

Abstract
Background
The major morbidity of hemophilia is bleeding induced hemophilic arthropathy (HA) which once established may not be interrupted completely even by prophylactic clotting factor replacement. Specific therapies to oppose inflammatory cytokines, including Interleukin 6 (IL-6) receptor antagonists, have become important in the management of inflammatory arthritides.
Objectives
We investigated combining therapy using MR16-1, a rat IgG antibody directed against mouse IL-6 receptor (anti-IL-6R), with factor VIII (FVIII) replacement to protect against bleeding induced arthropathy in hemophilia A mice.
Methods
Three recurrent hemarthroses were induced in the knee joint capsule of factor VIII knockout mice. Treatment at the time of each hemorrhage included either: No treatment; FVIII replacement given at the time of hemorrhage; FVIII replacement at hemorrhage plus anti-IL-6R as four weekly injections; FVIII replacement with non-specific control antibody (rat IgG); anti-IL-6R alone without FVIII replacement. Six weeks following the first hemarthosis joints were harvested and histopathology was scored for synovitis, for cartilage integrity and for macrophage infiltration.
Results
Animals that received anti-IL-6R as an adjunct to FVIII replacement demonstrated the best survival and the least acute joint swelling and pathology on histologic examination of synovium and cartilage (P<0.05 for each parameter). All histopathologic parameters in the mice receiving FVIII+anti-IL-6R were limited and were comparable to findings in injured hemostatically normal mice. The major benefits of adjunctive anti-IL-6R were decreasing synovial hyperplasia, hemosiderin deposition and macrophage infiltration.
Conclusions
Short-course specific inhibition of inflammatory cytokines as an adjunct to replacement hemostasis may be an approach to minimize hemophilic joint degeneration.
Keywords: IL-6, anti-IL-6, anti-cytokine, hemophilia, hemarthrosis, hemophilic arthropathy, MR16-1
Introduction
Hemophilia is an inherited bleeding disorder that results from deficient activity of blood clotting factor VIII (hemophilia A) or factor IX (hemophilia B) [1]. The major disease-related morbidity of hemophilia is hemophilic arthropathy (HA), a progressive destruction of joints that results from recurrent bleeding into the joint space [1, 2]. Pathological changes involving synovial hyperplasia, infiltration and proliferation of inflammatory cells, neoangiogenesis, and osteochondral destruction are its hallmarks. Extravasation of blood components into the joint space, in particular erythrocyte-derived heme iron and monocytes/macrophages, induces arthritis with both inflammatory and degenerative features [3]. Monocytes/macrophages recruited to the area along with accompanying inflammatory cytokines interleukin 6 (IL-6), interleukin 1(IL-1), tumor necrosis factor-alpha (TNF- α) increase inflammatory response in the joints [3, 4]. The hyperplastic synovium is at risk for recurrent cycles of target joint hemorrhage [5, 6].
Standard treatment of bleeding episodes is intravenous replacement of the deficient clotting factor. Prompt, early treatment with adequate dosage of clotting factor concentrate can effectively halt hemorrhage. Nevertheless, even without recurrent bleeding into the joint space, inflammatory processes are incited by intraarticular blood that continue degenerative changes for weeks following a bleeding episode; the inflammatory component of the disease may become chronically present [3, 5, 7, 8]. Once HA is established, the pathologic changes to cartilage and bone are irreversible [3]. Prophylaxis with clotting factor replacement starting at a young age may decrease the frequency of joint hemorrhage and the incidence of joint damage. However, recurrent/break-through joint bleeding and the possibility of degeneration of HA persist in some patients despite preventive prophylactic replacement [9–11]. Innovative therapies that can be used as an adjunct to clotting factor replacement to prevent this common and serious complication could play an important role.
IL-6 is a multifunctional cytokine that possesses several proinflammatory properties. It is central in the pathogenesis of several arthritis models [12, 13]. In rheumatoid arthritis (RA), IL-6 promotes synovitis by inducing neovascularization, infiltration of inflammatory cells, and synovial hyperplasia [14, 15]. It augments osteoclast formation and stimulates the production of matrix metalloproteinases (MMPs) resulting in degeneration of bone and cartilage [15]. IL-6, along with several cytokines and inflammatory mediators, including TNF-α, interferon-gamma (IFN-γ), vascular endothelial growth factor (VEGF), IL-1β, monocyte chemotactic protein-1 (MCP-1) have been implicated in blood-induced joint damage in hemophilia [4, 16]. Moreover, the production of IL-6 is significantly increased in synovium of HA patients [8].
MR16-1, a rat anti-mouse IL-6 receptor (anti-IL-6R) that blocks the IL-6 signaling pathway, has proved effective in decreasing inflammatory changes in mouse models of many autoimmune and inflammatory diseases [13, 17–19]. Preclinical efficacy of anti-IL-6R approaches led to the development of Tocilizumab (TCZ), a humanized anti-human IL-6R, which is presently used in RA patients and other inflammatory diseases [20]. However, the beneficial effects of opposing IL-6 have never been explored in hemophilia. This is the first report investigating combining hemostatic replacement factor with opposing inflammatory cytokine (IL-6) to protect against bleeding induced arthropathy in hemophilia A.
Materials and Methods
Mice
Eight- to fourteen-week-old factor VIII knock-out (FVIII −/−) mice generated by gene targeting (E16 FVIII B6;129S4-F8tm1kaz) were originally supplied by Dr. H. H. Kazazian Jr. (University of Pennsylvania, PA, USA) [21] and bred in house. Ten- to twelve-week-old C57BL/6J wild-type (WT) mice were purchased from Jackson Labs (Bar Harbor, ME). All investigations were performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill. Mice were anesthetized using Isoflurane/O2 for all procedures. All blood samples were collected from the retro-orbital plexus into 1:9 parts 3.2% citrated sodium, centrifuged to obtain platelet-poor plasma, and the plasma was stored at −80°C.
Treatment antibodies
MR16-1, rat anti-mouse IL-6R monoclonal IgG1 antibody, was a gift from Chugai Pharmaceutical (Tokyo, Japan). The characteristics of this antibody have been previously reported. [18, 22] Control IgG (purified from the serum of nonimmunized rats) was purchased from Jackson ImmunoResearch (West Grove, PA,). Dosage and regimen were chosen based on previous data generated in various mouse models by Mihara M et al. [13, 17].
Induction and treatment of recurrent hemarthrosis
Although the histopathologic features that develop within the hemophilic mouse joint following exposure to blood closely resemble those seen in human hemarthropathy, the FVIII −/− mice rarely bleed spontaneously in the joints [23]. Recurrent hemarthroses were produced by three sequential induced joint hemorrhages. Intra-articular hemorrhage was induced as previously described [24–26] by introducing a 30.5 gauge needle into the left knee joint capsule through the infra-patellar ligament; although not relevant therapeutically, 5 microliters of normal saline was introduced to maintain parameters identical to our previous report using this model [26]. Joint hemorrhages were induced on days 0, 7 and 14 to emulate recurrent hemarthrosis.
The dosage of FVIII treatment was selected based on a preliminary study demonstrating that plasma from FVIII knockout mice that receive a dose of human recombinant factor VIII 150 U kg−1 (Advate™; Baxter Bioscience, Deerfield, IL) via the retroorbital venous plexus has the same clotting time in seconds as does plasma from hemostatically normal wild type mice in an aPTT assay. Specifically, platelet poor plasma from 12 hemostatically normal WT mice was added to an equal volume aPTT reagent (Diagnostica Stago) and incubated for 3 minutes at 37 degrees C. Calcium chloride was then added and the time to form a clot was assayed on a ST-4 Coagulation Analyzer (Diagnostica Stago, Asniéres, France) and the time to clot was 32 seconds (range 31 to 33 seconds). We then injected separate groups of FVIII−/− mice with a variety of doses of factor VIII and collected plasma at 15 minutes after the dose to screen the clotting potential. The group of mice (N= 10) that received 150 IU/kg of human FVIII demonstrated a median clotting time in the aPTT assay of 30.9 seconds (range 26 seconds to 33 seconds). Additional hemophilic mice that received 150 U kg−1 FVIII as a preventive dose preceding needle injury survived the challenge with no significant joint swelling during 2 days follow-up (n=10). Of note, it has been previously shown that the circulating survival (half-life) of human factor VIII (Advate) is shorter in mice than in humans [27].
Experimental design
Treatments of FVIII −/− mice were assigned to 5 groups (Supplementary Fig. S1); Group 1) No Rx: No treatment after each injury; Group 2) FVIII alone: recombinant FVIII following induction of each injury; Group 3) FVIII + anti-IL-6R: FVIII following each injury and MR16-1 intraperitoneally 8 mg after 1st injury, 0.5 mg after 2nd, 3rd injury and 7 days after 3rd injury; Group 4) FVIII+control IgG: FVIII following each injury and intraperitoneal control IgG given intraperitoneally at the same dose and schedule as used for MR16-1; Group 5) Anti-IL-6R alone: mice received only MR16-1 without FVIII replacement. To simulate prompt “on demand” approach to clotting factor replacement, FVIII concentrate was given 2 minutes after bleeding was induced. MR 16-1 and control IgG were given immediately following FVIII. Treatment groups included equal numbers of male and female mice. The mean ages were equivalent for mice in each study group, varying from 10.6 ± 1.8 weeks in the FVIII + control IgG group to 11.2 ± 1.8 weeks in the Anti-Il-6R group at the time of the first injury. Groups were also balanced for mean weight, ranging from 23.2 ± 1.7 grams in the FVIII + anti-IL-6R group to 24.6 ± 1.6 grams in the FVIII alone group; doses of all drugs were calculated based on the weight of the individual mouse and not on mean weights of the study cohort.
Multiplex cytokine analysis
Peripheral blood was collected prior to injury and day 7. Plasma IL-1β, IL-6, interleukin 10 (IL-10), interleukin 17 (IL-17), TNF-α, MCP-1, vascular endothelial growth factor (VEGF), interleukin 4 (IL-4), IFN-γ concentration were measured on a Bio-Plex 200 system (Bio-Rad, Hercules, CA) using FMAP reagents from R&D Systems (Minneapolis, MN) according to manufacturer’s instructions. Curve-fitting for primary data concentration reporting was performed with the onboard Bio-Plex Manager v.5.0 software.
Joint diameter measurements
The diameters of the left knee joints were measured serially throughout the experiment. The measurements were performed using a digital caliper (Mitutoyu 547- 301, Kanagawa, Japan) from lateral to medial across the knee as previously described [25]. The diameter prior to the induction of first hemarthrosis was used as baseline to calculate the percentage change in joint diameter at later time points.
Histopathologic evaluation (Table S1)
Joint tissues were harvested on day 42. The knee joints were collected by sectioning the femur and tibia 1-cm from the joint, fixed, and decalcified using routine histological procedures [26]. The tissues were stained with hematoxylin and eosin (H&E) and Safranin-O. Histopathologic grading for synovitis used the previously validated murine hemophilic synovitis system of Valentino and Hakobyan [28]. Cartilage pathology was graded using the Modified Mankin’s score [29, 30]. At least three representative fields from an equatorial section of each knee were scored by two or more reviewers who were blinded to the experimental conditions and scores from each joint were averaged. The sum of the average synovitis and cartilage scores was calculated as the Global Score to represent the spectrum of bleeding-induced joint pathology. Macrophages were identified by immunostaining for CD68 with a rat anti-mouse F4/80 (Serotec, Raleigh, NC) at a 1:50 dilution, followed by biotinylated anti-rat IgG (Vector Labs, Burlingame, CA) at a 1:100 dilution, and scored for macrophage infiltration using Hoffman’s macrophage score, as previously described [31].
Anti-human FVIII Bethesda inhibitor assay
Plasma for FVIII Bethesda inhibitor antibody was evaluated in all FVIII treated groups on day 14 and 42 (prior to the 3rd injury and the day of sacrifice) using a START 4 Coagulation Analyzer, as previously described [32].
Statistical analysis
Data are expressed as mean ± SD for continuous variables and proportions for categorical variables. Differences between groups in continuous variables were examined for statistical significance using the Man-Whitney U test, while differences in proportions were examined by chi-square tests. Log-rank tests were used for overall comparison. Joint swelling in the percentage change of the diameters was compared using a generalized estimating equation (GEE) that counts dependence between outcomes in different follow-up times. P values less than 0.05 were considered significant. All of the analyses were conducted by IBM SPSS Statistics 19 (SPSS Inc., Chicago, IL) and SAS 9.2 (SAS Institute Inc., Cary, NC).
Results
Recurrent hemarthrosis were produced by three sequential induced joint hemorrhages. The repeated severe hemarthrosis resulted in a survival of 35% in untreated hemophilic mice, although hemostatically normal mice all survived. (Table 1) Treatment with anti-IL-6R alone (without hemostatic support) did not improve the survival (P=0.492, log rank test). Relative to the no treatment group, hemostatic support with FVIII significantly improved survival (P<0.01) to 88%. The addition of anti-IL-6R with FVIII normalized survival.
Table 1.
Survival rate
| Group | N | Survival (%) | P | |
|---|---|---|---|---|
| 1 | No treatment | 20 | 7 (35) | Reference |
| 2 | FVIII alone | 17 | 15 (88.2) | 0.001 |
| 3 | FVIII + anti-IL-6R | 15 | 15 (100) | <0.001 |
| 4 | FVIII + control IgG | 10 | 9 (90) | 0.008 |
| 5 | Anti-IL-6R alone | 20 | 8 (40) | 0.492 |
| 6 | Injured WT | 10 | 10 (100) | 0.002 |
Survival rate was evaluated on day 42 following the first injury. P values for significant difference compared to the “No treatment” group were calculated using log-rank test.
MR16-1 interference with IL-6 receptor increases circulating plasma IL-6
A panel of plasma cytokines were measured prior to injury and at day 7 prior to the second injury. Plasma from 16 uninjured and untreated FVIII −/− mice demonstrated a baseline circulating IL-6 level of 5.6 pg/ml. Hemophilic mice surviving a single induced hemarthrosis had significant elevations of plasma IL-6 at day 7 (to 88.4 ± 22.6 pg ml−1). This elevation was partially corrected if mice received FVIII replacement at the time of injury. (Fig. 1) The highest IL-6 levels were seen in mice treated with MR16-1, reflecting the action of the antibody to block the physiologic clearance of IL-6 via interaction with IL-6 receptor [33]. Compared to levels prior to hemarthrosis, there were no measurable differences at day 7 for any other cytokines evaluated, including IL-4, VEGF, and TNF-α (data not shown).
Figure 1. Plasma IL-6 levels are affected by intraarticular hemorrhage, hemostatic replacement and IL-6 receptor inhibition.
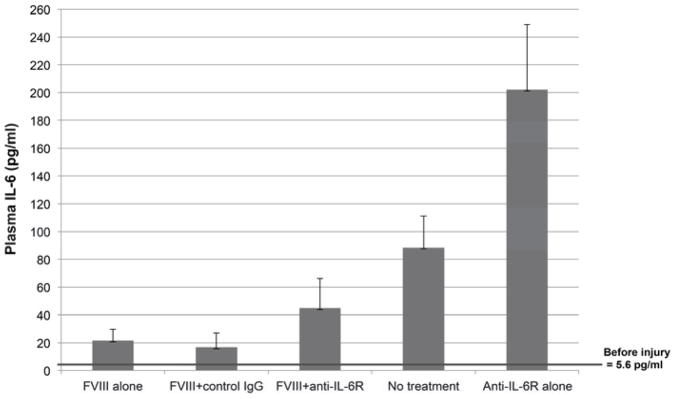
Plasma IL-6 increased significantly after injury. Plasma IL-6 was measured by multiplex cytokine analysis on day 7 prior to the second induced hemarthrosis. Each column and vertical line indicates mean and SD. Baseline plasma IL-6 levels in a group of uninjured hemophilia A mice were 5.6 pg ml−1 prior to the first injury and increased to 88.4 ± 22.6 pg ml−1 at day 7 if the mice received no treatment. Treatment with FVIII concentrate resulted in less severe IL-6 elevations. The higher levels of IL-6 in the anti-IL-6R alone group compared with no treatment and in the FVIII+anti-IL-6R group compared with FVIII alone reflect the effect of anti-IL-6R to inhibit IL-6R mediated IL-6 clearance, with the effect of increasing free plasma IL-6.
FVIII inhibitor formation
The E16 FVIII knockout mice reliably form neutralizing alloantibodies (inhibitors) following exposure to the xenoprotein human FVIII [23]. After two doses of recombinant human FVIII concentrate, one third of mice developed low titer FVIII inhibitors (less than 5 Bethesda Units) by day 14, regardless of treatment group. Following the three FVIII treatment doses essentially all mice developed high titer circulating inhibitors measured at the day 42 sacrifice (Table 2).
Table 2.
FVIII inhibitor formation
| Day 14 | Day 42 | |||
|---|---|---|---|---|
|
| ||||
| Incidence | Titer median (range) | Incidence | Titer median (range) | |
| FVIII + anti-IL-6R | 31.2% | 1.85 (1.4–8) | 93.75% | 126 (4.12–928) |
| FVIII alone | 29.4% | 1.6 (1.05–3.1) | 93.3% | 98.7 (1.46–448) |
| FVIII + control IgG | 33.3% | 0.6 (0.4–1.75) | 100% | 120 (20.6–264) |
| HA control | 44.4% | 3.35 (0.97–6.2) | 100% | 105.6 (7.8–240) |
FVIII inhibitor titers were reported in Bethesda Unit. There was no statistically significant difference among groups for either incidence or titer of inhibitors. HA control = Hemophilia A mice that received retroorbital FVIII 150 U kg−1weekly × 3 without inducing hemarthrosis (n=9). The nonparametric Mann-Whitney test was used to assess significance between groups.
Combined FVIII and anti-IL-6R therapy minimizes joint swelling following hemarthrosis
Hemostatically normal WT mice exhibited no measurable joint swelling at any time point (Fig. 2a). All hemophilic mice, regardless of treatment, developed markedly increased joint diameter after a single hemarthrosis, with the exception of mice that received both FVIII and anti-IL-6R. Following two hemorrhages, adjunctive anti-cytokine therapy with hemostatic support significantly prevented swelling when compared to FVIII support alone (P<0.001 versus FVIII alone, estimated by GEE method); 14/15 animals in the adjunctive therapy group had no measurable swelling following two hemorrhages. By the time of the day 14 hemarthrosis, 5/15 mice in the FVIII + anti-IL-6R group developed inhibitors and the response to the induced hemarthrosis was different for this subset of mice in that the third joint injury (induced following inhibitor development) was associated with marked swelling. The subset of mice that received adjunctive anti-cytokine therapy with FVIII and remained free of inhibitors at day 14 (10/15 mice) had no significant increase in joint diameter (Fig. 2b).
Figure 2. Joint swelling following hemarthrosis is affected by treatment choice.


Joint diameters were measured serially and reported as average percentage change in diameter compared to pre-injury baseline in hemostatically normal mice (orange line) and in hemophilic mice. On days 0, 7, and 14 measurements were collected immediately prior to induced hemarthrosisis 2a) Mice that received adjunctive anti-IL-6R with FVIII therapy (green dashed line) demonstrated less joint swelling than animals receiving either hemostatic therapy or anti-cytokine therapy alone. 2b) The total group of mice that received adjunctive anti-IL-6R + FVIII (green dashed line in Figure 2a and 2b) included a subset of mice with measurable circulating FVIII inhibitor at the time of the third joint hemorrhage. The swelling following hemarthrosis is shown separately for mice that were inhibitor positive on day 14 (red line) and those that were free of inhibitors at the time of all joint hemorrhages (inhibitor negative, blue line).
Adjunctive anti-IL-6R therapy reduces pathological sequelae of bleeding induced arthropathy on both synovium and cartilage
Following recurrent hemarthroses, all untreated hemophilia A mice that survived the bleeding challenges developed severe synovitis, as evaluated at six week following the first hemorrhage (Fig. 3a, 3b). The inter-reader correlation on the scoring of histopathology was good (R2 > 0.96). The average score for synovitis was 7.6 ± 0.5 out of a maximum score of 10 in the Valentino grading system. Microscopic findings demonstrated obvious proliferation of the synovial lining cells and subsynovial neoangiogenesis along with hemosiderin staining and synovial villous formation. (Fig. 3c) Pits and erosions of the articular cartilage surface were present in some mice. The measured changes in pathology score for cartilage were considerably smaller than those for the synovitis score; the average elevation in Mankin’s score for cartilage was 1.6 ± 0.6 and the average global score for whole joint evaluation was 9.1 ± 0.9. Treatment with FVIII replacement reduced the degree of synovitis. However, the pathological changes were still significantly different from the pathology in normal hemostatic mice (P<0.05). The Mankin’s score associated with FVIII treatment compared to no treatment was not statistically significant. We found no statistical difference in any histological parameters between FVIII alone and FVIII+control IgG, indicating that there was no protection from the development of pathologc outcome resulting from a non-specific action of IgG.
Figure 3. Pathological sequelae of bleeding induced arthropathy on synovium and cartilage and effect of therapy.
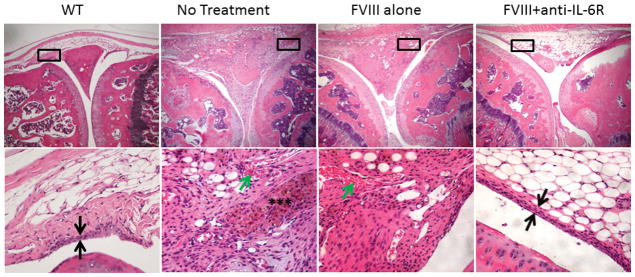


Histopathologic examination was performed at the time of sacrifice at six weeks following the first hemarthrosis.
a) H&E staining of injured knees; Original magnification of upper images 40×; black boxes indicate location of detail in corresponding lower image (200×). Black arrows indicate width of synovial lining layer, normally <4 cell layers. Green arrows: neoangiogenesis. Black asterixes: hemosiderin. Shown, Left to Right:WT) Wild type: no significant pathological changes, no synovitis or minimal synovitis; No Treatment) Joint space filled with proliferative, hemosiderin-stained, hypervascular synovium; FVIII alone) moderate subsynovial thickening and neoangiogenesis; FVIII+anti IL-6R) no significant pathological changes, no synovitis or minimal synovitis was observed.
b) Synovitis was scored using the Murine Hemophilic Synovitis grading system described by Valentino and Hakobyan. Cartilage pathology was scored using the Modified Mankin’s score. For each experimental animal these scores were summed to generate a Global Hemophilic Arthropathy score and the mean score from all animals in a treatment group that survived to day 42 is plotted (see also details in Supporting Table S1)
c) The individual defects that contribute to the composite Synovitis Grade are plotted for each treatment approach. The major effects of adjunctive anti-IL-6R therapy on synovium were reducing synovial hyperplasia and hemosiderin deposition. Each bar indicates mean and SD.*=P<0.05 versus injured WT mice by Mann-Whitney U test. ‡=P<0.05 comparing score in WT mice or FVIII+anti-IL-6R to FVIII therapy alone by Mann-Whitney U test
d) A subset of mice in each treatment group that included FVIII replacement demonstrated measurable circulating factor VIII inhibitor by the time of the third induced hemarthrosis. The synovitis graded at sacrifice (Valentino score) is plotted separately for the subsets of mice receiving each treatment approach based upon the presence (dark grey bars) or absence (light grey) of FVIII inhibitor at the time of the third joint hemorrhage. Each bar indicates mean and SD. * = P<0.05 by Mann Whitney U test
Despite equivalent rates of inhibitor formation between groups, combined therapy using FVIII+ anti-IL-6R significantly reduced pathological changes after hemarthrosis compared with treatment with FVIII alone. The average synovitis score, Mankin’s score and Global score were significantly lower: 2.67 ± 0.65, 0.27 ± 0.15 and 2.93 ± 0.68 respectively, for the combined therapy compared to 5.27 ± 0.71, 1.80 ± 0.68 and 7.07 ± 1.22 respectively, for the FVIII treatment alone (P<0.05). (Fig. 3b) All histological scores of FVIII+ anti-IL-6R group were comparable to hemostatically normal mice. Five of fifteen mice (33.3%) exhibited no sequelae at all on synovium (Valentino’s synovial score 0). The major effects of adjunctive anti-IL-6R therapy on synovium were decreasing synovial hyperplasia and diminishing hemosiderin deposition (P<0.05 versus FVIII alone) (Fig. 3c). Adjunctive anti-IL-6R therapy was associated with a significantly lower risk of cartilage pathology (OR=0.17 95%CI 0.03–0.85, P=0.032).
Another major effect of adjunctive anti-IL-6R therapy was reduced macrophage accumulation in synovium at the time of sacrifice (six weeks after first hemarthrosis). As shown in figure 4, CD68 staining demonstrated the lowest average macrophage score following combined therapy. There was no significant difference between the macrophage score of FVIII+anti-IL-6R and hemostatically normal mice. (P=0.43)
Figure 4. Macrophage accumulation in synovial tissues.


a) Macrophage accumulation was evaluated by CD68 staining and scored using a 4-point scale as described in “Material and Method”. Each column and vertical bar indicates mean and SD. * = P<0.05 versus injured WT by Mann Whitney U test.
b) Immunohistochemical staining of CD68+ macrophages in subsynovial space (staining brown) A) WT hemostatically normal (score 0); B), No treatment: multiple clusters/sheets of CD68+ cells (score 4); C) FVIII alone: macrophages scattered/touching (score 1) D) FVIII+antiIL-6R: No subsynovial macrophage accumulation (score 0)
Thirty to forty percent of mice in each treatment group that included human Factor VIII therapy developed inhibitors that were measurable immediately prior to the third joint hemorrhage; these mice would be expected to have impaired hemostatic potential of the infused FVIII, and so a separate analysis was performed to explore the influence of FVIII inhibitor antibody formation to the treatment outcomes. Regardless of whether an inhibitor was present or not, the mice that received FVIII alone or received FVIII with non-specific IgG developed a similar degree of synovitis (Fig. 3d), as did the mice that received FVIII plus anti-IL-6R but had Bethesda inhibitors. None of these groups of mice could be considered to have the benefit of restoration of hemostasis with inhibition of inflammatory cytokine. Only the subset that were free of factor VIII inhibitors and so could benefit from combined therapy FVIII with anti-IL-6R were protected from the development of synovitis. Almost no pathological changes were demonstrated; discoloration with hemosiderin was observed in only one mouse, and the average synovitis score, Mankin’s score and Global score were limited to 1.6 ± 0.72, 0.1 ± 0.1 and 1.7 ± 0.7 respectively. The pathological outcomes of the subset that benefitted from unimpaired FVIII replacement with adjunctive anti-cytokine therapy were equivalent to the outcome in non-hemophilic mice.
Discussion
The current study examines the hypothesis that the hemophilic joint that is exposed to recurrent hemorrhage will be optimally protected if, in addition to hemostatic therapy, specific adjunctive therapy is added that opposes inflammation. The hemophilic arthropathy that develops following exposure of the joints to blood has histopathologic features that resemble both inflammatory arthritis (e.g. rheumatoid arthritis) and degenerative arthritis (e.g. osteoarthritis). Upregulation of inflammatory cytokines as a hallmark of synovitis is exhibited in both hemophilic arthritis and rheumatoid arthritis [4]. In vitro studies have demonstrated comparable increases of catabolic cytokines such as IL-1, IL-6 and TNF-α in synovial tissue from patients with hemophilia arthropathy and RA [4]. The standard treatment of hemophilic synovitis addresses the congenital deficiency of clotting activity by replacing coagulation factors in response to bleeding and/or prophylactically. Although there is evidence that the synovitis in hemophilia is mediated by the action of inflammatory cytokines [3, 4], the current study is the first to take a pharmacologic approach to specifically oppose proinflammatory cytokine activity (IL-6 activity) as an adjunctive therapy to complement hemostatic support.
IL-6 plays several crucial roles in pathogenesis of inflammatory arthritis [12, 34]. IL-6 is secreted by a variety of cells, including T cells, B cells, monocytes, and fibroblasts and it possesses several biological activities that include regulation of immune response, inflammation, and hematopoiesis [12]. IL-6 exerts its biological activities through two molecules, an IL-6-specific receptor (IL-6R) and a membrane-bound signal transducer, gp130 [34]. When IL-6 binds to IL-6R (IL-6/IL-6R) and forms the complex with gp130, intracellular signaling pathways are activated that result in the promotion of T cell proliferation, B cells, megakaryocytes, macrophages differentiation and regulation of Th17/Treg cells balance [12, 34, 35]. Anti-IL-6R agents (MR16-1 or TCZ) inhibit the binding of IL-6 to IL-6R and thus block IL-6 intracellular signaling. The efficacy of TCZ to slow or prevent progressive joint degeneration in inflammatory arthritis has been demonstrated in multiple randomized clinical trials, in both adult and pediatric patients [36–39].
In the treatment of rheumatic spectrum disorders, cytokine inhibiting drugs are typically used on a chronic basis, including drugs that oppose TNF-α, IL1-β, or IL-6, [38]. We wished to examine the ability of short-course cytokine inhibition (without prolonged suppression) to alter the degenerative course that follows recurrent joint hemorrhage (target joint bleeding). Although factor VIII knockout mice are widely used in the study of hemophilia A, spontaneous joint bleeding is not commonly observed in this strain. Several groups have demonstrated, however, that if a bleeding episode is induced in the hemophilic mouse joint, histopathologic changes develop in the mouse joint that closely mimic those seen in human hemarthropathy [24–26, 40] and these changes are specifically associated with exposure of the joint to hemorrhage because they are not seen following a single joint capsule puncture in mice with normal clotting factor activity (wild type mice) [26].
IL-6 measurements in the MR16-1 treated mice gave a clear demonstration that the monoclonal antibody therapy had a biologic effect. Plasma IL-6 was higher in mice that received MR16-1, which is the expected result. Physiologic IL-6 clearance is triggered when IL-6 forms a complex with soluble IL-6R (IL-6/sIL-6R complex). MR16-1 competitively binds sIL-6R and thus inhibits IL-6 clearance [33]. Joint hemorrhage (reflected by joint swelling present throughout the first week after injury) resulted in increased plasma IL-6 levels, although the elevation was partly ameliorated by prompt factor VIII treatment. While the principal purpose of the cytokine analysis was to document, as was demonstrated, the measurable effect of MR16-1 upon IL-6, a panel of additional cytokines was measured at day 7 after injury. No detectable effect of MR16-1 administration upon circulating levels of other cytokines was measurable at that time point. It is likely that assaying at day 7 alone does not provide an informative picture of cytokine changes following joint hemorrhage and that assaying multiple timepoints would be required to develop a full profile of such events. Although such a profile of changes is beyond the scope of these investigations, Sen and colleagues have recently published just such an analysis of mRNA and cytokine protein changes at time points ranging from 1 hour to 60 days following this induced hemarthrosis challenge in hemophilic mice [40].
Hemostatic therapy, given promptly after bleeding, was required to avoid synovitis in this hemophilic model – cytokine inhibition alone as therapy for recurrent joint hemorrhage was associated with mortality, joint swelling and pathology that were no different from the untreated animals. Although FVIII replacement at the dose of 150 U kg−1 effectively rescued survival of the hemophilic mice, if anti-IL-6R was not administered, joint swelling was detected from day 3 onward (Fig. 2a,). The protection from joint swelling did not appear to result from enhanced initial hemostasis, i.e. by a smaller initial volume of extravasated blood immediately distending the joint, but rather by opposing the inflammatory response to hemorrhage. In separate studies performed in hemophilia B mice we have observed that on day 3 following a single joint capsular puncture, the red blood cells begin to disappear from the joint space; in contrast, synovial proliferation and macrophage infiltration begin at this timepoint. (Junjiang Sun, unpublished observation). In the current studies, only the combination of prompt hemostatic support and short-course anti-cytokine therapy resulted in normalization of swelling (days 1–13, Figure 2), synovitis pathology score (Figure 3b–d4a–c), and global score (Figure 3b) to values seen in non-hemophilic joints. Moreover, combining anti-IL-6R with factor replacement did not overcome the detrimental effect of inhibitor formation (Figures 2b, 3d). Mice with evidence of initial protection by FVIII combined with anti-IL-6R but with subsequent evidence of factor VIII neutralization by the time of the third injury were not protected from later joint distention. The development of this complication in a subset of mice further supports the conclusion that combination therapy is optimal, in that initial protection afforded by combination therapy was lost when effective hemostasis was lost.
Parallels that support this conclusion have been observed in hemophilia B mice exposed to the same joint bleeding challenge and treated with cytokine inhibition that targets TNF-α instead of IL-6 [41]. We have observed that hemophilia B mice (but not hemostatically normal mice) that receive three induced knee hemorrhages develop considerable synovitis despite episodic replacement with factor IX at the time of bleeding. The development of synovitis is much improved if a short course of TNF-α receptor blockade with Etanercept (ten doses given over 20 days) is initiated with the first hemarthrosis or prior to the second hemarthrosis. (Sun J, et al. manuscript submitted.) It is interesting to note that hemophilia B mice that were exposed to factor IX (FIX) during active hemarthrosis developed inhibitors at a low incidence that was eliminated in the groups receiving the combination of factor IX replacement with the anti-cytokine Etanercept [41]. Given the established property of IL-6 as a regulator of the balance between IL-17-producing Th17 cells and regulatory T cells [19, 35, 42], the current study recorded closely the development of factor VIII inhibitor antibodies in the study groups. The overall incidence, the rate of development, and the median titers of inhibitors did not differ between treatment groups, regardless of MR16-1 therapy. It must be acknowledged that human factor VIII is a strongly immunogenic xenoprotein in E16 hemophilic mice [23]. This is only one of a number of significant differences that complicates direct comparison of the results in the studies of Etanercept+FIX compared to the anti-IL-6R + FVIII studies; other differences from the FVIII studies include the significantly lower immunogenicity of human FIX in hemophilia B mice (C57Bl/6 strain) [23], the longer half-life of FIX, and the greater penetration of FIX into the joint space [43], where it may have local hemostatic action [26]. Nevertheless, the finding of improved protection from synovitis by combining hemostatic support with inflammatory cytokine inhibition in two different experimental systems supports further investigation of this strategy.
The effects of blood exposure upon the hemophilic joint may affect all three components of the joint: the synovium, the cartilage, and the subchondral bone. The cytokines IL-1 and TNF-α are known to be key inflammatory mediators of hemophilic arthropathy [5, 44]. These same cytokines, along with IL-17, have been demonstrated to stimulate RA synovial fibroblasts to produce large amounts of IL-6, and that IL-6 in turn stimulated further proliferation of synovial fibroblastic cells in the presence of sIL-6R [14, 15]. Furthermore, IL-6-sIL-6R trans-signaling is necessary to induce receptor activator of NF-κB ligand (RANKL) expression in synovial cells by TNF-α and IL-17 [15]. The central role of NF-κB-responsive inflammatory cytokine genes has recently been emphasized by other investigators using the same experimental model of hemophilic arthropathy as used in this study [40]. RANKL stimulates osteoclast activity, survival and bone resorption and enhances the hypertrophic process of synovium [44], putting the cartilage at risk for direct synovial invasion and replacement. Anti-IL-6R potentially may protect synovium, cartilage and bone via the inhibition of these biological activities of IL-6 and concurrently blocking the signaling of TNF-α, IL-1, and IL-17 that occurs through IL-6/sIL-6R. Of the components of the global joint evaluation performed in this study, the greatest improvements that were achieved by combining cytokine inhibition with hemostasis were in the areas of diminished synovial proliferation, hemosiderin deposition, macrophage infiltration, and to a lesser degree cartilage erosion. It is possible that if the global arthropathy evaluation were performed at different time points other differences would be seen. In particular, it is conceivable that if hemosiderin deposition within joints were heavy then synovial inflammation, although held in check during the course of the acute therapy with the anti-cytokine drug, might be stimulated to appear at later time points after discontinuation of the anti-IL-6R. With this in mind it is interesting to note that hemosiderin deposition was lower in the combined treatment group, so that the load of this potential chronic irritant was diminished. It is not possible to speculate whether hemosiderin load was decreased due to an effect upon the amount of acute hemorrhage, the amount of late rebleeding, or the efficiency of macrophage/monocyte scavenging of hemosiderin deposits.
Reactive infiltrating macrophages and activated macrophage-like synoviocytes, via pro-inflammatory cytokines including IL-1 and TNF-α, contribute to synovial and cartilage destruction in hemophilic arthropathy and fuel subacute and chronic intraarticular inflammation [5, 44]. It is worth noting that in another hemophilic mouse model of wound healing in hemophilia, the residence of macrophages in a dermal wound in hemophilic mice is greatly prolonged when compared to hemostatically normal mice receiving an identical injury [31]. The examination in our study was done at a subacute/early chronic period following recurrent hemarthroses, and if the hemophilic mice were not treated, abundant resident macrophages were detected in the subsynium at that stage, but not seen in wounded hemostatically normal mice. Combining anti-IL-6R with factor replacement therapy (but neither treatment alone) nearly normalized the degree and incidence of synovial macrophage accumulation. IL-6 has been shown to contribute to the perpetuation of chronic inflammation by augmenting continuous production of monocyte chemotactic protein-1 (MCP-1) from endothelial cells, mononuclear cells, and synovial cells [12, 34]. Ovlisen, et al, using a mouse model nearly identical to the model used in this study, showed elevated concentrations of IL-6 and monocyte chemotactic protein-1 (MCP-1) in the synovial fluid following induced hemarthrosis [45]. These data suggest that IL-6 promotes the local inflammatory reaction by amplifying inflammatory cell infiltration. Combined anti-IL-6R and factor replacement may normalize the macrophage accumulation.
As has been recently reviewed [46], there is limited human clinical trial data on the use of anti-inflammatory drugs (NSAID and corticosteroids) in combination with hemostasis for hemophilia. Direct inhibition of cytokines including TNF-α, IL-1, and IL-6 has proved efficacious in inflammatory arthritis that is resistant to other disease-modifying anti-rheumatoid drugs (DMARDS). TCZ in particular has several practical advantages for the type of short-term adjunctive treatment modeled in this study. Although previous experience with MR16-1 in mice guided the choice of a weekly dosing schedule for this study, the human analogue TCZ is dosed clinically on a monthly schedule (adults) or a twice monthly schedule (children). Efficacy and safety have been established in large scale clinical trials in inflammatory arthritis conducted in both adult and pediatric populations [36–39]. The factor VIII treatment in our study modeled an on-demand approach to treating hemophilic joint bleeding episodes and did not model the efficacy of regular prophylactic dosing of FVIII (either given alone or in combination with anti-cytokine). Institution of FVIII prophylaxis in the preschool years is the pediatric standard of care for prevention of hemarthropathy in most resource-rich countries. There exists data on the long-term use (at least 12 weeks and up to several years of therapy) in children as young as 2 years of age undergoing treatment for juvenile idiopathic arthritis, with a favorable risk-benefit profile [37, 39]. Safety and efficacy data in children less than 2 years old is not established. Clotting factor with adjunctive anti-IL-6R given in much shorter courses than reported in these human trials may deserve investigation for hemophilic children over 2 years of age who have breakthrough bleeding during FVIII prophylaxis as well as those receiving on demand therapy.
Critical to the appropriate application of adjunctive therapy will be an improved understanding of factors that distinguish which individuals (likely the majority of the severe hemophilia population) are well protected by prophylactic factor replacement regimens and at relatively low risk of rapid joint degeneration and which individuals are at increased risk of early or rapid deterioration following hemarthroses, and thus may benefit from opposing inflammation along with replacement of hemostasis. This distinction between these subsets of patients was recently underscored by the prospectively generated results of the Joint Outcome Study, in which some boys who reported multiple joint bleeding episodes remained free of MRI evidence of disease, while others with minimal or no reported hemarthroses nevertheless manifested synovial and osteochondral pathology [10]. Efforts to define predictors that distinguish individuals at increased risk of hemophilic joint deterioration are being pursued, including genetic markers or biomarkers of increased arthritis risk. It is possible that variable expression of polymorphic cytokine genes are among the underlying risk factors associated with joint deterioration, just as they have been associated with increased inhibitor risk in hemophilia [47, 48].
Acknowledgments
The authors would like to thank Dr. David Barrow of Cytokine Analysis Facility, School of Dentistry, University of North Carolina at Chapel Hill for making available the cytokine data.
Tragically, Masahiko Mahara died during the conduct of these investigations. The authors are grateful for the advice and for the support Dr. Mahara provided for this work. The authors also appreciate the contribution of enthusiasm and intellectual commitment from Dr. Linda Yamamoto.
Footnotes
Disclosure of Conflict of Interests
Masahiko Mahara was an employee of Chugai Pharmaceuticals. The other authors report no conflicts of interest related to this report.
References
- 1.Ahlberg A. Haemophilia in Sweden.7. Incidence Treatment and Prophylaxis of Arthropathy and Other Musculo-Skeletal Manifestations of Haemophilia a and B. Acta Orthop Scand. 1965;S:7. doi: 10.3109/ort.1965.36.suppl-77.01. [DOI] [PubMed] [Google Scholar]
- 2.Dunn AL. Pathophysiology, diagnosis and prevention of arthropathy in patients with haemophilia. Haemophilia. 2011;17:571–8. doi: 10.1111/j.1365-2516.2010.02472.x. [DOI] [PubMed] [Google Scholar]
- 3.Lafeber FPJG, Miossec P, Valentino LA. Physiopathology of haemophilic arthropathy. Haemophilia. 2008;14:3–9. doi: 10.1111/j.1365-2516.2008.01732.x. [DOI] [PubMed] [Google Scholar]
- 4.Roosendaal G, Vianen ME, Wenting MJ, van Rinsum AC, van den Berg HM, Lafeber FP, Bijlsma JW. Iron deposits and catabolic properties of synovial tissue from patients with haemophilia. J Bone Joint Surg Br. 1998;80:540–5. doi: 10.1302/0301-620x.80b3.7807. [DOI] [PubMed] [Google Scholar]
- 5.Roosendaal G, Lafeber FP. Pathogenesis of haemophilic arthropathy. Haemophilia. 2006;12:117–21. doi: 10.1111/j.1365-2516.2006.01268.x. [DOI] [PubMed] [Google Scholar]
- 6.Raffini L, Manno C. Modern management of haemophilic arthropathy. Br J Haematol. 2007;136:777–87. doi: 10.1111/j.1365-2141.2007.06490.x. [DOI] [PubMed] [Google Scholar]
- 7.Roosendaal G, TeKoppele JM, Vianen ME, van den Berg HM, Lafeber FP, Bijlsma JW. Blood-induced joint damage: a canine in vivo study. Arthritis Rheum. 1999;42:1033–9. doi: 10.1002/1529-0131(199905)42:5<1033::AID-ANR24>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 8.Roosendaal G, van Rinsum AC, Vianen ME, van den Berg HM, Lafeber FPJG, Bijlsma JWJ. Haemophilic arthropathy resembles degenerative rather than inflammatory joint disease. Histopathology. 1999;34:144–53. doi: 10.1046/j.1365-2559.1999.00608.x. [DOI] [PubMed] [Google Scholar]
- 9.Lofqvist T, Nilsson IM, Berntorp E, Pettersson H. Haemophilia prophylaxis in young patients--a long-term follow-up. J Intern Med. 1997;241:395–400. doi: 10.1046/j.1365-2796.1997.130135000.x. [DOI] [PubMed] [Google Scholar]
- 10.Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, Ingram JD, Manco-Johnson ML, Funk S, Jacobson L, Valentino LA, Hoots WK, Buchanan GR, DiMichele D, Recht M, Brown D, Leissinger C, Bleak S, Cohen A, Mathew P, Matsunaga A, Medeiros D, Nugent D, Thomas GA, Thompson AA, McRedmond K, Soucie JM, Austin H, Evatt BL. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–44. doi: 10.1056/NEJMoa067659. [DOI] [PubMed] [Google Scholar]
- 11.Pettersson H, Nilsson IM, Hedner U, Norehn K, Ahlberg A. Radiologic evaluation of prophylaxis in severe haemophilia. Acta Paediatr Scand. 1981;70:565–70. doi: 10.1111/j.1651-2227.1981.tb05741.x. [DOI] [PubMed] [Google Scholar]
- 12.Hashizume M, Mihara M. The roles of interleukin-6 in the pathogenesis of rheumatoid arthritis. Arthritis. 2011;2011:765624. doi: 10.1155/2011/765624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida H, Hashizume M, Mihara M. IL-6 blockade preferentially inhibits Th17 differentiation in collagen-induced arthritis. Rheumatol Int. 2011;31:127–31. doi: 10.1007/s00296-010-1552-9. [DOI] [PubMed] [Google Scholar]
- 14.Mihara M, Moriya Y, Kishimoto T, Ohsugi Y. Interleukin-6 (IL-6) induces the proliferation of synovial fibroblastic cells in the presence of soluble IL-6 receptor. Br J Rheumatol. 1995;34:321–5. doi: 10.1093/rheumatology/34.4.321. [DOI] [PubMed] [Google Scholar]
- 15.Hashizume M, Hayakawa N, Mihara M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology (Oxford) 2008;47:1635–40. doi: 10.1093/rheumatology/ken363. [DOI] [PubMed] [Google Scholar]
- 16.Acharya SS. Exploration of the pathogenesis of haemophilic joint arthropathy: understanding implications for optimal clinical management. Br J Haematol. 2012;156:13–23. doi: 10.1111/j.1365-2141.2011.08919.x. [DOI] [PubMed] [Google Scholar]
- 17.Mihara M, Shiina M, Nishimoto N, Yoshizaki K, Kishimoto T, Akamatsu K. Anti-interleukin 6 receptor antibody inhibits murine AA-amyloidosis. J Rheumatol. 2004;31:1132–8. [PubMed] [Google Scholar]
- 18.Takagi N, Mihara M, Moriya Y, Nishimoto N, Yoshizaki K, Kishimoto T, Takeda Y, Ohsugi Y. Blockage of interleukin-6 receptor ameliorates joint disease in murine collagen-induced arthritis. Arthritis Rheum. 1998;41:2117–21. doi: 10.1002/1529-0131(199812)41:12<2117::AID-ART6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 19.Hohki S, Ohguro N, Haruta H, Nakai K, Terabe F, Serada S, Fujimoto M, Nomura S, Kawahata H, Kishimoto T, Naka T. Blockade of interleukin-6 signaling suppresses experimental autoimmune uveoretinitis by the inhibition of inflammatory Th17 responses. Exp Eye Res. 2010;91:162–70. doi: 10.1016/j.exer.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 20.Navarro-Millan I, Singh JA, Curtis JR. Systematic review of tocilizumab for rheumatoid arthritis: a new biologic agent targeting the interleukin-6 receptor. Clin Ther. 2012;34:788–802. doi: 10.1016/j.clinthera.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10:119–21. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 22.Okazaki M, Yamada Y, Nishimoto N, Yoshizaki K, Mihara M. Characterization of anti-mouse interleukin-6 receptor antibody. Immunol Lett. 2002;84:231–40. doi: 10.1016/s0165-2478(02)00202-x. [DOI] [PubMed] [Google Scholar]
- 23.Sabatino DE, Nichols TC, Merricks E, Bellinger DA, Herzog RW, Monahan PE. Animal models of hemophilia. Progress in molecular biology and translational science. 2012;105:151–209. doi: 10.1016/B978-0-12-394596-9.00006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hakobyan N, Enockson C, Cole AA, Sumner DR, Valentino LA. Experimental haemophilic arthropathy in a mouse model of a massive haemarthrosis: gross, radiological and histological changes. Haemophilia. 2008;14:804–9. doi: 10.1111/j.1365-2516.2008.01689.x. [DOI] [PubMed] [Google Scholar]
- 25.Ovlisen K, Kristensen AT, Valentino LA, Hakobyan N, Ingerslev J, Tranholm M. Hemostatic effect of recombinant factor VIIa, NN1731 and recombinant factor VIII on needle-induced joint bleeding in hemophilia A mice. J Thromb Haemost. 2008;6:969–75. doi: 10.1111/j.1538-7836.2008.02954.x. [DOI] [PubMed] [Google Scholar]
- 26.Sun J, Hakobyan N, Valentino LA, Feldman BL, Samulski RJ, Monahan PE. Intraarticular factor IX protein or gene replacement protects against development of hemophilic synovitis in the absence of circulating factor IX. Blood. 2008;112:4532–41. doi: 10.1182/blood-2008-01-131417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elm T, Karpf DM, Ovlisen K, Pelzer H, Ezban M, Kjalke M, Tranholm M. Pharmacokinetics and pharmacodynamics of a new recombinant FVIII (N8) in haemophilia A mice. Haemophilia. 2012;18:139–45. doi: 10.1111/j.1365-2516.2011.02608.x. [DOI] [PubMed] [Google Scholar]
- 28.Valentino LA, Hakobyan N. Histological changes in murine haemophilic synovitis: a quantitative grading system to assess blood-induced synovitis. Haemophilia. 2006;12:654–62. doi: 10.1111/j.1365-2516.2006.01348.x. [DOI] [PubMed] [Google Scholar]
- 29.Mankin HJ, Dorfman H, Lippiello L, Zarins A. Biochemical and metabolic abnormalities in articular cartilage from osteo-arthritic human hips. II. Correlation of morphology with biochemical and metabolic data. J Bone Joint Surg Am. 1971;53:523–37. [PubMed] [Google Scholar]
- 30.Kuroki H, Nakagawa Y, Mori K, Ohba M, Suzuki T, Mizuno Y, Ando K, Takenaka M, Ikeuchi K, Nakamura T. Acoustic stiffness and change in plug cartilage over time after autologous osteochondral grafting: correlation between ultrasound signal intensity and histological score in a rabbit model. Arthritis Res Ther. 2004;6:R492–504. doi: 10.1186/ar1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffman M, Harger A, Lenkowski A, Hedner U, Roberts HR, Monroe DM. Cutaneous wound healing is impaired in hemophilia B. Blood. 2006;108:3053–60. doi: 10.1182/blood-2006-05-020495. [DOI] [PubMed] [Google Scholar]
- 32.Waters B, Qadura M, Burnett E, Chegeni R, Labelle A, Thompson P, Hough C, Lillicrap D. Anti-CD3 prevents factor VIII inhibitor development in hemophilia A mice by a regulatory CD4+CD25+-dependent mechanism and by shifting cytokine production to favor a Th1 response. Blood. 2009;113:193–203. doi: 10.1182/blood-2008-04-151597. [DOI] [PubMed] [Google Scholar]
- 33.Uchiyama Y, Yoshida H, Koike N, Hayakawa N, Sugita A, Nishimura T, Mihara M. Anti-IL-6 receptor antibody increases blood IL-6 level via the blockade of IL-6 clearance, but not via the induction of IL-6 production. Int Immunopharmacol. 2008;8:1595–601. doi: 10.1016/j.intimp.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Fonseca JE, Santos MJ, Canhao H, Choy E. Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun Rev. 2009;8:538–42. doi: 10.1016/j.autrev.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 35.Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. 2010;40:1830–5. doi: 10.1002/eji.201040391. [DOI] [PubMed] [Google Scholar]
- 36.Hashimoto J, Garnero P, van der Heijde D, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Yoshikawa H, Nishimoto N. Humanized anti-interleukin-6-receptor antibody (tocilizumab) monotherapy is more effective in slowing radiographic progression in patients with rheumatoid arthritis at high baseline risk for structural damage evaluated with levels of biomarkers, radiography, and BMI: data from the SAMURAI study. Mod Rheumatol. 2011;21:10–5. doi: 10.1007/s10165-010-0325-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yokota S, Imagawa T, Mori M, Miyamae T, Aihara Y, Takei S, Iwata N, Umebayashi H, Murata T, Miyoshi M, Tomiita M, Nishimoto N, Kishimoto T. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;371:998–1006. doi: 10.1016/S0140-6736(08)60454-7. [DOI] [PubMed] [Google Scholar]
- 38.Nishimoto N, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Azuma J. Long-term safety and efficacy of tocilizumab, an anti-IL-6 receptor monoclonal antibody, in monotherapy, in patients with rheumatoid arthritis (the STREAM study): evidence of safety and efficacy in a 5-year extension study. Ann Rheum Dis. 2009;68:1580–4. doi: 10.1136/ard.2008.092866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Benedetti F, Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, Cuttica R, Ravelli A, Schneider R, Woo P, Wouters C, Xavier R, Zemel L, Baildam E, Burgos-Vargas R, Dolezalova P, Garay SM, Merino R, Joos R, Grom A, Wulffraat N, Zuber Z, Zulian F, Lovell D, Martini A. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385–95. doi: 10.1056/NEJMoa1112802. [DOI] [PubMed] [Google Scholar]
- 40.Sen D, Chapla A, Walter N, Daniel V, Srivastava A, GRJ Nuclear Factor (NF)-kappaB and its associated pathways are major molecular regulators of blood induced joint damage in a murine model of hemophilia. J Thromb Haemost. 2012 Dec 11; doi: 10.1111/jth.12101. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 41.Sun J. TNF-alpha antagonists augment factor replacement to prevent arthropathy in hemophilic mice. J Thromb Haemost. 2009;7(Suppl 2):225 (abstract). [Google Scholar]
- 42.Yoshida H, Hashizume M, Mihara M. IL-6 blockade preferentially inhibits Th17 differentiation in collagen-induced arthritis. Rheumatol Int. 2011;31:127–31. doi: 10.1007/s00296-010-1552-9. [DOI] [PubMed] [Google Scholar]
- 43.Chang P, Aronson DL, Borenstein DG, Kessler CM. Coagulant proteins and thrombin generation in synovial fluid: a model for extravascular coagulation. Am J Hematol. 1995;50:79–83. doi: 10.1002/ajh.2830500202. [DOI] [PubMed] [Google Scholar]
- 44.Valentino LA. Blood-induced joint disease: the pathophysiology of hemophilic arthropathy. J Thromb Haemost. 2010;8:1895–902. doi: 10.1111/j.1538-7836.2010.03962.x. [DOI] [PubMed] [Google Scholar]
- 45.Ovlisen K, Kristensen AT, Jensen AL, Tranholm M. IL-1 beta, IL-6, KC and MCP-1 are elevated in synovial fluid from haemophilic mice with experimentally induced haemarthrosis. Haemophilia. 2009;15:802–10. doi: 10.1111/j.1365-2516.2008.01973.x. [DOI] [PubMed] [Google Scholar]
- 46.Forsyth AL, Rivard GE, Valentino LA, Zourikian N, Hoffman M, Monahan PE, Van Meegeren MER, Forriol F. Consequences of intra-articular bleeding in haemophilia: science to clinical practice and beyond. Haemophilia. 2012;18:112–9. doi: 10.1111/j.1365-2516.2012.02835.x. [DOI] [PubMed] [Google Scholar]
- 47.Lopez-Jimenez J, Soto-Padilla J. Association of Hemophilic Arthropathy with Genetic Markers Related to Bleeding Tendency, Joint Inflammation and Structural Cartilage. Haemophilia. 2012;18:116 (Abstract). [Google Scholar]
- 48.Pavlova A, Delev D, Lacroix-Desmazes S, Schwaab R, Mende M, Fimmers R, Astermark J, Oldenburg J. Impact of polymorphisms of the major histocompatibility complex class II, interleukin-10, tumor necrosis factor-alpha and cytotoxic T-lymphocyte antigen-4 genes on inhibitor development in severe hemophilia A. J Thromb Haemost. 2009;7:2006–15. doi: 10.1111/j.1538-7836.2009.03636.x. [DOI] [PubMed] [Google Scholar]