

Abstract
The use of antipsychotics is hindered by the frequent occurrence of metabolic and cardiovascular side effects, resulting in worsened quality of life and greater mortality as a result of cardiovascular and cerebrovascular disorders in schizophrenia patients than the comparable general population. The various antipsychotics induce extrapyramidal symptoms, impaired glucose and lipid metabolism, weight gain, hypertension and arrhythmias, with variable frequency. Second-generation antipsychotics appear to have several advantages over first-generation antipsychotics, including a claimed better action on cognitive function and the negative symptoms of schizophrenia, and lower frequency of extrapyramidal side effects; however, their use is associated with a greater frequency of metabolic and cardiovascular disturbances. The mechanisms of these important side effects are not well understood, and generic approaches (psychoeducational programmes and symptomatic therapies) have been proposed to limit their severity. Extensive data from the literature indicate that autonomic nervous system dysfunction—intrinsic to schizophrenia and strongly exacerbated by antipsychotic treatment—is the cause of the pervasive metabolic and vascular dysfunctions associated with schizophrenia. In this article, we marshal further literature data to argue that the metabolic and cardiovascular side effects of antipsychotics are primarily mediated by their ability to block peripheral dopamine receptors, which physiologically modulate sympathetic activity. We also propose that these effects might be overcome by providing peripheral dopaminergic stimulation.
Introduction
Compared with first-generation antipsychotics (FGAs), second-generation antipsychotics (SGAs) seem characterized by reduced risk of extrapyramidal motor side effects but high frequency of adverse metabolic events [1]. Insulin resistance, weight gain with abdominal obesity, impaired glucose tolerance, diabetes mellitus, dyslipidaemia, essential hypertension—major diagnostic criteria for metabolic syndrome[2]—all occur frequently in patients treated with SGAs [1]. Metabolic syndrome is a major cardiovascular risk factor in schizophrenia patients, and is associated with greater mortality due to cardiovascular and cerebrovascular diseases than the comparable general population [3]. Furthermore, all antipsychotics (FGAs and SGAs) can cause severe and sometimes fatal cardiac arrhythmias [4].
Although the metabolic derangement and cardiovascular disorders associated with antipsychotic use are well recognized, a comprehensive mechanism able to account for all these effects has not been proposed, so it is time to examine new pathogenetic mechanisms that might account for them. We propose a new pathogenetic mechanism for these effects supported by a MEDLINE search using the search terms metabolic syndrome, insulin resistance, diabetes, dyslipidaemia, hyperglycemia, weight gain, sympathetic, histamine, serotonin, dopamine, triglycerides, cholesterol, hypertension, cardiovascular disorders and sudden cardiac death, cross-referenced with the term schizophrenia. We examined papers published in English between 1980 and 2012 and checked references in relevant papers to identify additional studies.
Schizophrenia as an Independent Risk Factor for Metabolic Syndrome
Impaired glucose metabolism was described in psychiatric patients before antipsychotics were introduced in the 1950s [5]. Although disputed by some authors [6, 7], drug-naïve and first-episode patients with schizophrenia seem to have impaired fasting glucose tolerance, more insulin resistance and higher levels of plasma glucose, insulin and cortisol than healthy control subjects. [8] Moreover, antipsychotic-naive and antipsychotic-free schizophrenia patients have significantly greater visceral adiposity than age- and sex-matched controls, despite similar subcutaneous adiposity [9–11]. Intra-abdominal adiposity is much more strongly linked to insulin resistance and type 2 diabetes than excess subcutaneous fat [12].
These metabolic disturbances in patients with schizophrenia have been attributed to sedentary lifestyle, poor diet and smoking [1]. However, increased sympathetic-adrenal activity [13], with high plasma levels of catecholamines and cortisol [14], has also been observed in untreated schizophrenia patients. Increased sympathetic-adrenal activity could be a consequence of psychosis-related stress [15], but could also be an effect of nicotine, as these patients are often heavy smokers. Nicotine acts on nicotinic (cholinergic) receptors in sympathetic ganglia, activating postganglionic neurons and stimulating the secretion of catecholamines from their terminals [16]. Smoking is associated with insulin resistance [17], dyslipidaemia [18] and an increased propensity for type 2 diabetes [19, 20] through a body mass index-independent mechanism [21]. Contrary to what one might expect, stopping smoking does not reduce these metabolic abnormalities but, by mechanisms not well understood, is often accompanied by increased weight and a further increase in the risk of diabetes [22, 23].
Metabolic and Cardiovascular Effects of Sympathetic Overactivity
High circulating levels of noradrenaline (norepinephrine), adrenaline (epinephrine) and cortisol oppose the anabolic effects of insulin and can induce metabolic changes. Thus, by activating α2-adrenoreceptors, noradrenaline and adrenaline promote the transformation of liver and muscle glycogen to free glucose, enhance glucagon secretion and inhibit insulin release from the pancreatic islets [24], inducing hyperglycaemia; they also act on β2-adrenoreceptors in muscle and adipose tissue to reduce glucose uptake and utilization by these tissues, promoting insulin resistance [25]. In addition, noradrenaline and adrenaline stimulate adipose lipolysis, promoting the release of glycerol, free fatty acids, leptin, adiponectin and proinflammatory cytokines into the circulation [26]. The latter three substances are implicated in obesity [27] (see Sect. 3). Increased fatty acid levels contribute to the development of insulin resistance [27] and stimulate the assembly and secretion of very low density and low-density lipoproteins (LDLs) by the liver, so that plasma levels of triglycerides and LDL-cholesterol increase, and high-density lipoprotein (HDL)-cholesterol decreases [28].
Thus, sympathetic overactivity correlates with hypercholesterolaemia and hypertriglyceridaemia. It is noteworthy that serum cholesterol levels are significantly reduced in animals treated with 6-hydroxydopamine, a sympathetic nervous system toxin [29].
Cortisol also increases insulin resistance, further contributing to high levels of circulating glucose and lipids; and in the long term, cortisol promotes abdominal fat accumulation and stimulates the intake of carbohydrates and fat [30].
Finally, excessive sympathetic tone has a major effect on the cardiovascular system: it is implicated in cardiovascular hypertrophy and vulnerability to arrhythmias, and plays a pathogenetic role in essential hypertension by raising cardiac output and heart rate, and increasing peripheral resistance and renin production [31]. Cardiac effects of adrenergic activity are mediated mainly by β1-receptors, although β2- and α-adrenoceptors are also involved. β-Adrenoceptor activation increases both normal (sinoatrial node) and abnormal (Purkinje fibres) pacemaker activity.
Worsening of Metabolic Syndrome by Antipsychotics
The metabolic syndrome that characterizes many untreated patients diagnosed with schizophrenia can worsen after a few months’ treatment with antipsychotics [1, 11, 32]. This happens more frequently with SGAs than FGAs, and with greater probability for some SGAs than others [32, 33]. The prevalence of metabolic syndrome increases with duration of treatment [32].
SGAs are a heterogeneous class of drugs, each agent having differing pharmacological properties and a unique binding profile to multiple receptors, which determine differences in efficacy and side effects [34].
Increased body weight is usually linked to insulin resistance (major cause of metabolic syndrome) [35] and to changes in plasma glucose and lipid levels, but obesity cannot be the causative factor for most cases of diabetes arising during antipsychotic treatment [36]. Dose-dependent insulin resistance occurs following acute clozapine and olanzapine administration in rats [37], indicating an immediate effect of these agents on metabolic variables. Some [38, 39], but not all [40], studies have found that antipsychotics have a similar short-term effect in humans. In particular, rapid onset of diabetes has been documented after the initiation of antipsychotic therapy, together with rapid disappearance of diabetes after discontinuation [41]—too rapid to be explained by changes in body weight.
Clozapine and olanzapine use appears to be associated with greatest risk of metabolic syndrome; iloperidone, quetiapine, risperidone, paliperidone, sertindole and zotepine are associated with intermediate risk; while amisulpride, aripiprazole, asenapine, lurasidone and ziprasidone are associated with only a small increase in risk [11, 42]. One study found that the increased risk of diabetes was identical in users of olanzapine, quetiapine and risperidone [43]. No antipsychotic is completely free of metabolic side effects [44]; even those usually without such effects may cause them in specific situations. Thus, although aripiprazole does not increase body weight in adults, weight gain has been reported in paediatric patients following aripiprazole administration [45, 46]. Furthermore, long-term side effects tend to come to light only after a drug has been on the market for some time [47], and most data on the side effects of antipsychotics have been obtained from trials on patients with many years of prior drug exposure, and this could lead to the underestimation of side effects in the more recently introduced antipsychotics such as aripiprazole, asenapine, lurasidone and ziprasidone [48]. Finally, metabolic alterations appear to be an intrinsic effect of antipsychotics, since they appear when these drugs are employed to treat diseases other than schizophrenia [49, 50].
Previously Proposed Mechanisms for Metabolic Side Effects of Antipsychotics
Despite a large body of research, no specific neurotransmitter or receptor has been identified as the main cause of the metabolic derangement often induced by antipsychotics [51]. Since antipsychotic-induced obesity seems to be related more to increased calorie intake than reduced thermogenesis [52], the antagonistic action of antipsychotics on receptors involved in the control of satiety, feeding and glucose metabolism, and their interactions with orexigenic and anorexigenic peptides have been extensively investigated [53]. Antipsychotic-induced weight gain has been proposed as due to antagonistic effects at, among the others, the histamine H1 [54, 55], serotonin 5-HT2C [56] and dopamine D2 [57] receptors; while the diabetogenic effect has been correlated with binding affinity to H1 [55], 5-HT2C and muscarinic acetylcholine receptors [34]. In fact, antagonism at 5-HT2C receptors increases insulin resistance and reduces glucose uptake by skeletal muscle, increasing the risk of diabetes [55]. However, antipsychotics such as clozapine, olanzapine, risperidone, sertindole and zotepine have different propensities for inducing metabolic effects, even though they have similar and only moderate binding affinity for 5-HT2C receptors; moreover ziprasidone, which has the most potent binding [58], is apparently largely free of metabolic side effects. It therefore appears difficult to reconcile the variable ability of antipsychotics to induce metabolic changes with their binding to the 5-HT2C receptor.
The receptor binding profiles involved in dyslipidaemia have not yet been identified [34].
Turning to consider acetylcholine receptors, antagonism at muscarinic M3 receptors on pancreatic β cells (as shown by olanzapine and clozapine), with consequent suppression of cholinergic-stimulated insulin secretion, has also been suggested as a cause of the metabolic effects of some SGAs [45, 59]. However, when selective M3 antagonists are used to treat patients with urinary incontinence they have not been associated with increased risk of diabetes. Furthermore, insulin and glucose clearance responses to the oral glucose tolerance test in vagotomized humans do not differ substantially from those of healthy controls [60]. It is likely therefore that M3 antagonism is only a precipitating factor for diabetes in patients treated with antipsychotics.
Leptin, ghrelin and adiponectin—peptide hormones involved in the regulation of appetite and body weight—might be involved in the metabolic syndrome induced by antipsychotics [61]. Leptin is a cytokine-like molecule produced by subcutaneous adipocytes and actively transported into the hypothalamus where it acts to limit food intake. However, most patients on antipsychotics have increased, not reduced, blood levels of leptin, suggesting that leptin increase is a consequence (not a cause) of increased body weight, or that leptin resistance has developed [61]. Ghrelin is a hunger-inducing hormone produced in the stomach that regulates feeding behaviour through central activity. Plasma levels of ghrelin have been reported increased, unchanged and reduced in various studies on schizophrenia patients treated with antipsychotics [61]. Adiponectin is secreted by adipose tissue and circulating levels correlate inversely with body weight and insulin resistance. As with studies on ghrelin, studies on adiponectin levels in antipsychotic-induced weight gain have produced conflicting results [61]. For more data on these peptides and antipsychotic-induced metabolic side effects see the following review articles [45, 48, 61, 62].
The appearance of metabolic side effects in some but not all patients treated with a given antipsychotic suggests the involvement of genetic factors. Gene polymorphisms may occur at the level of drug transporters, metabolizing enzymes or the protein targets (receptors) of the drug. Antipsychotic-induced weight gain is associated with a polymorphism of the promoter region of the 5-HT2C receptor gene and a functional polymorphism of the leptin gene [63]. Associations of increased risk of metabolic syndrome with polymorphisms of the MTHFR gene [64] and the D2 receptor gene [65] have been suggested in patients receiving SGAs.
From the above survey, it is evident that a convincing account of the mechanisms responsible for the frequent and severe metabolic side effects of SGAs is not available. We suggest that the main mechanism for these effects is deranged autonomic function due first to the ability of these agents to block peripheral dopamine receptors, which increases sympathetic activity (see Sect. 5 for the proposed mechanism); and secondly to the ability of some of these agents to also block peripheral muscarinic receptors, which reduces vagal parasympathetic activity.
The Missing Piece of the Puzzle: The Peripheral Dopaminergic System
Outside the CNS, dopamine has been traditionally viewed as an inactive precursor of noradrenaline and adrenaline. However, dopamine receptors are widespread peripherally, being present for example in heart, kidneys, adrenal glands, liver, endocrine pancreas and resistance vessels such as renal, coronary, pulmonary, mesenteric and cerebral arteries [66]. D1 and D5 (D1-like), and D2, D3 and D4 (D2-like) receptor subtypes have not been extensively investigated in peripheral organs, hence for convenience we apply the D1/D2 classification of Kebabian and Calne [67].
Stimulation of D1 receptors, located postsynaptically in renal, mesenteric and splenic arteries, causes direct vasodilatation; and by reducing vascular resistance reduces blood pressure [68]. D2 receptors are located presynaptically on postganglionic sympathetic neurons and on adrenal chromaffin cells: their activation inhibits noradrenaline release from nerve endings, and adrenaline secretion from the adrenal medulla [66, 69], resulting in indirect vasodilatation, reduced cardiac contractility and anabolic effects through inhibition of sympathetic tone. These data suggest that the peripheral dopaminergic system is an important modulator of the endocrine and cardiovascular systems [68, 69]. In fact, during intense sympathetic stimulation, endogenous dopamine (in addition to noradrenaline and adrenaline) is secreted and serves to modulate sympathetic discharge [70]. It is not surprising, therefore, that full D2 receptor agonists have proven effective in reducing blood pressure and plasma glucose and lipids, thereby ameliorating some aspects of metabolic syndrome [68, 71, 72]. Moreover, partial dopamine agonists employed in the treatment of psychosis (e.g. aripiprazole) do not seem to cause metabolic syndrome [42], probably thanks to their stimulating action on extracerebral presynaptic D2 receptors.
Metabolic Effects of Peripheral Dopamine D2 Receptor Blockade
Although the various antipsychotics have differing effects, attributable to differences in their binding to dopamine, serotonin, histamine, glutamate, muscarinic and α-adrenergic receptors, all effective antipsychotics block D2 receptors—including peripheral D2 receptors—with varying potency and dose-dependent effects [34]. Even antipsychotics such as clozapine, generally believed to have very low affinity for D2 receptors, probably occupy more than 70 % of these receptors, although more loosely than FGAs [73]. When antipsychotics bind to the D2 receptor they prevent activation by endogenous dopamine, so the modulating effect of dopamine on the sympathetic-adrenal system is inhibited. This effect is illustrated by the peripheral D2 antagonist domperidone: administration of this agent, which does not cross the blood-brain barrier except in rare cases of blood-brain barrier immaturity or disruption [74], significantly increases the release of both adrenal noradrenaline and adrenaline [75], and reverses the antihypertensive effect of dopaminergic drugs [76].
Thus, we arrive at the central hypothesis of the present paper: chronic D2 receptor blockade by antipsychotics abolishes peripheral dopaminergic modulation, resulting in chronically increased sympathetic tone, in turn leading to impaired control of glucose and lipid metabolism, chronic increase in blood pressure, and increased propensity for cardiac arrhythmias (Fig. 1).
Fig. 1.
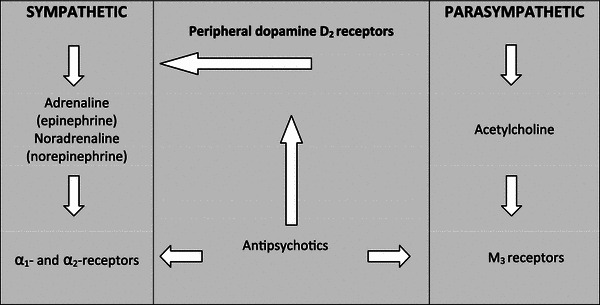
Schematic outline of proposed effects of antipsychotics on sympathetic and parasympathetic outflow. Antipsychotics block peripheral dopamine D2, α-adrenergic, and cholinergic M3 receptors. Physiologically, D2 receptors modulate the release of catecholamines from sympathetic terminals and the adrenal glands. D2 receptor blockade causes increased release of catecholamines. If the α-adrenergic receptors are not completely occupied by an antipsychotic, sympathetic activity is increased, and would normally be balanced by increased parasympathetic tone. If, however, M3 receptors are also blocked by the antipsychotic, sympathetic activity is no longer countered by parasympathetic activity so the former exerts its well known clinical effects leading to metabolic syndrome and arrhythmia
Two findings provide strong circumstantial support for this sequence of events: First, catecholamine levels are increased in the plasma and cerebrospinal fluid of schizophrenia patients on long-term antipsychotic treatment—and are reduced after these drugs are withdrawn [77, 78]. Second, the hyperglycaemic response to acute administration of high doses of antipsychotics in the mouse is completely blocked by pretreatment with yohimbine (an α2-adrenoceptor antagonist), suggesting that the hyperglycaemic effect is due, at least in experimental animals, to increased sympathetic nervous system activity [79].
Cardiovascular Effects of Peripheral D2 Receptor Blockade
The hypertension sometimes present in patients being treated with antipsychotics [80] can be controlled by β-blockers [81], suggesting that adrenergic hyperactivity is involved. Increased sympathetic tone is known to play a major role in the genesis of essential hypertension [31]. In this context, it is noteworthy that catecholamine release is increased in isolated cat adrenal glands perfused with antipsychotics, and reduced by apomorphine (dopaminergic agonist), confirming the existence, in adrenal glands, of a dopaminergic system that inhibits catecholamine release and can be inactivated by the administration of antipsychotics, in turn suggesting the adrenal medulla as a possible site of action for the hypertensive response evoked by antipsychotics [82].
Use of both FGAs and SGAs is also associated with a dose-related increased risk of sudden death due to cardiac arrhythmia [4]. Antipsychotics prolong the electrocardiogram QT interval and this has been identified as an important trigger for torsades de pointes, a transient and unpredictable form of polymorphic ventricular tachycardia [83]. The magnitude of the sudden death risk varies between studies—often just case reports—but seems to be at least as high for SGAs as for FGAs [83]. Strikingly, it took around 50 years of use before the US FDA issued its warning (September 2007) regarding the association of haloperidol with QT prolongation and arrhythmias. We suggest that cardiac D2 receptor blockade by antipsychotics is responsible for the increased sudden deaths due to cardiac arrhythmias in FGA and SGA users. In fact, increased sudden death for this cause has also been reported after intravenous administration of the selective peripheral D2 antagonist domperidone [84]. Cardiac D2 receptor blockade by antipsychotics would increase this risk by inhibiting the modulator effect that endogenous dopamine exerts on sympathetic activity. The resulting enhanced stimulation of adrenergic receptors reduces refractory period duration, and may lead to the initiation of torsades de pointes [85].
Additional Mechanisms
If D2 receptor blockade by antipsychotics is the main reason why metabolic derangements are common in users of these drugs, then haloperidol and ziprasidone, the most potent D2 blockers [34], should be most strongly associated with these derangements. This is not the case however since, as noted above, clozapine and olanzapine, followed by quetiapine, appear mainly responsible for these effects [42]. This conflict can be explained by considering that D2 blockade increases the availability of noradrenaline and adrenaline (sympathetic activity) but these catecholamines can exert their effects only if the following occurs:
Adrenergic receptors are free. We know, for example, that antipsychotics block the α1- and α2-adrenoceptors with variable affinity, so the increased availability of an adrenergic neurotransmitter does not produce any metabolic or cardiovascular effect if receptor occupancy is already high and the proportion of receptors remaining active is low. Net adrenergic activity can even be reduced if receptor occupancy by antipsychotics is high. In fact, orthostatic hypotension may occur—instead of hypertension—as a side effect of antipsychotic use, due to α1-adrenoceptor antagonism [86].
Parasympathetic activity does not counteract their actions. Parasympathetic activity opposes sympathetic effects on the pancreatic islets [24] and the cardiovascular system [87] by acting on cholinergic M3 receptors. If these receptors are inhibited by antipsychotics, the increased sympathetic activity is no longer counterbalanced by parasympathetic activity and the net result is sympathetic overactivity. For this reason, clozapine, olanzapine and quetiapine (moderate binding at D2 but potent binding at M3 receptors) are strong inducers of metabolic side effects, while haloperidol and ziprasidone—potent binding at D2 but weak at M3 receptors—are much less so [32, 42].
Antipsychotics also bind to a range of non-dopaminergic targets, such as serotonin and histamine receptors, which also play a role in weight gain and obesity [34], so the ability of each antipsychotic to induce metabolic derangements would be a result of its actions on all the receptor systems involved in the maintenance of metabolic homeostasis.
Findings in Parkinson’s Disease
Our hypothesis that D2 receptor blockade by antipsychotics is responsible for metabolic syndrome, and ultimately contributes to increased mortality in schizophrenia, is reinforced by observations in Parkinson’s disease (PD), an illness in which sympathetic degeneration and dopaminergic treatment produce effects that are opposite to those found in schizophrenia. One neuropathological hallmark of PD is generalized sympathetic denervation, which is known to be present even in patients without clinically evident autonomic failure [88]. Reduced sympathetic activity accounts for the reduced frequencies of hypertension, type 2 diabetes and dyslipidaemia in untreated PD patients [89].
The main aim of pharmacological treatment in PD is to stimulate D2 receptors, principally in the striatum. Levodopa is still the most effective drug in this respect. However, the quantities of levodopa-derived dopamine present in the periphery (notwithstanding use of a peripheral decarboxylase inhibitor) are sufficient to further reduce sympathetic activity—via actions on peripheral D2 receptors—further contributing to lowered frequencies of diabetes, hypertension and dyslipidaemia in PD patients taking levodopa [90].
Overcoming the Metabolic and Cardiovascular Side Effects of Antipsychotics
The arguments presented above make a strong case that the metabolic and cardiovascular side effects of antipsychotics hinge on their action on peripheral (extracerebral) dopaminergic, cholinergic and adrenergic receptors (although a central component cannot be ruled out). The inhibitory action of these antipsychotics on D2 receptors plays a major role in this mechanism. It is therefore conceivable that by displacing the antipsychotic from peripheral D2 receptors by means of a dopaminergic drug, the balance between sympathetic and parasympathetic activity may be restored, at least in part.
The administration of dopamine agonists to patients with psychotic disorders may raise concerns, as the stimulation of cerebral D2 receptors can exacerbate psychotic symptoms. However, if the dopaminergic medication is unable to cross the blood-brain barrier this risk is very low.
The dopaminergic drug that has these features is levodopa. Levodopa itself is inactive, but is decarboxylated in the body to active dopamine. However, over 90 % of administered levodopa is decarboxylated outside the CNS, and this dopamine does not cross the blood–brain barrier. To overcome this problem when levodopa is administered to parkinsonian patients, it is combined with a peripheral dopa-decarboxylase inhibitor (carbidopa or benserazide) so that more levodopa passes into the brain to increase the amount of dopamine available there. By contrast, when levodopa is used in the absence of a peripheral decarboxylase inhibitor, its central action is minimized in favour of its peripheral action. This property of levodopa is unique, and its use with or without a peripheral decarboxylase inhibitor allows differential targeting of dopamine to the CNS and the periphery. SGAs such as clozapine, olanzapine and quetiapine bind the D2 receptor more weakly, and have a higher dissociation constant, than dopamine [91]. It is therefore theoretically possible to displace these antipsychotics from peripheral D2 receptors using relatively low doses of levodopa, so that the probability of exacerbating psychotic symptoms would be low.
In fact, it is possible that very low levels of central levodopa-derived dopamine might mimic the therapeutic effects of partial dopamine agonists thought to act by stimulating presynaptic D2 receptors. In vitro experiments have shown that it is possible to produce a partial agonistic effect by combining a full direct receptor agonist with a full antagonist [92]. Slightly elevated central dopamine levels might also promote quick dissociation of antipsychotics from D2 receptors [91], improving their pharmacological profile.
In a review of 30 studies involving more than 700 patients, Jaskiw and Popli reported that administration of high doses of levodopa to patients with schizophrenia caused clinical worsening (measured with various psychiatric rating scales) in 20 % of cases when not associated with antipsychotics, but a significant clinical improvement occurred in 50 % of cases when antipsychotics were also administered. The authors concluded that add-on levodopa may be beneficial in patients already on antipsychotics [93]. It is noteworthy that the beneficial effects of levodopa prevailed over its negative effects even though high doses of levodopa, associated with a peripheral decarboxylase inhibitor, were given. It is noteworthy, also, that levodopa has proved capable of attenuating peripheral sympathetic activity in experimental animals [94], and has been successfully used (without a decarboxylase inhibitor) to treat congestive heart failure [95].
Levodopa has been widely used (and studied) for the treatment of PD for over 50 years. It may cause nausea when first given due to stimulation of dopaminergic receptors in the area postrema, but this effect usually disappears after a week or two. It is unlikely that this effect will occur when combined with antipsychotic use, first, because of the low doses proposed and, secondly, because antipsychotics have an antagonistic effect on the dopaminergic receptors of the area postrema. The long-term side effects of levodopa treatment in PD (‘on-off’, psychiatric disturbances) are closely associated with progression of the disease itself [96], and it has been shown that chronic levodopa is not toxic for dopaminergic neurons [97].
Conclusions
Many cerebral and peripheral factors are involved in the metabolic and cardiovascular side effects of antipsychotics. We propose a new unifying pathogenetic hypothesis for these effects in which autonomic nervous system dysfunction—triggered by the disease and exacerbated by antipsychotic treatment—is at the root of the pervasive metabolic and cardiovascular disorders that characterize patients with schizophrenia. Our suggestion that levodopa may prevent these side effects is both attractive and easily testable.
Acknowledgements
No sources of funding were received to prepare this article. Dr. Scigliano has no competing financial interests in relation to the work described. He conceived and planned the work that led to the manuscript, played an important role in the analysis and interpretation of the data, wrote the paper and approved the final submitted version. Dr. Ronchetti has no competing financial interests in relation to the work described. He played an important role in the acquisition, analysis and interpretation of the data and made substantive suggestions for revision and approved the final paper.
References
- 1.McEvoy JP, Meyer JM, Goff DC, et al. Prevalence of the metabolic syndrome in patients with schizophrenia: baseline results from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) schizophrenia trial and comparison with national estimates from NHANES III. Schizophr Res. 2005;80(1):19–32. doi: 10.1016/j.schres.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 2.Miranda PJ, DeFronzo RA, Califf RM, et al. Metabolic syndrome: definition, pathophysiology, and mechanisms. Am Heart J. 2005;149(1):33–45. doi: 10.1016/j.ahj.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 3.Osby U, Correia N, Brandt L, et al. Mortality and causes of death in schizophrenia in Stockholm County, Sweden. Schizophr Res. 2000;45(1–2):21–28. doi: 10.1016/S0920-9964(99)00191-7. [DOI] [PubMed] [Google Scholar]
- 4.Ray WA, Chung CP, Murray KT, et al. Atypical antipsychotic drugs and the risk of sudden cardiac death. N Engl J Med. 2009;360(3):225–235. doi: 10.1056/NEJMoa0806994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasanin J. The blood sugar curve in mental disease: II. The schizophrenia (dementia praecox) group. Arch Neurol Psychiatry. 1926;16(4):414–419. doi: 10.1001/archneurpsyc.1926.02200280022002. [DOI] [Google Scholar]
- 6.Sengupta S, Parrilla-Escobar MA, Klink R, et al. Are metabolic indices different between drug-naïve first-episode psychosis patients and healthy controls? Schizophr Res. 2008;102(1–3):329–336. doi: 10.1016/j.schres.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Padmavati R, McCreadie RG, Tirupati S. Low prevalence of obesity and metabolic syndrome in never-treated chronic schizophrenia. Schizophr Res. 2010;121(1–3):199–202. doi: 10.1016/j.schres.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 8.Ryan MC, Collins P, Thakore JH. Impaired fasting glucose tolerance in first-episode, drug-naive patients with schizophrenia. Am J Psychiatry. 2003;160(2):284–289. doi: 10.1176/appi.ajp.160.2.284. [DOI] [PubMed] [Google Scholar]
- 9.Thakore JH, Mann JN, Vlahos I, et al. Increased visceral fat distribution in drug-naive and drug-free patients with schizophrenia. Int J Obes Relat Metab Disord. 2002;26(1):137–141. doi: 10.1038/sj.ijo.0801840. [DOI] [PubMed] [Google Scholar]
- 10.Thakore JH. Metabolic disturbance in first-episode schizophrenia. Br J Psychiatry Suppl. 2004;47:S76–S79. doi: 10.1192/bjp.184.47.s76. [DOI] [PubMed] [Google Scholar]
- 11.De Hert M, Detraux J, van Winkel R, et al. Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat Rev Endocrinol. 2011;8:114–126. doi: 10.1038/nrendo.2011.156. [DOI] [PubMed] [Google Scholar]
- 12.Kissebah AH, Krakower GR. Regional adiposity and morbidity. Physiol Rev. 1994;74(4):761–811. doi: 10.1152/physrev.1994.74.4.761. [DOI] [PubMed] [Google Scholar]
- 13.Zahn TP, Pickar D. Autonomic activity in relation to symptom ratings and reaction time in unmedicated patients with schizophrenia. Schizophr Res. 2005;79(2–3):257–270. doi: 10.1016/j.schres.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 14.Kemali D, Maj M, Ariano MG, et al. 24-hour plasma levels of prolactin, cortisol, growth hormone and catecholamines in schizophrenic patients. Neuropsychobiology. 1985;14(3):109–114. doi: 10.1159/000118214. [DOI] [PubMed] [Google Scholar]
- 15.Dinan TG. Stress and the genesis of diabetes mellitus in schizophrenia. Br J Psychiatry Suppl. 2004;47:S72–S75. doi: 10.1192/bjp.184.47.s72. [DOI] [PubMed] [Google Scholar]
- 16.Watts DT. The effect of nicotine and smoking on the secretion of epinephrine. Ann NY Acad Sci. 1960;90:74–80. doi: 10.1111/j.1749-6632.1960.tb32619.x. [DOI] [PubMed] [Google Scholar]
- 17.Facchini FS, Hollenbeck CB, Jeppesen J, et al. Insulin resistance and cigarette smoking. Lancet. 1992;339:1128–1130. doi: 10.1016/0140-6736(92)90730-Q. [DOI] [PubMed] [Google Scholar]
- 18.Muscat JE, Harris RE, Haley NJ, et al. Cigarette smoking and plasma cholesterol. Am Heart J. 1991;121(1 Pt 1):141–147. doi: 10.1016/0002-8703(91)90967-M. [DOI] [PubMed] [Google Scholar]
- 19.Willi C, Bodenmann P, Ghali WA, et al. Active smoking and the risk of type 2 diabetes: a systematic review and meta-analysis. JAMA. 2007;298(22):2654–2664. doi: 10.1001/jama.298.22.2654. [DOI] [PubMed] [Google Scholar]
- 20.Chiolero A, Faeh D, Paccaud F, et al. Consequences of smoking for body weight, body fat distribution, and insulin resistance. Am J Clin Nutr. 2008;87(4):801–809. doi: 10.1093/ajcn/87.4.801. [DOI] [PubMed] [Google Scholar]
- 21.Cullen MW, Ebbert JO, Vierkant RA, et al. No interaction of body mass index and smoking on diabetes mellitus risk in elderly women. Prev Med. 2009;48(1):74–78. doi: 10.1016/j.ypmed.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filozof C, Fernández Pinilla MC, Fernández-Cruz A. Smoking cessation and weight gain. Obes Rev. 2004;5(2):95–103. doi: 10.1111/j.1467-789X.2004.00131.x. [DOI] [PubMed] [Google Scholar]
- 23.Iino K, Iwase M, Tsutsu N, et al. Smoking cessation and glycaemic control in type 2 diabetic patients. Diabetes Obes Metab. 2004;6(3):181–186. doi: 10.1111/j.1462-8902.2004.00329.x. [DOI] [PubMed] [Google Scholar]
- 24.Ahren B. Autonomic regulation of islet hormone secretion: implications for health and disease. Diabetologia. 2000;43(4):393–410. doi: 10.1007/s001250051322. [DOI] [PubMed] [Google Scholar]
- 25.Jamerson KA, Julius S, Gudbrandsson D, et al. Reflex sympathetic activation induces acute insulin resistance in the human forearm. Hypertension. 1993;21(5):618–623. doi: 10.1161/01.HYP.21.5.618. [DOI] [PubMed] [Google Scholar]
- 26.Arner P. Human fat cell lipolysis: biochemistry, regulation and clinical role. Best Pract Res Clin Endocrinol Metab. 2005;19:471–482. doi: 10.1016/j.beem.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 27.Kahn SE, Hull RL, Utzschneider KM. Mechanism linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 28.Ginsberg HN, Zhang YL, Hernandez-Ono A. Regulation of plasma triglycerides in insulin resistance and diabetes. Arch Med Res. 2005;36(3):232–240. doi: 10.1016/j.arcmed.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 29.O’Meara NM, Devery RA, Owens D, et al. Alterations in cellular cholesterol metabolism following administration of 6-hydroxydopamine to rabbits. Br J Pharmacol. 1992;105(2):495–499. doi: 10.1111/j.1476-5381.1992.tb14282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosmond R, Dallman MF, Björntop P. Stress related cortisol secretion in men: relationship with abdominal obesity and endocrine, metabolic and hemodynamic abnormalities. J Clin Endocrinol Metab. 1998;83(6):1853–1859. doi: 10.1210/jc.83.6.1853. [DOI] [PubMed] [Google Scholar]
- 31.Amerena J, Julius S. The role of the autonomic nervous system in hypertension. Hypertens Res. 1995;18(2):99–110. doi: 10.1291/hypres.18.99. [DOI] [PubMed] [Google Scholar]
- 32.De Hert M, van Winkel R, Van Eyck D, et al. Prevalence of diabetes, metabolic syndrome and metabolic abnormalities in schizophrenia over the course of the illness: a cross-sectional study. Clin Pract Epidemol Ment Health. 2006;27(2):14. doi: 10.1186/1745-0179-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rummel-Kluge C, Komossa K, Schwarz S, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2010;123(2–3):225–233. doi: 10.1016/j.schres.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nasrallah HA. Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol Psychiatry. 2008;13(1):27–35. doi: 10.1038/sj.mp.4002066. [DOI] [PubMed] [Google Scholar]
- 35.Lann D, LeRoith D. Insulin resistance as the underlying cause for the metabolic syndrome. Med Clin North Am 2007 Nov;91(6):1063-77, viii. [DOI] [PubMed]
- 36.Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs. 2005;19(Suppl. 1):1–93. doi: 10.2165/00023210-200519001-00001. [DOI] [PubMed] [Google Scholar]
- 37.Houseknecht KL, Robertson AS, Zavadoski W, et al. Acute effects of atypical antipsychotics on whole-body insulin resistance in rats: implications for adverse metabolic effects. Neuropsychopharmacology. 2007;32(2):289–297. doi: 10.1038/sj.npp.1301209. [DOI] [PubMed] [Google Scholar]
- 38.Vidarsdottir S, de Leeuw van Weenen JE, Frölich M, et al. Effects of olanzapine and haloperidol on the metabolic status of healthy men. J Clin Endocrinol Metab. 2010;95(1):118–125. doi: 10.1210/jc.2008-1815. [DOI] [PubMed] [Google Scholar]
- 39.Albaugh VL, Singareddy R, Mauger D, et al. A double blind, placebo-controlled, randomized crossover study of the acute metabolic effects of olanzapine in healthy volunteers. PLoS One. 2011;6(8):e22662. doi: 10.1371/journal.pone.0022662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sowell M, Mukhopadhyay N, Cavazzoni P, et al. Evaluation of insulin sensitivity in healthy volunteers treated with olanzapine, risperidone, or placebo: a prospective, randomized study using the two-step hyperinsulinemic, euglycemic clamp. J Clin Endocrinol Metab. 2003;88(12):5875–5880. doi: 10.1210/jc.2002-021884. [DOI] [PubMed] [Google Scholar]
- 41.Koller E, Schneider B, Bennett K, et al. Clozapine-associated diabetes. Am J Med. 2001;111(9):716–723. doi: 10.1016/S0002-9343(01)01000-2. [DOI] [PubMed] [Google Scholar]
- 42.American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596–601. [DOI] [PubMed]
- 43.Lambert BL, Cunningham FE, Miller DR, et al. Diabetes risk associated with use of olanzapine, quetiapine, and risperidone in Veterans Health Administration patients with schizophrenia. Am J Epidemiol. 2006;164(7):672–681. doi: 10.1093/aje/kwj289. [DOI] [PubMed] [Google Scholar]
- 44.Citrome L. Risk-benefit analysis of available treatments for schizophrenia. Psychiatric Times. 2007;1:27–30. [Google Scholar]
- 45.Coccurello R, Moles A. Potential mechanisms of atypical antipsychotic-induced metabolic derangement: clues for understanding obesity and novel drug design. Pharmacol Ther. 2010;127(3):210–251. doi: 10.1016/j.pharmthera.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 46.Marcus RN, Owen R, Manos G, et al. Safety and tolerability of aripiprazole for irritability in pediatric patients with autistic disorder: a 52-week, open-label, multicenter study. J Clin Psychiatry. 2011;72(9):1270–1276. doi: 10.4088/JCP.09m05933. [DOI] [PubMed] [Google Scholar]
- 47.De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733–759. doi: 10.2165/11634500-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 48.Reynolds GP, Kirk SL. Metabolic side effects of antipsychotic drug treatment: pharmacological mechanisms. Pharmacol Ther. 2010;125(1):169–179. doi: 10.1016/j.pharmthera.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 49.Guo JJ, Keck PE, Jr, Corey-Lisle PK, et al. Risk of diabetes mellitus associated with atypical antipsychotic use among medicaid patients with bipolar disorder: a nested case-control study. Pharmacotherapy. 2007;27(1):27–35. doi: 10.1592/phco.27.1.27. [DOI] [PubMed] [Google Scholar]
- 50.Zheng L, Mack WJ, Dagerman KS, et al. Metabolic changes associated with second-generation antipsychotic use in Alzheimer’s disease patients: the CATIE-AD study. Am J Psychiatry. 2009;166(5):583–590. doi: 10.1176/appi.ajp.2008.08081218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Newcomer JW. Metabolic syndrome and mental illness. Am J Managed Care. 2007;13(11):S170–S177. [PubMed] [Google Scholar]
- 52.Basson BR, Kinon BJ, Taylor CC, et al. Factors influencing acute weight change in patients with schizophrenia treated with olanzapine, haloperidol, or risperidone. J Clin Psychiatry. 2001;62:231–238. doi: 10.4088/JCP.v62n0404. [DOI] [PubMed] [Google Scholar]
- 53.Panariello F, De Luca V, de Bartolomeis A. Weight gain, schizophrenia and antipsychotics: new findings from animal model and pharmacogenomic studies. Schizophr Res Treat. 2011;2011:459284. doi: 10.1155/2011/459284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kroeze WK, Hufeisen SJ, Popadak BA, et al. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology. 2003;28(3):519–526. doi: 10.1038/sj.npp.1300027. [DOI] [PubMed] [Google Scholar]
- 55.Matsui-Sakata A, Ohtani H, Sawada Y. Receptor occupancy-based analysis of the contributions of various receptors to antipsychotics-induced weight gain and diabetes mellitus. Drug Metab Pharmacokinet. 2005;20(5):368–378. doi: 10.2133/dmpk.20.368. [DOI] [PubMed] [Google Scholar]
- 56.Huang XF, Han M, Huang X, et al. Olanzapine differentially affects 5-HT2A and 2C receptor mRNA expression in the rat brain. Behav Brain Res. 2006;171:355–362. doi: 10.1016/j.bbr.2006.03.040. [DOI] [PubMed] [Google Scholar]
- 57.Baptista T, Parada M, Hernandez L. Long term administration of some antipsychotic drugs increases body weight and feeding in rats: are D2 dopamine receptors involved? Pharmacol Biochem Behav. 1987;27(3):399–405. doi: 10.1016/0091-3057(87)90340-6. [DOI] [PubMed] [Google Scholar]
- 58.Miyamoto S, Duncan GE, Marx CE, et al. Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry. 2005;10(1):79–104. doi: 10.1038/sj.mp.4001556. [DOI] [PubMed] [Google Scholar]
- 59.Silvestre JS, Prous J. Research on adverse drug events: I. Muscarinic M3 receptor binding affinity could predict the risk of antipsychotics to induce type 2 diabetes. Methods Find Exp Clin Pharmacol. 2005;27(5):289–304. doi: 10.1358/mf.2005.27.5.908643. [DOI] [PubMed] [Google Scholar]
- 60.Fabris SE, Thorburn A, Litchfield A, et al. Effect of parasympathetic denervation of liver and pancreas on glucose kinetics in man. Metabolism. 1996;45(8):987–991. doi: 10.1016/S0026-0495(96)90268-1. [DOI] [PubMed] [Google Scholar]
- 61.Jin H, Meyer JM, Mudaliar S, et al. Impact of atypical antipsychotic therapy on leptin, ghrelin, and adiponectin. Schizophr Res. 2008;100(1–3):70–85. doi: 10.1016/j.schres.2007.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roerig JL, Steffen KJ, Mitchell JE. Atypical antipsychotic-induced weight gain: insights into mechanisms of action. CNS Drugs. 2011;25(12):1035–1059. doi: 10.2165/11596300-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 63.Reynolds GP, Hill MJ, Kirk SL. The 5-HT2C receptor and antipsychotic induced weight gain: mechanisms and genetics. J Psychopharmacol. 2006;20(4 Suppl.):15–18. doi: 10.1177/1359786806066040. [DOI] [PubMed] [Google Scholar]
- 64.van Winkel R, Rutten BP, Peerbooms O, et al. MTHFR and risk of metabolic syndrome in patients with schizophrenia. Schizophr Res. 2010;121(1–3):193–198. doi: 10.1016/j.schres.2010.05.030. [DOI] [PubMed] [Google Scholar]
- 65.Hong CJ, Liou YJ, Bai YM, et al. Dopamine receptor D2 gene is associated with weight gain in schizophrenic patients under long-term atypical antipsychotic treatment. Pharmacogenet Genomics. 2010;20(6):359–366. doi: 10.1097/FPC.0b013e3283397d06. [DOI] [PubMed] [Google Scholar]
- 66.Missale C, Nash SR, Robinson SW, et al. Dopamine receptors: from structure to function. Physiol Rev. 1998;78(1):189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 67.Kebabian JW, Calne DB. Multiple receptors for dopamine. Nature. 1979;277(5692):93–96. doi: 10.1038/277093a0. [DOI] [PubMed] [Google Scholar]
- 68.Murphy MB. Dopamine: a role in the pathogenesis and treatment of hypertension. J Hum Hypertens. 2000;14(Suppl. 1):S47–S50. doi: 10.1038/sj.jhh.1000987. [DOI] [PubMed] [Google Scholar]
- 69.Mannelli M, Delitala G, De Feo ML, et al. Effects of different dopaminergic antagonists on bromocriptine-induced inhibition of norepinephrine release. J Clin Endocrinol Metab. 1984;59(1):74–78. doi: 10.1210/jcem-59-1-74. [DOI] [PubMed] [Google Scholar]
- 70.Mannelli M, Ianni L, Lazzeri C, et al. In vivo evidence that endogenous dopamine modulates sympathetic activity in man. Hypertension. 1999;34(3):398–402. doi: 10.1161/01.HYP.34.3.398. [DOI] [PubMed] [Google Scholar]
- 71.Kok P, Roelfsema F, Frölich M, et al. Activation of dopamine D2 receptors simultaneously ameliorates various metabolic features of obese women. Am J Physiol Endocrinol Metab. 2006;291:E1038–E1043. doi: 10.1152/ajpendo.00567.2005. [DOI] [PubMed] [Google Scholar]
- 72.Cincotta AH, Meier AH, Cincotta M., Jr Bromocriptine improves glycaemic control and serum lipid profile in obese type 2 diabetic subjects: a new approach in the treatment of diabetes. Expert Opin Investig Drugs. 1999;8(10):1683–1707. doi: 10.1517/13543784.8.10.1683. [DOI] [PubMed] [Google Scholar]
- 73.Seeman P, Tallerico T. Antipsychotic drugs which elicit little or no parkinsonism bind more loosely than dopamine to brain D2 receptors, yet occupy high levels of these receptors. Mol Psychiatry. 1998;3(2):123–134. doi: 10.1038/sj.mp.4000336. [DOI] [PubMed] [Google Scholar]
- 74.Brogden RN, Carmine AA, Heel RC, et al. Domperidone: a review of its pharmacological activity, pharmacokinetics and therapeutic efficacy in the symptomatic treatment of chronic dyspepsia and as an antiemetic. Drugs. 1982;24(5):360–400. doi: 10.2165/00003495-198224050-00002. [DOI] [PubMed] [Google Scholar]
- 75.Mannelli M, Pupilli C, Fabbri G, et al. Endogenous dopamine (DA) and DA2 receptors: a mechanism limiting excessive sympathetic-adrenal discharge in humans. J Clin Endocrinol Metab. 1988;66(3):626–631. doi: 10.1210/jcem-66-3-626. [DOI] [PubMed] [Google Scholar]
- 76.Luchsinger A, Grilli M, Velasco M. Metoclopramide and domperidone block the antihypertensive effect of bromocriptine in hypertensive patients. Am J Ther. 1998;5(2):81–88. doi: 10.1097/00045391-199803000-00005. [DOI] [PubMed] [Google Scholar]
- 77.See RE, Fido AA, Maurice M, et al. Risperidone-induced increase of plasma norepinephrine is not correlated with symptom improvement in chronic schizophrenia. Biol Psychiatry. 1999;45(12):1653–1656. doi: 10.1016/S0006-3223(98)00199-1. [DOI] [PubMed] [Google Scholar]
- 78.Zander KJ, Fischer B, Zimmer R, et al. Long-term neuroleptic treatment of chronic schizophrenic patients: clinical and biochemical effects of withdrawal. Psychopharmacology (Berl) 1981;73(1):43–47. doi: 10.1007/BF00431099. [DOI] [PubMed] [Google Scholar]
- 79.Savoy YE, Ashton MA, Miller MW, et al. Differential effects of various typical and atypical antipsychotics on plasma glucose and insulin levels in the mouse: evidence for the involvement of sympathetic regulation. Schizophr Bull. 2010;36(2):410–418. doi: 10.1093/schbul/sbn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goff DC, Sullivan LM, McEvoy JP, et al. A comparison of ten-year cardiac risk estimates in schizophrenia patients from the CATIE study and matched controls. Schizophr Res. 2005;80(1):45–53. doi: 10.1016/j.schres.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 81.Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic: a double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789–796. doi: 10.1001/archpsyc.1988.01800330013001. [DOI] [PubMed] [Google Scholar]
- 82.Montiel C, Artalejo AR, Bermejo PM, et al. A dopaminergic receptor in adrenal medulla as a possible site of action for the droperidol-evoked hypertensive response. Anesthesiology. 1986;65(5):474–479. doi: 10.1097/00000542-198611000-00004. [DOI] [PubMed] [Google Scholar]
- 83.Haddad PM, Anderson IM. Antipsychotic-related QTc prolongation, torsade de pointes and sudden death. Drugs. 2002;62(11):1649–1671. doi: 10.2165/00003495-200262110-00006. [DOI] [PubMed] [Google Scholar]
- 84.Roussak JB, Carey P, Harry H. Cardiac arrest after treatment with intravenous domperidone. BMJ (Clin Res Ed) 1984;289(6458):1579. doi: 10.1136/bmj.289.6458.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abildskov JA, Lux RL. Mechanisms in adrenergic dependent onset of torsades de pointes. Pacing Clin Electrophysiol. 1997;20(1 Pt 1):88–94. doi: 10.1111/j.1540-8159.1997.tb04816.x. [DOI] [PubMed] [Google Scholar]
- 86.Gugger JJ. Antipsychotic pharmacotherapy and orthostatic hypotension: identification and management. CNS Drugs. 2011;25(8):659–671. doi: 10.2165/11591710-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 87.Julius S. Abnormalities of autonomic nervous control in human hypertension. Cardiovasc Drugs Ther. 1994;8(Suppl. 1):11–20. doi: 10.1007/BF00877080. [DOI] [PubMed] [Google Scholar]
- 88.Goldstein DS, Holmes CS, Dendi R, et al. Orthostatic hypotension from sympathetic denervation in Parkinson’s disease. Neurology. 2002;58(8):1247–1255. doi: 10.1212/WNL.58.8.1247. [DOI] [PubMed] [Google Scholar]
- 89.Scigliano G, Musicco M, Soliveri P, et al. Reduced risk factors for vascular disorders in Parkinson’s disease patients: a case-control study. Stroke. 2006;37(5):1184–1188. doi: 10.1161/01.STR.0000217384.03237.9c. [DOI] [PubMed] [Google Scholar]
- 90.Scigliano G, Ronchetti G, Girotti F, et al. Sympathetic modulation by levodopa reduces vascular risk factors in Parkinson disease. Parkinsonism Relat Disord. 2009;15(2):138–143. doi: 10.1016/j.parkreldis.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 91.Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47(1):27–38. [PubMed] [Google Scholar]
- 92.Ebert B, Wafford KA, Whiting PJ, et al. Molecular pharmacology of gamma-aminobutyric acid type A receptor agonists and partial agonists in oocytes injected with different alpha, beta, and gamma receptor subunit combinations. Mol Pharmacol. 1994;46(5):957–963. [PubMed] [Google Scholar]
- 93.Jaskiw GE, Popli AP. A meta-analysis of the response to chronic L-dopa in patients with schizophrenia: therapeutic and heuristic implications. Psychopharmacology (Berl) 2004;171(4):365–374. doi: 10.1007/s00213-003-1672-y. [DOI] [PubMed] [Google Scholar]
- 94.Whitsett TL, Halushka PV, Goldberg LI. Attenuation of postganglionic sympathetic nerve activity by L-dopa. Circ Res. 1970;27(4):561–570. doi: 10.1161/01.RES.27.4.561. [DOI] [PubMed] [Google Scholar]
- 95.Rajfer SI, Anton AH, Rossen JD, et al. Beneficial hemodynamic effects of oral levodopa in heart failure: relation to the generation of dopamine. N Engl J Med. 1984;310(21):1357–1362. doi: 10.1056/NEJM198405243102105. [DOI] [PubMed] [Google Scholar]
- 96.Marsden CD, Parkes JD. “On-off” effects in patients with Parkinson’s disease on chronic levodopa therapy. Lancet. 1976;1(7954):292–296. doi: 10.1016/S0140-6736(76)91416-1. [DOI] [PubMed] [Google Scholar]
- 97.Parkkinen L, O’Sullivan SS, Kuoppamäki M, et al. Does levodopa accelerate the pathologic process in Parkinson disease brain? Neurology. 2011;77(15):1420–1426. doi: 10.1212/WNL.0b013e318232ab4c. [DOI] [PubMed] [Google Scholar]