

Abstract
We attempt to understand the evolutionary origin of protein folds by simulating their divergent evolution with a three-dimensional lattice model. Starting from an initial seed lattice structure, evolution of model proteins progresses by sequence duplication and subsequent point mutations. A new gene's ability to fold into a stable and unique structure is tested each time through direct kinetic folding simulations. Where possible, the algorithm accepts the new sequence and structure and thus a “new protein structure” is born. During the course of each run, this model evolutionary algorithm provides several thousand new proteins with diverse structures. Analysis of evolved structures shows that later evolved structures are more designable than seed structures as judged by recently developed structural determinant of protein designability, as well as direct estimate of designability for selected structures by thermodynamic sampling of their sequence space. We test the significance of this trend predicted on lattice models on real proteins and show that protein domains that are found in eukaryotic organisms only feature statistically significant higher designability than their prokaryotic counterparts. These results present a fundamental view on protein evolution highlighting the relative roles of structural selection and evolutionary dynamics on genesis of modern proteins.
The wealth of data emerging from fully sequenced genomes and structural proteomics provide major insight into reconstruction of evolutionary history of protein domains (1). In particular, it was found that distributions of many properties observed in protein universe can be well fit by power law (2-4). We showed in our recent work that the observed power-law distribution stemming from domain structure comparison can be explained by evolutionary dynamics that models all proteins as diverging from one or few precursors (4). Our model succeeded in the quantitative description of power-law distribution in the degree similarity of protein domains (4). We make use of this recent success as a starting point for thinking about more concrete models describing the origins of modern protein domains. Many current models describing divergent evolution are formulated in abstract terms of protein domains or sequences as nodes of dynamically evolving graphs; as such, they tend to assign the observed inequalities in fold and sequence family size to pure evolutionary chance. It is therefore hard to evaluate how realistic these models are because they do not take into account the physical constraints imposed by the thermodynamics of the sequence-structure relationship in real proteins. Other researchers motivated mostly by arguments from protein physics proposed that structures of existing proteins are highly nonrandom. It was suggested (5-9) that one of the possible factors determining evolutionary success of a structure in evolutionary selection is its designability, i.e., its ability to accommodate numerous sequences that can fold stably into that structure.
Some have argued that the designability hypothesis implicitly assumes convergence as a major mechanism by suggesting that various sequences may converge to the same highly designable structures irrespective of their evolutionary history. Although the two views (evolutionary dynamics by divergence and the designability hypothesis) make valid points, both lack the necessary detail needed for evaluation of their relative correspondence with real domains. Attempts to reconcile the two views have been made in the past (9, 10). For example, Taverna and Goldstein (9), using a two-dimensional lattice model where all sequences and conformations of short chains can be exhaustively enumerated, found that for some evolutionary scenarios, where stability conditions were imposed, the ensemble of evolved lattice proteins was indeed enriched by more designable structures.
In this work, we address the question of the relative roles of chance and selection in protein evolution by simulating a more realistic divergent model of protein structure evolution. This version is based on a three-dimensional lattice model representation of protein structure. In the past, lattice models were instrumental in gaining fundamental insights into protein folding (9, 11-16). Despite their approximate character, they feature a unique sequence-structure relationship akin to that of real proteins (17). The major benefit of such models is that they are computationally tractable so that it is feasible to run a realistic evolutionary scenario that includes testing by direct kinetic simulations the ability of emerging proteins to fold and be stable. More details of the evolutionary algorithm are provided in Methods.
Methods
Lattice Model. We employ standard cubic lattice model where lattice amino acids occupy lattice sites and each site can be occupied by no more than one amino acid (15-17). Sequence neighbors occupy neighboring lattice sites. Only lattice amino acids that are in spatial contact, but are not sequence neighbors, can interact. Energy of each contact interaction is determined by the types of amino acids involved. We use the model with 20 types of amino acid and Miyazawa-Jernigan group potentials from table 6 of ref. 18. The Monte-Carlo folding algorithm is as described (17, 19) with move set including end moves, corner flips, and crankshaft moves. Every attempt to move a monomer is counted as a time step.
Evolutionary Algorithm. Our evolutionary model uses a cubic lattice of 36-mer as a basic model. It proceeds as follows:
Start from initial structure and design a sequence that stably folds into that structure with Monte-Carlo design in sequence space (17, 20). Check, with folding Monte-Carlo simulation in conformational space (17, 19), that the designed sequence does indeed stably fold into the target native structure.
Keeping target structure fixed, perform Monte-Carlo in sequence space (in the form of swaps as elementary move, to preserve amino acid composition). This step runs at a certain evolutionary temperature, Tevol, that has to be carefully selected (see below). This step creates sequence families providing divergence in sequence but not structural space.
Randomly select several evolved proteins and make gene duplication and point mutation attempts for each. Fold each of the new gene sequences several (10) times each, startingfrom a randomly generated random-coil conformation for a specified number of Monte-Carlo steps (106) each to determine whether a new sequence consistently and stably folds into any conformation within the fixed duration of folding run (foldability criterion). A sequence folds stably if in each folding run it ends in the same (native) lowest-energy conformation, and in each run it spends >15% of time in this conformation at T = 0.28 (stability criterion). The chosen temperature is quite high from the point of view of stability but makes folding particularly fast. (The requirement of stability was relaxed in one of the runs presented here.)
If the new sequence folds (and is stable as required in most but not all runs of evolutionary algorithm) to its native structure, as defined in step 3, the gene duplication attempt is accepted: a new protein is born; the next step proceeds from step 2.
If the new sequence fails to fold stably, the gene duplication attempt is rejected, and the new step proceeds from step 3, i.e., a new set of gene duplications is attempted.
Selection of Tevol that generates sequence families in step 2 is a delicate aspect of the evolutionary algorithm. Too low Tevol results in very stable sequences for the native structures; mutations at step 3 are very often accepted but new sequences would mostly fold into the same native structure providing very little or no structural divergence. On the contrary, very high Tevol generates essentially random sequences, so that attempted point mutations rarely result in a stably foldable sequence; no mutations are accepted at step 3 and the algorithm does not proceed. However, selecting Tevol in a certain range (0.1-0.12, with Miyazawa-Jernigan parameters for 20 amino acids from table 6 of ref. 18, as well as parameters from ref. 21) results in efficient evolutionary process that generates numerous novel stable proteins. In all cases, the amino acid compositions of evolved proteins are close to that of real proteins (see Figs. 5-8 and Table 1, which are published as supporting information on the PNAS web site). We also note that introduction of the sequence family creation Monte-Carlo sequence design step is motivated by the need to facilitate structural and sequence divergence; it may be not necessary if more computing power is available so that folding of each duplicated and mutated gene sequence can be tested.
Calculation of Designability by Thermodynamic Sampling of Sequence Space. To avoid an impossible task of exhaustive sampling of sequence space, we used thermodynamic approach to evaluate designability of structures presented in Fig. 3. To this end, we carried out long Monte-Carlo design simulations [keeping amino acid composition constant, i.e., with swap as elementary move (20)] in a range of temperatures starting from high initial temperature, T2 = 3.0, and decreasing temperature with increment 0.01 until a final low temperature 0.01 is reached. At each temperature, 5 × 106 Monte-Carlo design moves are made. Average energy E(T) is calculated at each temperature by direct averaging of energy of all sequences found at that temperature. Entropy in sequence space at a final temperature T1 is obtained from a general thermodynamic relation (17, 22, 23),
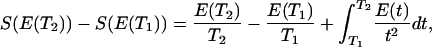 |
[1] |
in which the high-temperature value S(E(T2)) is close to the entropy of random sequences because at high T2 = 3 the algorithm generated essentially random sequences. Entropy of random sequences depends on composition only; it is given by equation 1 of ref. 22. Finally, from E(T) and S(T) temperature can be excluded, giving S(E) dependence. Designability, i.e., the number of sequences that fold into a structure with energy equal or lower than a given energy E, is obtained from S(E) by exponentiation.
Fig. 3.

(a) Initial (Left) and one of the evolved (Right) conformations. The evolved conformation is not maximally compact but has much greater higherorder normalized contact traces: TrC4/36 = 4.72(4.0), TrC6/36 = 34.79(16.39), and TrC8/36 = 234.2(62.3) (the numbers in parentheses correspond to starting structure shown in Left). (b) Designability: number of sequences that fit initial (diamonds) and one of the evolved (circles) structures with or below a given energy. The details of the calculation are given in Methods.
Calculation of Contact Density and Higher-Order Traces of Contact Maps. We adopt the definition of contact in proteins whereby two groups are in contact if the distance between their Cβ atoms (Cα for GLY) does not exceed 7.5 Å. The contact matrix (C) of a protein is defined such that Cij = 1 if groups i and j are in contact and 0 otherwise. The higher-order traces of C are obtained by recursive relation 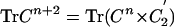 ,, where
,, where 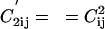 for off-diagonal elements and 0 otherwise. Making diagonal elements of C′2 zero allows to eliminate trivial contributions (like
for off-diagonal elements and 0 otherwise. Making diagonal elements of C′2 zero allows to eliminate trivial contributions (like 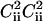 , etc.) into diagonal elements of Cn.
, etc.) into diagonal elements of Cn.
Results and Discussion
Our model is constructed to realistically simulate the “Big Bang” scenario of protein morphogenesis (4). Each run of the evolutionary algorithm starts from a single or few (no >10) seed compact proteins and proceeds to generate several thousand (3,000-15,000) “new” proteins. Many runs of the algorithm, starting from different seed proteins, were repeated and the results are similar between runs, so that here we present the analysis of two evolutionary runs each starting from the same seed structure. Proteins that evolved in the first run were required to fold and be stable in their native conformations (see Methods), whereas in the second run the stability requirement was relaxed; instead a new gene sequence was required to fold consistently to the same lowest-energy structure but no requirement was imposed on how long it should stay in the native conformation.
The runs started from one of the maximally compact 36-mer structures studied earlier in our work (24). Not surprisingly, during the first run of evolution under the stability requirement for the evolved sequences the algorithm generated many proteins with enhanced (compared to the initial seed structure) stability of their native states. For comparison, in the second run when the stability requirement was relaxed, evolution generated many structurally diverse proteins with higher native energies (Fig. 1a).
Fig. 1.

(a) Evolution of energy of the native conformations of proteins. Each data point represents average over 75 consecutive evolved structures. Squares correspond to the first run of evolutionary selection where stability condition is applied (see Methods); circles correspond to the second evolution run that starts from the same seed structure but where sequences are required to fold but not necessarily be stable in their native conformations. (b) Evolution of compactness of evolved structures as measured by TrC2. Thirty-six, the total number of monomers of the chain, normalize this quantity. TrC2 is double the total number of contacts. The data averaging and the meaning of symbols is as in a.(c) Evolution of TrC8 normalized by total number of monomers. This quantity reflects topological properties of a structure related to the number of uninterrupted closed long loops of length 8 that can be drawn on the system of contacts. It was calculated as trace of 8th power of contact matrix from which reduced elements, such as TrC4TrC4, etc., were subtracted (see Methods). The data averaging and the meaning of symbols are as in a.
We set out to determine the characteristics of evolved structures that separate them from initial and seed structures. A logical characteristic to probe is designability, the number of sequences that can fold into a structure. The reasoning is that the more designable structures would have a higher chance to be “found” by our algorithm because they can accommodate more sequences. To check our hypothesis, we need to define a structural determinant of protein designability because it is impractical (although possible in principle; see ref. 22 and Methods) to determine directly, by sequence space sampling, the designability of every evolved structure. England and Shakhnovich (25) found that for a large class of amino acid interaction potentials, B, the free energy per monomer, f, in sequence space for a protein structure defined by its contact matrix (C) can be presented as expansion in their contact traces:
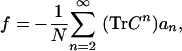 |
[2] |
where N is the length of the chain. The weights ai are all positive functions that depend on the amino acid interaction energies, B. The contact matrix, C, is defined as Cij = 1 if amino acids i and j are in contact and 0 otherwise (see Methods for contact definition). The trace of the n order or contact matrix, TrCn, is a sum of all diagonal elements of the nth power of contact matrix, C; we call this quantity the nth-order contact trace (CT) (23). Structures that can accommodate sequences with lower free energy in sequence space, f, are more designable because they provide higher entropy, i.e., allow more sequences to fold into the structure with or below a given energy, E (22, 26). The lowest-order contribution to designability in Eq. 1 is proportional to TrC2, and corresponds to contact density (CD), which is just the number of contacts per residue (a measure of the compactness of a structure). Earlier studies indicated that compactness may indeed be a factor contributing to protein designability (6, 22, 23, 26).
Compactness of evolved structures is lower than that of initial structure in both evolutionary runs, although the relaxed stability requirement in the second evolutionary run resulted in less compact structures overall (Fig. 1b). This finding is not surprising because the initial structure is maximally compact so that evolution could proceed only via decrease in compactness. Nevertheless, this finding might indicate that evolved structures appear to be less designable at least to the first approximation of designability. However, one can imagine that structures under entropic pressure to decrease their compactness can compensate loss in designability by evolving structures with greater values of higher-order CT such as, e.g., TrC8 (Fig. 1c), as well as TrC4 and TrC6 (data not shown). This surprising discovery is robust between evolutionary runs, and does not dependent on whether evolution generates stable or not so stable proteins (Fig. 1c).
One possible reason why evolution progressed to higher TrC8 is that we started from a structure with atypically low value of that parameter so that the majority of compact lattice 36-mers would have higher TrC8. Assuming this, evolution would relax toward more typical structures, with higher TrC8. Another possibility is that proteins with higher designability (reflected in greater values of higher-order traces of contact matrix in Eq. 1) have some selective advantage so that evolution would press toward more designable structures. Evolution toward more designable structures will be entropically counterbalanced by the difficulty of finding them in the ensemble of all compact structures so that finally a certain distribution of higher-order traces of contact matrix C (e.g., TrC4, TrC6, and TrC8) will be achieved as a compromise between these two factors. Comparison of the ensemble of evolved structures with set of randomly collapsed ones (Figs. 2 and 5-8 and Table 1) shows that the second possibility is the more likely one; i.e., evolution effectively exerted pressure to select more designable proteins as reflected in the clear shifts of distribution of higher-order traces of contact matrix toward higher, compared to random ensemble values, despite the fact that such structures are not readily available in the unbiased ensemble of compact 36-mers. As an additional control, we ran the evolutionary algorithm with a different set of parameters; the ones from a more recent publication (21). The results (Figs. 2 and 5-8 and Table 1) show that selection of more designable proteins is independent of the parameter set used.
Fig. 2.

Distribution of eight-order normalized contact trace in evolved (black and red curves) and random (green curves) ensembles. Random ensemble consists of 3,000 randomly collapsed 36-mers. Red lines correspond to the structures that evolved in the first run of the evolutionary algorithm; black curves correspond to control evolutionary run, which used another set of parameters (21). The data for these histograms are binned into bins of size 15. To account for the effect of possible variation in contact density (TrC2) on higher-order trace due to correlation between them (see Figs. 5- 8), here we normalized higher-order traces, both for random and evolved structures, by their contact densities.
What is the reason for evolutionary pressure on somewhat esoteric structural parameters such as higher traces of model protein contact matrices? Comparison of two curves in Fig. 1c clearly rules out that structures with higher TrC8 evolved in response to pressure of creating structures with higher stability. Furthermore, higher-order traces of contact matrix are correlated with contact density (TrC2) in random ensemble (see Figs. 5-8), so that evolution toward lower TrC2 (Fig. 1b) could cause only decrease of higher-order traces, while we observe that the structures evolve with higher values of these quantities (Figs. 1c and 2). The only feasible explanation, apparent from theory of protein designability (25) (see Eq. 1) is that evolved structures have a higher chance of “being found” if they have higher sequence space entropy, i.e., if the structure can accommodate more sequences, then the chance for a new sequence to fold stably into that structure is higher. Although direct comparison of designabilities for all 3,724 evolved structures is computationally impractical, we can make a direct comparison of designabilities between initial and one of the evolved structures. It is quite clear from Fig. 3 that the initial structure is dramatically less designable than one of the evolved structures of higher TrC8. For example, at energy -17 [in units of Miyazawa-Jernigan parameters used in this study (18)] characteristic of native states for most sequences evolved in the first evolutionary run (Fig. 1a), the evolved structure can accommodate ≈1024 more sequences than the initial one. It is important to note that the evolved structure shown in Fig. 3 is more designable despite its lower CD (TrC2). However, the decrease of designability due to lower CD is compensated by much greater values of higher-order contact traces (e.g., TrC8/36 = 210 for evolved structure shown in Fig. 3a and is 62 for the starting maximally compact conformation).
A possible reason for correlation between contact traces (a structural feature) and sequence entropy (i.e., designability) is that contact traces reflect topological characteristics of the network of contacts within the structure. For example, fourth-order contact trace (TrC4) reflects the number of length-4 closed loops in the system of contacts. We can point out that certain closed sets of contacts allow optimal placement of amino acids that interact favorably. For example, if four amino acids that strongly attract each other are folded into a structure where they all interact favorably (i.e., being placed in a closed loop of length 4), then this formation represents a greater contribution to the stability of the overall structure than configurations in which the same four amino acids are arranged linearly or in cases where one of the contacts is out of the contact range. Such optimal placement of several strongly interacting key amino acids allows more sequences to be folded into the structure by relaxing energy constraints for the rest of the sequence. Thus, structures that have certain important features, such as availability of long closed uninterrupted loops of interactions and higher density of contacts per residue, are expected to be capable of accommodating a wider variety of different sequences. This qualitative argument is similar in spirit to the derivation of Boltzmann distribution in statistical mechanics (27) and similar to the justification for the “Boltzmann device” used in the derivation of knowledge-based potentials (6, 28) for the study of protein folding and prediction of ligand binding energies.
The time course of evolution shown in Fig. 1 b and c suggests that later-evolved proteins may be less compact (CD) but nevertheless more designable (as proxied by higher-order contact traces, e.g., TrC8) than their more ancient counterparts. This intriguing possibility can be tested on real proteins. Indeed, earlier we observed (B.E.S., E. Deeds, C. Delisi, and E.I.S., unpublished data) that earliest domains belonging to last universal common ancestor (LUCA) have statistically higher CD than later diverged domains. Ideally, one would use full phylogeny to distinguish between earlier- and later-evolved proteins. However, the fundamental task of creating a reliable domain-based phylogeny is not complete despite several fruitful efforts (29). Our approach here is based on the fact that eukaryotic cells evolved after prokaryotic ones and thus protein domains that exist only in eukaryotes (eukaryotic innovation domains) can serve as representatives of later-evolved protein structures, to be compared with domains that are exclusive to prokaryotes. Sequence analysis with the ELISA database (30) (http://romi.bu.edu/elisa) yielded 817 eukaryotic innovation Dali domains and 1,775 prokaryotic-only Dali domains. In accord with predictions from model evolution, we found that eukaryotic innovation domains are indeed statistically less compact that prokaryotic-only domains (Fig. 4a). Importantly, this trend is not a consequence of possible differences in length distribution between eukaryotic innovation and prokaryotic-only domains; it persists in all length windows (see Figs. 5-8).
Fig. 4.

(a) Distribution of CD in eukaryotic innovation domains (squares) and prokaryotic-only domains (circles). The data for these histograms are binned into bins of size 0.2. The CD is calculated as explained in Methods. (b) Distribution of TrC8 normalized by domain length in prokaryotic-only Dali domains (circles) and eukaryotic innovation domains (squares). The data were binned with bin size 4,000. Only domains with CD in the range between 3 and 4 are taken; 262 eukaryotic innovation domains and 843 prokaryotic-only domains fall in this range. The Kolmogorov-Smirnov (KS) P value for the null hypothesis that these two datasets were drawn from the same distribution is 0.001. For control, we randomly split prokaryotic-only domains ensemble into two equal parts and compared their distributions of TrC8. In contrast to comparison between eukaryotic innovation and prokaryotic-only domains, these two sets appear to be identical: KS P value for the same null hypothesis is 0.647.
To complete the analogy with our modeling, we turn to the analysis of higher-order contact traces of eukaryotic innovation domains and prokaryotic domains. In doing so one has to keep in mind that there is positive correlation between traces of second-order (CD) and higher-order contact traces. To this end, to make appropriate comparison between higher-order contact traces, we select only domains that fall into a narrow range of CDs, namely between 3 and 4. This range corresponds to domains of lower than average compactness (23) consistent with our expectations that selection mechanism based on higher-order contact traces is likely to work on domains of relatively low compactness. Comparison between eukaryotic innovation domains and prokaryotic-only domains (Fig. 4b) shows a statistically significant shift toward higher TrC8 in eukaryotic innovation domains The difference in distribution of TrC8 between the two groups of protein domains is highly statistically significant: KS P value for the null hypothesis of no difference in distributions is 0.001. Comparison of eukaryotic innovation and prokaryotic-only domains shows that in this case possible loss of designability in eukaryotes due to their statistically lower contact density is compensated by contributions of the higher-order traces of contact matrices of their domains, similar to the effect observed in model evolution. This phenomenon is more complex than earlier observation that domains from thermophilic organisms are more designable (23). In the latter cases, greater designability of thermophilic proteins is observed at the level of contact density, i.e., in the lowest order of expansion in Eq. 1. Higher-order contact traces show similar trend in thermophiles vs. mesophiles. However, in the case of thermophilic organisms it is hard to say whether this is an independent trend or a consequence of correlation between traces of different order (see Figs. 5-8).
Our results clearly show that (i) increasing protein designability was certainly a factor in evolution of structures and (ii) that existing theory (25) correctly predicts the structural determinants of protein designability.
Why did model (and real) evolution optimize designability? More designable proteins were not selected through optimization of foldability (and stability) applied in model evolution. Nevertheless, divergent evolution came up with more designable structures. This is clearly an example of collective selection pressure where evolution is controlled not only by measures of fitness for an individual protein and its functionality inside an organism, but also by the accessibility of new structure for innovation. In this sense, designability represents a selective advantage for ensembles of evolving proteins.
Our simulations represent a rigorous albeit minimalist model of protein evolution where only the basic physical characteristics of proteins (their ability to fold and their stability) were chosen for selection. Whereas this requirement represents a bare-bones, necessary condition for proteins to survive, in reality, other requirements such as functional selection and ability to participate in protein-protein interactions were most likely factors in selection of specific protein structures (1, 31). Nevertheless, we see that even such minimal requirements on selection lead to significant consequences for the evolution of the protein structure universe.
Our analysis presents a rare example when a very specific prediction derived from theory appears to directly affect protein evolution, both in model and real proteins. Although earlier studies (23) suggested that this may be the case, they focused exclusively on comparison of CD of proteins from various proteomes. Although designability appeared to be the most plausible explanation for observed differences between mesophilic and thermophilic proteomes in ref. 23, other, perhaps related, factors such as stability and/or aggregation could not be ruled out. Indeed as Fig. 1b shows, the less stable proteins evolve with lower compactness in general. The present study goes much further as it demonstrates that less obvious structural characteristics were evolutionarily selected. Furthermore, as model evolution suggests (Fig. 1c), such selection is unrelated to protein stability.
The relation between properties of contact matrices and protein topology points out to a possible reason for remarkable symmetry observed in proteins. Indeed, our analysis shows that availability of uninterrupted closed loops of intraprotein contacts may be beneficial for structure designability. One way to achieve this is to form regular structures, in particular with symmetric open interiors that are not interrupted by the crossing chain. This is one of the most common structural features of globular proteins, most of which have contiguous hydrophobic cores.
Finally, our findings highlight the interplay between selection and chance in protein evolution. While evolutionary pressure is applied directly to select for proteins that can stably fold, it is countered by the difficulty in finding a proper structure- sequence match. The result of this balance between selection and effective entropic factors in structure space is the emergence of more designable structures, whereas in sequence space, evolution selects sequences that have pronounced, but not extreme, energy gaps in their native conformations (32, 33). This represents close analogy with statistical mechanics where temperature serves as a rough equivalent of the strength of selective pressure.
This study shows that a divergent model of protein structure morphogenesis is able not only to reproduce global power laws observed in protein universe (3, 4) but also to capture the unexpected selection of special structures that we observe to be predominant in the protein universe.
Supplementary Material
Acknowledgments
We are grateful to Eric Deeds and Richard Goldstein for useful discussions. This work was supported by the National Institutes of Health (E.I.S.). N.V.D. acknowledges support from a University of North Carolina Research Council grant.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Ponting, C. P. & Russell, R. R. (2002) Annu. Rev. Biophys. Biomol. Struct. 31, 45-71. [DOI] [PubMed] [Google Scholar]
- 2.Qian, J., Luscombe, N. M. & Gerstein, M. (2001) J. Mol. Biol. 313, 673-681. [DOI] [PubMed] [Google Scholar]
- 3.Koonin, E. V., Wolf, Y. I. & Karev, G. P. (2002) Nature 420, 218-223. [DOI] [PubMed] [Google Scholar]
- 4.Dokholyan, N. V., Shakhnovich, B. & Shakhnovich, E. I. (2002) Proc. Natl. Acad. Sci. USA 99, 14132-14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finkelstein, A. V. & Ptitsyn, O. B. (1987) Prog. Biophys. Mol. Biol. 50, 171-190. [DOI] [PubMed] [Google Scholar]
- 6.Finkelstein, A. V., Gutin, A. M. & Badretdinov, A. (1995) Subcell. Biochem. 24, 1-26. [DOI] [PubMed] [Google Scholar]
- 7.Govindarajan, S. & Goldstein, R. A. (1996) Proc. Natl. Acad. Sci. USA 93, 3341-3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li, H., Helling, R., Tang, C. & Wingreen, N. (1996) Science 273, 666-669. [DOI] [PubMed] [Google Scholar]
- 9.Taverna, D. M. & Goldstein, R. A. (2000) Biopolymers 53, 1-8. [DOI] [PubMed] [Google Scholar]
- 10.Xia, Y. & Levitt, M. (2002) Proc. Natl. Acad. Sci. USA 99, 10382-10387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirny, L. & Shakhnovich, E. (2001) Annu. Rev. Biophys. Biomol. Struct. 30, 361-396. [DOI] [PubMed] [Google Scholar]
- 12.Klimov, D. K. & Thirumalai, D. (2001) Proteins 43, 465-475. [DOI] [PubMed] [Google Scholar]
- 13.Socci, N. D., Onuchic, J. N. & Wolynes, P. G. (1998) Proteins 32, 136-158. [PubMed] [Google Scholar]
- 14.Pande, V. S., Grosberg, A. Y. & Tanaka, T. (1997) Biophys. J. 73, 3192-3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sali, A., Shakhnovich, E. & Karplus, M. (1994) Nature 369, 248-251. [DOI] [PubMed] [Google Scholar]
- 16.Chan, H. S. & Dill, K. A. (1996) Proteins 24, 335-344. [DOI] [PubMed] [Google Scholar]
- 17.Shakhnovich, E. I. (1994) Phys. Rev. Lett. 72, 3907-3910. [DOI] [PubMed] [Google Scholar]
- 18.Miyazawa, S. & Jernigan, R. L. (1985) Macromolecules 18, 534-552. [Google Scholar]
- 19.Sali, A., Shakhnovich, E. & Karplus, M. (1994) J. Mol. Biol. 235, 1614-1636. [DOI] [PubMed] [Google Scholar]
- 20.Shakhnovich, E. I. & Gutin, A. M. (1993) Protein Eng. 6, 793-800. [DOI] [PubMed] [Google Scholar]
- 21.Miyazawa, S. & Jernigan, R. L. (1996) J. Mol. Biol. 256, 623-644. [DOI] [PubMed] [Google Scholar]
- 22.Shakhnovich, E. I. (1998) Fold. Des. 3, R45-R58. [PubMed] [Google Scholar]
- 23.England, J. L., Shakhnovich, B. E. & Shakhnovich, E. I. (2003) Proc. Natl. Acad. Sci. USA 100, 8727-8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abkevich, V. I., Gutin, A. M. & Shakhnovich, E. I. (1994) Biochemistry 33, 10026-10036. [DOI] [PubMed] [Google Scholar]
- 25.England, J. L. & Shakhnovich, E. I. (2003) Phys. Rev. Lett. 90, 218101. [DOI] [PubMed] [Google Scholar]
- 26.Wolynes, P. G. (1996) Proc. Natl. Acad. Sci. USA 93, 14249-14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landau, L. D., Lifshitz, E. M. & Pitasevskii, L. P. (1978) Statistical Physics (Pergamon, Oxford).
- 28.Grzybowski, B. A., Ishchenko, A. V., Shimada, J. & Shakhnovich, E. I. (2002) Acc. Chem. Res. 35, 261-269. [DOI] [PubMed] [Google Scholar]
- 29.Mirkin, B. G., Fenner, T. I., Galperin, M. Y. & Koonin, E. V. (2003) BMC Evol. Biol. 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shakhnovich, B. E., Harvey, J. M., Comeau, S., Lorenz, D., DeLisi, C. & Shakhnovich, E. (2003) BMC Bioinformatics 4, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teichmann, S. A., Chothia, C. & Gerstein, M. (1999) Curr. Opin. Struct. Biol. 9, 390-399. [DOI] [PubMed] [Google Scholar]
- 32.Mirny, L. A., Abkevich, V. I. & Shakhnovich, E. I. (1998) Proc. Natl. Acad. Sci. USA 95, 4976-4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gutin, A. M., Abkevich, V. I. & Shakhnovich, E. I. (1995) Proc. Natl. Acad. Sci. USA 92, 1282-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.