

Abstract
Nonsubstrate interactions of thrombin with fibrin play an important role in modulating its procoagulant activity. To establish the structural basis for these interactions, we crystallized d-Phe-Pro-Arg-chloromethyl ketone-inhibited human thrombin in complex with a fragment, Eht, corresponding to the central region of human fibrin, and solved its structure at 3.65-Å resolution. The structure revealed that the complex consists of two thrombin molecules bound to opposite sides of the central part of Eht in a way that seems to provide proper orientation of their catalytic triads for cleavage of fibrinogen fibrinopeptides. As expected, binding occurs through thrombin's anion-binding exosite I. However, only part of it is involved in forming an interface with the complementary negatively charged surface of Eht. Among residues constituting the interface, Phe-34, Ser-36A, Leu-65, Tyr-76, Arg-77A, Ile-82, and Lys-110 of thrombin and the Aα chain Trp-33, Phe-35, Asp-38, Glu-39, the Bβ chain Ala-68 and Asp-69, and the γ chain Asp-27 and Ser-30 of Eht form a net of polar contacts surrounding a well defined hydrophobic interior. Thus, despite the highly charged nature of the interacting surfaces, hydrophobic contacts make a substantial contribution to the interaction.
Conversion of fibrinogen to fibrin occurs after removal by thrombin of two pairs of fibrinopeptides, termed FPA and FPB, respectively (1). Thrombin is a trypsin-like enzyme with a very high specificity toward fibrinogen, in which it cleaves only 4 of 376 trypsin-sensitive bonds (1, 2). Thrombin binds to its substrate, fibrinogen, but unlike trypsin and most other serine proteases, it remains bound to the product, fibrin, after fibrinopeptides are removed (3–6). Fibrin-bound thrombin retains proteolytic activity toward synthetic and physiological substrates including fibrinogen (4, 7, 8) and is largely protected from inactivation by a specific thrombin inhibitor, heparin-antithrombin III complex (9). At the same time, in the absence of heparin, the protective effect is modest because the catalytic site of fibrin-bound thrombin remains accessible to antithrombin III (9, 10). In addition to these effects, thrombin binding to fibrin during blood clotting serves to sequester thrombin and reduce the thrombin feedback reaction that enhances thrombin generation, an activity that is known as “antithrombin I” (11). Thus, localization of thrombin to sites of vascular injury where fibrin is deposited plays an important physiological role, which is emphasized by the observations that patients with abnormal fibrinogens New York I and Naples I, both characterized by defective thrombin binding, suffer severe thromboembolic disease (12–16).
Thrombin is a multifunctional enzyme. Besides being critical for conversion of fibrinogen into fibrin, thrombin activates other proteins involved in blood coagulation, factors V, VIII, XI, and XIII, and stimulates platelets and other types of cells (ref. 17 and references therein). When bound to thrombomodulin, thrombin rapidly activates protein C, thus initiating an anticoagulant pathway (18). Such multifunctional characteristics of thrombin are connected to its ability to interact with various proteins and cell receptors through a number of binding sites on its surface (17). These include the anion-binding exosite, which interacts with various anionic compounds and proteins and is involved in fibrin(ogen) binding (19, 20). The crystal structure of thrombin revealed two positively charged patches on its surface (21, 22). One of these patches, located ≈20 Å from the catalytic triad, corresponds to the fibrinogen binding exosite and was termed exosite I, whereas another one containing a heparin binding site was termed exosite II (22). Structural studies of thrombin complexed with certain inhibitors and substrate analogs revealed how thrombin interacts with FPA through its active site cleft and provided some information about its interaction with other fibrinogen regions via exosite I (17, 23–27).
Fibrinogen is a chemical dimer consisting of two identical subunits, each composed of three polypeptide chains, Aα, Bβ, and γ held together by disulfide bonds (28). The disulfide-linked NH2-terminal portions of all six chains form the central E region, whereas COOH-terminal portions form two terminal D regions and two αC domains (28–30). Recent x-ray studies of fibrinogen and its fragments established the high-resolution structure of most of the molecule (31–34). Specifically, it was found that, in the central region, the NH2-terminal portions of the Aα/Bβ chains and the γ chain form the funnel-shaped domain on one side of the molecule and the γN-domain on the other side, respectively, while the remaining portions of all three chains in both subunits form triple helical coiled coils (33, 34). Fibrin contains two types of thrombin binding sites, termed “low” and “high” affinity (35). The high-affinity site was localized in the COOH-terminal portion of a γ chain splice variant, γ′, which occurs in a small proportion of fibrinogen molecules (≈15%) (11, 35). The low-affinity sites, which are complementary to thrombin exosite I, were localized in the central E region. Based on the stoichiometry (35), there are two such sites per E region. The interaction of thrombin with fibrinogen via these sites plays a major role in substrate recognition and the cleavage of FPA (residues Aα1–16) and FPB (residues Bβ1–14) from the NH2-terminal portions of the Aα and Bβ chains, respectively, thus triggering conversion of fibrinogen into fibrin.
Numerous studies with fibrinogen fragments implicated the Aα27–50 and Bβ15–42 portions of the E region in interaction with thrombin (reviewed in refs. 1, 6, and 36). The Bβ chain Ala-68 was also suggested to be involved in this interaction because its mutation to Thr in fibrinogen Naples I results in reduced thrombin binding (16). Crystal structures of the bovine fibrinogen E5 fragment (33) and chicken fibrinogen (34) revealed the spatial positioning of BβAla-68 and most of the residues in the thrombin binding portion of the Aα chain, enabling the prediction of the location of the thrombin binding sites on the surface of the E region (33, 34). In the present study, we crystallized thrombin in complex with human fibrinogen fragment Eht, which is devoid of FPA and FPB and established the structure of the complex at 3.65-Å resolution. The structure further localized the complementary binding sites and revealed structural details of the interaction between these molecules.
Methods
Proteins. Human α-thrombin (Enzyme Research Laboratories, South Bend, IN) was dissolved in 50 mM sodium citrate buffer, pH 6.5, containing 0.2 M NaCl and 0.1% polyethylene glycol (PEG) 8000 to a final concentration of 2.5 mg/ml and rapidly inhibited by the addition of a 5-fold molar excess of d-Phe-Pro-Arg-chloromethyl ketone (PPACK) (Calbiochem).
Fragment Eh was prepared from human fibrinogen by digestion with the proteolytic enzyme hementin obtained from the anterior glands of the giant Amazon leech (a generous gift from Andrei Budzynski) as described in detail (37). Hementin cleaves selectively through the coiled coil connector regions of fibrinogen (38) and, thus, Eh contained intact NH2 termini including FPA and FPB. The fragment was deglycosylated by incubation with PNGase F (New England Biolabs, Ipswich, MA) at an enzyme/substrate ratio of 1:100 (wt/wt)for2hat37°Cin20mM phosphate, pH 7.4, with 0.1 M NaCl. The mixture was then treated with thrombin (1 NIH unit per mg of fragment) for 30 min at 37°C to cleave FPA and FPB. Digestion was stopped by addition of a 5-fold molar excess of PPACK followed by overnight dialysis against the same buffer. NH2-terminal sequence analysis of the final fragment, denoted as Eht, revealed three sequences starting at AαGly-17, BβGly-15, and γTyr-1, thus confirming that both fibrinopeptides had been removed.
Preparation and Crystallization of the Eht Fragment in Complex with Thrombin. The complex was formed by mixing PPACK-inhibited thrombin and Eht at 2 mg/ml, both in the above buffers, at a 2:1 molar ratio. Selection of this ratio was based on the measured stoichiometry (35). The mixture was then dialyzed overnight against 15 mM imidazole buffer, pH 6.5, during which the complex was precipitated. The precipitate was pelleted by centrifugation at 16,000 × g for 5 min and then dissolved in 15 mM imidazole buffer, pH 6.5, containing 0.25 M NaCl, to a final concentration of 7–10 mg/ml.
Crystallization of the complex was performed by the micro-dialysis technique using 15-μl buttons (Hampton Research, Laguna Niguel, CA). The complex at 7–10 mg/ml was dialyzed against a solution containing 100 mM Tris (pH 7.9), 0.2 M (NH4)2PO4, and 20% polyethylene glycol (PEG) 3350. Diamond-shaped crystals appeared after 5–6 weeks and grew to the final size of 0.2 × 0.25 × 0.25 mm. The crystals were transferred into cryoprotectant solution containing 30% PEG 3350 and 15% glycerol and then frozen directly in the nitrogen cryostream at 100 K before x-ray data collection.
Data Collection and Processing. A complete set of x-ray data were collected at ≈100 K on a MAR 345 image-plate detector mounted on Siemens M18X rotating anode x-ray generator. The d*Trek program system (39) was used for x-ray data processing. The unit cell parameters and data processing statistics are summarized in Table 1.
Table 1. Crystallographic parameters and refinement statistics for the complex of thrombin with the Eht fragment.
| Space group | P31 | ||
| Unit cell dimensions, Å | |||
| a, b | 76.2 | ||
| c | 192.4 | ||
| No. of observations | 56,645 | ||
| No. of reflections used | 25,847 | ||
| Data statistics by resolution shells | |||
| Shell, Å | I/σ1 | Completeness, % | Rmerge*, % |
| 20.0–7.73 | 6.7 | 97.7 | 0.085 |
| 7.73–6.19 | 4.3 | 97.2 | 0.154 |
| 6.19–5.43 | 3.6 | 96.7 | 0.193 |
| 5.43–4.94 | 3.5 | 95.7 | 0.195 |
| 4.94–4.59 | 3.4 | 94.5 | 0.205 |
| 4.59–4.32 | 3.2 | 95.0 | 0.225 |
| 4.32–4.11 | 2.8 | 92.5 | 0.255 |
| 4.11–3.93 | 2.2 | 88.9 | 0.325 |
| 3.93–3.78 | 2.1 | 88.5 | 0.350 |
| 3.78–3.65 | 1.9 | 87.6 | 0.389 |
| All reflections | 3.4 | 93.4 | 0.191 |
| Solvent content, % | 55.9 | ||
| No. of protein atoms | 6,836 | ||
| R factor†, % | 22.2 | ||
| Rfree‡, % | 28.9 | ||
| rms deviations from ideal values: | |||
| Bond length, Å | 0.014 | ||
| Bond angles, ° | 1.94 | ||
| Luzatti coordinate error§, Å | 0.42 | ||
| Average B value, Å | 27.5 | ||
Rmerge = Σ|Ij – 〈I〉|/ΣIi; where Ij is the measured and 〈I〉 is the mean intensity of reflection hkl
Crystallographic R + factor = Σ||Fobs| – |Fcalc||/Σ|Fobs|, where Fobs and Fcalc are observed and calculated structure factors of reflection hkl, respectively
Rfree is the R factor calculated over 10% of randomly selected reflections not included in the refinement
At this resolution, the estimation of errors of atomic coordinates is problematic; the Luzatti estimate is provided to give a sense of the positional accuracy
Structure Determination and Model Building. The structure was solved by molecular replacement using the cns program package (40). Crystal structures of human α-thrombin (PDB ID 1PPB; ref. 22) and the bovine fibrinogen E5 fragment (PDB ID 1JY2; ref. 33) were taken as probe models. The rotation search was conducted in the resolution range of 15–3.65 Å, independently for each model. The orientation of the molecules was identified, and the structure of the complex, composed of two thrombin molecules bound to the central part of the E5 fragment, was reconstructed, giving the correlation coefficient of 0.478 and the R factor of 46%.
The analysis of the diffraction data revealed the presence of hemihedral twinning (www.doe-mbi.ucla.edu/Services/Twinning, ref. 41). The twin fraction was calculated at 38.2% corresponding to the h, -h-k, -l twinning operator. This information was taken into account in subsequent steps of the refinement and in creating the set of reflections for cross-validation. The refinement was carried out initially by cns, and at the latter stages by shelx-97 (42). At the first step, the position of the complex was optimized by the rigid-body refinement procedure, followed by the rigid-body refinement of each of the individual molecules. After this step, the value of the R factor dropped to 36%. At this point, the structure was subjected to visual examination to eliminate possible steric overlaps between individual molecules and to make the necessary amino acid substitutions in the structure of the E5 fragment according to the sequence of human fibrinogen. To reduce the bias from the starting model, the first cycles of the refinement were performed omitting from the structure those residues for which no density was detected on an annealed composite omit map. In the subsequent cycles, the omitted residues were added to the model as interpretable electron density appeared. A total of 16 cycles of conventional least-squares refinement of atomic coordinates interspersed with model building were conducted. Initial tight noncrystallographic symmetry (NCS) restraints were applied to thrombin molecules and individual chains of Eht. These were loosened near the end of the refinement process without any dramatic changes in the agreement (rms) between the NCS-related parts of the complex. The model building was performed with XtalView program package (43) employing σA weighted 2Fo - Fc and Fo - Fc electron density maps contoured at 1.0σ and 2.0σ levels, respectively. The refinement statistics are presented in Table 1.
Illustrations were prepared with the program pymol (DeLano Scientific, San Francisco) and ds viewepro (Accelrys, San Diego).
Results
Overall Structure of the Complex Between Eht Fragment and Thrombin. The complex comprises two thrombin molecules bound to the central part of the rod-shaped Eht fragment (Fig. 1). This finding is consistent with the measured stoichiometry (35) and the predicted arrangement of thrombin-binding sites in the bovine fibrinogen E5 fragment (33). Thrombin molecules are located symmetrically with respect to the noncrystallographic two-fold axis, on opposite sides of the Eht fragment at an ≈70° angle with respect to the three-stranded coiled coils, making an X-shaped structure with the approximate dimensions of 127 × 88 Å. When viewed along the axial projection of the coiled coil domains, the complex appears as a shallow 127 × 67 Å V-shaped structure with the γN-domain of Eht at the base and the thrombin molecules as extended arms (Fig. 1C). Each thrombin molecule is bound to the outer wall of the funnel-shaped domain of the Eht fragment, burying ≈1,200 Å2 of its surface area. The binding involves exosite I and occurs in such a way that the active sites of both thrombin molecules, which are occupied by PPACK (shown in magenta), are facing in the same direction, toward the side of the Eht molecule where both pairs of fibrinogen fibrinopeptides would be located, and thus are well positioned for thrombin cleavage.
Fig. 1.

Two different projections of the three-dimensional structure of the complex of thrombin with the Eht fragment. (A and B) Ribbon diagram and solvent-accessible surface of the complex viewed along a noncrystallographic twofold symmetry axis perpendicular to the plane of the page, respectively. (C) The complex viewed along the axial projection of the coiled-coil domains. Aα, Bβ, and γ chains in A are in blue, green, and red, respectively. NH2-terminal portions of the Aα and Bβ chains and that of the γ chain forming the funnel-shaped and the γN-domains, respectively (33), in B and C have the same color scheme as in A, whereas their remaining portions forming the coiled coil domains are in gray. Thrombin molecules in all panels are in beige, whereas the PPACK inhibitor bound to the active sites is in magenta and residues included in exosite I (16) are in orange. The distance between the active sites is ≈70 Å. The broken curved lines in B indicate the S' groove in the active site cleft of each thrombin molecule connecting the active site with exosite I.
Structure of Individual Components in the Complex. In our structure, each thrombin molecule includes nearly complete light and heavy chain components except for four residues at the NH2 terminus of the light chain and one residue at the COOH terminus of the heavy chain, which were not identified because of lack of electron density. The structure of the Eht fragment includes only those residues whose positions had previously been established in the structure of the bovine fibrinogen E5 fragment used here as the starting model, namely, residues 32–73 of the Aα chains, 56–105 of the Bβ chains, and 2–45 and 6–45 of the two γ chains. The structure of remaining portions of Eht, including residues Aα17–31 and Bβ15–55, were not identified. Although some electron density near the central region that may be connected with these portions was observed, it was not possible to interpret it reliably. Therefore, no attempt to model these portions was made.
Direct structural comparison indicates that despite a number of subtle structural changes, the overall folds of the individual components of the complex are in a good agreement with those of the starting model. The rms differences between positions of Cα atoms in the final structures of the two thrombin molecules and their starting model are 0.74 and 0.75 Å; the overall rms difference between corresponding Cα atoms of Eht and its starting model, E5, is 1.05 Å. Comparison of Eht with the corresponding region of chicken fibrinogen (1M1J, which replaced the original entry 1JFE; ref. 34) also reveals a high similarity between the two structures (rms is 1.13 Å). Thus, the overall fold of the central region in human, bovine, and chicken fibrinogens is virtually the same.
Analysis of the Interface Between the Eht Fragment and Thrombin in the Complex. Although the structure of the complex was solved at 3.65 Å, the electron densities for most side chains were clearly interpreted. This is illustrated by a section of electron density map, which is published as supporting information on the PNAS web site. At the same time, it should be realized that, at such resolution, the accuracy of defining specific atomic interactions, particularly those between different molecules, is limited.
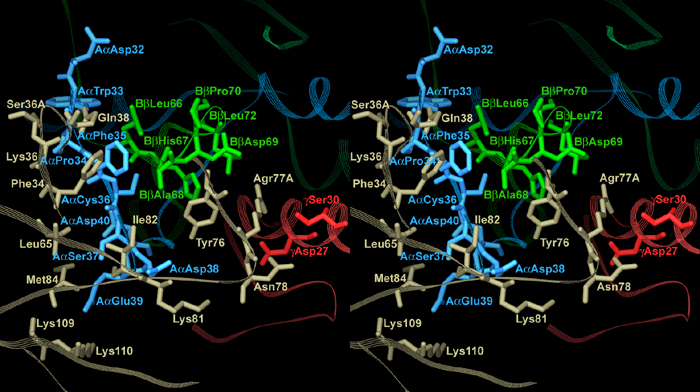
Analysis of side chain conformation in the interface area revealed a number of polar and nonpolar contacts (Fig. 2). There are six hydrophobic residues, four in thrombin and two in Eht, that cluster in two hydrophobic patches, one on each molecule. The patch on thrombin includes Phe-34, Leu-65, Tyr-76, and Ile-82, whereas the one on the Eht fragment is formed by AαPhe-35 and BβAla-68. These patches face each other to form the hydrophobic core-like interior of the interface. A network of polar contacts revealed on the periphery of the hydrophobic interior includes three hydrogen bonds and three salt bridges. Thrombin Lys-110 forms one salt bridge with the Aα chain Glu-39, whereas its Arg-77A forms two bridges with BβAsp-69 and γAsp-27. In addition, the Nh1 atom of the Arg-77A guanidine group is located within the distance of a hydrogen bond from the Oγ atom of γ chain Ser-30, thus making Arg-77A an important contributor to the network of polar contacts. The other residues that contribute to the network are the Aα chain Asp-38 and thrombin Ser-36A. The Oδ1 atom of AαAsp-38 carboxyl group and the Oγ atom of Ser-36A hydroxyl group form hydrogen bonds with the main chain nitrogen of Ile-82 and the main chain carbonyl oxygen of AαTrp-33, respectively. At the same time, because the distance between the latter pair of atoms is close to the upper limit suggested for a hydrogen bond, this contact could be rather weak.
Fig. 2.

Stereoview of the hydrophobic interior of the interface between thrombin and the Eht fragment and the surrounding network of polar interactions. Residues forming the interior are covered with wire mesh van der Waals atomic surfaces and colored as their parent chains, beige for thrombin and blue, green, and red for the Aα,Bβ and γ chains of Eht, respectively. Residues involved in the polar interactions are colored by atom types: blue for nitrogens, red for oxygens, and white for carbons; interatomic contacts are represented with dashed lines. The colors of the labels coincide with those of the corresponding chains. A view of the whole interface region is published as supporting information on the PNAS web site.
It should be noted that, in addition to the above interactions, other polar contacts could potentially be formed. First, although the distances between the main chain carbonyl oxygen (O) of Ile-82 and Oγ of AαSer-37, and between Oh of Tyr-76 and Oδ2 of AαAsp-38 exceed that for a hydrogen bond, the excess in each case is within the estimated error of the atomic coordinates and therefore one cannot exclude that they form weak hydrogen bonds. Second, despite the fact that Lys-36 and Lys-109 cannot be traced completely because of lack of electron density, the orientations of their peptide groups suggest that these residues could form salt bridges with AαAsp-40 and AαGlu-39, respectively.
Discussion
The interaction between thrombin and the central region E of fibrin(ogen) has attracted much attention because of its importance in the conversion of fibrinogen into fibrin and in the in vivo regulation of thrombin activity. Although previous reports (17, 26) had provided structural details on binding of FPA to the active site cleft of thrombin, the exact nature of the interaction between fibrin(ogen) and thrombin exosite I could only be surmised. This was partially clarified by molecular modeling of thrombin in complex with the NH2-terminal regions of the fibrinogen Aα and Bβ chains (44). In this study, we solved the crystal structure of thrombin in complex with the fibrinogen-derived thrombin-treated Eht fragment and established the mode of their interaction via thrombin exosite I.
The structure revealed that the interaction between thrombin and Eht in the complex occurs through a hydrophobic interior surrounded by a network of at least three hydrogen bonds and three salt bridges (Fig. 2). Among the seven thrombin residues forming contacts with the fragment, Phe-34, Leu-65, Tyr-76, Arg-77A, Ile-82, and Lys-110 belong to exosite I, which was described by Bode et al. (17, 22). The remaining Ser36A is located at the exosite border (Fig. 3) and could also be included in it. Interestingly, all of these residues comprise only a portion of exosite I, whereas the remaining part is solvent-accessible in the complex (Figs. 1 B and C and 3). From the Eht side, four of the eight residues involved in thrombin binding, Trp-33, Phe-35, Asp-38 and Glu-39, come from the Aα chain, two residues, Ala-68 and Asp-69, come from the Bβ chain, and Asp-27 and Ser-30 come from the γ chain (Fig. 3). Because all of these residues, except γAsp-27 and γSer-30, belong to the funnel-shaped domain, the latter could be regarded as the thrombin-binding domain.
Fig. 3.

Location of the complementary binding sites in the Eht fragment and thrombin. (Upper) Residues of Eht involved in the interaction with thrombin, Aα chain Trp-33, Phe-35, Asp-38, Glu-39, Bβ chain Ala-68 and Asp-69 of the thrombin-binding domain, and γ chain Asp-27 and Ser-30 of the coiled-coil domain (in gray). AαSer-37, which could also participate in the interaction (see text), and AαAsp-40 and BβAsp-71, of the proposed negatively charged platform (34), are presented as well. The residues are presented in a projection similar to that for the corresponding residues in Fig. 2. (Lower) Thrombin residues interacting with Eht, Phe-34, Ser-36A, Leu-65, Ile-82, Tyr-76, Arg-77A, and Lys-110; residues included in exosite I (16) are also highlighted. In both panels, residues are colored according to their properties: hydrophobic residues are in yellow, positively and negatively charged residues are in blue and red, respectively, and polar residues as well as AαTrp-33 forming polar contact are in pink. The complementary binding surfaces in both molecules are contoured with triangles; the repeating end arrows connecting the vertices show their mutual orientations in the complex.
Although the surface of thrombin exosite I is highly positively charged because of the presence of nine basic residues (Fig. 3), the present study revealed that only two of them, Arg-77A and Lys-110, are involved in formation of salt bridges with complementary residues of the Eht fragment. Similarly, among five negatively charged residues of Eht (Fig. 3), which were suggested to form the negatively charged thrombin binding platform (34), only BβAsp-69 and AαGlu-39 form salt bridges with the above mentioned thrombin Arg77A and Lys-110, respectively. Furthermore, these salt bridges represent only part of the major interactions between E1 and thrombin, which, as mentioned above, also involve a number of hydrogen bonds and hydrophobic contacts in the interface. These results suggest that contribution of the charged surfaces to the major interactions between fibrin(ogen) and thrombin is not as significant as one would expect based on the charge distribution. These surfaces may be more important for initial electrostatic attraction and proper orientation of the two molecules to facilitate complex formation.
The structure of the complex provides a reasonable explanation for the results of previous mutagenesis studies. At least five fibrinogen residues forming the interface were mutated in the whole molecule or in its recombinant Aα chain fragment to evaluate their involvement in the binding of thrombin (36, 45, 46). Among them, mutation of BβAla-68 to Thr, which mimics the naturally occurring mutation in fibrinogen Naples I, resulted in a dramatic decrease of thrombin binding and thrombin-catalyzed fibrinopeptide release (16, 46). The other mutations, AαTrp33Ser, AαPhe35Leu, AαAsp38Gly, and AαGlu39Gly, had a less severe effect on thrombin interaction (36, 45). The effect of the BβAla68Thr mutation is not surprising because BβAla-68 contributes to the hydrophobic interior of the interface (Fig. 2) and its substitution by Thr places a polar group in the nonpolar interface environment, which, in addition to possible steric problems, can interact with water, further disrupting thrombin binding. This highlights the importance of hydrophobic contacts in the fibrin(ogen)/thrombin interaction. It is also not surprising that the AαTrp33Ser and AαPhe35Leu mutations caused only a moderate decrease in interaction with thrombin (36, 45). Indeed, AαPhe-35 is involved in hydrophobic interaction (Fig. 2), and its replacement with nonpolar Leu might preserve hydrophobic contacts. Similarly, substitution of Ser (or any other residue) for AαTrp-33, whose main chain carbonyl oxygen may form a hydrogen bond with Oγ atom of Ser-36A (Fig. 2), should not result in disruption of this bond. At the same time, one would expect a more substantial inf luence of the AαAsp38Gly and AαGlu39Gly mutation because these residues form a hydrogen bond with main chain nitrogen of Ile-82 and the salt bridge with Lys-110, respectively (Fig. 2). However, because the interactions are on the periphery of the binding site, compensating structural changes may occur to minimize the disruption. In addition, analysis of the crystal packing revealed that formation of the salt bridge between AαGlu-39 and Lys-110 could be promoted by crystal packing forces, suggesting that, in solution, this bridge may be rather weak and its contribution to the energetics of complex formation could be less than one would expect from electrostatic interaction.
In Ala-scanning mutagenesis studies, mutations of Tyr-76 and Arg-77A of thrombin resulted in a severe drop of fibrinogen clotting and fibrin-binding activities (47, 48). The structure of the complex reveals that these two residues indeed play an important role in the interface formation. Namely, Tyr-76 is involved in hydrophobic contacts and its hydroxyl group forms a hydrogen bond with the Aα chain Asp-38, whereas Arg-77A forms hydrogen bond with the γSer-30 and two salt bridges with BβAsp-69 and γAsp-27 (Fig. 2). In the same studies, the effects of substituting Ala for two other residues, Ser-36A and Lys-110, were less dramatic. The most probable explanation is that Ser-36A forms the hydrogen bond that, as noted in Results, could be rather weak, and the salt bridge formed by Lys-110 could also be weak, as discussed. It should be noted that Lys36Ala, Lys81Ala, and Lys109Ala mutants also exhibited reduced fibrin binding, in agreement with their possible contribution to the contacts formation (see Results).
As mentioned above, the locations of the 17–31 portion of the Aα chain and the 15–55 portion of the Bβ chain in the Eht fragment were not established. There is no experimental evidence for the involvement of the Aα17–31 portion in thrombin binding, although a recent molecular modeling study suggests that it interacts with the S′ groove of thrombin connecting the active site with exosite I (44). On the other hand, the Bβ15–55 region is clearly involved because fibrin lacking Bβ15–42 residues shows reduced thrombin binding (35, 49). The exact contribution of this portion to the interaction with thrombin needs to be further clarified. It should also be noted that, in the Eht fragment, both pairs of fibrinopeptides were removed before complex formation. Therefore, this fragment most probably mimics the corresponding region in fibrin and the interactions described here would reflect those between fibrin and thrombin, if the latter interacts with fibrinogen in a different manner. In this connection, it was reported that thrombin, in a ternary complex with fibrin and heparin, preserves its catalytic activity toward fibrinogen (50, 51), suggesting that its active site cleft remains unoccupied. This is in agreement with the structure of our complex in which the active site and the S′ groove of thrombin appear to be accessible to a substrate (Fig. 1B). Thus, it is tempting to speculate that the interactions identified in this complex may indeed mimic those in the catalytically active complex of thrombin with fibrin.
Supplementary Material
Acknowledgments
We thank Dr. Jane Ladner for help with the crystallographic studies and Dr. Kenneth Ingham for helpful discussion. This work was supported by National Institutes of Health Grants HL-56051 (to L.M.) and HL-70627 (to M.W.M.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: FPA and FPB, fibrinopeptides A and B, respectively; PPACK, d-Phe-Pro-Arg-chloromethyl ketone; Eht, thrombin-treated fragment E from fibrinogen prepared by digestion with hementin.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PBD ID code 1QVH).
References
- 1.Blomback, B. (1996) Thromb. Res. 83, 1-75. [DOI] [PubMed] [Google Scholar]
- 2.Stubbs, M. T. & Bode, W. (1993) Semin. Thromb. Hemost. 19, 344-351. [DOI] [PubMed] [Google Scholar]
- 3.Liu, C. Y., Nossel, H. L. & Kaplan, K. L. (1979) J. Biol. Chem. 254, 10421-10425. [PubMed] [Google Scholar]
- 4.Francis, C. W., Markham, R. E., Jr., Barlow, G. H., Florack, T. M., Dobrzynski, D. M. & Marder, V. J. (1983) J. Lab. Clin. Med. 102, 220-230. [PubMed] [Google Scholar]
- 5.Kaminski, M. & McDonagh, J. (1983) J. Biol. Chem. 258, 10530-10535. [PubMed] [Google Scholar]
- 6.Mosesson, M. W. (1993) Semin. Thromb. Hemost. 19, 361-367. [DOI] [PubMed] [Google Scholar]
- 7.Kaminski, M. & McDonagh, J. (1987) Biochem. J. 242, 881-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beguin, S. & Kumar, R. (1997) Thromb. Haemost. 78, 590-594. [PubMed] [Google Scholar]
- 9.Hogg, P. J. & Jackson, C. M. (1989) Proc. Natl. Acad. Sci. USA 86, 3619-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bock, P. E., Olson, S. T. & Bjork, I. (1997) J. Biol. Chem. 272, 19837-19845. [DOI] [PubMed] [Google Scholar]
- 11.Mosesson, M. W. (2003) Thromb. Haemost. 89, 9-12. [PubMed] [Google Scholar]
- 12.Al-Mondhiry, H., Bilezikian, S. B. & Nossel, H. L. (1975) Blood 45, 607-619. [PubMed] [Google Scholar]
- 13.Liu, C. Y., Koehn, J. A. & Morgan, F. J. (1985) J. Biol. Chem. 260, 4390-4396. [PubMed] [Google Scholar]
- 14.Haverkate, F., Koopman, J., Kluft, C., D'Angelo, A., Cattaneo, M. & Mannucci, P. M. (1986) Thromb. Haemost. 55, 131-135. [PubMed] [Google Scholar]
- 15.Di Mino, G., Martinez, J., Cirillo, F., Cerbone, A. M., Silver, M. J., Colucci, M., Margaglione, M., Tauro, R., Semeraro, N., Quattrone, A. & Mancini, M. (1991) Arterioscler. Thromb. 11, 785-796. [DOI] [PubMed] [Google Scholar]
- 16.Koopman, J., Haverkate, F., Lord, S. T., Grimbergen, J. & Mannucci, P. M. (1992) J. Clin. Invest. 90, 238-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bode, W. & Stubbs, M. T. (1993) Semin. Thromb. Hemost. 19, 321-333. [DOI] [PubMed] [Google Scholar]
- 18.Esmon, C. T., Esmon, N. L. & Harris, K. W. (1982) J. Biol. Chem. 257, 7944-7947. [PubMed] [Google Scholar]
- 19.Fenton, J. W., 2nd. (1986) Ann. N.Y. Acad. Sci. 485, 5-15. [DOI] [PubMed] [Google Scholar]
- 20.Fenton, J. W., 2nd, Olson, T. A., Zabinski, M. P. & Wilner, G. D. (1988) Biochemistry 27, 7106-7112. [DOI] [PubMed] [Google Scholar]
- 21.Bode, W., Mayr, I., Baumann, U., Huber, R., Stone, S. R. & Hofsteenge, J. (1989) EMBO J. 8, 3467-3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bode, W., Turk, D. & Karshikov, A. (1992) Protein Sci. 1, 426-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ni, F., Konishi, Y., Frazier, R. B., Scheraga, H. A. & Lord, S. T. (1989) Biochemistry 28, 3082-3094. [DOI] [PubMed] [Google Scholar]
- 24.Rydel, T. J., Ravichandran, K. G., Tulinsky, A., Bode, W., Huber, R., Roitsch, C. & Fenton, J. W., 2nd. (1990) Science 249, 277-280. [DOI] [PubMed] [Google Scholar]
- 25.Grutter, M. G., Priestle, J. P., Rahuel, J., Grossenbacher, H., Bode, W., Hofsteenge, J. & Stone, S. R. (1990) EMBO J. 9, 2361-2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stubbs, M. T., Oschkinat, H., Mayr, I., Huber, R., Angliker, H., Stone, S. R. & Bode, W. (1992) Eur. J. Biochem. 206, 187-195. [DOI] [PubMed] [Google Scholar]
- 27.Martin, P. D., Robertson, W., Turk, D., Huber, R., Bode, W. & Edwards, B. F. (1992) J. Biol. Chem. 267, 7911-7920. [PubMed] [Google Scholar]
- 28.Henschen, A. & McDonagh, J. (1986) in Blood Coagulation, eds. Zwaal, R. F. A. & Hemker, H. C. (Elsevier Science, Amsterdam), pp. 171-241.
- 29.Doolittle, R. F. (1984) Annu. Rev. Biochem. 53, 195-229. [DOI] [PubMed] [Google Scholar]
- 30.Weisel, J. W. & Medved, L. (2001) Ann. N.Y. Acad. Sci. 936, 312-327. [DOI] [PubMed] [Google Scholar]
- 31.Yee, V. C., Pratt, K. P., Cote, H. C., Trong, I. L., Chung, D. W., Davie, E. W., Stenkamp, R. E. & Teller, D. C. (1997) Structure (London) 5, 125-138. [DOI] [PubMed] [Google Scholar]
- 32.Spraggon, G., Everse, S. J. & Doolittle, R. F. (1997) Nature 389, 455-462. [DOI] [PubMed] [Google Scholar]
- 33.Madrazo, J., Brown, J. H., Litvinovich, S., Dominguez, R., Yakovlev, S., Medved, L. & Cohen, C. (2001) Proc. Natl. Acad. Sci. USA 98, 11967-11972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang, Z., Kollman, J. M., Pandi, L. & Doolittle, R. F. (2001) Biochemistry 40, 12515-12523. [DOI] [PubMed] [Google Scholar]
- 35.Meh, D. A., Siebenlist, K. R. & Mosesson, M. W. (1996) J. Biol. Chem. 271, 23121-23125. [DOI] [PubMed] [Google Scholar]
- 36.Binnie, C. G. & Lord, S. T. (1993) Blood 81, 3186-3192. [PubMed] [Google Scholar]
- 37.Mosesson, M. W., Siebenlist, K. R., Hernandez, I., Wall, J. S. & Hainfeld, J. F. (2002) Thromb. Haemost. 87, 651-658. [PubMed] [Google Scholar]
- 38.Malinconico, S. M., Katz, J. B. & Budzynski, A. Z. (1984) J. Lab. Clin. Med. 104, 842-854. [PubMed] [Google Scholar]
- 39.Pflugrath, J. W. (1999) Acta Crystallogr. D 55, 1718-1725. [DOI] [PubMed] [Google Scholar]
- 40.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 41.Yeates, T. O. (1997) Methods Enzymol. 276, 344-358. [PubMed] [Google Scholar]
- 42.Sheldrick, G. M. & Schneider, T. (1997) Methods Enzymol. 277, 319-343. [PubMed] [Google Scholar]
- 43.McRee, D. E. (1999) in Practical Protein Crystallography (Academic, San Diego), pp. 271-328.
- 44.Rose, T. & Di Cera, E. (2002) J. Biol. Chem. 277, 18875-18880. [DOI] [PubMed] [Google Scholar]
- 45.Lord, S. T. & Binnie, C. G. (1990) Blood Coagul. Fibrinolysis 1, 461-463. [DOI] [PubMed] [Google Scholar]
- 46.Lord, S. T., Strickland, E. & Jayjock, E. (1996) Biochemistry 35, 2342-2348. [DOI] [PubMed] [Google Scholar]
- 47.Tsiang, M., Janin, A. K., Dunn, K. E., Rojas, M. E., Leung, L. L. K. & Gibbs, C. S. (1995) J. Biol. Chem. 270, 16854-16863. [DOI] [PubMed] [Google Scholar]
- 48.Hall, S. W., Gibbs, C. S. & Leung, L. L. K. (2001) Thromb. Haemost. 86, 1466-1474. [PubMed] [Google Scholar]
- 49.Siebenlist, K. R., DiOrio, J. P., Budzynski, A. Z. & Mosesson, M. W. (1990) J. Biol. Chem. 265, 18650-18655. [PubMed] [Google Scholar]
- 50.Hogg, P. J. & Jackson, C. M. (1990) J. Biol. Chem. 265, 248-255. [PubMed] [Google Scholar]
- 51.Hogg, P. J., Jackson, C. M., Labanowski, J. K. & Bock, P. E. (1996) J. Biol. Chem. 271, 26088-26095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}