (VEH; aCSF), the full 5-HT1AR agonist ±8-OH-DPAT (DPAT; 7.5 mM), or combined DPAT (7.5 mM) + the 5-HT1AR antagonist WAY100635 (WAY; 4.6 mM), followed 10 min later by systemic treatment injections of Vehicle (VEH; 20% DMSO, 0.9% NaCl) or SKF. ALO AIMs and rotations were observed during this time. Treatment groups consist of n = 5–9/treatment group. (A) Lines depict the means (expressed as % baseline ± SEM) of nigral GABA. (B) Bars depict the means (expressed as % baseline ± SEM) of nigral GABA during the 120 min of vehicle (VEH (aCSF) + VEH) and 120 min of drug treatments. Effects over time were determined by two-way mixed design ANOVAs, main effects for treatment collapsed across time were analyzed with one-way ANOVAs, and LSD post hoc tests were utilized. *p < 0.05 vs VEH + SKF; +p < 0.05 vs DPAT (7.5 mM) + WAY (4.6 mM) + SKF. #p < 0.05 vs VEH + VEH; ×p < 0.05 vs DPAT (7.5 mM) + VEH.
The reasons for this lack of effect remain unclear but may highlight a potential difference between L-DOPA and D1R agonist-mediated dyskinesia. Given that L-DOPA is converted into DA and can thus impact all DA receptors, it is possible that L-DOPA’s effect is due to stimulation of multiple DA receptor subtypes or a synergistic response of D1R/D2R activation. However, recent work has indicated that striatal D1/5R, and not D2/3R, antagonism attenuated LID and the concurrent enhancement of nigral GABA.33 It also appears that striatal D1R stimulation in particular may be responsible, as nigral D1R antagonism did not completely abolish nigral L-DOPA-induced GABA. Furthermore, non-DA mediated mechanisms might contribute, including a purported increase in nigral glutamate.33 Finally, it is important to note that D1R-dependent increases in striatal GAD mRNA found in Experiment 1 did not translate into enhanced nigral GABA here in Experiment 2. However, GAD67 mRNA, which is more strongly associated with LID than GAD65,28 was not significantly different between acute and chronic SKF81297 administration in the DA-depleted striatum (Figure 2E). Thus, as all animals in Experiment 2 received several days of D1R agonist treatment prior to microdialysis, nigral GABA levels may have reached a ceiling where further increases could not be detected with SKF81297 injection alone.
The effects on striatal 5-HT1AR stimulation on nigral GABA release were diverse. Interestingly, striatal ±8-OH-DPAT combined with systemic SKF81297 moderately augmented (overall ∼40%) extracellular nigral GABA levels (Figure 6B), an effect that was reversed with local antagonist coadministration. This increase in nigral GABA from combined 5-HT1AR/D1R agonism coincides with the increases in striatal GAD mRNA found in Experiment 1. Conversely, intrastriatal ±8-OH-DPAT alone resulted in mild rotational activity and a gradual reduction of nigral GABA (overall ∼30%), followed by a return to baseline when ±8-OH-DPAT was removed from the striatum. The results appear somewhat conflicting but also support evidence demonstrating that both agonists and antagonists of GABA receptors into the SNpr can trigger contralateral rotations in 6-OHDA-lesioned rats59 and that unilateral microinjection of a GABAA receptor antagonist into the SNpr can induce rotational behavior in monkeys.60 Therefore, a possible mechanism for the increase in rotational activity observed with 5-HT1AR agonists alone8,9 and in combination with D1R agonism36,37 (Figures 2B and 4B) includes the modulation of nigral GABA.
Receptor Specificity and Off-Target Effects of ±8-OH-DPAT Administration
While it has the highest affinity for 5-HT1AR, ± 8-OH-DPAT also exhibits moderate affinity for the 5-HT transporter, 5-HT1B/1D/2C/7 receptors, and D2R.61 Indeed, higher doses/concentrations of ±8-OH-DPAT, like the ones utilized in the current study, can have off-target effects on these other receptor subtypes and may also increase extracellular levels of 5-HT and DA.62 D2R are especially important as their presence is abundant within the striatum, D2R agonism can lead to rotations in hemiparkinsonian rats,36 and stimulation of D2R may reduce glutamatergic transmission.63 Furthermore, increasing striatal DA could lead to stimulation of both D1R and D2R, which may explain the increases in rotations and striatal GAD65/67 mRNA found in Experiment 1; however, it is inconsistent with the reduction of AIMs as D1R or D2R agonism induces AIMs.35,36,64 In addition, the same doses of ±8-OH-DPAT reduced L-DOPA-induced striatal PPD and GAD mRNA, extracellular striatal DA efflux, AIMs, and rotations,7,15,20,32,36 suggesting the current results may not be due to increases in striatal DA or stimulation of D2R and implicates the interaction between stimulation of D1R and 5-HT1AR.
Increasing 5-HT and the possible activity at other 5-HT receptor subtypes could account for a reduction of AIMs. For instance, 5-HT1B receptor expression is more prevalent in the striatum59 and stimulation of these receptors can diminish LID and DA agonist-induced AIMs.1,16,65,66 However, the decrease in D1R agonist-induced AIMs by 5-HT1B receptor agonism was moderate and rotational activity was not modified.65 Importantly, in previous studies7,20,37 and the current one (see Figures 4–6), the effects on AIMs and rotations were reversed with coadministration of the 5-HT1AR antagonist WAY100635, further proposing the participation of this receptor subtype in our molecular and neurochemical effects. Nonetheless, future studies may wish to utilize lower concentrations of ±8-OH-DPAT in order to improve receptor specificity. Additionally, it would be interesting to investigate the effects of 5-HT1AR stimulation on D2R pathway activity.
Conclusions
Due to the prevalence of LID and the lack of effective antidyskinetic treatments, discovering unique, safe, and efficacious therapies that combat dyskinesia without aggravating parkinsonian features is a central goal in PD research. The use of 5-HT1AR agonists to treat LID has shown much promise, and one such compound, sarizotan, successfully reached phase III clinical trials (Merck KGaA). However, sarizotan’s successful translation was limited by antidyskinetic efficacy (<25% LID reduction) and exacerbation of parkinsonian features.7 Despite this setback, interest in 5-HT1AR agonists remains high and the importance of discovering the underlying mechanisms that convey their potential therapeutic profile and deleterious side effects is thus paramount for furthering their use in the clinic.
Our laboratory was the first to show that striatal 5-HT1AR stimulation diminishes D1R-mediated dyskinetic behaviors and improves motor performance in hemiparkinsonian rats.37 In the current set of experiments, we probed possible neural mechanisms of action for these effects and consequently have revealed important similarities and differences between the effects of 5-HT1AR stimulation on L-DOPA versus D1R agonism. When given with L-DOPA, 5-HT1AR agonists may reduce LID and rotations by attenuating raphe-striatal DA release, corticostriatal glutamate levels, and postsynaptic striatal factors associated with LID (i.e., PPD and GAD mRNA). While attenuating raphe-striatal DA release likely reduces LID, it may also explain reports concerning the reduced efficacy of L-DOPA and worsening of PD symptoms. By employing a D1R agonist, the raphe-striatal DA release mechanism is avoided, allowing for better investigation into the effects on striatal glutamate and postsynaptic factors. Similar to use with L-DOPA, 5-HT1AR agonists may reduce D1R-mediated dyskinesia in part by diminishing corticostriatal glutamate release. In contrast, 5-HT1AR agonists appear to increase rotations by maintaining or enhancing striatonigral factors (i.e., striatal PPD and GAD mRNA and nigral GABA release), which could represent a beneficial, motor-enhancing effect of D1/5-HT1AR costimulation. Additionally, by themselves 5-HT1AR agonists can reduce striatal glutamate and nigral GABA, possibly explaining the mild rotational activity and varied findings concerning their potential antiparkinsonian effects. In sum, 5-HT1AR agonists for the treatment of LID in PD can produce several behavioral outcomes that may be explained by their multiple mechanisms of action. Their impact on D1R signaling may prove to be an important therapeutic action and should be considered in future experiments that investigate the use of 5-HT1AR agonists in the treatment of LID.
Methods
Animals
Adult male Sprague–Dawley rats were used in all experiments (225–250 g upon arrival; Taconic Farms, Hudson, NY). Rats were kept in plastic cages (22 cm high, 45 cm deep and 23 cm wide) and given free access to food (Rodent Diet 5001; Lab Diet, Brentwood, MO) and water. The colony room was kept on a 12 h light/dark cycle (light on at 0700 h) and maintained at 22–23 °C. The guidelines of the Institutional Animal Care and Use Committee of Binghamton University and the “Guide for the Care and Use of Laboratory Animals” (Institute of Laboratory Animal Resources, National Academic Press 1996; NIH publication number 85-23, revised 1996) were maintained throughout experiments.
6-Hydroxydopamine Lesions and Microdialysis Guide Cannulae Implantation Surgeries
One week after arrival, all rats (N = 69) in Experiments 1 and 2 received unilateral DA lesions of the left medial forebrain bundle (MFB). Each rat was administered desipramine HCl (25 mg/kg, ip; Sigma, St. Louis, MO) 30 min prior to surgery in order to protect norepinephrine neurons. Rats were anesthetized with inhalant isoflurane (2–3%; Sigma) in oxygen (2.5 L/min) and placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). The following coordinates relative to bregma were used for the site of injection: AP, −1.8 mm; ML, +2.0 mm; DV, −8.6 mm, with the incisor bar positioned at 5.0 mm below the interaural line.67 After drilling a small hole in the skull above the site of injection, a 10 μL Hamilton syringe attached to a 26 gauge needle was lowered into the MFB. At that point, 4 μL of 6-hydroxydopamine hydrobromide (6-OHDA; 3 μg/μL; Sigma), dissolved in 0.9% NaCl + 0.1% ascorbic acid, was injected at a rate of 2 μL/min and the needle was withdrawn 1 min later. Stainless steel wound clips were used to close the surgical site for rats in Experiment 1. These animals were pair-housed in clean cages, allowed to recover with ad libitum food and water, and wound clips were removed 1 week postsurgery.
During the same 6-OHDA lesion surgery, rats in Experiment 2 (n = 26) were also fitted with two plastic microdialysis guide cannulae (CMA 12 Elite; Stockholm, Sweden), where one cannula targeted the dorsal striatum ipsilateral to the lesioned side (AP, +1.2 mm; ML, +2.5 mm; DV, −3.7 mm; relative to bregma) and the second targeted the SNpr ipsilateral to the lesioned side (AP: −5.3 mm, ML: +2.2 mm, DV: −7.8 mm).67 Cannulae were positioned and affixed to the skull with screws and liquid and powder dental acrylic (Lang Dental, Wheeling, IL). At the completion of surgery, these animals were single-housed in clean cages, allowed to recover with ad libitum food and water, and soft chow was provided when necessary. Rats from both experiments were monitored and handled twice per week for 3 weeks postsurgery in order to ensure full recovery and acclimation to experimenters.
Pharmacological Treatments and Test Day Procedures
Experiment 1: Effects of 5-HT1AR Stimulation on D1R Agonist-Mediated Striatal PPD and GAD65/67 mRNA
During surgery and for both Experiments 1 and 2, dosing solutions for all drugs were prepared at the concentrations indicated without correcting for the weight of the salts. Two weeks following 6-OHDA lesion surgery, rats in Experiment 1 (N = 43) were assigned to four equally lesioned groups based on the forepaw adjusting steps test.37 One week later, three of the groups of rats (n = 33) received the D1R agonist R(+)-SKF-81297 hydrobromide (SKF81297; 0.8 mg/kg, sc;) on three separate occasions 2–3 days apart, while the final group of rats (n = 10) received Vehicle (20% DMSO in 0.9% NaCl, sc). Immediately following injections, all rats were individually placed into cylinders in the same testing room and remained there for 2 h. Those that received SKF81297 were rated for axial/limb/orolingual abnormal involuntary movements (ALO AIMs) and rotations were observed (see description below). Vehicle-treated animals were not rated for AIMs or rotations during the priming process. SKF81297 was chosen over SKF38393 as it has shown a higher intrinsic activity of DA in in vitro studies, 88% compared to 45–70%.68,69 Moreover, the dose of SKF81297 and priming regimen have been used in our lab to produce stable ALO AIMs expression that is similar to those induced by L-DOPA treatment (12 mg/kg, + Benserazide, 15 mg/kg, sc).20,36,37 Responding rats in the current study, included in the final analyses, were required to have an ALO AIMs ≥ 20 (n = 32). This criterion is consistent with our previous reports20,36,37 and corresponds with at least 95% DA depletion upon HPLC analysis of striatal tissue samples.
At the end of priming, adjustments to SKF81297-primed groups were made in order to ensure three equally dyskinetic groups based on ALO AIMs median scores (overall ALO AIMs median score = 58.5 (±4); Group I = 55.5 (±3); Group II = 56.5 (±4.5); Group III = 60.5 (±3.5); see Figure 1 for experimental design). Two days following priming, SKF81297-primed rats received one of three treatments: Vehicle (0.9% NaCl, sc) + Vehicle (20% DMSO, 0.9% NaCl, sc), Vehicle (0.9% NaCl, sc) + SKF81297 (0.8 mg/kg, sc), or the full 5-HT1AR agonist (±)-8-hydroxy-2-(dipropylamino)tetralin hydrobromide (±8-OH-DPAT; 1.0 mg/kg, sc;) + SKF81297 (0.8 mg/kg, sc). All Vehicle-primed rats received Vehicle (0.9% NaCl, sc) + Vehicle (20% DMSO, 0.9% NaCl, sc). The second treatment was given immediately following the first treatment injection, and doses were chosen based on previously published work.36,37 All rats were individually placed into cylinders in the same testing room and observed for ALO AIMs and rotations every 10 min for 2 h immediately following the second treatment injection. After 2 h of behavioral ratings, rats were killed by decapitation and left (lesioned) and right (intact) striata were dissected and placed into RNALater (Qiagen) for real time RT-PCR (see description). The 2 h time point was chosen based on previously published real time RT-PCR work in our laboratory.7 During this time, ALO AIMs are reduced and rotations are enhanced with ±8-OH-DPAT treatment prior to SKF81297.20,36
Experiment 2: Effects of Striatal 5-HT1AR Stimulation on D1R Agonist-Mediated Extracellular Striatal Glutamate and Nigral GABA Levels
Three weeks following surgery, all rats (n = 26) in Experiment 2 received SKF81297 (0.8 mg/kg, sc) on three separate occasions 2–3 days apart, and ALO AIMs were observed immediately after injections. At the end of SKF81297-priming, responding rats (ALO AIMs ≥ 20; n = 26) were split into one of four equal treatment groups (overall ALO AIMs median score = 54 (±6); Group I = 59 (±9.5); Group II = 52 (±5); Group III = 58 (±10.5); Group IV = 50 (±5); see Figure 1 for experimental design). The microdialysis procedure was conducted similarly to methods previously described.20 On the day of testing, striatal probes (CMA 12 Elite; membrane length = 3 mm; 20 000 Da; Stockholm, Sweden) and nigral probes (CMA 12 Elite; membrane length = 1 mm; 20 000 Da) were inserted into rats’ guide cannulae and locked into place so that the dialysis membrane extended −3.7 to −6.7 ventral to bregma within the striatum and −7.8 to −8.8 mm ventral to bregma within the SNpr. After 60 min of probe stabilization (2.0 μL artificial cerebral spinal fluid [aCSF]/min), striatal and nigral dialysis samples were collected every 20 min for 40 min to determine baseline levels of amino acids. At this point, rats in each group received a systemic treatment injection of Vehicle (20% DMSO in 0.9% NaCl, sc) and sample fractions were collected every 20 min for 2 h to determine any changes in amino acid levels due to systemic injection. Following this, rats received one of three striatal treatments: Vehicle (aCSF), ± 8-OH-DPAT (7.5 mM), or combined ±8-OH-DPAT (7.5 mM) + the 5-HT1AR antagonist, N-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-N-2-pyridinylcyclohexanecarboxamide maleate salt (WAY100635, 4.6 mM). These striatal drug treatments were administered via reverse-phase in vivo microdialysis for a total of 2 h. All reverse dialysis solutions were adjusted to pH 7.4 (±0.02) using o-phosphoric acid and/or NaOH. Concentrations were chosen primarily based on prior microdialysis work,20 and a pilot study including various concentrations of ±8-OH-DPAT (3, 7.5, and 15 mM) demonstrated that 7.5 mM produced similar behavioral effects as previous experiments.20,36,37 In addition, similar concentrations of ±8-OH-DPAT and WAY100635 have been previously utilized in rodent in vivo microdialysis and microiontophoresis studies.70−72
Ten minutes following the beginning of striatal reverse-phase microdialysis (corresponding to when drug has reached the striatum), rats received a systemic treatment injection with either Vehicle (20% DMSO, 0.9% NaCl, sc) or SKF81297 (0.8 mg/kg, sc). Striatal and nigral sample fractions were collected every 20 min for 2 h for analysis of glutamate and GABA via high performance liquid chromatography coupled to electrochemical detection (HPLC-ED; see description). Concurrently, ALO AIMs were observed every 10 min immediately following the systemic injection. Following 2 h of reverse-phase microdialysis, 1 final h of collection occurred, where only aCSF was infused, and was considered post-treatment sampling. Each rat underwent this microdialysis procedure for 2 consecutive days as no significant differences in behavior or neurochemistry were found between test days (data not shown).20
Abnormal Involuntary Movements
Rats were monitored for AIMs and rotations using a similar procedure previously described.36 Following treatment, rats were individually placed in plastic cylinders (22.2 cm diameter, 25.4 cm height; Thermo Fisher Scientific, Waltham, MA) and a trained observer blind to treatment condition assessed each rat for exhibition of axial, limb, and orolingual (ALO) AIMs (inter-rater reliability = 0.98). In addition, both contralateral rotations (defined as complete 360° turns away from the lesioned side of the brain) and ipsilateral rotations (defined as completed 360° turns toward the lesioned side of the brain) were tallied and reported as the difference between contralateral and ipsilateral rotations. “Axial” AIMs were referred to as dystonic posturing, represented by a twisting of the neck and torso directed toward the side of the body contralateral to the lesion. “Limb” AIMs were defined as rapid, purposeless movements of the forelimb located on the side of the body contralateral to the lesion. “Orolingual” AIMs were composed of repetitive openings and closings of the jaw and tongue protrusions and considered abnormal as they occur at times when the rats were not chewing or gnawing on food or other objects. Rats were observed for ALO AIMs and rotations for 1 min every 10th min over 120 min (Experiment 1) or 180 min (Experiment 2). For AIMs, a severity score of 0–4 was assigned for each category: 0, not present; 1, present for <50% of the observation period; 2, present for >50% or more of the observation period; 3, present for the entire observation period and interrupted by a loud stimulus (a tap on the plastic cylinder); or 4, present for the entire observation period but not interrupted by a loud stimulus. Scores for axial, limb, and orolingual were combined to create a single ALO AIMs score for data analysis. The theoretical maximum ALO AIMs score was 12 per time period (4 × 3 AIMs subcategories) with an overall maximum score of 144 for 120 min (12 × 12 time periods) and 216 for 180 min (12 × 18 time periods).
Real-Time Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
Two hours after treatment, rats in Experiment 1 were killed and left and right striata were removed and placed in RNAlater (Qiagen, Valencia, CA) for subsequent analyses of PPD, GAD65, GAD67, and the housekeeper gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Tissue samples were homogenized into Trisol using TissueLyser 2 (Qiagen), extracted with RNeasy mini protocol (Qiagen), normalized, and converted into cDNA using the QuantiTect Reverse Transcription Kit (Qiagen), which included a DNase treatment step.73 Probed cDNA amplification was performed in a 10 μL reaction consisting of 5 μL of IQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA), 0.5 μL of primer (final concentration 250 nM), 0.5 μL of cDNA template, and 4 μL of RNase-free water run in triplicate in a 384 well plate (BioRad) and captured in real-time using the CFX Real-Time PCR detection system (BioRad). Relative gene expression was quantified using the 2–ΔCT method, with expression values normalized to 100% of ultimate control values. Gene sequences were obtained from GenBank at the National Center for Biotechnology Information, and primer specificity was verified by the Basic Local Alignment Search Tool (http://www.ncbi.nlm.nih.gov/). The primer sequences used were for PPD (5′-GGGTTCGCTGGATTCAAATA-3′/5′-TGTGTGGAGAGGGACACTCA-3′); GAD65 (5′-CAGCCTGTGAAGGAGAAAGG-3′/5′-TACAGGGGCGATCTCATAGG-3′); GAD67 (5′-CACAAACTCAGCGGCATAGA-3′/5′-GACCAGGATGGCAGAACACT-3′); and GAPDH (5′-GCCATCTCTTGCTCGAAGTC-3′/5′-ATGACTCTACCCACGGCAAG-3′).
Tissue Dissection and Cryostat Sectioning
When Experiment 2 was completed, a subset of rats (n = 8) were sacrificed by decapitation and brains were immediately removed. Anterior and central striata from these rats were visually examined for verification of striatal microdialysis probe placements. Posterior striata from these rats were freshly dissected, frozen at −80 °C, and subjected to monoamine analysis via HPLC-ED,2 which revealed 98.9% and 99.9% reductions in striatal DOPAC and DA, respectively, in the lesioned striata compared to intact. Striata from all other rats (n = 18) were examined for verification of striatal microdialysis probe placements using a cryostat. During dissection, posterior brains from the subset of rats (n = 8) and whole brains from all other rats (n = 18) were removed and rapidly frozen in methylbutane (−30 °C) and stored at −20 °C. Cresyl violet (FD Neurotechnologies, Baltimore, MD) staining was used to determine perfusion sites and extent of gliosis from cryostat-generated 20 μm coronal sections containing striatal and nigral probe sites that were postfixed with 4% paraformaldehyde (PFA; Fisher Scientific, Hanover Park, IL). All rats that completed Experiment 2 were found to have probe placements within the dorsocentral or dorsolateral aspects of the striatum (Figure 3A) and the SNpr (Figure 3B).
High-Performance Liquid Chromatography for Glutamate and GABA Dialysate Samples
Dialysate samples from all rats in Experiment 2 were analyzed via HPLC-ED for extracellular glutamate and GABA levels, according to a previously described protocol.20 The OPA/β-ME derivatizing reagent (15 μL) reacted with striatal dialysate samples (30 μL) and 20 μL of the derivatized sample was analyzed for abundance of glutamate and GABA. Peak areas were oxidized at the second electrode and eluting amino acid derivatives were analyzed by EZChrom Elite software via a Scientific Software Inc. module (SS420x). The oxidation current values were converted to masses (ng of amino acid) using standard curves (2 × 10–6 – 1 × 10–8 M). The determination of striatal probe recovery (±22%) and nigral probe recovery (±18%) were performed in vitro by perfusing standard solutions (2 × 10–6–1 × 10–8 M). Absolute basal striatal glutamate and nigral GABA levels (M [±SEM]) were 1.9 × 10–6 [±2 × 10–7] and 7.0 × 10–8 [±2 × 10–8], respectively. All values utilized for final analyses (and ultimately expressed as % baseline) existed within the standard curve (2 × 10–6–1 × 10–8 M). Outlier values were considered to be 2 standard deviations above or below the mean and were not included in final analyses.
Data Analyses
Treatment effects for ALO AIMs (expressed as medians ± median absolute deviation [MAD]) for both experiments were analyzed by employing nonparametric Kruskal–Wallis tests and significant differences between treatments were determined by Mann–Whitney post hoc comparisons. Rotations (expressed as means ± SEM), relative mRNA (expressed as mean percent ± SEM from control), and amino acid time course data (expressed as mean % baseline ± SEM) were analyzed by using mixed design two-way ANOVAs. Treatment effects (expressed as means ± SEM) for striatal glutamate and nigral GABA (% baseline) were analyzed with between-subjects one-way ANOVAs. Significant differences between groups were determined by LSD post hoc tests for rotations, mRNA, glutamate, and GABA. Analyses for both experiments were performed with the use of Statistica software ’98 (Statsoft Inc., Tulsa, OK), and alpha was set at p < 0.05.
Acknowledgments
The authors would especially like to thank Thomas Button and Nancy Montieth for their technical assistance.
Glossary
Abbreviations
- 5-HT
serotonin
- 5-HT1AR
serotonin 1A receptor
- 6-OHDA
6-hydroxydopamine hydrobromide
- aCSF
artificial cerebral spinal fluid
- ALO AIMs
axial, limb, and orolingual Abnormal Involuntary Movements
- Aq
aqueduct (Sylvius)
- β-ME
β-mercaptoethanol
- Cc
corpus callosum
- CPU
caudate putamen
- D1R
D1 receptor
- D2R
D2 receptor
- DA
dopamine
- DMSO
dimethyl sulfoxide
- DOPAC
3,4-dihydroxyphenylacetic acid
- DPAT
±8-OH-DPAT, (±)-8-hydroxy-2-(dipropylamino)tetralin hydrobromide
- GABA
γ-aminobutyric-acid
- GAD
glutamic acid decarboxylase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HPLC-ED
high performance liquid chromatography coupled to electrochemical detection
- LID
L-DOPA-induced dyskinesia
- LV
lateral ventricle
- MAD
median absolute deviation
- MFB
medial forebrain bundle
- NaCl
sodium chloride
- OPA
o-phthaldialdehyde
- PD
Parkinson’s disease
- PFA
paraformaldehyde
- PPD
preprodynorphin
- PPE
preproenkephalin
- SKF
SKF81297, R(+)-SKF-81297 hydrobromide
- SNpr
substantia nigra pars reticulata
- VEH
vehicle
- WAY
WAY100635, N-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-N-2-pyridinylcyclohexanecarboxamide maleate salt
This work was supported by NIHF31NS066684 (K.B.D.), NIHNS059600 (C.B.), and the Center for Development and Behavioral Neuroscience at Binghamton University.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Carta M.; Carlsson T.; Kirik D.; Bjorklund A. (2007) Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain 130, 1819–1833. [DOI] [PubMed] [Google Scholar]
- Eskow K. L.; Gupta V.; Alam S.; Park J. Y.; Bishop C. (2007) The partial 5-HT1A agonist buspirone reduces the expression and development of l-DOPA-induced dyskinesia in rats and improves L-DOPA efficacy. Pharmacol., Biochem. Behav. 87, 306–314. [DOI] [PubMed] [Google Scholar]
- Eskow K. L.; Dupre K. B.; Barnum C. J.; Dickinson S. O.; Park J. Y.; Bishop C. (2009) The role of the dorsal raphe nucleus in the development, expression, and treatment of L-DOPA-induced dyskinesia in hemiparkinsonian rats. Synapse 63, 610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostock C. Y.; Dupre K. B.; Eskow Jaunarajs K. L.; Walters H.; George J.; Krolewski D.; Walker P. D.; Bishop C. (2011) Role of the primary motor cortex in L-DOPA-induced dyskinesia and its modulation by 5-HT1A receptor stimulation. Neuropharmacology 61, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow C. W.; Damier P.; Goetz C. G.; Mueller T.; Nutt J.; Rascol O.; et al. (2004) Multicenter, open-label, trial of sarizotan in Parkinson disease patients with levodopa-induced dyskinesias (the SPLENDID Study). Clin. Neuropharmacol. 27, 58–62. [DOI] [PubMed] [Google Scholar]
- Bara-Jimenez W.; Bibbiani F.; Morris M. J.; Dimitrova T.; Sherzai A.; Mouradian M. M.; et al. (2005) Effects of serotonin 5-HT1A agonist in advanced Parkinson’s disease. Mov. Disord. 20, 932–936. [DOI] [PubMed] [Google Scholar]
- Bishop C.; Krolewski D. M.; Eskow K. L.; Barnum C. J.; Dupre K. B.; Deak T.; Walker P. D. (2009) Contribution of the striatum to the effects of 5-HT1A receptor stimulation in L-DOPA-treated hemiparkinsonian rats. J. Neurosci. Res. 87, 1645–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara K.; Shimizu K.; Suno M.; et al. (2006) Tandospirone, a 5-HT1A agonist, ameliorates movement disorder via non-dopaminergic systems in rats with unilateral 6-hydroxydopamine-generated lesions. Brain Res. 1112, 126–133. [DOI] [PubMed] [Google Scholar]
- Mignon L.; Wolf W. A. (2007) Postsynaptic 5-HT(1A) receptor stimulation increases motor activity in the 6-hydroxydopamine-lesioned rat: Implications for Parkinson’s disease. Psychopharmacology (Berlin, Ger.) 192, 49–59. [DOI] [PubMed] [Google Scholar]
- Iravani M. M.; Tayarani-Binazir K.; Chu W. B.; Jackson M. J.; Jenner P. (2006) In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primates, the selective 5-hydroxytryptamine 1A agonist (R)-(+)-8-OHDPAT inhibits levodopa-induced dyskinesia but only with increased motor disability. J. Pharmacol. Exp. Ther. 319, 1225–1234. [DOI] [PubMed] [Google Scholar]
- Carey R. J.; DePalma G.; Damianopoulos E.; Muller C. P.; Huston J. P. (2004) The 5-HT1A receptor and behavioral stimulation in the rat: Effects of 8-OHDPAT on spontaneous and cocaine-induced behavior. Psychopharmacology 177, 46–54. [DOI] [PubMed] [Google Scholar]
- Goetz C. G.; Damier P.; Hicking C.; Laska E.; Muller T.; Olanow C. W.; Rascol O.; Russ H. (2007) Sarizotan as a treatment for dyskinesias in Parkinson’s disease: a double-blind placebo-controlled trial. Mov. Disord. 22, 179–186. [DOI] [PubMed] [Google Scholar]
- Carta M.; Lindgren H.; Lundblad M.; Stancampiano R.; Fadda F.; Cenci M. A. (2006) Role of striatal L-DOPA in the production of dyskinesia in 6-hydroxydopamine lesioned rats. J. Neurochem. 96, 1718–1727. [DOI] [PubMed] [Google Scholar]
- Cenci M. A.; Ohlin K. E.; Rylander D. (2009) Plastic effects of L-DOPA treatment in the basal ganglia and their relevance to the development of dyskinesia. Parkinsonism Relat. Disord. 15, S59–93. [DOI] [PubMed] [Google Scholar]
- Kannari K.; Yamato H.; Shen H.; Tomiyama M.; Suda T.; Matsunaga M. (2001) Activation of 5-HT(1A) but not 5-HT(1B) receptors attenuates an increase in extracellular dopamine derived from exogenously administered L-dopa in the striatum with nigrostriatal denervation. J. Neurochem. 76, 1346–1353. [DOI] [PubMed] [Google Scholar]
- Lindgren H. S.; Andersson D. R.; Lagerkvist S.; Nissbrandt H.; Cenci M. A. (2010) L-DOPA-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: temporal and quantitative relationship to the expression of dyskinesia. J. Neurochem. 112(6), 1465–1476. [DOI] [PubMed] [Google Scholar]
- Sgambato-Faure V.; Cenci M. A. (2012) Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog. Neurobiol. 96, 69–86. [DOI] [PubMed] [Google Scholar]
- Robelet S.; Melon C.; Guillet B.; Salin P.; Kerkerian-LeGoff L. (2004) Chronic l-DOPA treatment increases extracellular glutamate levels and GLT1 expression in the basal ganglia in a rat model of Parkinson’s disease. Eur. J. Neurosci. 20, 1255–1266. [DOI] [PubMed] [Google Scholar]
- Samadi P.; Gregoire L.; Morissette M.; Calon F.; Tahar A. H.; Dridi M.; et al. (2008) mGluR5 metabotropic glutamate receptors and dyskinesias in MPTP monkeys. Neurobiol. Aging 29, 1040–1051. [DOI] [PubMed] [Google Scholar]
- Dupre K. B.; Ostock C. Y.; Eskow Jaunarajs K. L.; Button T.; Savage L.; Wolf W.; Bishop C. (2011) Local modulation of striatal glutamate efflux by 5-HT1A receptor stimulation in dyskinetic, hemiparkinsonian rats. Exp. Neurol. 229, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon F.; Rajput A. H.; Hornykiewicz O.; Bedard P. J.; Di Paolo T. (2003) Levodopa-induced motor complications are associated with alterations of glutamate receptors in Parkinson’s disease. Neurobiol. Dis. 14(3), 404–416. [DOI] [PubMed] [Google Scholar]
- Ouattara B.; Gregoire L.; Morissette M.; Gasparini F.; Vranesic I.; Bilbe G.; Johns D. R.; Rajput A.; Hornykiewicz O.; Rajput A. H.; Gomez-Mancilla B.; Di Paolo T. (2011) Metabotropic glutamate receptor type 5 in levodopa-induced motor complications. Neurobiol. Aging 32(7), 1286–1295. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Conn P. J.; Niswender C. M. (2009) Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 8(6), 475–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci M. A. (2007) Dopamine dysregulation of movement control in L-DOPA-induced dyskinesia. Trends Neurosci. 30, 236–243. [DOI] [PubMed] [Google Scholar]
- Westin J. E.; Vercammen L.; Strome E. M.; Konradi C.; Cenci M. A. (2007) Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA–induced dyskinesia and the role of dopamine D1 receptors. Biol. Psychiatry 62, 800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen C. R. (1992) The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu. Rev. Neurosci. 15, 285–320. [DOI] [PubMed] [Google Scholar]
- Fink J. S.; Weaver D. R.; Rivkees S. A.; Peterfreund R. A.; Pollack A. E.; Adler E. M.; et al. (1992) Molecular cloning of the rat A2 adenosine receptor: selective coexpression with D2 dopamine receptor in rat striatum. Mol. Brain Res. 14, 186–95. [DOI] [PubMed] [Google Scholar]
- Cenci M. A.; Lee C. S.; Bjorklund A. (1998) L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur. J. Neurosci. 10, 2694–2706. [PubMed] [Google Scholar]
- Mela F.; Marti M.; Dekundy A.; Danysz W.; Morari M.; Cenci M. A. (2007) Antagonism of metabotropic glutamate receptor type 5 attenuates L-DOPA-induced dyskinesia and its molecular and neurochemical correlates in a rat model of Parkinson’s disease. J. Neurochem. 101, 483–497. [DOI] [PubMed] [Google Scholar]
- Wang H.; Katz J.; Dagostino P.; Soghomonian J. J. (2007) Unilateral 6-hydroxydopamine lesion of dopamine neurons and subchronic L-DOPA administration in the adult rat alters the expression of the vesicular GABA transporter in different subsets of striatal neurons and in the substantia nigra, pars reticulata. Neurosci. 145, 727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprade N.; Soghomonian J. J. (1999) Gene expression of the GAD67 and GAD65 isoforms of glutamate decarboxylase is differentially altered in subpopulations of striatal neurons in adult rats lesioned with 6-OHDA as neonates. Synapse 33, 36–48. [DOI] [PubMed] [Google Scholar]
- Tomiyama M.; Kimura T.; Maeda T.; Kannari K.; Matsunaga M.; Baba M. (2005) A serotonin 5-HT1A receptor agonist prevents behavioral sensitization to L-DOPA in a rodent model of Parkinson’s disease. Neurosci. Res. 52, 185–194. [DOI] [PubMed] [Google Scholar]
- Mela F.; Marti M.; Bido S.; Cenci M. A.; Morari M. (2012) In vivo evidence for a differential contribution of striatal and nigral D1 and D2 receptors to L-DOPA induced dyskinesia and the accompanying surge of nigral amino acid levels. Neurobiol. Dis. 573–582. [DOI] [PubMed] [Google Scholar]
- Rascol O.; Nutt J. G.; Blin O.; Goetz C. G.; Trugman J. M.; Soubrouillard C.; et al. (2001) Induction by dopamine D1 receptor agonist ABT-431 of dyskinesia similar to levodopa in patients with Parkinson disease. Arch. Neurol. 58, 249–254. [DOI] [PubMed] [Google Scholar]
- Delfino M.; Kalisch R.; Czisch M.; Larramendy C.; Ricatti J.; Taravini I. R.; et al. (2007) Mapping the effects of three dopamine agonists with different dyskinetogenic potential and receptor selectivity using pharmacological functional magnetic resonance imaging. Neuropsychopharmacology 32, 1911–1921. [DOI] [PubMed] [Google Scholar]
- Dupre K. B.; Eskow K. L.; Negron G.; Bishop C. (2007) The differential effects of 5-HT1A receptor stimulation on dopamine receptor-mediated abnormal involuntary movements and rotations in the primed hemiparkinsonian rat. Brain Res. 1158, 135–143. [DOI] [PubMed] [Google Scholar]
- Dupre K. B.; Eskow K. L.; Barnum C. J.; Bishop C. (2008) Striatal 5-HT1A receptor stimulation reduced D1 receptor-induced dyskinesia and improves movement in the hemiparkinsonian rat. Neuropharmacology 55, 1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breger L. S.; Dunnett S. B.; Lane E. L. (2013) Comparison of rating scales used to evaluate L-DOPA-induced dyskinesia in the 6-OHDA lesioned rat. Neurobiol. Dis. 50, 142–150. [DOI] [PubMed] [Google Scholar]
- Pahwa R.; Factor S. A.; Lyons K. E.; Ondo W. G.; Gronseth G.; Bronte-Stewart H.; Hallett M.; Miyasaki J.; Stevens J.; Weiner W. J. (2006) Practice parameter: treatment of Parkinson disease with motor fluctuations and dyskinesia (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurol. 66, 983–995. [DOI] [PubMed] [Google Scholar]
- You Z. B.; Herrera-Marschitz M.; Nylander I.; Goiny M.; O’Connor W. T.; Ungerstedt U.; Terenius L. (1994) The striatonigral dynorphin pathway of the rat studied with in vivo microdialysis: II. Effects of dopamine D1 and D2 receptor agonists. Neurosci. 63, 427–434. [DOI] [PubMed] [Google Scholar]
- Laprade N.; Soghomonian J. J. (1995) Regulation of mRNA levels encoding for two isoforms of glutamate decarboxylase and preproenkephalin in rat striatum by dopaminergic receptors. Mol. Brain Res. 34, 65–74. [DOI] [PubMed] [Google Scholar]
- Laprade N.; Soghomonian J. J. (1997) Glutamate decarboxylase (GAD65) gene expression is increased by dopamine receptor agonists in a subpopulation of rat striatal neurons. Mol. Brain Res. 48, 333–345. [DOI] [PubMed] [Google Scholar]
- Yamamoto N.; Soghomonian J. J. (2008) Timecourse of SKF-81297-induced increase in glutamic acid decarboxylase 65 and 65 mRNA levels in striatonigral neurons and decrease in GABA A receptor α1 subunit mRNA levels in the substantia nigra, pars reticulata, in adult rats with unilateral 6-hydroxydopamine lesion. Neuroscience 154, 1088–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta A. R.; Fenu S.; Pala P.; Tronci E.; Morelli M. (2003) Selective modifications in GAD67 mRNA levels in striatonigral and striatopallidal pathways correlate to dopamine agonist priming in 6-hydroxydopamine-lesioned rats. Eur. J. Neurosci. 18, 2563–2572. [DOI] [PubMed] [Google Scholar]
- Antonelli T.; Fuxe K.; Tomasini M. C.; Bartoszyk G. D.; Seyfried C. A.; Tanganelli S.; Ferraro L. (2005) Effects of sarizotan on the corticostriatal glutamate pathways. Synapse 58, 193–199. [DOI] [PubMed] [Google Scholar]
- Mignon L. J.; Wolf W. A. (2005) 8-Hydroxy-2-(di-n-propylamino)tetralin reduces striatal glutamate in an animal model of Parkinson’s disease. NeuroReport 16, 699–703. [DOI] [PubMed] [Google Scholar]
- Mengod G.; Vilaro M. T.; Raurich A.; Lopez-Gimenez J. F.; Cortes R.; Palacios J. M. (1996) 5-HT receptors in mammalian brain: receptor autoradiography and in situ hybridization studies of new ligands and newly identified receptors. Histochem. J. 28, 747–758. [DOI] [PubMed] [Google Scholar]
- Kindlundh-Hogberg A. M. S.; Svenningsson P.; Schioth H. B. (2006) Qunatitative mapping shows that serotonin rather than dopamine receptor mRNA expressions are affected after repeated intermittent administration of MDMA in rat brain. Neuropharmacology 51, 838–847. [DOI] [PubMed] [Google Scholar]
- Numan S.; Lundgren K. H.; Wright D. E.; Herman J. P.; Seroogy K. B. (1995) Increased expression of 5HT2 receptor mRNA in rat striatum following 6-OHDA lesions of the adult nigrostriatal pathway. Mol. Brain Res. 29, 391–396. [DOI] [PubMed] [Google Scholar]
- Frechilla D.; Cobreros A.; Saldise L.; Moratalla R.; Insausti R.; Luquin M.; et al. (2001) Serotonin 5-HT(1A) receptor expression is selectively enhanced in the striosomal compartment of chronic parkinsonian monkeys. Synapse 39, 288–296. [DOI] [PubMed] [Google Scholar]
- Huot P.; Johnston T. H.; Koprich J. B.; Winkelmolen L.; Fox S. H.; Brotchie J. M. (2012) Regulation of cortical and striatal 5-HT(1A) receptors in the MPTP-lesioned macaque. Neurobiol. Aging 33(207), e9–207.e19. [DOI] [PubMed] [Google Scholar]
- Huot P.; Johnston T. H.; Visanji N. P.; Darr T.; Pires D.; Hazrati L.; Brotchie J. M.; Fox S. H. (2012) Increased levels of 5-HT1A receptor binding in ventral visual pathways in Parkinson’s disease. Mov. Disord. 27, 735–742. [DOI] [PubMed] [Google Scholar]
- Hioki H.; Nakamura H.; Ma Y. F.; Konno M.; Hayakawa T.; Nakamura K. C.; Fujiyama F.; Kaneko T. (2010) Vesicular glutamate transporter 3-expressing nonserotonergic projection neurons constitute a subregion in the rat midbrain raphe nuclei. J. Comp. Neurol. 518, 668–686. [DOI] [PubMed] [Google Scholar]
- Calcagno E.; Carli M.; Invernizzi R. W. (2006) The 5-HT(1A) receptor agonist 8-OH-DPAT prevents prefrontocortical glutamate and serotonin release in response to blockade of cortical NMDA receptors. J. Neurochem. 96, 853–860. [DOI] [PubMed] [Google Scholar]
- Calabresi P.; Giacomini P.; Centonze D.; Bernardi G. (2000) Levodopa-induced dyskinesia: A pathological form of striatal synaptic plasticity?. Ann. Neurol. 47, S60–S68. [PubMed] [Google Scholar]
- Picconi B.; Centonze D.; Hakansson K.; Bernardi G.; Greengard P.; Fisone G.; Cenci M. A.; Calabresi P. (2003) Loss of bidirectional striatal synaptic plasticity in LDOPA-induced dyskinesia. Nat. Neurosci. 6, 501–506. [DOI] [PubMed] [Google Scholar]
- Yamamoto N.; Pierce R. C.; Soghomonian J. J. (2006) Subchronic administration of L-DOPA to adult rats with a unilateral 6-hydroxydopamine lesion of dopamine neurons results in a sensitization of enhanced GABA release in the substantia nigra, pars reticulata. Brain Res. 1123, 196–200. [DOI] [PubMed] [Google Scholar]
- Rangel-Barajas C.; Silva I.; Garcıa-Ramırez M.; Sanchez-Lemus E.; Floran L.; Aceves J.; et al. (2008) 6-OHDA-induced hemiparkinsonism and chronic L-DOPA treatment increase dopamine D1-stimulated [3H]-GABA release and [3H]-cAMP production in substantia nigra pars reticulata of the rat. Neuropharmacology 55, 704–711. [DOI] [PubMed] [Google Scholar]
- Kozlowski M. R.; Marshall J. F. (1980) Rotation induced by intranigral injections of GABA agonists and antagonists: Zone-specific effects. Pharmacol., Biochem. Beh. 561–567. [DOI] [PubMed] [Google Scholar]
- Burbaud P.; Bonnet B.; Guehl D.; Lagueny A.; Bioulac B. (1998) Movement disorders induced by gamma-aminobutyric agonist and antagonist injections into the internal globus pallidus and substantia nigra pars reticulata of the monkey. Brain Res. 780, 102–107. [DOI] [PubMed] [Google Scholar]
- Van Wijngaarden I.; Tulp M.; Soudijn W. (1990) The concept of selectivity in 5-HT receptor research. Eur. J. Pharmacol. 188, 301–312. [DOI] [PubMed] [Google Scholar]
- Lorrain D. S.; Matuszewich L.; Hull E. M. (1998) 8-OH-DPAT influences extracellular levels of serotonin and dopamine in the medial preoptic area of male rats. Brain Res. 790, 217–223. [DOI] [PubMed] [Google Scholar]
- Cepeda C.; Hurst R. S.; Altemus K. L.; Flores-Hernandez J.; Calvert C. R.; Jokel E. S.; Grandy D. K.; Low M. J.; Rubinstein M.; Ariano M. A.; Levine M. S. (2001) Facilitated glutamatergic transmission in the striatum of D2 dopamine receptor-deficient mice. J. Neurophysiol. 85, 659–670. [DOI] [PubMed] [Google Scholar]
- Monville C.; Torres E. M.; Dunnett S. B. (2005) Validation of the L-DOPA-induced dyskinesia in the 6-OHDA model and evaluation of the effects of selective dopamine receptor agonists and antagonists. Brain Res. Bull. 68, 16–23. [DOI] [PubMed] [Google Scholar]
- Jaunarajs K. L.; Dupre K. B.; Steiniger A.; Klioueva A.; Moore A.; Kelly C.; Bishop C. (2009) Serotonin 1B receptor stimulation reduces D1 receptor agonist-induced dyskinesia. NeuroReport 20, 1265–1269. [DOI] [PubMed] [Google Scholar]
- Muñoz A.; Li Q.; Gardoni F.; Marcello E.; Qin C.; Carlsson T.; Kirik D.; Di Luca M.; Björklund A.; Bezard E.; Carta M. (2009) Combined 5-HT1A and 5-HT1B receptor agonists for the treatment of L-DOPA-induced dyskinesia. Brain 131, 3380–3394. [DOI] [PubMed] [Google Scholar]
- Paxinos G., and Watson W. (1998) The Rat Brain in Stereotaxic Coordinates, fourth ed., Academic Press, San Diego. [Google Scholar]
- Andersen P. H.; Jansen J. A. (1990) Dopamine receptor agonists: Selectivity and dopamine D1 receptor efficacy. Eur. J. Pharmacol. 188, 335–347. [DOI] [PubMed] [Google Scholar]
- Arnt J.; Hyttel J.; Sanchez C. (1992) Partial and full dopamine D1 receptor agonists in mice and rats: Relation between behavioural effects and stimulation of adenylate cyclase activity in vitro. Eur. J. Pharmacol. 213, 259–267. [DOI] [PubMed] [Google Scholar]
- Adell A.; Carceller A.; Artigas F. (1993) In vivo brain dialysis study of the somatodendritic release of serotonin in the Raphe nuclei of the rat: Effects of 8-hydroxy-2-(di-n-propylamino)tetralin. J. Neurochem. 60, 1673–1681. [DOI] [PubMed] [Google Scholar]
- Hajós M.; Hajós-Korcsok E.; Sharp T. (1999) Role of the medial prefrontal cortex in 5-HT1A receptor-induced inhibition of 5-HT neuronal activity in the rat. Br. J. Pharmacol. 126, 1741–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaiyakul P.; Reidman D.; Pilipovic L.; Maher T.; Ally A. (2001) Further evidence that extracellular serotonin in the rostral ventrolateral medulla modulates 5-HT(1A) receptor-mediated attenuation of exercise pressor reflex. Brain Res. 900, 186–194. [DOI] [PubMed] [Google Scholar]
- Hueston C. M.; Barnum C. J.; Eberle J. A.; Ferraioli F. J.; Buck H. M.; Deak T. (2011) Stress-dependent changes in neuroinflammatory markers observed after common laboratory stressors are not seen following acute social defeat of the Sprague Dawley rat. Phys. Behav. 104, 187–198. [DOI] [PubMed] [Google Scholar]