

Abstract
There is no high-resolution structure of the human P-glycoprotein (P-gp, ABCB1) drug pump. Homology models based on the crystal structures of mouse and Caenorhabditis elegans P-gps show extensive contacts between intracellular loop 2 (ICL2, in the first transmembrane domain) and the second nucleotide-binding domain. Human P-gp modeled on these P-gp structures yields different ICL2 structures. Only the model based on the C. elegans P-gp structure predicts the presence of a salt bridge. We show that the Glu256–Arg276 salt bridge was critical for P-gp folding.
The P-glycoprotein drug pump (P-gp, ABCB1) catalyzes the ATP-dependent efflux of a wide range of hydrophobic compounds.1−3 The 1280 amino acids of human P-gp4 are organized as two tandem repeats that are joined by a linker region. Each repeat consists of an NH2-terminal transmembrane domain (TMD) containing six transmembrane (TM) segments followed by a nucleotide-binding domain (NBD).
There is a considerable degree of cross talk between the TMDs and NBDs during the reaction cycle as drug binding activates ATPase activity and ATP hydrolysis leads to efflux of the drug from the TMDs. Although there is no high-resolution crystal structure of human P-gp, homology models5−7 based on the crystal structures of P-gps from Caenorhabditis elegans,5 mouse,8 or bacterial Sav18669 showed that the TMDs were connected to the NBDs by four intracellular loops (ICLs). The ICLs were proposed to act as “ball-and-socket” joints to connect the TMDs to the NBDs5 and act as transmission interfaces.9 ICL1 (TMD1–NBD1), ICL2 (TMD1–NBD2), ICL3 (TMD2–NBD2), and ICL4 (TMD1–NBD2) mediate linkages between domains. The crystal structure of C. elegans P-gp predicted that the most important connections between the TMDs and NBDs in human P-gp would involve ICL2 as 14 amino acids in this loop were predicted to connect TMD1 to NBD2 via salt bridges, hydrogen bonds, or van der Waals interactions. The structure of ICL2 appears to be critical for biosynthesis of P-gp as point mutations in this region inhibit maturation.10
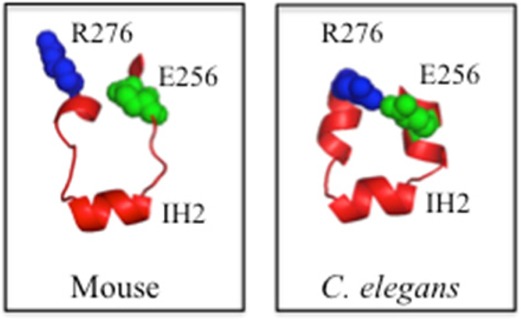
An accurate model of human P-gp is important for understanding its mechanism and for in silico docking studies for the discovery of novel inhibitors.6,11,12 Homology models of human P-gp based on the mouse and C. elegans crystal structures generally yielded similar structures.5,6 There was, however, a significant difference in the structure of ICL2. ICL2 connects TM segments 4 and 5 (Figure 1A). ICL2 appears to play a key role in coupling NBD1–TMD2 interactions because cysteines introduced into ICL2 (A266C) and NBD2 (F1086C) could be cross-linked and the F1086C change abolished activity.13 While both structures predict that residues 261–267 form an interhelical loop (IH2) (forms the ball portion of the ball-and-socket ICL2–NBD connection), adjacent amino acids were predicted to adopt loop or α-helical structures in the mouse or C. elegans structures, respectively. The difference is significant because only the C. elegans model suggests the presence of a salt bridge between Glu256 and Arg276. Here, we tested whether the Glu256–Arg276 salt bridge plays an important role in P-gp folding.
Figure 1.
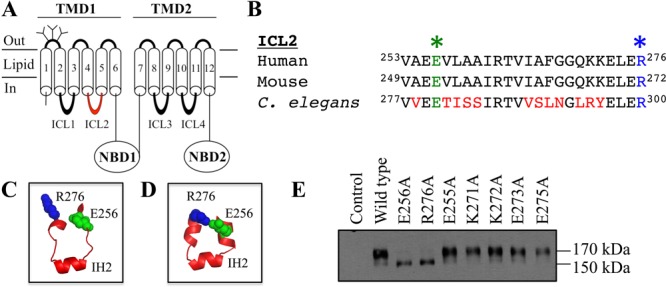
Mutations to the predicted ICL2 Glu256–Arg276 salt bridge inhibit maturation. (A) Model of human P-gp. (B) Alignment of ICL2 segments showing human Glu256 (green star) and Arg276 (blue star). Homology models of the human P-gp ICL2 segment based on the crystal structures of mouse (C)8 or C. elegans (D).5 (E) Immunoblot analysis of P-gp mutants.
The amino acid sequences of human, mouse, and C. elegans are similar (Figure 1B). Homology models of human P-gp based on the mouse or C. elegans structure, however, predict quite different structures for the ICL2 segments (residues Val253–Ala259 and Gly269–Lys272) that flank IH2 (residues Arg262–Phe267) (Figure 1C,D). The mouse-based homology model predicts that the Val253–Ala259 and Gly269–Lys272 segments would be loops (Figure 1C), whereas the C. elegans-based model predicts that these segments would be present as α-helices (Figure 1D). One critical structural difference between the two models is that only the C. elegans-based model predicts that Glu256 forms a salt bridge with Arg276 (Figure 1D).
We predicted that a salt bridge between residues Glu256 and Arg276 might be an important structural determinant for folding of P-gp because mutational analysis of ICL2 suggests that it plays a key role in folding of human P-gp. For example, mutation of glycine residues in the ICLs10 showed that Gly to Val changes at positions 251, 268, and 269 in ICL2 inhibited folding and maturation of P-gp from the core-glycosylated form (150 kDa) in the endoplasmic reticulum (ER) to the mature (170 kDa) protein containing complex carbohydrates. By contrast, none of the Gly mutations in ICL1, -3, or -4 inhibited maturation.10
To test if the charged amino acids Glu256 and Arg276 were important for maturation, we characterized mutants E256A and R276A. Figure 1E shows that E256A or R276A mutations inhibited maturation. While the mature 170 kDa protein was the major product of wild-type P-gp, the immature 150 kDa protein was the major product of mutants E256A and R276A.
We then tested whether mutations of other charged amino acids in the ICL2 segment between Val253 and Arg276 also inhibited maturation of P-gp (Glu255, Lys271, Lys272, Glu273, and Glu275). It was found that the E255A, K271A, K272A, E273A, and E275A mutants were different from the E256A and R276A mutants because they yielded mature 170 kDa P-gp as their major product (Figure 1E). The results in Figure 1E suggest that interactions between Glu256 and Arg276 were important for maturation of P-gp.
If a salt bridge between Glu256 and Arg276 were important for the maturation of human P-gp, it would be expected that an E256R/R276E mutant would still mature. Accordingly, we reversed the positions of the glutamic acid and arginine residues (E256R/R276E) and tested the mutant’s ability to undergo maturation. The mature 170 kDa P-gp protein was the major product in mutant E256R/R276E (Figure 2A). The 170 kDa protein was resistant to treatment with endoglycosidase H but sensitive to treatment with PNGase F (Figure 2B). The E256R or R276E mutation, however, inhibited maturation of P-gp (Figure 2A). For example, Figure 2B shows that the 150 kDa E256R protein was sensitive to treatment with endoglycosidase H or PNGase F. Similar results were obtained when mutant R276E was treated with endoglycosidase H or PNGase F (data not shown). These results suggest a salt bridge between residues 256 and 276 promoted P-gp maturation.
Figure 2.

Reversal of the Glu256 and Arg276 positions yields a mature and active P-gp. (A) Whole cell sodium dodecyl sulfate (SDS) extracts of wild-type P-gp and mutants E256R, R276E, and E256R/R276E. (B) Whole cell SDS extracts described above treated with endoglycosidase H (H) or PNGase F (F) and samples subjected to immunoblot analysis. (C) ATPase activities measured in the presence of various concentrations of verapamil.
To test if mutant E256R/R276E yielded an active protein, the histidine-tagged mutant was expressed in HEK293 cells, isolated by nickel chelate chromatography, and assayed for verapamil-stimulated ATPase activity. Verapamil was used because it is transported by P-gp14 and strongly stimulates (>10-fold) the ATPase activity of human P-gp.15 The E256R/R276E mutant showed verapamil-stimulated ATPase activity that was similar to that of the wild-type protein (Figure 2C). The results suggest that the structure of the regions flanking IH2 is consistent with the structure of C. elegans P-gp (Figure 1D) and that interaction between residues 256 and 276 is critical for the synthesis of the protein.
Is a salt bridge between residues 256 and 276 essential for activity? To address this question, we first tested whether it was possible to promote maturation of the E256A or R276A mutant. We previously showed that P-gp was inactive when it was trapped in the ER as a core-glycosylated intermediate.16 Drug substrates (e.g., cyclosporine A) can promote maturation of P-gp processing mutants.17
Mutants E256A and R276A were expressed in HEK293 cells in the presence or absence of 10 μM cyclosporine A. Samples of whole cell sodium dodecyl sulfate extracts were subjected to immunoblot analysis. It was observed that the presence of 10 μM cyclosporine A promoted maturation of both mutants to yield the mature form of P-gp as the major product (Figure S1A of the Supporting Information). The putative Glu256–Arg276 salt bridge was not essential for activity as both the E256A and R276A mutants showed wild-type verapamil-stimulated ATPase activity (Figure S1B of the Supporting Information).
In this study, we provide biochemical evidence that the structure of ICL2 in human P-gp resembles the crystal structure of C. elegans, rather than that of mouse P-gp. The presence of a salt bridge between Glu256 and Arg276 was found to promote maturation of human P-gp. Only the C. elegans P-gp structure predicted that these residues would be close enough to interact. The presence of a Glu256–Arg276 salt bridge in ICL2 may promote enhanced folding of P-gp during synthesis in the ER.
Residues Glu256 and Arg276 in human P-gp are also conserved in mouse (Glu252 and Arg272, respectively) and C. elegans (Glu280 and Arg300, respectively) P-gps. The C. elegans P-gp structure shows that Glu280 and Arg300 could form a salt bridge (Figure 1D). Although the side chains of mouse P-gp (Glu252 and Arg272) are not in the proper orientation (Figure 1C) to form a salt bridge, they may still form a salt bridge under more dynamic, physiological conditions. The distance between the α-carbons of Glu252 and Arg272 in mouse P-gp8 is actually shorter (11.4 Å) than the distance between the α-carbons of Glu280 and Arg300 in C. elegans P-gp (12.3 Å).
How might a Glu256–Arg276 salt bridge promote folding of human P-gp? One possibility is that it promotes packing of the TM segments because ICL2 links TM segments 4 and 5. In support of this hypothesis, we previously reported that a W232R mutation in TM4 appears to act as a suppressor mutation to rescue P-gp processing mutants through hydrogen bond interactions with Asn296 in TM5.21 Processing mutations likely trap polytopic membrane proteins like P-gp and CFTR in protease-sensitive conformations with incomplete packing of the TM segments.18,19 Suppressor mutations in the TMDs can rescue P-gp processing mutants by promoting packing of the TM segments.21,20
The Sav1866 bacterial drug pump contains the equivalent glutamate (Glu205), but the arginine residue is not conserved (Asn225).9 Although the distances between the α-carbons of Glu205 and Asn225 (11.4 A) are similar to those of mammalian P-gp, a salt bridge may not be necessary for folding of Sav1866. This may be due to differences in the bacterial and eukaryotic folding pathways.
Supporting Information Available
Details of experimental procedures and Figure S1. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by a grant from the Canadian Institutes of Health Research (25043). D.M.C. is the recipient of the Canada Research Chair in Membrane Biology.
The authors declare no competing financial interest.
Supplementary Material
References
- Sharom F. J. (2006) Biochem. Cell Biol. 84, 979–992. [DOI] [PubMed] [Google Scholar]
- Ambudkar S. V.; Dey S.; Hrycyna C. A.; Ramachandra M.; Pastan I.; Gottesman M. M. (1999) Annu. Rev. Pharmacol. Toxicol. 39, 361–398. [DOI] [PubMed] [Google Scholar]
- Eckford P. D.; Sharom F. J. (2009) Chem. Rev. 109, 2989–3011. [DOI] [PubMed] [Google Scholar]
- Chen C. J.; Chin J. E.; Ueda K.; Clark D. P.; Pastan I.; Gottesman M. M.; Roninson I. B. (1986) Cell 47, 381–389. [DOI] [PubMed] [Google Scholar]
- Jin M. S.; Oldham M. L.; Zhang Q.; Chen J. (2012) Nature 490, 566–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikadi Z.; Hazai I.; Malik D.; Jemnitz K.; Veres Z.; Hari P.; Ni Z.; Loo T. W.; Clarke D. M.; Hazai E.; Mao Q. (2011) PLoS One 6, e25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globisch C.; Pajeva I. K.; Wiese M. (2008) ChemMedChem 3, 280–295. [DOI] [PubMed] [Google Scholar]
- Aller S. G.; Yu J.; Ward A.; Weng Y.; Chittaboina S.; Zhuo R.; Harrell P. M.; Trinh Y. T.; Zhang Q.; Urbatsch I. L.; Chang G. (2009) Science 323, 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson R. J.; Locher K. P. (2006) Nature 443, 180–185. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (1994) J. Biol. Chem. 269, 7243–7248. [PubMed] [Google Scholar]
- Pajeva I. K.; Globisch C.; Wiese M. (2009) FEBS J. 276, 7016–7026. [DOI] [PubMed] [Google Scholar]
- Klepsch F.; Chiba P.; Ecker G. F. (2011) PLoS Comput. Biol. 7, e1002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2008) J. Biol. Chem. 283, 28190–28197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omote H.; Al-Shawi M. K. (2002) J. Biol. Chem. 277, 45688–45694. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2003) J. Biol. Chem. 278, 50136–50141. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (1999) J. Biol. Chem. 274, 24759–24765. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (1997) J. Biol. Chem. 272, 709–712. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2011) Biochemistry 50, 672–685. [DOI] [PubMed] [Google Scholar]
- Chen E. Y.; Bartlett M. C.; Loo T. W.; Clarke D. M. (2004) J. Biol. Chem. 279, 39620–39627. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2002) J. Biol. Chem. 277, 27585–27588. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. (2007) J. Biol. Chem. 282, 32043–32052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.