

Abstract
The recently determined crystal structures of the sarcoplasmic reticulum Ca2+-ATPase show that in the E1Ca2 form, domain A is almost isolated from the other cytoplasmic domains, P and N, whereas in E2, domain A has approached domains P and N, with E183 of the highly conserved P-type ATPase signature sequence TGES in domain A now being close to the phosphorylated aspartate in domain P, thus raising the question whether E183 acquires a catalytic role in E2 and E2P conformations. This study compares the partial reactions of mutant E183A and wild-type Ca2+-ATPase, using transient and steady-state kinetic measurements. It is demonstrated that dephosphorylation of the E2P phosphoenzyme intermediate, as well as reverse phosphorylation of E2 with Pi, is severely inhibited in the mutant. Furthermore, the apparent affinity of E2 for the phosphoryl transition state analog vanadate is reduced by three orders of magnitude, consistent with a destabilization of the transition state complex, and the mutant displays reduced apparent affinity for Pi in the E2 form. The E1Ca2 conformation, on the other hand, shows normal phosphorylation with ATP and normal Ca2+ binding properties, and the rates of the conformational transitions E1PCa2 → E2P and E2 → E1Ca2 are only 2- to 3-fold reduced, relative to wild type. These results, which likely can be generalized to other P-type ATPases, indicate that E183 is critical for the phosphatase function of E2 and E2P, possibly interacting with the phosphoryl group or attacking water in the transition state complex, but is of little functional importance in E1 and E1P.
The Ca2+-ATPase of sarcoplasmic reticulum (EC 3.6.1.38) is a member of the P-type ATPase family that encompasses ion pumps of crucial importance for cell function, such as Ca2+-ATPases, Na+,K+-ATPases, H+-ATPases, and heavy metal-transporting ATPases (1–5). These enzymes share a unique reaction cycle (Scheme 1) in which the hydrolysis of ATP occurs through phosphorylation and dephosphorylation of a conserved aspartic acid residue (D351 in the sarcoplasmic reticulum Ca2+-ATPase). The catalytic site exists in at least two conformational states, E1 and E2, with “kinase activity” and “phosphatase activity,” respectively (6). Hence, the E1P phosphoenzyme intermediate can donate the phosphoryl group back to ADP, forming ATP, whereas E2P reacts with water, liberating Pi (1). The structural basis for this difference in catalytic specificity is not well understood. The conformational rearrangement that changes the catalytic specificity directs the translocation of ions in the membrane domain some 40 Å away from the catalytic site, thus constituting an important link between ATP utilization and ion transport.
Scheme 1.

The recently published high-resolution crystal structures of the sarcoplasmic reticulum Ca2+-ATPase in the Ca2+ bound E1Ca2 and thapsigargin-bound E2-TG dephosphoenzyme forms (7, 8), and the model for the vanadate-bound E2-V structure originating from analysis of bidimensional membrane crystals (7, 9) provide some clues to the understanding of the structural changes occurring during the catalytic cycle, although crystal structures of the phosphorylated conformations are still missing. The cytoplasmic part of the enzyme consists of three separate domains, the phosphorylation (P), nucleotide binding (N), and “actuator” (A) domains. In E1Ca2, domain A is rather isolated from domains P and N, whereas in the E2-TG and E2-V crystals, domain A has approached domains P and N, resulting in a more closed configuration of the cytoplasmic “head” of the enzyme. The movement of domain A in connection with the transition between E1 and E2 is consistent with previous biochemical measurements on native enzyme of accessibility to proteolytic cleavage (in the Ca2+-ATPase) (10, 11) and reaction with antibody (in H+-ATPase) (12), as well as the pattern of Fe2+ catalyzed oxidative cleavage (in Na+,K+-ATPase) (13). According to the E2-TG and E2-V structures and the earlier prediction from oxidative cleavage patterns, the highly conserved TGES motif of domain A is transposed into the catalytic site close to the phosphorylated aspartic acid residue and another P-type ATPase signature sequence TGDGVND of domain P, raising the question of whether the glutamate of TGES (E183 in Ca2+-ATPase) actually acquires a catalytic role in E2/E2P in connection with the phosphatase reaction or is of structural importance for the conformational change docking domain A into domain P. This glutamate is conserved in all known P-type ATPases and is critical for Ca2+-ATPase (14), Na+,K+-ATPase (15), and H+-ATPase (16, 17) overall pump function. The previous mutagenesis study of E183 in sarcoplasmic reticulum Ca2+-ATPase revealed that this residue is important for the conformational change between E1P and E2P (14). Because mutation of the corresponding glutamate in Na+,K+-ATPase and yeast H+-ATPase is incompatible with cell viability (15, 17), further information on its functional role was not obtained for these enzymes.
In the present study, the importance of E183 of sarcoplasmic reticulum Ca2+-ATPase for the various partial reactions of the cycle shown as Scheme 1 has been reexamined in equilibrium and transient kinetic studies of mutant E183A. We present evidence for a critical catalytic role of E183 during phosphorylation/dephosphorylation in E2/E2P forms, whereas this residue plays only a minor role in the E1PCa2 → E2P and E2 → E1Ca2 conformational changes and is unimportant for catalysis in E1/E1P.
Experimental Procedures
All methods have been described (18–22), including expression of the cDNA (rabbit fast twitch muscle Ca2+-ATPase, SERCA1a isoform) in COS-1 cells, isolation of microsomes containing expressed enzyme, and assays of partial reaction steps of the pump cycle by measurement of the acid-stable phosphorylated intermediate, using manual mixing and quench-flow techniques. The cDNA encoding the Ca2+-ATPase mutant E183A was the same as used previously (14), but was shuttled to vector pMT2 (23) to enhance the expression level, and sequenced throughout, verifying the mutation and the absence of undesired mutations. All experiments were conducted at least twice, and the data were analyzed by nonlinear regression and computation with simzyme software (21). The analysis of ligand concentration dependence was based on the Hill function. Monoexponential functions were fitted to the phosphorylation and dephosphorylation time courses.
Results
Expression, Overall Activity, and Phosphorylation Capacity of E183A. The E183A mutant Ca2+-ATPase was expressed in COS-1 cells and was devoid of 45Ca2+ transport activity (determined at 37°C, 3 μM Ca2+, and 5 mM MgATP) and ATPase activity (at 37°C, 100 μM Ca2+, and 5 mM MgATP). E183A was, however, able to undergo phosphorylation, both in the forward direction of normal turnover in presence of [γ-32P]ATP and Ca2+, and in the backward direction in presence of 32Pi and absence of Ca2+, giving rise to phosphoenzyme levels similar to those seen with the wild type. These results agree with what has been found previously for this mutant (14).
The E1PCa2 → E2P Transition. The complete loss of Ca + transport and ATPase activity observed with E183A, and at the same time preserved ability to undergo phosphorylation from ATP, suggests that a partial reaction step subsequent to the phosphoryl transfer from ATP to Asp-351 is affected in the mutant. The further processing of the phosphoenzyme involves the E1PCa2 → E2P transition (reaction 2 in Scheme 1), followed by the hydrolysis of E2P (reaction 3 in Scheme 1). To study these reaction steps, the enzyme was phosphorylated with [γ-32P]ATP at conditions where E1PCa2 accumulates as the major steady-state intermediate in the wild type (0°C, neutral pH, presence of K+) (19), and the phosphoenzyme decay was subsequently studied by a chase with EGTA (to remove Ca2+ and thereby terminate phosphorylation) and excess nonradioactive ATP. As seen in Fig. 1A (open symbols), a marked 40-fold slowing of dephosphorylation was found with E183A relative to wild type, demonstrating a block in the E1PCa2 → E2P → E2 reaction sequence, thus accounting for the loss of overall activity in E183A.
Fig. 1.

Phosphoenzyme processing. (A) Phosphorylation was carried out for 15 s at 0°C in 40 mM Mops [3-(N-morpholino)propanesulfonic acid]/Tris (pH 7.0), 80 mM KCl, 5 mM MgCl2, 1 mM EGTA, 0.955 mM CaCl2 (10 μM free Ca2+), 10 μM calcium ionophore A23187, and 5 μM[γ-32P]ATP. To measure dephosphorylation, the phosphoenzyme was chased at 0°C by addition of 10 mM EGTA with either 1 mM nonradioactive ATP (open symbols) or 1 mM ADP (filled symbols), and acid quenching was performed at the indicated time intervals. The lines for ATP chase show the best fits of a monoexponential decay function, giving the rate constants indicated in parentheses: open circles, wild type (0.41 s-1); open triangles, E183A (0.010 s-1). For ADP chase, the filled circle corresponds to wild type and the filled triangle corresponds to E183A. (B) Phosphorylation was performed for the indicated time intervals at 0°Cin40mMTES{N-[tris(hydroxymethyl)methyl]-2-aminoethane-sulfonic acid}/Tris (pH 7.8), 80 mM LiCl, 10 mM MgCl2, 50 μM CaCl2, 10 μM calcium ionophore A23187, and 5 μM [γ-32P]ATP, followed by addition of an equal volume of 40 mM TES/Tris (pH 7.8), 80 mM LiCl, 10 mM EGTA, and 2 mM ADP, and acid quenching 4 s later. The lines show the best fits of a monoexponential function, giving the rate constants indicated in parentheses: circles, wild type (0.24 s-1); triangles, E183A (0.13 -1). In each case, the 100% value corresponds to the phosphorylation level reached at infinite time, as deduced from the fit.
The two phosphoenzyme intermediates, E1PCa2 and E2P, can be distinguished by their different reactivities toward ADP, E1PCa2 being able to transfer the phosphate back to ADP forming ATP, and E2P being insensitive to ADP, dephosphorylating only by hydrolysis (1). Thus, when in the wild-type enzyme the accumulated E1PCa2 state is mixed with ADP, virtually all phosphoenzyme disappears rapidly, as seen in Fig. 1 A (filled circle). In contrast, 43% phosphoenzyme remained after a 5-s incubation of phosphorylated E183A with ADP (Fig. 1 A, filled triangle), suggesting accumulation of ADP-insensitive E2Pinthe mutant.
To further examine the E1PCa2 → E2P transition, we studied the reaction sequence E1Ca2 → E1PCa2 → E2P by phosphorylating the enzyme with [γ-32P]ATP under conditions (0°C, alkaline pH, high Mg2+/Ca2+ ratio, substitution of K+ with Li+) where E2P hydrolysis is rather slow for the wild type, and E2P therefore accumulates as the major steady-state intermediate. The phosphorylation reaction itself (E1Ca2 → E1PCa2) is relatively fast under these conditions, reaching steady state within <2 s for both wild type and mutant (data not shown). At varying time intervals after the initiation of phosphorylation, the amount of ADP-insensitive E2P accumulated was tested by a 4-s chase with ADP, removing any E1PCa2. As seen in Fig. 1B, E2P accumulated at a rate that was only 2-fold lower for the mutant as compared with wild type. This moderate effect contrasts with the 40-fold reduced rate of the E1PCa2 → E2P → E2 reaction sequence seen for E183A in Fig. 1 A, implying a rather severe block of E2P hydrolysis in the mutant.
Properties of the E2P State. To study the rate of E2P hydrolysis more directly, E2P formed by phosphorylation of Ca2+-deprived enzyme with 32Pi (reverse reaction 3 in Scheme 1) was subjected to a dephosphorylation chase by dilution into a medium containing nonradioactive Pi. The organic solvent dimethyl sulfoxide was included in the phosphorylation medium to increase the affinity for Pi (24). Dephosphorylation was either performed under conditions mimicking those in Fig. 1 A for chase of E1PCa2 (Fig. 2A) or under conditions identical to those for formation of the 32Pi phosphorylated enzyme, except for a reduction of the concentration of dimethyl sulfoxide from 30% to 15%, dilution of 32Pi, and removal of Mg2+ to terminate phosphorylation (Fig. 2B). As seen in Fig. 2, E183A displayed a conspicuous slowing of dephosphorylation compared with the wild type under both conditions (126- and 15-fold for Fig. 2 A and B, respectively), demonstrating that E2P hydrolysis is effectively blocked in the mutant. The large difference between wild type and E183A with respect to the sensitivity to the change in dephosphorylation conditions is also noteworthy. For the wild type, acidic pH and the presence of dimethyl sulfoxide inhibits E2P hydrolysis (25), whereas the presence of K+ enhances this reaction, accounting for the 20-fold higher rate seen for the wild type in Fig. 2 A compared with Fig. 2B, despite the lower temperature in the experiment corresponding to Fig. 2 A. In contrast, E183A appeared much less sensitive to the changes in dephosphorylation conditions, displaying only a 2-fold rate difference between the two assays.
Fig. 2.

Dephosphorylation of phosphoenzyme formed from 32Pi. Phosphorylation was performed at 25°C for 10 min in 100 mM Mes/Tris (pH 6.0), 2 mM EGTA, 10 mM MgCl2, 30% (vol/vol) dimethyl sulfoxide, and 0.5 mM 32Pi. Dephosphorylation was studied at 0°C by a 19-fold dilution of the phosphorylated (and precooled) sample into 40 mM Mops/Tris (pH 7.0), 2 mM EGTA, 5 mM MgCl2, 80 mM KCl, and 0.5 mM nonradioactive Pi (A)orat25°C by a 19-fold dilution of the phosphorylated sample into 100 mM Mes/Tris (pH 6.0), 2 mM EGTA, 10 mM EDTA, 15% (vol/vol) dimethyl sulfoxide, and 0.5 mM nonradioactive Pi (B), followed by acid quenching at the indicated time intervals. The lines show the best fits of a monoexponential decay function, giving the rate constants (corresponding to A and B, respectively) indicated in parentheses: circles, wild type (0.63 s-1; 0.03 s-1); triangles, E183A (0.005 s-1; 0.002 s-1).
The interaction of the E2 form with Pi can be described by Scheme 2. The dissociation constant for the enzyme–Pi complex is KP = k-1/k1, and k2 and k-2 are the rate constants for formation and hydrolysis, respectively, of the covalent bond. The markedly reduced rate of dephosphorylation found for E183A, relative to wild type (Fig. 2), could in principle be explained by a decrease of k-1 or k-2, or an increase of k2. In the wild type, the presence of dimethyl sulfoxide reduces both k-1 and k-2 (25). At pH 7.0 and in the absence of dimethyl sulfoxide (as in Fig. 2 A), k-1 is, however, much higher than k2 and k-2 (25), and therefore the effect of the mutation on the rate of dephosphorylation can be ascribed to a change in the covalent step.
Scheme 2.

To further investigate the functional properties of the E2/E2P forms of mutant E183A, we studied the Pi concentration dependence of phosphorylation with Pi. Under conditions similar to those of the dephosphorylation assay in Fig. 2B (15% dimethyl sulfoxide present), the wild type displayed a K0.5 (ligand concentration giving half-maximum effect) for Pi of 54 μM, whereas a 4-fold reduction of apparent affinity (increase of K0.5) was seen for E183A (Fig. 3A, open symbols). In the presence of 30% dimethyl sulfoxide, the difference was more pronounced: 8- to 9-fold reduction of apparent affinity for E183A relative to wild type (Fig. 3A, filled symbols). These findings are surprising in light of the above-described increased stability of E2P in E183A (Fig. 2). From Scheme 2, it can be deduced that the Pi concentration giving half-maximum phosphorylation is
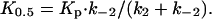 |
[1] |
Changes to these constants that, as discussed above, can explain the markedly reduced rate of dephosphorylation found for E183A (Fig. 2) are predicted to result in a decrease of K0.5 for Pi. The fact that the opposite is found, as shown in Fig. 3A, suggests that in addition to a decrease of k-2, there is also a reduced rate of formation of the covalent bond between Asp-351 and the phosphate (i.e., reduced k2) in E183A, and/or a reduction of the true affinity for Pi (increase of KP). This was further examined by studying the time course of phosphorylation with 32Pi. As seen in Fig. 3B, the phosphorylation with 32Pi proceeded at least 15-fold slower for E183A as compared with wild type. Under similar conditions, but at equilibrium (Fig. 3A; dimethyl sulfoxide concentration 15%), there was only a small difference between the saturation of mutant and wild-type enzyme with Pi, both being >50% saturated. An increase in dimethyl sulfoxide concentration to 30% did not increase the rate of phosphorylation further (data not shown), suggesting that Pi binding is not rate limiting in either case. Overall, the results are strongly suggestive that the mutation inhibits both forward and reverse covalent steps (k2 and k-2).
Fig. 3.

Phosphorylation from 32Pi. The reaction was performed at 25°Cin100 mM Mes/Tris (pH 6.0), 2 mM EGTA, 10 mM MgCl2, 15% (vol/vol) (open symbols) or 30% (vol/vol) (filled symbols) dimethyl sulfoxide, and either varying concentrations of 32Pi for 10 min (A)orat0.5mM 32Pi for the indicated time intervals (B). In A, the lines show the best fits of the Hill equation, EP = EPmax · [Pi]n/(K0.5 + [Pi]n) to the data, giving the K0.5 values and Hill numbers, respectively, indicated in parentheses: open circles, wild type (54 μM; 0.95); open triangles, E183A (222 μM; 0.67); filled circles, wild type (11 μM; 0.93); filled triangles, E183A (87 μM; 0.70). In each case, the 100% value corresponds to the phosphorylation level at infinite Pi concentration, as deduced from the fit. In B, the lines show the best fits of a monoexponential function, giving the rate constants indicated in parentheses: circles, wild type (0.73 s-1); triangles, E183A (0.048 s-1). In each case, the 100% value corresponds to the phosphorylation level reached at infinite time, as deduced from the fit.
Rate of Phosphoenzyme Formation from ATP and the Ca2+ Binding Properties. To study the kinetics of phosphorylation of E1Ca2 with ATP, quench flow methodology (21, 22) was applied. Fig. 4A shows the time course of phosphorylation from [γ-32P]ATP of enzyme preequilibrated with Ca2+ at 25°C. As demonstrated (21), the data obtained with the wild type can be reproduced by a computer simulation of the simplified reaction cycle shown in Scheme 1, using the following rate coefficients: k1 = 35 s-1, k2–3 = 5 s-1, and k4 = 25 s-1 (numbering of reaction steps as indicated in Scheme 1 with the E1PCa2 → E2P transition and the hydrolysis of E2P being combined into a single dephosphorylation step with rate coefficient k2–3). For E183A, the data could be satisfactorily fitted by using the rate coefficients k1 = 30 s-1, k2–3 = 1 s-1, and k4 = 7 s-1, i.e., in accordance with the reduced rate of forward dephosphorylation seen in Fig. 1 A, and suggesting a reduced rate of the E2 → E1Ca2 transition and a rather wild-type-like rate of the phosphorylation of E1Ca2 with ATP.
Fig. 4.

Time course of phosphorylation at 25°C from [γ-32P]ATP of enzyme preincubated with (A) and without (B)Ca2+, and of Ca2+ dissociation determined by loss of ability to phosphorylate (C). The experiments were performed by use of quench-flow instrumentation with mixing protocols as described (21, 22). (A) Enzyme preincubated in 40 mM Mops/Tris (pH 7.0), 80 mM KCl, 5 mM MgCl2, and 100 μM CaCl2 was mixed with an equal volume of the same buffer containing 10 μM [γ-32P]ATP, followed by acid quenching at the indicated time intervals. The lines show computer simulations based on Scheme 1, giving rate constants for the E1Ca2 → E1PCa2 reaction step of 35 s 1 for the wild type and 30 s-1 for E183A (see text for simulation details). (B) Enzyme preincubated in 80 mM KCl, 5 mM MgCl2, 2 mM EGTA, and either 40 mM Mops/Tris (pH 7.0; open symbols) or 40 mM Mes/Tris (pH 6.0; filled symbols) was mixed with an equal volume of 80 mM KCl, 5 mM MgCl2, and 2.2 mM CaCl2,10 μM[γ-32P]ATP, and either 40 mM Mops/Tris (pH 7.0; open symbols) or 40 mM Mes/Tris (pH 6.0; filled symbols), followed by acid quenching at the indicated time intervals. The lines show the best fits of a monoexponential function, giving the rate constants indicated in parentheses: open circles, wild type at pH 7 (20.9 s-1); filled circles, wild type at pH 6 (6.3 s-1); open triangles, E183A at pH 7 (7.7 s-1); filled triangles, E183A at pH 6 (3.2 s-1). (C) Microsomes preincubated in 40 mM Mes/Tris (pH 6.0), 80 mM KCl, 5 mM MgCl2, and 100 μM CaCl2 were mixed with an equal volume of 40 mM Mes/Tris (pH 6.0), 80 mM KCl, 5 mM MgCl2, and 4 mM EGTA. At the indicated time intervals, the double volume of 40 mM Mes/Tris (pH 6.0), 80 mM KCl, 5 mM MgCl2, 2 mM EGTA, and 10 μM [γ-32P]ATP was added, followed by acid quenching 34 ms later. To obtain the point corresponding to zero time, 4 mM EGTA was replaced by 100μM CaCl2. The lines show the best fit of a monoexponential decay function, giving the rate constants indicated in parentheses: circles, wild type (2.9 s-1); triangles, E183A (3.0 s-1).
The slight “overshoot” of phosphorylation seen for the wild type in Fig. 4A arises from the accumulation of a significant amount of E2 at steady state, owing to rate limitation by the E2 → E1Ca2 transition. The fact that a similar overshoot is apparent for E183A is surprising in light of the blocked dephosphorylation reaction, requiring inhibition of the E2 → E1Ca2 step for E2 to be accumulated. To examine E2 → E1Ca2 more directly, the time course of phosphorylation was also studied after simultaneous addition of [γ-32P]ATP and Ca2+ to Ca2+-deprived enzyme. This experiment was conducted both at pH 6 and 7 (Fig. 4B, filled and open symbols, respectively). In the wild type, the E2 → E1Ca2 transition is the rate-limiting step in the E2 → E1Ca2 → E1PCa2 reaction sequence, and more so at pH 6 than at pH 7, owing to the low rate of proton dissociation from E2 (26). Therefore, the apparent rate constant for phosphorylation determined for the wild type from the data in Fig. 4B is significantly lower (6 and 21 s-1 at pH 6 and 7, respectively) than the rate constant of 35 s-1 determined for E1Ca2 → E1PCa2 as shown in Fig. 4A. For E183A, the apparent rate of phosphorylation of the Ca2+-deprived enzyme was lower (2–3-fold) than that of the wild type at both pH values (Fig. 4B), demonstrating that the E2 → E1Ca2 transition is moderately slowed in the mutant.
To assess the properties of the Ca2+ binding sites of E1, we first determined the Ca2+ dependence of steady-state phosphorylation from ATP. In good agreement with previous data for the same mutant (14), we found only a slight difference in apparent Ca2+ affinity for wild type and E183A (K0.5 values of 0.95 μM and 0.77 μM, respectively, 0°C, pH 7, 5 μM [γ-32P]ATP). As demonstrated previously by computer simulation (27), a slight decrease of the K0.5 should indeed be expected when phosphoenzyme turnover is reduced, simply because lower Ca2+ concentrations are required for the phosphoenzyme to accumulate under these conditions. The rate of Ca2+ dissociation from E1Ca2 was then determined by using a method that takes advantage of the dependence of the reaction with ATP on the Ca2+ occupancy of the sites (22, 27). Because of this dependence, the ability to phosphorylate from ATP disappears at a rate corresponding to the rate of Ca2+ dissociation, when EGTA is added to remove Ca2+. As seen in Fig. 4C, similar Ca2+ dissociation rates were obtained for wild type and E183A. Thus, the function of the Ca2+ binding sites seems rather unaffected by the E183A mutation.
Vanadate Binding. Because the dephosphorylation of E2P and the reverse phosphorylation of E2 with Pi were both slow in E183A, we considered the possibility that E183 plays a catalytic role and that the phosphoryl transition state complex might be less tightly bound in the mutant relative to wild type. This hypothesis was tested by measuring the affinity for vanadate, which is an analog of the pentacoordinated transition state of the phosphoryl group, binding to the E2 form (28). This was accomplished by using an assay for vanadate inhibition of phosphorylation from [γ-32P]ATP (22). The enzyme is preequilibrated with varying concentrations of orthovanadate in the absence of a phosphorylating substrate and Ca2+, and the level of phosphorylatable vanadate-free enzyme is subsequently tested by addition of excess Ca2+ and [γ-32P]ATP at 0°C, where the dissociation of vanadate is very slow in the wild type. As seen in Fig. 5, the K0.5 value for vanadate inhibition of phosphorylation is almost three orders of magnitude higher in E183A as compared with wild type.
Fig. 5.

Vanadate inhibition of phosphorylation from [γ-32P]ATP. The enzyme was incubated for 1 h at 25°C and subsequently 15 min at 0°C in 40 mM Mops/Tris (pH 7.0), 80 mM KCl, 5 mM MgCl2, 2 mM EGTA, and the indicated concentration of orthovanadate. Phosphorylation was then carried out by sequential addition of 2.5 mM CaCl2 and 5 μM[γ-32P]ATP at 0°C, followed by acid quenching 15 s later. The maximum level of phosphorylation obtained in the absence of vanadate was taken as 100%. The lines show the best fits of the equation EP = EPmax · (1 - [vanadate]n/(K0.5 + [vanadate]n)), giving the K0.5 values indicated in parentheses: circles, wild type (0.17 μM); triangles, E183A (≈250 μM).
Discussion
Comparison of the E1Ca2 and E2 crystal structures of the Ca2+-ATPase shows large rearrangements of both cytoplasmic and transmembrane sectors. In the E1Ca2 crystal structure, domain A is isolated from the other cytoplasmic domains, P and N, whereas in E2, domain A has approached domains P and N, with E183 of the highly conserved TGES motif in domain A now being close to the phosphorylated aspartate (D351) and other catalytically important residues of domain P (see, e.g., figure 1 of ref. 8). This movement poses the intriguing question of the functional role of E183. On the basis of the results presented here, it is clear that the presence of E183 at the catalytic site is required for the enzyme to exert its phosphatase function, i.e., the hydrolysis of E2P, as demonstrated by the data in Fig. 2, showing a conspicuous block of dephosphorylation of E2P in mutant E183A. On the other hand, E183 is not required for the kinase function of the enzyme, as seen from the normal rate of phosphorylation from ATP, i.e., the E1Ca2 → E1PCa2 reaction (Fig. 4A). These findings suggest that a major rearrangement of domain A, similar to that deduced from the E1Ca2 and E2 crystal structures, occurs also in relation to the E1PCa2 → E2P transition of the phosphoenzyme, bringing E183 in position for participation in catalysis in E2/E2P forms. The idea that phosphatase activity of P-type ATPases depends on the glutamate of the highly conserved TGES motif in domain A (previously denoted “β-strand domain”), in fact, dates back to the first mutagenesis studies conducted with proton pumping yeast ATPase (16), but it is not until now that it has been substantiated by direct measurement of a low rate of E2P hydrolysis in mutant enzyme. The reduced rate of E2P hydrolysis was not detected in a previous Ca2+-ATPase mutagenesis study, in which it was concluded that a slow E1PCa2 → E2P transition causes the lack of transport activity in E183A (14). The demonstration of a block of E2P hydrolysis in the present study prompted a more detailed characterization of the E1PCa2 → E2P transition, and in addition to the measurements illustrated in Fig. 1 A, the assay illustrated in Fig. 1B was introduced, directly demonstrating a modest, 2-fold reduction of the rate of E1PCa2 → E2P. Likewise, the rate of the E2 → E1Ca2 transition was found moderately (2- to 3-fold) reduced in the present study (Fig. 4B). From this we conclude that E183 is of some, but not crucial, importance for the docking and release of domain A at the catalytic site, and that the role of E183 in the phosphatase function is far more critical.
Importantly, we found that in mutant E183A not only is the dephosphorylation of E2P blocked but also the reverse reaction, the phosphorylation of E2 with Pi (Fig. 3B). Furthermore, a three orders of magnitude reduction of the affinity for vanadate was detected (Fig. 5). The latter is not caused by a displacement of the E1–E2 equilibrium in favor of E1, away from the vanadate-reactive E2 state, as our rapid kinetic studies of the E2 → E1Ca2 transition indicated the opposite: accumulation of E2 in the absence of Ca2+ and a slow transition to E1Ca2 (Fig. 4B), with normal Ca2+ binding properties of E1Ca2 (Fig. 4C). According to classic transition-state theory (29, 30), the catalytic rate of an enzyme depends on the stability of the enzyme–transition state complex, increasing with the affinity of the enzyme for the transition state complex and decreasing in cases where the transition state complex is bound less tightly. Because vanadate is considered an analog of the presumed pentacoordinated phosphoryl transition state complex in the hydrolysis/formation of E2P, the low affinity for vanadate is consistent with the hypothesis that the reduced rate of catalysis in E183A is caused by defective interaction of the enzyme with one of the oxygens of the transition state complex. The deduced atomic structure of E2–V (9) suggests that the side chain of E183 is close to an apical oxygen of the vanadate on the opposite side to the phosphorylated D351. Further insights into the transition-state structure may be obtained from examination of the atomic structures of related enzymes. The active site of P-type ATPases shares structural similarities with other phosphotransferases, such as phosphoserine phosphatase, which have a Rossmann fold corresponding to domain P but lack domain A with the TGES sequence present in P-type ATPases. The atomic structure of a phosphoserine phosphatase has been determined with the transition-state analogue AlF3. It reveals a pentacoordinated Al atom with an attacking water molecule and carboxyl oxygen of the phosphorylated aspartate at each apex (31). The water molecule is hydrogen-bonded to another aspartate (equivalent to T353 of Ca2+-ATPase) and a water molecule, held in position by a glutamate, E20, from a domain outside the Rossmann fold. In the Ca2+-ATPase, E183 may perform a function similar to E20, fixing a critical water molecule that precisely positions the attacking water. Alternatively, if closer to the latter, E183 may act as the general acid/base to activate the water, or, if further away, may hold other critical amino acid side chains, such as neighboring T353. The very large effect of E183A on the apparent affinity for vanadate could suggest that E183 directly coordinates the apical oxygen, in line with E183 directly interacting with the attacking water.
As an alternative possibility for explaining the importance of E183 for catalysis of hydrolysis/formation of E2P it might also be suggested that in E2/E2P, but not in E1/E1P, this residue ligates the Mg2+ ion involved in the catalysis (32).
Recently, the reaction cycle of a Na+,K+-ATPase mutant with substitution of the threonine of the conserved TGES segment for alanine was characterized (15). Interestingly, this mutation caused changes in the catalytic function of E2/E2P qualitatively very similar to those reported here for Ca2+-ATPase mutant E183A, although much less marked (5-fold reduced rate of E2P dephosphorylation, 4-fold reduced intrinsic affinity of E2 for vanadate), and also with no effect on catalysis in E1/E1P (15). These findings could indicate that also the threonine plays a role, although minor relative to the glutamate, in stabilization of the transition state complex in E2/E2P.
Acknowledgments
We thank Karin Kracht and Lene Jacobsen for expert technical assistance. Dr. David H. MacLennan kindly provided the cDNA encoding mutant E183A. This work was supported in part by grants from the Danish Medical Research Council, the Lundbeck Foundation, the Novo Nordisk Foundation, and the Research Foundation of Aarhus University.
Abbreviations: A, actuator; K0.5, ligand concentration giving half-maximum effect; Mops, 3-(N-morpholino)propanesulfonic acid; N, nucleotide binding; P, phosphorylation.
References
- 1.de Meis, L. & Vianna, A. L. (1979) Annu. Rev. Biochem. 48, 275-292. [DOI] [PubMed] [Google Scholar]
- 2.Inesi, G., Sumbilla, C. & Kirtley, M. E. (1990) Physiol. Rev. 70, 749-776. [DOI] [PubMed] [Google Scholar]
- 3.MacLennan, D. H., Rice, W. J. & Green, N. M. (1997) J. Biol. Chem. 272, 28815-28818. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan, J. H. (2002) Annu. Rev. Biochem. 71, 511-535. [DOI] [PubMed] [Google Scholar]
- 5.Morsomme, P., Slayman, C. W. & Goffeau, A. (2000) Biochim. Biophys. Acta 1469, 133-157. [DOI] [PubMed] [Google Scholar]
- 6.Serrano, R. (1988) Biochim. Biophys. Acta 947, 1-28. [DOI] [PubMed] [Google Scholar]
- 7.Toyoshima, C., Nakasako, M., Nomura, H. & Ogawa, H. (2000) Nature 405, 647-655. [DOI] [PubMed] [Google Scholar]
- 8.Toyoshima, C. & Nomura, H. (2002) Nature 418, 605-611. [DOI] [PubMed] [Google Scholar]
- 9.Xu, C., Rice, W. J., He, W. & Stokes, D. L. (2002) J. Mol. Biol. 316, 201-211. [DOI] [PubMed] [Google Scholar]
- 10.Andersen, J. P., Vilsen, B., Collins, J. H. & Jørgensen, P. L. (1986) J. Membr. Biol. 93, 85-92. [DOI] [PubMed] [Google Scholar]
- 11.Danko, S., Yamasaki, K., Daiho, T., Suzuki, H. & Toyoshima, C. (2001) FEBS Lett. 505, 129-135. [DOI] [PubMed] [Google Scholar]
- 12.Seto-Young, D., Bandell, M., Hall, M. & Perlin, D. S. (1998) J. Biol. Chem. 273, 18282-18287. [DOI] [PubMed] [Google Scholar]
- 13.Goldshleger, R. & Karlish, S. J. D. (1997) Proc. Natl. Acad. Sci. USA 94, 9596-9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clarke, D. M., Loo, T. W. & MacLennan, D. H. (1990) J. Biol. Chem. 265, 14088-14092. [PubMed] [Google Scholar]
- 15.Toustrup-Jensen, M. & Vilsen, B. (2003) J. Biol. Chem. 278, 11402-11410. [DOI] [PubMed] [Google Scholar]
- 16.Portillo, F. & Serrano, R. (1988) EMBO J. 7, 1793-1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Portillo, F. (1997) FEBS Lett. 402, 136-140. [DOI] [PubMed] [Google Scholar]
- 18.Maruyama, K. & MacLennan, D. H. (1988) Proc. Natl. Acad. Sci. USA 85, 3314-3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vilsen, B., Andersen, J. P., Clarke, D. M. & MacLennan, D. H. (1989) J. Biol. Chem. 264, 21024-21030. [PubMed] [Google Scholar]
- 20.Sørensen, T., Vilsen, B. & Andersen, J. P. (1997) J. Biol. Chem. 272, 30244-30253. [DOI] [PubMed] [Google Scholar]
- 21.Sørensen, T. L., Dupont, Y., Vilsen, B. & Andersen, J. P. (2000) J. Biol. Chem. 275, 5400-5408. [DOI] [PubMed] [Google Scholar]
- 22.Clausen, J. D. & Andersen, J. P. (2003) Biochemistry 42, 2585-2594. [DOI] [PubMed] [Google Scholar]
- 23.Kaufman, R. J., Davies, M. V., Pathak, V. K. & Hershey, J. W. (1989) Mol. Cell. Biol. 9, 946-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Meis, L., Martins, O. B. & Alves, E. W. (1980) Biochemistry 19, 4252-4261. [DOI] [PubMed] [Google Scholar]
- 25.Seekoe, T., Peall, S. & McIntosh, D. B. (2001) J. Biol. Chem. 276, 46737-46744. [DOI] [PubMed] [Google Scholar]
- 26.Forge, V., Mintz, E. & Guillain, F. (1993) J. Biol. Chem. 268, 10961-10968. [PubMed] [Google Scholar]
- 27.Andersen, J. P., Sørensen, T. L., Povlsen, K. & Vilsen, B. (2001) J. Biol. Chem. 276, 23312-23321. [DOI] [PubMed] [Google Scholar]
- 28.Cantley, L. C., Jr., Cantley, L. G. & Josephson, L. (1978) J. Biol. Chem. 253, 7361-7368. [PubMed] [Google Scholar]
- 29.Pauling, L. (1946) Chem. Eng. News 24, 1375-1377. [Google Scholar]
- 30.Fersht, A. (1985) Enzyme Structure and Mechanism (Freeman, New York), 2nd Ed., pp. 1-475.
- 31.Wang, W., Cho, H. S., Kim, R., Jancarik, J., Yokota, H., Nguyen, H. H., Grigoriev, I. V., Wemmer, D. E. & Kim, S.-H. (2002) J. Mol. Biol. 319, 421-431. [DOI] [PubMed] [Google Scholar]
- 32.Patchornik, G., Goldshleger, R. & Karlish, S. J. D. (2000) Proc. Natl. Acad. Sci. USA 97, 11954-11959. [DOI] [PMC free article] [PubMed] [Google Scholar]