

Abstract
Transferrin receptor 2 (TfR2) is a member of the transferrin receptor-like family of proteins. Mutations in TfR2 can lead to a rare form of the iron overload disease, hereditary hemochromatosis. TfR2 is proposed to sense body iron levels and increase the level of expression of the iron regulatory hormone, hepcidin. Human TfR2 (hTfR2) contains four potential Asn-linked (N-linked) glycosylation sites on its ectodomain. The importance of glycosylation in TfR2 function has not been elucidated. In this study, by employing site-directed mutagenesis to remove glycosylation sites of hTfR2 individually or in combination, we found that hTfR2 was glycosylated at Asn 240, 339, and 754, while the consensus sequence for N-linked glycosylation at Asn 540 was not utilized. Cell surface protein biotinylation and biotin-labeled Tf indicated that in the absence of N-linked oligosaccharides, hTfR2 still moved to the plasma membrane and bound its ligand, holo-Tf. However, without N-linked glycosylation, hTfR2 did not form the intersubunit disulfide bonds as efficiently as the wild type (WT). Moreover, the unglycosylated form of hTfR2 could not be stabilized by holo-Tf. We further provide evidence that the unglycosylated hTfR2 behaved in manner different from that of the WT in response to holo-Tf treatment. Thus, the putative iron-sensing function of TfR2 could not be achieved in the absence of N-linked oligosaccharides. On the basis of our analyses, we conclude that unlike TfR1, N-linked glycosylation is dispensable for the cell surface expression and holo-Tf binding, but it is required for efficient intersubunit disulfide bond formation and holo-Tf-induced stabilization of TfR2.
Disorders in the regulation of iron homeostasis constitute an important class of human genetic diseases. Specifically, patients with the iron overload disorder, hereditary hemochromatosis (HH), have excess iron that accumulates in the body that can lead to liver cirrhosis, diabetes, arthritis, and heart failure.1 One form, HH type 3, is caused by mutations in the TfR2 gene.2,3 Transferrin receptor 2 (TfR2) is a member of the transferrin receptor-like family of proteins.4 It is strongly expressed in hepatocytes, which are the primary site of iron accumulation in HH. The precise role of TfR2 in establishing iron homeostasis is not known, although TfR2 has been postulated to sense the level of iron-loaded Tf (holo-Tf) in the blood. High levels of holo-Tf induce the transcription of hepcidin, a hormone secreted by hepatocytes, which limits the uptake of iron into the body as well as the recycling of iron.5,6 Determining the structural features of proteins is essential to understanding the basis of how they function. As an important aspect of this structural analysis, the role of glycosylation in the function of TfR2 was investigated.
Asn-linked glycosylation (N-linked) is a common modification of membrane proteins. It occurs cotranslationally with the transfer of a presynthesized high-mannose oligosaccharide chain from a lipid precursor to an Asn residue. The canonical motif for N-linked glycosylation is Asn-X-Ser/Thr and in some cases Asn-X-Cys, where X is any amino acid except Pro.7,8 Addition of oligosaccharide side chains adds branched and mobile polar domains, generating proteins of greater complexity.9 N-Linked oligosaccharides can serve as a signal for intracellular sorting and cell–cell interactions, participate in protein folding and trafficking, promote resistance to proteases, prevent protein aggregation, and/or maintain protein stability. Thus, removal of the consensus glycosylation sequence or inhibition of glycosylation often results in misfolding or aggregation.9 Aggregated proteins either are rapidly degraded or remain as large complexes that disrupt cell function and decrease cell viability.10
TfR2 is a paralog of the well-characterized Tf receptor, TfR1. The ectodomain of TfR2 is 55% identical and 65% similar to the ubiquitous TfR1. The N-linked oligosaccharides of TfR1 play an essential role in TfR1 folding and trafficking to the cell surface. Without N-linked oligosaccharides, TfR1 shows a reduced level of iron-bound transferrin (holo-Tf) binding and a decreased level of cell surface expression.11−13 Like TfR1, TfR2 is a type II membrane protein with a single-pass transmembrane domain and a short N-terminal cytoplasmic domain. The large extracellular region of human TfR2 (hTfR2) has four potential consensus sequences for N-linked glycosylation. However, neither the actual glycosylation sites nor the functional consequences of glycosylation of hTfR2 have been investigated. In this study, we first identified which glycosylation sites were utilized and then used site-directed mutagenesis to delete each individually and in combination. We found that N-linked oligosaccharides are not required for the binding of holo-Tf to hTfR2 or for the trafficking of hTfR2 to the cell surface, but they are required for efficient disulfide bond formation and holo-Tf-induced stabilization of hTfR2.
Experimental Procedures
Construction of Mutant TfR2 Plasmids
The four predicted N-linked glycosylation sites of hTfR2 are at Asn 240, 339, 540, and 754. The codons for each Asn (N) were mutated individually or in combination to Ala (A) (Table 1). Site-directed mutagenesis was performed by using the QuikChange Lightning Kit (Stratagene). In brief, 100 ng of double-stranded DNA template (pcDNA3-hTFR2 with a FLAG tag at the N-terminus) was mixed with the primers [forward and reverse primers, 125 ng each (Table 1 of the Supporting Information)], 10 mM dNTPs, 1× reaction buffer, and Pfu DNA polymerase. The mixture was amplified by polymerase chain reaction (PCR). Initially, the reaction mix was incubated at 95 °C for 30 s. The following cycles were used: denaturation for 30 s at 95 °C, annealing for 1 min at 55 °C, and extension synthesis at 68 °C for 7 min for 18 cycles. PCR products were digested with the DpnI enzyme to remove the parental strands. The digested DNA mixture was transformed into Escherichia coli XL1-blue cells by heat shock at 42 °C. Mutagenesis products were all verified by DNA sequencing.
Table 1. Mutants of hTfR2.
| residues | motifs | mutant name | |
|---|---|---|---|
| single-asparagine mutants | N240 | NVT | N240A |
| N339 | NQT | N339A | |
| N540 | NHS | N540A | |
| N554 | NPS | N554A | |
| N754 | NSS | N754A | |
| triple-asparagine mutants | N240/339/754 | NVT/NQT/NSS | 3-Mut |
| quadruple-asparagine mutants | N240/339/540/754 | NVT/NQT/NHS/NSS | 4-Mut |
Cell Culture, Transfection, and Stable Cell Lines
All cells were maintained in an incubator at 37 °C and 5% CO2. HEK 293 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Sigma) with 4.5 g/L glucose, 4 mM l-glutamine, 1 mM sodium pyruvate, and 10% fetal bovine serum (FBS, Atlanta Biologicals). HepG2 and Hep3B cells were maintained in Minimum Essential Medium (MEM, Sigma) with 1× nonessential amino acids (NEAA) and 10% FBS. Effectene transfection reagent (Qiagen) was used for transient transfection. Briefly, cells were seeded at 40% confluency in six-well plates or 100 mm culture dishes. Transfection began 24 h after the plates had been seeded with 0.4 or 2 μg of plasmid DNA, 3.2 or 16 μL of enhancer, and 10 or 50 μL of Effectene reagent. Transfection was conducted for 48 h before further analysis. For establishing stable cell lines, Hep3B cells were transfected on day 1 by using Fugene HD transfection reagent (Roche), and on day 3, cells were selected with MEM containing 600 μg/mL G418 (Geneticin). Selected clones were screened by Western blot analysis with the M2 anti-FLAG antibody. Postselection cells were maintained in MEM supplemented with 10% FBS, 1× NEAA, and 400 μg/mL G418.
Enzymatic Digestion and Transferrin Binding Assay
For deglycosylation, HEK 293 cells were transiently transfected with pcDNA3/hTfR2-FLAG and harvested 48 h after transfection. Solubilized cell lysates were used for enzymatic digestions. Five micrograms of lysate was incubated with PNGase F or Endo Hf (New England Biolabs) according to the manufacturer’s protocol before Western analysis. The same volume of buffer without the enzyme was added to the control samples. To examine the binding of iron-loaded Tf (holo-Tf) to hTfR2, Hep3B cells were transiently transfected with wild-type or mutant hTfR2. After 24 h, cells were solubilized in NETT buffer [150 mM NaCl, 5 mM EDTA, 10 mM Tris, 1% Triton X-100, and 1× Complete Mini protease inhibitor Mixture (Roche) (pH 7.4)], and the lysates were cleared by centrifugation at 10000g for 10 min. Cleared cell lysates were then incubated with 1 μM NHS-SS-biotin-labeled holo-Tf at 4 °C for 1 h prior to incubation with NeutrAvidin gel (Sigma) for an additional hour. Proteins bound to the NeutrAvidin gel were eluted with 50 mM DTT in water. Eluted fractions together with 10% of the input (lysates) were analyzed by Western blotting for hTfR2. To compare the binding affinity, Hep3B/WT hTfR2 or Hep3B/3-Mut hTfR2 stable cells were lysed in NETT buffer. Cleared cell lysates were then incubated with 10, 30, or 100 nM holo-Tf at 4 °C for 1 h prior to incubation with M2-FLAG gel (Sigma) for an additional hour. Bound proteins were eluted under native conditions by using 100 μg/mL 3× FLAG peptide (Sigma-Aldrich) in TBS.
Western Blot Analysis and Immunoprecipitation
Cells were washed with cold phosphate-buffered saline (PBS) twice and lysed in NETT buffer. Protein concentrations of the cell lysates were measured by using the RC DC Protein Assay (Bio-Rad). Samples were mixed with 1× Laemmli buffer and incubated for 30 min at 37 °C. Proteins were separated electrophoretically on an SDS–10% polyacrylamide gel, transferred to nitrocellulose, and incubated for 1 h in blocking buffer [5% nonfat dry milk in Tris-buffered saline with Tween 20 (TBST)]. Blots were incubated for 1 h at room temperature in blocking buffer containing mouse anti-FLAG, M2 (1:10000, Sigma), mouse anti-TfR2 (9F81C11, 1:5000), or mouse anti-TfR1 (Thermo Scientific, 1:5000). After four washes with TBST, blots were incubated with a 1:5000 goat anti-mouse secondary antibody conjugated to horseradish peroxidase (HRP, Millipore). To confirm equivalent loading, blots were stripped for 15 min in Restore PLUS Western Blot Stripping Buffer (Thermo Scientific), blocked for 1 h in blocking buffer, and reprobed with mouse anti-actin (Millipore, 1:10000) or rabbit anti-tubulin (Rockland, 1:5000) followed by HRP-conjugated goat anti-mouse (Millipore) or donkey anti-rabbit (GE Healthcare) secondary antibody. After two washes with TBST and TBS, bands were visualized by using enhanced chemiluminescence (SuperSignal West Pico, Thermo Scientific) and X-ray film. To immunoprecipitate FLAG-tagged hTfR2s, supernatants from Hep3B/WT hTfR2 or Hep3B/3-Mut hTfR2 stable cells were incubated for 1 h at 4 °C with M2-FLAG gel. The immunoprecipitated materials were washed three times with cold NETT buffer and eluted with TBS containing 100 μg/mL 3× FLAG peptide. Eluted fractions together with 20% of the input (lysates) were analyzed by Western blotting for hTfR2s and Tf with the addition of goat anti-Tf (1:10000) and HRP-conjugated donkey anti-goat (Millipore) antibodies.
Isolation of Plasma Membrane Proteins by Cell Surface Biotinylation
Wild-type hTfR2 and the nonglycosylated mutant (3-Mut) constructs were expressed in HEK 293 cells by transient transfection as described above. Twenty-four hours after transfection, the medium was removed, and the cells were washed twice with ice-cold PBS. Plasma membrane proteins were labeled by using the membrane-impermeant cleavable biotinylation reagent, NHS-SS-biotin (Thermo Scientific). The culture dishes were kept on ice, and all solutions were ice-cold. Briefly, each dish of cells was incubated with 10 mL of the NHS-SS-biotin solution (0.25 mg/mL in PBS) for 30 min while being gently shaken. After biotinylation, 500 μL of quenching solution was added to each dish to block the unreacted NHS-SS-biotin. Cells were collected and lysed in 500 μL of NETT lysis buffer with 1× protease inhibitors followed by centrifugation at 10000g for 10 min at 4 °C. The clarified supernatant was added to a spin column containing prewashed immobilized NeutrAvidin gel and incubated for 60 min at room temperature. After four washes, biotinylated samples were incubated with 50 mM DTT in 1× sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer for 60 min at room temperature to cleave the disulfide bond and release biotinylated proteins. Western analysis was used to detect cell surface TfR2 and Na+,K+-ATPase with mouse anti-FLAG, M2 antibody and mouse anti-Na+,K+-ATPase antibody (Santa Cruz, 1:2000), followed by HRP-conjugated secondary antibodies.
Immunofluorescence
Immunofluorescence microscopy was used to determine if WT hTfR2 and 3-Mut hTfR2 were on the plasma membrane. The constructs were expressed in HEK 293 cells. Cells seeded on poly-l-lysine (Sigma)-coated coverslips were washed twice with PBS+/+ (PBS with 1 mM MgCl2 and 0.1 mM CaCl2) and fixed with 2% paraformaldehyde for 15 min at room temperature. After being fixed, cells were washed three times with PBS and blocked with 1% bovine serum albumin (BSA) for 30 min. For TfR2 labeling, cells were incubated with mouse anti-TfR2 (9F81C11, 1:500) primary antibody for 30 min at room temperature. The secondary antibody was donkey anti-mouse IgG Alexa Fluor-594 (Invitrogen, 1:500). To compare the response of cells to holo-Tf, Hep3B cells were transiently transfected with WT or 3-Mut hTfR2 for 24 h, followed by treatment with 10 μM holo-Tf or PBS for 12 h. Fixed cells were washed three times with PBS, permeabilized with 0.1% saponin for 10 min, and washed three times with PBS before being blocked in 1% BSA for 30 min. TfR2 was detected with mouse anti-FLAG, M2 (Sigma, 1:500) primary antibody and Alexa Fluor-594-conjugated donkey anti-mouse IgG secondary antibody. Lysosomes were visualized with rabbit polyclonal anti-lysosome-associated membrane protein 1 (LAMP1, 1:500, Santa Cruz), followed by goat anti-rabbit IgG Alexa Fluor-488 secondary antibody (Invitrogen, 1:500). To stain the nuclei, cells were washed three times with PBS and incubated for 5 min with 10 μg/mL 4′,6-diamidino-2-phenylindole (DAPI). After three washes with PBS, coverslips were mounted on microscope slides with mounting medium (Invitrogen) and sealed with nail polish. Images were captured with a Zeiss LSM 710 confocal microscope with a 63× oil objective (Oregon Health & Science University core facility).
Statistical Analysis
Data were analyzed by an unpaired Student’s t test with GraphPad Prism, version 5. P values of <0.05 were considered to be statistically significant.
Results
Analysis of the Glycosylation Pattern of hTfR2 by Deglycosylation Enzymes
We initially wanted to determine the extent of high-mannose and complex oligosaccharides on TfR2 by digesting TfR2 with either of the endoglycosidases, endoglycosidase H (Endo Hf), which cleaves high-mannose and hybrid oligosaccharides, or peptide N-glycosidase F (PNGase F), which cleaves all forms of N-linked oligosaccharides. To accomplish this, a FLAG epitope was inserted at the N-terminus of TfR2, because we were not sure whether our monoclonal antibody to TfR2 would recognize deglycosylated TfR2 and for ease of isolation using anti-FLAG-coupled agarose. Previous studies demonstrated that the FLAG epitope does not interfere with proper functions of TfR2 in terms of Tf binding and iron delivery.14,15 To analyze the glycosylation status and to determine the extent of high-mannose and complex N-linked oligosaccharides in hTfR2, HEK 293 cells were transiently transfected with N-terminally FLAG-tagged hTfR2 for 24 h. Cell lysates were incubated with Endo Hf or PNGase F. Cell lysates from both HEK 293 cells transiently expressing hTfR2 (Figure 1A) and HepG2 cells that endogenously express hTfR2 were analyzed (Figure 1B). The upper hTfR2 band was completely shifted to a nonglycosylated form after treatment with PNGase F, indicating that the mature form of hTfR2 contains N-linked oligosaccharides as previously shown in SK-Hep1 cells.14 Only a small amount of hTfR2 is sensitive to Endo Hf digestion, suggesting that majority of TfR2 is composed of complex oligosaccharides in both cell types. Unlike that of TfR1, which contains two complex oligosaccharides and one high-mannose oligosaccharide,16 digestion of hTfR2 with Endo Hf showed no intermediate migrating bands, indicating that the majority of TfR2 contains all complex oligosaccharides with a small amount of TfR2 that has only high-mannose oligosaccharides.16
Figure 1.

Enzymatic deglycosylation of hTfR2. (A) HEK 293 cells were transiently transfected with pcDNA3/hTfR2-FLAG. Cells were harvested 48 h after transfection. (B) HepG2 cells were harvested, and lysates were used for enzymatic digestion. Five micrograms of protein was incubated with or without PNGase F or Endo Hf before Western analysis. Overexpression of hTfR2 in HEK 293 cells results in two bands by Western blotting. The upper band shifts down with both PNGase F and Endo Hf treatments. Endogenous TfR2 in HepG2 cells is also glycosylated as the TfR2 band shifted down with deglycosylation enzyme treatments.
Identification of the Utilized N-Linked Glycosylation Sites in hTfR2
We employed both bioinformatic prediction and experimental validation approaches to examine the glycosylation sites on hTfR2. NetNGlyc 1.0 and NetOGlyc 3.1 were initially used to predict the existence of N-linked and O-linked glycosylation sites.17 Human TfR2 is predicted to be glycosylated at four potential Asn sites at amino acids 240, 339, 540, and 754. It has no predicted O-linked glycosylation sites. Asn 554 served as a negative control; as part of a NPS motif, it is predicted not to be utilized for glycosylation (Figure 2A). To identify which predicted Asn is indeed glycosylated, the four potential Asn (N) residues as well as Asn 554 were replaced with Ala (A) individually. The effect of these mutations on the electrophoretic mobility of hTfR2 was examined by transiently expressing the wild type (WT) or mutants in HEK 293 cells, followed by Western analysis of the collected cell lysates. We observed that single replacements of Asn with Ala at positions 240, 339, and 754 all led to decreases in the molecular mass of the hTfR2 band, suggesting that each site is glycosylated, whereas mutation of Asn540 did not affect the migration of TfR2 N540A via SDS–PAGE compared with the WT or TfR2 N554A (Figure 2B). With deletion of all four N-linked glycosylation sites (240, 339, 540, and 754) or three N-linked glycosylation sites (240, 339, and 754), migration of the triple and quadruple TfR2 mutations on SDS–PAGE gels was the same, indicating that Asn 240, Asn 339, and Asn 754 are glycosylated whereas Asn 540 is not (Figure 2C). We further confirmed that Asn 540 is not utilized for glycosylation by digesting the WT and 3-Mut (with Asn 240, Asn 339, and Asn 754 mutated to Ala) with PNGase F. The results indicate that there is no additional molecular mass shift of 3-Mut after PNGase F treatment and that 3-Mut migrates like WT hTfR2 digested with PNGase F on SDS–PAGE gels (Figure 2D).
Figure 2.
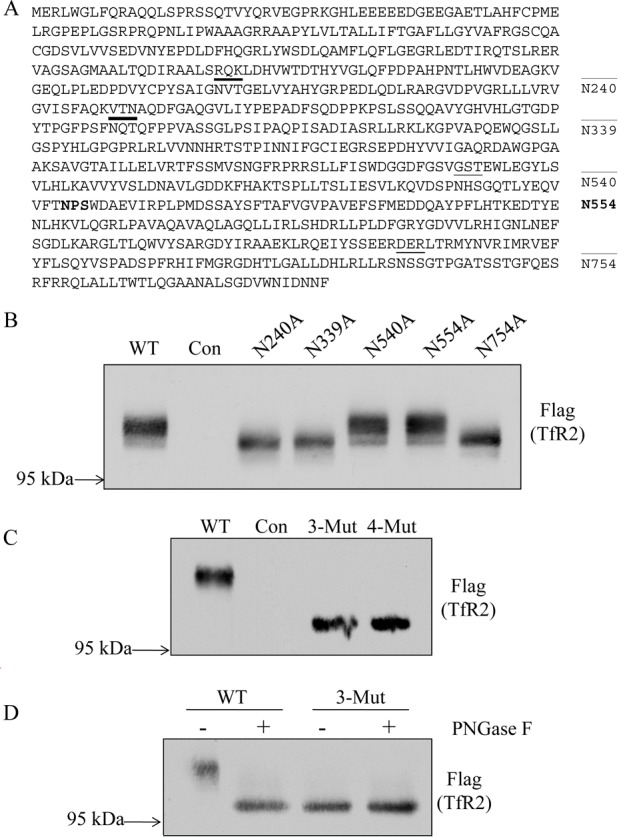
Identification of N-linked glycosylation sites in hTfR2. (A) Schematic representation of the N-linked glycosylation sites on hTfR2 protein sequence. Asn 240, 339, 540, and 754 are predicted to be glycosylated (underlined). Asn 554 (bold) has an NPS/T motif and was used as a negative control because this Asn could not be glycosylated. (B) Western blot analysis of cell lysates from HEK 293 cells transiently transfected with empty vector (pcDNA3, Con) or hTfR2-FLAG expression vectors encoding either the wild type (WT) or N-glycosylation site mutants (N240A, N339A, N540A, and N754A). Positions 240, 339, and 754 are identified as being glycosylated, but position 540 is not. N554A was used as a negative control. (C) Western blot analysis of cell lysates from HEK 293 cells transiently transfected with empty vector (pcDNA3, Con), WT hTfR2 (WT), the hTfR2 nonglycosylated triple mutant (N240/339/754A, 3-Mut), or the quadruple mutant (N240/339/540/754A, 4-Mut). (D) HEK 293 cells transfected with empty vector (pcDNA3, Con), WT hTfR2 (WT), or the hTfR2 nonglycosylated mutant (N240/339/754A, 3-Mut) were harvested, and cell lysates were incubated with or without PNGase F before Western blotting. The samples were electrophoresed on a 12 cm 10% polyacrylamide gel for 24 h to ensure greater separation, transferred to nitrocellulose, and probed with anti-Flag antibody for TfR2. The data represent three independent experiments.
N-Linked Glycosylation Does Not Affect Plasma Membrane Localization of hTfR2
The ability of the glycosylation mutant TfR2 to fold correctly and move to the plasma membrane was detected by cell surface biotinylation and by fluorescence microscopy. Previous studies indicated that WT TfR2 is both present on the plasma membrane and in endosomal compartments.18,19 To determine the role of N-linked glycosylation on the localization of hTfR2, HEK 293 cells were transiently transfected with wild-type hTfR2 (WT) or the triple mutant (3-Mut, which has all three glycosylated Asn residues mutated to Ala). Total cell lysates and cell surface proteins biotinylated with a membrane-impermeable reagent, NHS-SS-biotin, were analyzed on Western blots. Similar to WT hTfR2, a strong signal for the nonglycosylated mutant could be detected on the cell surface (Figure 3A). The cell surface localization of WT hTfR2 and 3-Mut hTfR2 was also examined by confocal microscopy using the antibody against the ectodomain of TfR2 under nonpermeabilized conditions. Both WT and 3-Mut hTfR2 could be detected on the plasma membrane (Figure 3B). These results indicate that N-linked glycosylation of hTfR2 is not required for the efficient movement of the protein to the cell surface.
Figure 3.
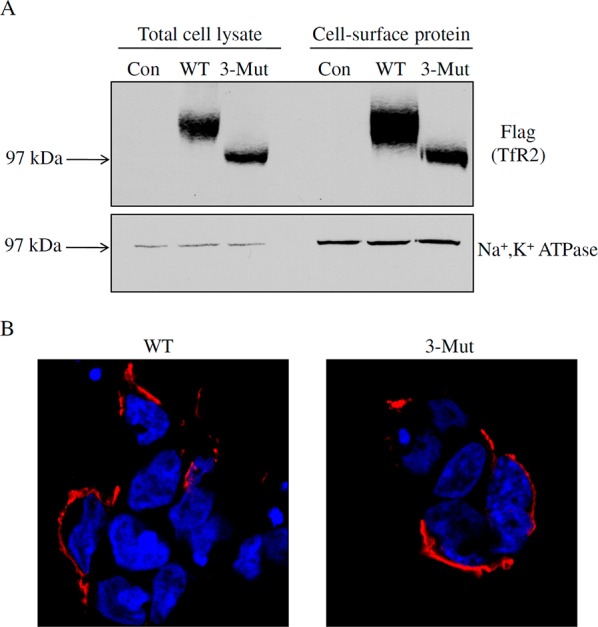
N-Linked glycosylation does not affect plasma membrane localization of hTfR2. (A) HEK 293 cells were transiently transfected with empty pcDNA3 vector (Con), wild-type hTfR2-FLAG (WT), or its mutant with all three glycosylated Asn residues replaced with alanines (3-Mut). After 24 h, total cell lysates were harvested by using NETT cell lysis buffer, while cell surface proteins were labeled with cell membrane-impermeable NHS-SS-biotin. Samples were analyzed by Western blotting for TfR2. After being stripped, blots were reprobed for Na+,K+-ATPase as a marker for plasma membrane proteins. (B) For immunofluorescence, HEK 293 cells were transiently transfected with WT or 3-Mut hTfR2-FLAG. Twenty-four hours after transfection, WT or 3-Mut TfR2 was detected by using mouse anti-TfR2 antibody followed by Alexa Fluor-594-conjugated secondary antibody. Images show that both WT hTfR2 and the nonglycosylated form of hTfR2 (3-Mut) were detected at the plasma membrane. DAPI was used to stain nuclei. Data are representative of one of three independent experiments.
N-Linked Glycosylation Is Required for Holo-Tf-Induced Stabilization of hTfR2
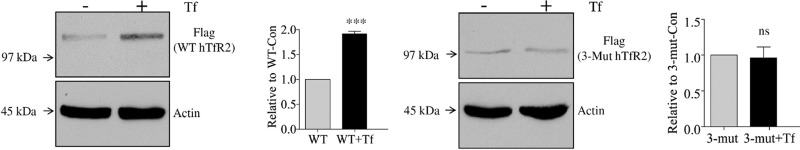
In the presence of an elevated level of holo-Tf, TfR2 becomes more stable.5,6 Presumably, this stabilization increases the level of signaling by TfR2. However, the binding of holo-Tf to TfR2 is not adequate for TfR2 to be stabilized, as supported by observations that only hepatoma or primary hepatic cells respond to the Tf stimulus.6,18,20−22 To test whether the N-linked glycosylation is required, Hep3B cells, a hepatoma cell line, were transiently transfected with WT or 3-Mut hTfR2. Hep3B cells do not express detectable TfR2. Cells were then treated with PBS (Con) or 30 μM holo-Tf (+Tf) for 12 h before Western blotting analysis. The level of WT hTfR2 approximately doubled after holo-Tf treatment, whereas the level of nonglycosylated hTfR2 did not change (Figure 4A–D). Thus, N-linked glycosylation is required for holo-Tf-induced stabilization of hTfR2. To determine the role of each glycosylation site in holo-Tf-induced stabilization, Hep3B cells were first transfected with single-Asn hTfR2 mutants (N240A, N339A, and N754A) and then treated with holo-Tf. Western analysis indicated that TfR2 expressing a single mutation in N-linked glycosylation could be stabilized by holo-Tf, showing that the ablation of a single glycosylation site does not affect the holo-Tf-induced stabilization of hTfR2 (Figure 4E). The observed effect was not due to differences in transient transfection efficiency. Hep3B cells stably transfected with WT or 3-Mut hTfR2 (Figure 4F,G) showed similar results. These findings indicate that N-linked oligosaccharides are required for holo-Tf-induced stabilization of hTfR2 and that no single N-linked site is responsible for the loss of responsiveness to Tf. Only removal of multiple glycosylation sites has functional consequences.
Figure 4.

N-Linked glycosylation is required for holo-Tf-induced stabilization of hTfR2. Hep3B cells were transiently transfected with WT or 3-Mut hTfR2 in 100 mm dishes. Twenty-four hours later, cells from each transfection were split into a six-well plate and cultured for an additional 1 day. Then cells were treated with PBS (Con) or 10 μM holo-Tf (+Tf) for 12 h before being harvested and analyzed by Western blotting. (A and C) Western blotting results indicate that WT hTfR2 could be stabilized by holo-Tf, while 3-Mut hTfR2, which lacks N-linked glycosylation, could not be stabilized by holo-Tf. (B and D) Quantification of band densities (***, p < 0.0001; ns, not statistically significant). (E) Hep3B cells were transiently transfected with single-Asn mutants (N240A, N339A, N540A, and N754A) in 60 mm dishes. Twenty-four hours later, cells from each transfection were split into a six-well plate and cultured for an additional 1 day. Cells were then treated with PBS or 10 μM holo-Tf (+Tf) for 12 h before being harvested and analyzed by Western blotting. (F and G) Hep3B/WT hTfR2 or Hep3B/3-Mut hTfR2 stable cells were cultured for 24 h, and then cells were treated with PBS (Con) or 10 μM holo-Tf (+Tf) for 12 h before Western analysis. The data represent three independent experiments.
N-Linked Glycosylation Does Not Affect the Binding of Holo-Tf to hTfR2
N-Linked glycosylation affects the ligand affinity of some receptors.23−25 To examine whether glycosylation of hTfR2 is required for its binding to holo-Tf, Hep3B cells were transiently transfected with empty pcDNA3 vector (Con), WT hTfR2 (WT), or the nonglycosylated triple mutant (3-Mut). Biotin-labeled holo-Tf was capable of pulling down both WT and nonglycosylated forms of hTfR2, indicating that hTfR2 does not need N-linked oligosaccharides for holo-Tf binding (Figure 5A,B). The lack of biotinylated actin indicates that the cells remained intact during the labeling. To compare the relative binding affinities of holo-Tf for hTfR2, cell lysates from Hep3B cells that stably express WT or 3-Mut hTfR2 were used. Incubation of cell lysates with 10–100 nM holo-Tf, followed by isolation of hTfR2s with FLAG resin, demonstrated that the affinity of nonglycosylated hTfR2 for holo-Tf was not lower than that of WT hTfR2 (Figure 5C). In fact, we observed a slightly higher binding affinity of the unglycosylated TfR2 for holo-Tf. This may be due to steric hindrance induced by glycosylation, because other studies have also observed that glycosylation can decrease the affinity of the receptor for its ligand.26,27 Therefore, the loss of holo-Tf sensitivity associated with nonglycosylated hTfR2 is not due to defects in ligand binding.
Figure 5.

N-Linked glycosylation is not required for Tf binding of hTfR2. (A and B) Hep3B cells were transiently transfected with WT or 3-Mut hTfR2. After 24 h, total cell lysates were harvested by using NETT cell lysis buffer, and then cell lysates were incubated with 1 μM biotin-labeled holo-Tf at 4 °C for 1 h. The lysates were incubated with NeutrAvidin gel for an additional 1 h and eluted with 50 mM DTT in water. WT or 3-mut hTfR2-transfected cell lysates without giving biotin-labeled holo-Tf were used as a control to eliminate the possibility that TfR2 itself binds to the NeutrAvidin gel. Bound fractions together with 10% of the input (lysates) were analyzed by Western blotting for TfR2. After being stripped, blots were reprobed for actin to demonstrate the absence of proteins that could bind nonspecifically to the NeutrAvidin gel. (C) Hep3B/WT hTfR2 or Hep3B/3-Mut hTfR2 stable cells were lysed in NETT buffer. Equal amounts of lysates were incubated with 10, 30, or 100 nM holo-Tf for 1 h and then incubated with FLAG gel for an additional 1 h. Bound proteins were eluted from the gel by incubation with 100 μg/mL 3× FLAG peptide in TBS. Eluted fractions together with 20% of the input (lysates) were analyzed by Western blotting for hTfR2. After being stripped, blots were reprobed for Tf and tubulin. Data are representative of one of three independent experiments.
N-Linked Glycosylation Affects Dimerization of hTfR2
Many transmembrane receptors function as dimers.28,29 Dimerization of TfR2 was described more than 10 years ago by the ability of TfR2 to form intersubunit disulfide bonds.15 However, the possibility that N-glycosylation might regulate receptor dimerization has not yet been explored for hTfR2. Cell lysates from the cells stably transfected with the FLAG-tagged WT and 3-Mut hTfR2 were subjected to nonreducing SDS gel electrophoresis to detect intersubunit disulfide bonds. An ∼200 kDa protein was detected for both WT and 3-Mut hTfR2, indicating the formation of intersubunit disulfide bonds. In 3-Mut hTfR2-transfected cells, a band with a similar intensity around 97 kDa was seen under nonreducing conditions, indicating that 3-Mut hTfR2 was inefficient in forming intersubunit disulfide bonds (Figure 6A,B). To test whether Tf stabilized the fraction of unglycosylated hTfR2 that formed intersubunit disulfide bonds, cells were incubated with holo-Tf before being subjected to solubilization and nonreducing SDS–PAGE. Without N-linked glycosylation, neither the hTfR2 dimer nor the monomer could be stabilized by holo-Tf (Figure 6C), even though both WT and 3-Mut hTfR2 dimer forms were present at the cell surface (Figure 6D). The lack of intersubunit disulfide bond formation of the unglycosylated hTfR2 therefore does not account for its lack of holo-Tf sensitivity.
Figure 6.

N-Linked glycosylation affects dimerization of hTfR2. (A) Hep3B/WT hTfR2 or Hep3B/3-Mut hTfR2 stable cells were harvested and solubilized with NETT lysis buffer, and protein concentrations were measured before Western analysis. (B) Band intensities were expressed as percentages of total levels (D, dimer; M, monomer). (C) Hep3B/3-Mut hTfR2 stable cells were treated with PBS or 10 μM holo-Tf (+Tf) for 12 h before being harvested. Equal amounts of proteins were incubated with sample loading buffer, which does not contain dithiothreitol (DTT), before being analyzed by Western blotting. (D) Cell surface proteins from Hep3B/WT hTfR2 or Hep3B/3-Mut hTfR2 stable cells were labeled with cell membrane-impermeable NHS-SS-biotin at 4 °C for 1 h, and then cell lysates were incubated with FLAG beads for an additional 1 h. Bound proteins were eluted with 100 μg/mL 3× FLAG peptide in TBS. Half of the eluted fraction together with 10% of the input (lysates) was analyzed by Western blotting for hTfR2s, and the other half was probed with Avidin-HRP to detect cell surface hTfR2s under nonreducing conditions. The data represent three independent experiments.
N-Linked Glycosylation Affects Tf-Induced Redirection of hTfR2 Subcellular Localization
To gain further mechanistic insight, we performed immunofluorescence experiments. Holo-Tf delivered through TfR2 was initially observed at the plasma membrane and in early endosomes, and then at a later stage in multivesicular bodies (MVB),20 suggesting that, unlike TfR1, which recycles back to the plasma membrane after internalization, TfR2 could be targeted to lysosomes for degradation. This hypothesis is supported by the observation that TfR2 moves through endocytic, recycling, and degradative pathways, as TfR2 partially colocalized with early, recycling, and late endosomal markers. Indeed, other studies show that holo-Tf stabilizes TfR2 by redirecting it from a degradative pathway to a recycling pathway, because when treated with holo-Tf, less TfR2 colocalizes with a lysosomal marker, LAMP1.18,30 Consistent with previous results, immunofluorescence data from the study show less colocalization of WT hTfR2 with LAMP1 after holo-Tf incubation, indicating that less WT hTfR2 exploits the degradation pathway with holo-Tf stimulation (Figure 7A). In contrast, incubation of Tf with the 3-Mut hTfR2-transfected cells does not affect the colocalization of this nonglycosylated hTfR2 with LAMP1, suggesting that the unglycosylated hTfR2 is still targeted to lysosomes for degradation upon holo-Tf treatment (Figure 7B).
Figure 7.
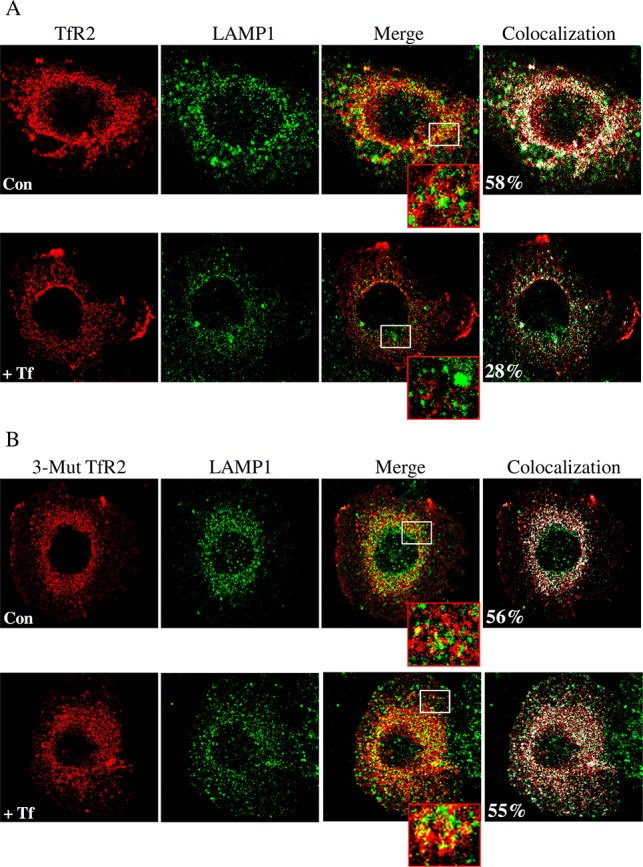
Subcellular distribution of hTfR2 and 3-Mut hTfR2 in the presence and absence of Tf. (A and B) Hep3B cells were transiently transfected with WT or 3-Mut hTfR2 in 100 mm dishes. After 24 h, each set of transfected cells was split into a six-well plate containing poly-l-lysine-coated coverslips and cultured for 1 day. Cells were then treated with PBS (Con) or 10 μM holo-Tf (+Tf) for 12 h before being fixed. Fixed and permeabilized cells were analyzed for TfR2 by using the mouse anti-FLAG antibody followed by the Alexa Fluor-594-conjugated secondary antibody (red). Colocalization of WT or 3-Mut hTfR2 with LAMP1 was assessed by using rabbit anti-LAMP1 primary antibodies followed by the Alexa Fluor-488-labeled secondary antibody (green). Merged images show the colocalization (yellow) of either WT or hTfR2 with LAMP1. The lower image in each row shows an enlarged detail of the white boxed area of each merged picture. All images were obtained by using a Zeiss laser scanning LSM 710 confocal microscope. Colocalization (designated by white) was determined by using the colocalization tool provided with the Zeiss software.
Discussion
TfR2 is strongly expressed in hepatocytes, the primary site of iron accumulation in hemochromatosis. A variety of mutations in the TfR2 gene result in type 3 HH, which is a genetic iron overload disease. The function of TfR2 in iron metabolism is not clear. The majority of hepatic Tf-mediated iron uptake under normal conditions occurs through TfR1.31 TfR1 expression is downregulated with increased cellular iron levels.32 As a result, TfR1 expression in the liver is not detectable in HH patients.33 In contrast, TfR2 is stabilized by holo-Tf, and its level of expression increases in the liver of hemojuvelin knockout mice, a mouse model of juvenile hemochromatosis (unpublished data). In addition, TfR2 is capable of binding and internalizing holo-Tf in vitro.15,34 However, cellular iron uptake may not be the major function of TfR2, because both human and mouse mutations in TfR2 as well as the mouse knockout lead to increased rather than decreased levels of liver iron deposition.2,35 Moreover, in mice lacking functional TfR2, the rate of Tf-mediated iron uptake by hepatocytes was only slightly decreased compared to that of wild-type mice with a similar amount of iron loading.31
TfR2 is more likely to be a modulator of hepcidin expression rather than playing a role in cellular iron uptake.36 The iron regulatory hormone, hepcidin, is synthesized mainly by hepatocytes. Hepcidin levels in mice lacking functional Tfr2 remain lower than those of wild-type mice with comparable iron loads, resulting in constantly increased levels of absorption of iron from the intestine and elevated levels of release of iron from macrophages.37,38 How TfR2 senses iron levels and regulates hepcidin expression remains elusive. In contrast to TfR1, TfR2 does not vary in response to changes in intracellular iron levels. TfR2 levels rise in response to an increased level of Tf saturation, which is usually a consequence of an increased level of iron in the body. This response is proposed to increase the rate of hepcidin synthesis, which in turn decreases further the rate of iron uptake.
Since the discovery of TfR2 in 1999, much effort has been focused on examining the proteins that interact with it. Few studies have investigated the structural features of TfR2 involved in establishing its proper function. Co- and post-translational modifications are often essential for protein function. These modifications regulate the trafficking of proteins within the cell, interaction with other cellular molecules such as proteins, lipids, and nucleic acids, and protein stability. Glycosylation of membrane proteins is by far the most abundant modification. More than half of the proteins in humans possess oligosaccharides.39,40 Glycoproteins are components of extracellular matrices and cellular surfaces where the carbohydrate side chains are implicated in cell–cell and cell–matrix recognition events.41 N-Linked oligosaccharides can affect protein trafficking and/or proper function.42,43 For example, the lutropin receptor and the norepinephrine transporter require N-linked oligosaccharides for cell surface localization, but not for the high-affinity ligand binding or substrate transport activity.44,45 In some cases, N-linked oligosaccharides appear to be dispensable for proper cell surface localization. The organic solute transporter subunit α and the breast cancer resistance protein can still travel to the plasma membrane and be fully functional in the absence of N-linked oligosaccharides.46,47
Potential N-linked glycosylation sites can be identified by the presence of the Asn-X-Ser/Thr consensus sequence; however, not all such motifs are glycosylated, and the glycosylated sites for some proteins vary by tissue.48 In the study presented here, the gel electrophoresis data in combination with PNGase F treatment indicate that only three of the four potential N-linked glycosylation sites in the ectodomain of hTfR2 are glycosylated. We further showed that mutation of all three glycosylation sites does not block the plasma membrane trafficking or the binding of holo-Tf to hTfR2 in contrast to TfR1 where elimination of the N-linked glycosylation sites prevented the movement of TfR1 to the cell surface or secretion of a truncated version of TfR1.12,49 However, removing all of the N-linked oligosaccharides from hTfR2 has functional consequences. Without N-linked glycosylation, TfR2 is no longer sensitive to holo-Tf-induced stabilization and has a decreased level of intersubunit disulfide bond formation. The latter could indicate a weaker interaction along the TfR2 dimer interface.
Holo-Tf binds to TfR2, but binding alone is not sufficient to stabilize TfR2. TfR2 is stabilized by holo-Tf in a tissue- and cell-type-specific manner. In hepatic cell lines (HepG2, Huh7, and Hep3B cells transfected with TfR2), holo-Tf does stabilize TfR2.5,6,21 Consistent with the in vitro results, mice with higher levels of Tf saturation also have increased hepatic TfR2 levels.6 However, in K562 cells, an erythroleukemia cell line, which endogenously expresses TfR2 or non-hepatic cell lines transfected with TfR2, TfR2 cannot be stabilized by holo-Tf.1 These results suggest that the stabilization of TfR2 by Tf is hepatocyte-specific and may involve hepatocyte-specific protein interactions. The mechanism by which an elevated level of holo-Tf stabilizes TfR2 is not well understood except that the cytoplasmic domain of TfR2 is responsible for its stabilization by holo-Tf.21 In the study presented here, our data indicate that N-linked oligosaccharides are essential for the stabilization of hTfR2 by holo-Tf. We found that mutation of individual glycosylation sites at N240, N339, or N754 does not affect holo-Tf sensitivity. However, the steady state levels of cells expressing 3-Mut hTfR2, which lacks N-linked glycosylation, do not increase with holo-Tf treatment. A change in the subcellular localization of proteins usually involves alterations in protein–protein, protein–lipid, or protein–carbohydrate interactions.50 The removal of N-linked oligosaccharides could disturb the interaction between hTfR2 and a carbohydrate binding protein, which is required for conveying hTfR2 from a degradative to a recycling pathway after holo-Tf treatment. Further research is needed to test this hypothesis.
TfR2 is proposed to be an iron sensor, and the response of TfR2 to holo-Tf could play a role in signaling to increase the level of transcription of hepcidin.5,6,21 Loss of functional TfR2 leads to a decreased level of hepcidin and iron overload in the body.2,35,38 A TfR2 lacking N-linked glycosylation may fail to sense increased holo-Tf levels, providing a possible explanation for iron overload seen in patients with genetic disorders of glycosylation. Congenital disorders of glycosylation are rare, but the biological processes involved are widespread. Defects could involve the activation, appearance, and transport of carbohydrate precursors, the glycosidases and glycosyltransferases for synthesizing glycans, the proteins that control the glycosylation machinery.48 Hovinga et al. reported two cases of severe hepatic iron overload in patients with congenital dyserythropoietic anemia (CDA) (a disease with N-linked glycosylation synthesis defects).51 In fact, hepatic iron overload is a frequent complication in CDA patients.52 Abnormal glycosylation of proteins involved in iron homeostasis is likely to be a contributing factor for the excess iron loading in the liver.
Our results highlight the importance of N-linked oligosaccharides in the function of hTfR2 protein. Studies have shown that N-linked glycosylation can generate various tissue-specific glycoforms, contributing to the structural or functional diversity of certain proteins.53,54 Whether different tissues generate different glycoforms of TfR2 is unknown. In different tissues, whether varied numbers of N-glycosylation sites are used or N-linked oligosaccharides are processed differently for TfR2 remains to be investigated.
Acknowledgments
We thank Dr. An-Sheng Zhang and Dr. Anne B. Mason for critical reading of the manuscript and helpful comments.
Supporting Information Available
Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Institutes of Health Grants DK072166 and DK054488 (C.A.E.), an Oregon Health & Science University Tartar Trust Award (N.Z.), and a Collins Medical Trust Award (N.Z.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ganz T.; Nemeth E. (2011) Hepcidin and disorders of iron metabolism. Annu. Rev. Med. 62, 347–360. [DOI] [PubMed] [Google Scholar]
- Camaschella C.; Roetto A.; Cali A.; De Gobbi M.; Garozzo G.; Carella M.; Majorano N.; Totaro A.; Gasparini P. (2000) The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 25, 14–15. [DOI] [PubMed] [Google Scholar]
- Roetto A.; Totaro A.; Piperno A.; Piga A.; Longo F.; Garozzo G.; Cali A.; De Gobbi M.; Gasparini P.; Camaschella C. (2001) New mutations inactivating transferrin receptor 2 in hemochromatosis type 3. Blood 97, 2555–2560. [DOI] [PubMed] [Google Scholar]
- Stimpson H. E.; Lewis M. J.; Pelham H. R. (2006) Transferrin receptor-like proteins control the degradation of a yeast metal transporter. EMBO J. 25, 662–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. B.; Enns C. A. (2004) Diferric transferrin regulates transferrin receptor 2 protein stability. Blood 104, 4287–4293. [DOI] [PubMed] [Google Scholar]
- Robb A.; Wessling-Resnick M. (2004) Regulation of transferrin receptor 2 protein levels by transferrin. Blood 104, 4294–4299. [DOI] [PubMed] [Google Scholar]
- Miletich J. P.; Broze G. J. Jr. (1990) Beta protein C is not glycosylated at asparagine 329. The rate of translation may influence the frequency of usage at asparagine-X-cysteine sites. J. Biol. Chem. 265, 11397–11404. [PubMed] [Google Scholar]
- Mellquist J. L.; Kasturi L.; Spitalnik S. L.; Shakin-Eshleman S. H. (1998) The amino acid following an asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry 37, 6833–6837. [DOI] [PubMed] [Google Scholar]
- Helenius A.; Aebi M. (2001) Intracellular functions of N-linked glycans. Science 291, 2364–2369. [DOI] [PubMed] [Google Scholar]
- Helenius A. (1994) How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol. Biol. Cell 5, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A. M.; Enns C. A. (1991) A mutated transferrin receptor lacking asparagine-linked glycosylation sites shows reduced functionality and an association with binding immunoglobulin protein. J. Biol. Chem. 266, 17648–17654. [PubMed] [Google Scholar]
- Byrne S. L.; Leverence R.; Klein J. S.; Giannetti A. M.; Smith V. C.; MacGillivray R. T.; Kaltashov I. A.; Mason A. B. (2006) Effect of glycosylation on the function of a soluble, recombinant form of the transferrin receptor. Biochemistry (Moscow, Russ. Fed.) 45, 6663–6673. [DOI] [PubMed] [Google Scholar]
- Yang B.; Hoe M. H.; Black P.; Hunt R. C. (1993) Role of oligosaccharides in the processing and function of human transferrin receptors. Effect of the loss of the three N-glycosyl oligosaccharides individually or together. J. Biol. Chem. 268, 7435–7441. [PubMed] [Google Scholar]
- Kawabata H.; Germain R. S.; Vuong P. T.; Nakamaki T.; Said J. W.; Koeffler H. P. (2000) Transferrin receptor 2-α supports cell growth both in iron-chelated cultured cells and in vivo. J. Biol. Chem. 275, 16618–16625. [DOI] [PubMed] [Google Scholar]
- Kawabata H.; Yang R.; Hirama T.; Vuong P. T.; Kawano S.; Gombart A. F.; Koeffler H. P. (1999) Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J. Biol. Chem. 274, 20826–20832. [DOI] [PubMed] [Google Scholar]
- Hayes G. R.; Williams A.; Costello C. E.; Enns C. A.; Lucas J. J. (1995) The critical glycosylation site of human transferrin receptor contains a high-mannose oligosaccharide. Glycobiology 5, 227–232. [DOI] [PubMed] [Google Scholar]
- Julenius K.; Molgaard A.; Gupta R.; Brunak S. (2005) Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 15, 153–164. [DOI] [PubMed] [Google Scholar]
- Johnson M. B.; Chen J.; Murchison N.; Green F. A.; Enns C. A. (2007) Transferrin receptor 2: Evidence for ligand-induced stabilization and redirection to a recycling pathway. Mol. Biol. Cell 18, 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace D. F.; Summerville L.; Crampton E. M.; Subramaniam V. N. (2008) Defective trafficking and localization of mutated transferrin receptor 2: Implications for type 3 hereditary hemochromatosis. Am. J. Physiol. 294, C383–C390. [DOI] [PubMed] [Google Scholar]
- Robb A. D.; Ericsson M.; Wessling-Resnick M. (2004) Transferrin receptor 2 mediates a biphasic pattern of transferrin uptake associated with ligand delivery to multivesicular bodies. Am. J. Physiol. 287, C1769–C1775. [DOI] [PubMed] [Google Scholar]
- Chen J.; Enns C. A. (2007) The cytoplasmic domain of transferrin receptor 2 dictates its stability and response to holo-transferrin in Hep3B cells. J. Biol. Chem. 282, 6201–6209. [DOI] [PubMed] [Google Scholar]
- Gao J.; Chen J.; Kramer M.; Tsukamoto H.; Zhang A. S.; Enns C. A. (2009) Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 9, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goke R.; Just R.; Lankat-Buttgereit B.; Goke B. (1994) Glycosylation of the GLP-1 receptor is a prerequisite for regular receptor function. Peptides 15, 675–681. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Austin S. C.; Smyth E. M. (2001) Glycosylation of the human prostacyclin receptor: Role in ligand binding and signal transduction. Mol. Pharmacol. 60, 480–487. [PubMed] [Google Scholar]
- Park S. J.; Kleffmann T.; Hessian P. A. (2011) The G82S polymorphism promotes glycosylation of the receptor for advanced glycation end products (RAGE) at asparagine 81: Comparison of wild-type rage with the G82S polymorphic variant. J. Biol. Chem. 286, 21384–21392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Sun S.; Li Z. (2012) Two glycosylation sites in H5N1 influenza virus hemagglutinin that affect binding preference by computer-based analysis. PLoS One 7, e38794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D.; Shi X.; Arledge K. C.; Song D.; Jiang L.; Fu L.; Gong X.; Zhang S.; Wang X.; Zhang L. (2012) A single residue within the V5 region of HIV-1 envelope facilitates viral escape from the broadly neutralizing monoclonal antibody VRC01. J. Biol. Chem. 287, 43170–43179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin C. H. (1995) Dimerization of cell surface receptors in signal transduction. Cell 80, 213–223. [DOI] [PubMed] [Google Scholar]
- Bain D. L.; Heneghan A. F.; Connaghan-Jones K. D.; Miura M. T. (2007) Nuclear receptor structure: Implications for function. Annu. Rev. Physiol. 69, 201–220. [DOI] [PubMed] [Google Scholar]
- Chen J.; Wang J.; Meyers K. R.; Enns C. A. (2009) Transferrin-directed internalization and cycling of transferrin receptor 2. Traffic 10, 1488–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua A. C.; Delima R. D.; Morgan E. H.; Herbison C. E.; Tirnitz-Parker J. E.; Graham R. M.; Fleming R. E.; Britton R. S.; Bacon B. R.; Olynyk J. K.; Trinder D. (2010) Iron uptake from plasma transferrin by a transferrin receptor 2 mutant mouse model of haemochromatosis. J. Hepatol. 52, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert N.; Lescoat G.; Sciot R.; Moirand R.; Jego P.; Leroyer P.; Brissot P. (1993) Regulation of ferritin and transferrin receptor expression by iron in human hepatocyte cultures. J. Hepatol. 18, 301–312. [DOI] [PubMed] [Google Scholar]
- Fleming R. E.; Migas M. C.; Holden C. C.; Waheed A.; Britton R. S.; Tomatsu S.; Bacon B. R.; Sly W. S. (2000) Transferrin receptor 2: Continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc. Natl. Acad. Sci. U.S.A. 97, 2214–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R. M.; Reutens G. M.; Herbison C. E.; Delima R. D.; Chua A. C.; Olynyk J. K.; Trinder D. (2008) Transferrin receptor 2 mediates uptake of transferrin-bound and non-transferrin-bound iron. J. Hepatol. 48, 327–334. [DOI] [PubMed] [Google Scholar]
- Fleming R. E.; Ahmann J. R.; Migas M. C.; Waheed A.; Koeffler H. P.; Kawabata H.; Britton R. S.; Bacon B. R.; Sly W. S. (2002) Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc. Natl. Acad. Sci. U.S.A. 99, 10653–10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N.; Enns C. A. (2012) Iron transport machinery of human cells: Players and their interactions. Curr. Top. Membr. 69, 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace D. F.; Summerville L.; Subramaniam V. N. (2007) Targeted disruption of the hepatic transferrin receptor 2 gene in mice leads to iron overload. Gastroenterology 132, 301–310. [DOI] [PubMed] [Google Scholar]
- Nemeth E.; Roetto A.; Garozzo G.; Ganz T.; Camaschella C. (2005) Hepcidin is decreased in TFR2 hemochromatosis. Blood 105, 1803–1806. [DOI] [PubMed] [Google Scholar]
- Apweiler R.; Hermjakob H.; Sharon N. (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8. [DOI] [PubMed] [Google Scholar]
- Ben-Dor S.; Esterman N.; Rubin E.; Sharon N. (2004) Biases and complex patterns in the residues flanking protein N-glycosylation sites. Glycobiology 14, 95–101. [DOI] [PubMed] [Google Scholar]
- Rudd P. M.; Elliott T.; Cresswell P.; Wilson I. A.; Dwek R. A. (2001) Glycosylation and the immune system. Science 291, 2370–2376. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Miller L. J.; Dong M. (2010) Role of N-linked glycosylation in biosynthesis, trafficking, and function of the human glucagon-like peptide 1 receptor. Am. J. Physiol. 299, E62–E68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H.; Yamashita Y. (2012) Role of N-glycosylation in cell surface expression and protection against proteolysis of the intestinal anion exchanger SLC26A3. Am. J. Physiol. 302, C781–C795. [DOI] [PubMed] [Google Scholar]
- Nguyen T. T.; Amara S. G. (1996) N-linked oligosaccharides are required for cell surface expression of the norepinephrine transporter but do not influence substrate or inhibitor recognition. J. Neurochem. 67, 645–655. [DOI] [PubMed] [Google Scholar]
- Liu X.; Davis D.; Segaloff D. L. (1993) Disruption of potential sites for N-linked glycosylation does not impair hormone binding to the lutropin/choriogonadotropin receptor if Asn-173 is left intact. J. Biol. Chem. 268, 1513–1516. [PubMed] [Google Scholar]
- Mohrmann K.; van Eijndhoven M. A.; Schinkel A. H.; Schellens J. H. (2005) Absence of N-linked glycosylation does not affect plasma membrane localization of breast cancer resistance protein (BCRP/ABCG2). Cancer Chemother. Pharmacol. 56, 344–350. [DOI] [PubMed] [Google Scholar]
- Soroka C. J.; Xu S.; Mennone A.; Lam P.; Boyer J. L. (2008) N-Glycosylation of the α subunit does not influence trafficking or functional activity of the human organic solute transporter α/β. BMC Cell Biol. 9, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze H. H., and Schachter H. (2009) Genetic Disorders of Glycosylation. In Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., et al., Eds.) Chapter 42, Cold Spring Harbor Laboratory Press, Plainview, NY. [PubMed] [Google Scholar]
- Williams A. M.; Enns C. A. (1993) A region of the C-terminal portion of the human transferrin receptor contains an asparagine-linked glycosylation site critical for receptor structure and function. J. Biol. Chem. 268, 12780–12786. [PubMed] [Google Scholar]
- Farina A.; Hattori M.; Qin J.; Nakatani Y.; Minato N.; Ozato K. (2004) Bromodomain protein Brd4 binds to GTPase-activating SPA-1, modulating its activity and subcellular localization. Mol. Cell. Biol. 24, 9059–9069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovinga J. A.; Solenthaler M.; Dufour J. F. (2003) Congenital dyserythropoietic anaemia type II (HEMPAS) and haemochromatosis: A report of two cases. Eur. J. Gastroenterol. Hepatol. 15, 1141–1147. [DOI] [PubMed] [Google Scholar]
- Heimpel H.; Anselstetter V.; Chrobak L.; Denecke J.; Einsiedler B.; Gallmeier K.; Griesshammer A.; Marquardt T.; Janka-Schaub G.; Kron M.; Kohne E. (2003) Congenital dyserythropoietic anemia type II: Epidemiology, clinical appearance, and prognosis based on long-term observation. Blood 102, 4576–4581. [DOI] [PubMed] [Google Scholar]
- Parekh R. B.; Tse A. G.; Dwek R. A.; Williams A. F.; Rademacher T. W. (1987) Tissue-specific N-glycosylation, site-specific oligosaccharide patterns and lentil lectin recognition of rat Thy-1. EMBO J. 6, 1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder S.; Lindenthal S.; Ehrenfeld J. (2001) Tissue-specific N-glycosylation of the ClC-3 chloride channel. Biochem. Biophys. Res. Commun. 286, 635–640. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.