

Abstract
A central axiom of ligand-receptor theory is that agonists bind more tightly to active than to inactive receptors. However, measuring agonist affinity in inactive receptors is confounded by concomitant activation. We identified a cysteine substituted mutant γ-aminobutyric acid type A (GABAA) receptor with unique characteristics allowing the determination of allosteric agonist site occupancy in both inactive and active receptors. Etomidate, the allosteric agonist, is an anesthetic that activates or modulates α1β2γ2L GABAA receptors via transmembrane sites near β2M286 residues in M3 domains. Voltage-clamp electrophysiology studies of α1β2M286Cγ2L receptors show that GABA is an efficacious agonist and that etomidate modulates GABA-activated activity, but direct etomidate agonism is absent. Quantitative analysis of mutant activity using an established Monod-Wyman-Changeux (MWC) allosteric model indicates that the intrinsic efficacy of etomidate, defined as its relative affinity for active versus inactive receptors, is lower than in wild-type receptors. Para-chloromercuribenzene sulfonate covalently modifies β2M286C side-chain sulfhydryls, irreversibly altering GABA-induced currents. Etomidate concentration dependently reduces the apparent rate of β2M286C-pCMBS bond formation, tracked electrophysiologically. High etomidate concentrations completely protect the β2M286C suflhydryl from covalent modification, suggesting close steric interactions. The 50% protective etomidate concentration (PC50) is 14 μM in inactive receptors and 1.1 to 2.2 μM during GABA-activation, experimentally demonstrating that activated receptors bind etomidate more avidly than do inactive receptors. The experimental PC50 values are remarkably close to, and therefore validate, MWC model predictions for etomidate dissociation constants in both inactive and active receptors. Our results support MWC models as valid frameworks for understanding the agonism, coagonism, and modulation of ligand-gated ion channels.
Introduction
Functional models of ligand binding and efficacy at receptors such as ligand-gated ion channels incorporate the proposition that agonism is a feature of ligands that bind with higher affinity to active receptors relative to inactive receptors and therefore shift the inactive⇔active equilibrium toward active (Colquhoun, 1998; Kenakin, 2004; Buxton, 2006; Auerbach, 2012). This concept applies to both orthosteric and allosteric agonists. Consequently, the apparent affinity of an agonist for receptors in assays of both function and ligand-binding depends on its intrinsic efficacy (Colquhoun, 1998). Monod-Wyman-Changeux (MWC) allosteric models of receptor function define agonist efficacy as the ratio of affinities for active versus inactive states (Changeux, 2012) and provide a framework for estimating agonist affinity and intrinsic efficacy parameters in ligand-gated ion channels, which are the only receptors in which signals directly proportional to receptor activity (voltage-clamped currents) can be readily obtained (Galzi et al., 1996; Chang and Weiss, 1999; Rüsch et al., 2004; Auerbach, 2012; Ruesch et al., 2012). Nonetheless, experimental validation of model-derived estimates for agonist binding affinities at inactive receptors is confounded by agonist induction of receptor activation, which simultaneously alters agonist-receptor interactions. One rare example of this achievement used complex rapid mixing and fluorescent detection of the nicotinic agonist dansyl-C6-choline binding to Torpedo nicotinic acetylcholine receptors (Raines and Krishnan, 1998). In the present study, we identified a mutant γ-aminobutyric acid type A (GABAA) receptor with unique functional characteristics that enable experimental determination of binding site occupancy for an allosteric agonist or modulator, etomidate, in both inactive and active receptors.
Synaptic GABAA receptors are pentameric membrane complexes typically formed from two α, two β, and one γ subunit symmetrically surrounding a chloride-conducting ion channel (Olsen and Sieghart, 2008). Each subunit contains a large N-terminal extracellular domain and four transmembrane domains (M1–M4), with M2 domains from each subunit surrounding the ion channel. The M1, M3, and M4 domains form an outer ring of helical domains between the M2 domains and membrane lipids. Two GABA sites per receptor are formed at the extracellular interfaces between α and β subunits.
The allosteric agonist used in this study is etomidate, a potent general anesthetic that acts by modulating GABAA receptors. In wild-type α1β2γ2L GABAA receptors, low etomidate concentrations enhance GABA-mediated receptor activation, whereas high etomidate concentrations (≥10 μM) directly activate receptors in the absence of GABA. The gating effects of both GABA and etomidate are quantitatively described by a Monod-Wyman-Changeux (MWC) two-state allosteric coagonist mechanism with two equivalent sites for etomidate (Rüsch et al., 2004; Guitchounts et al., 2012). The etomidate sites are located within transmembrane interfaces between α-M1 and β-M3 domains based on labeling with photoreactive etomidate derivatives (Li et al., 2006; Chiara et al., 2012).
The receptors we used in this study incorporate cysteine mutations at one photolabeled residue in etomidate binding sites: β2M286. Studies in rat α1β2M286Cγ2L GABAA receptors demonstrate that the β2M286C sulfhydryl is covalently modified by a water-soluble reagent, para-choromercuribenzene sulfonate (pCMBS) and that GABA activation accelerates this reaction (Bali and Akabas, 2004, 2012). We conducted a detailed electrophysiological analysis of heterologously expressed human α1β2M286Cγ2L GABAA receptor function, which revealed the absence of etomidate agonism. However, etomidate modulation of GABA-mediated activation is maintained in the mutant receptors, albeit weaker than in wild-type receptros. These properties allowed us to determine experimentally the etomidate affinity in both inactive and GABA-activated mutant channels, based on etomidate-dependent inhibition of covalent bond formation between β2M286C and pCMBS. By tracking the electrophysiological effects of this reaction in mutant receptors, we quantified the apparent rate of pCMBS-β2M286C bond formation in both inactive (no GABA) and GABA-activated receptors while varying the etomidate concentration. High concentrations of etomidate completely protected β2M286C from covalent modification. Etomidate-dependent receptor occupancy estimates based on cysteine protection studies were compared with those predicted by fitting functional data with the MWC allosteric coagonist model for etomidate and GABA (Rüsch et al., 2004).
Materials and Methods
Animal Use.
Female Xenopus laevis were housed in a veterinary-supervised environment in accordance with local and federal guidelines. Frogs were anesthetized by immersion in 0.2% tricaine (Sigma-Aldrich, St. Louis, MO) before undergoing minilaparotomy to harvest oocytes.
Chemicals.
R(+)-etomidate was obtained from Bedford Laboratories (Bedford, OH). The clinical preparation in 35% propylene glycol was diluted directly into buffer. Previous studies have shown that propylene glycol at the dilutions used for these studies has no effect on GABAA receptor function (Rüsch et al., 2004). Picrotoxin (PTX) was purchased from Sigma-Aldrich, and a 2 mM solution in an electrophysiology buffer was made by prolonged gentle shaking. Alphaxalone was purchased from MP Biomedical (Solon, OH) and prepared as a 2 mM stock solution in dimethylsufoxide. During the experiments with alphaxalone, dimethylsufoxide was present at 0.1%, which had no effect on GABAA receptor function. p-Chloromercuribenzenesulfonic acid sodium salt was purchased from Toronto Research Chemicals (North York, ON, Canada) and maintained in a cold, dry environment. Fresh solutions of pCMBS were prepared frequently, and sulfhydryl modifying activity was assayed using colorimetric titration of dithionitrobenzoic acid, as described by Karlin and Akabas (1998).
Molecular Biology.
cDNAs for human GABAA receptor α1, β2, and γ2L subunits were cloned into pCDNA3.1 vectors (Invitrogen, Carlsbad, CA). The β2M286C mutation was created with oligonucleotide-directed mutagenesis using a QuikChange kit (Stratagene, La Jolla, CA). Clones from the mutagenesis reaction were subjected to DNA sequencing through the entire cDNA region to confirm the presence of the mutation and the absence of stray mutations.
Oocyte Electrophysiology.
Messenger RNA synthesis and Xenopus oocyte expression were performed as previously described (Stewart et al., 2008). Experiments were performed at room temperature (21–23°C) in ND96 buffer (in mM: 96 NaCl, 2 KCl, 0.8 MgCl2, 1.8 CaCl2, 5 HEPES, pH 7.5). GABAA receptor responses to GABA were assessed in Xenopus oocytes using two-microelectrode voltage-clamp electrophysiology, as previously described (Rüsch et al., 2004). GABA pulses were from 5 to 20 seconds, depending on the concentration of GABA used and the time to peak current. Normalizing GABA responses at maximal GABA (2–10 mM) were recorded every second or third sweep. Oocyte currents were low-pass filtered at 1 kHz (Model OC-725B; Warner Instruments, Hamden, CT) and digitized at 2 kHz using commercial digitizer hardware (Digidata 1200; Molecular Devices, Sunnyvale, CA) and software (Clampex 7; Molecular Devices) for offline analysis.
Cysteine Modification with pCMBS and Etomidate Protection in Xenopus Oocytes.
The pCMBS concentrations were chosen so that control modification (with or without 2 mM GABA) appeared complete after 40–60 seconds of pCMBS exposure. In modification experiments, GABA responses to both low GABA (30 μM, approximately EC5) and high GABA (1–2 mM) were measured before and after each exposure to pCMBS ± GABA. Additional control modification experiments included 2 μM alphaxalone with GABA to maximize the fraction of activated receptors. In protection experiments, etomidate was present throughout the experiment and was thus added to background buffer, pCMBS solutions, and GABA solutions. Because etomidate enhances GABA responses in the mutant receptors, functional testing after modification was performed with etomidate plus low GABA (combined efficacy about 5% of maximum) and etomidate plus 1 mM GABA. In most experiments, modification exposures to pCMBS of 5–10 seconds each were performed, followed by at least 2 minutes of wash for pCMBS alone and longer washes (5–10 minutes) when GABA or etomidate was present. Up to 10 modification and post-test cycles were performed on each oocyte. The ratio of low GABA versus high GABA current responses was calculated, and all ratios were normalized to the control ratio before pCMBS exposure.
Normalized response ratios were plotted as a function of total pCMBS exposure (seconds or seconds × [pCMBS]). For some data sets, exponential time constants were derived from single exponential fits to data. Initial modification rates were also calculated as slopes (M-1s−1) of linear least-squares fits to the initial three or four normalized ratio points (baseline and two or three postmodification points).
Electrophysiology in Human Embryonic Kidney (HEK)293 Cell Membrane Patches.
HEK293 cell maintenance and transfection for functional studies were performed as previously described (Scheller and Forman, 2002). Cells transfected with plasmids encoding GABAA receptor subunits were maintained in culture medium for 24–48 hours before electrophysiology experiments. Current recordings from excised outside-out membrane patches were performed at room temperature (21–23°C) under −50 mV voltage clamp as previously described (Scheller and Forman, 2002). Bath and superfusion solutions contained (in mM) 145 NaCl, 5 KCl, 10 HEPES, 2 CaCl2, and 1 MgCl2 at pH 7.4 (pH adjusted with N-methyl glucosamine). The intracellular (pipette) fluid contained (in mM) 140 KCl, 10 HEPES, 1 EGTA, and 2 MgCl2 at pH 7.3 (pH adjusted with KOH). Currents were stimulated using pulses of GABA delivered via a quad (2 × 2) superfusion pipette coupled to piezoelectric elements that switched superfusion solutions in less than 1 millisecond. Currents were filtered at 5 kHz and digitized at 10 kHz for offline analysis.
Data Analysis.
Quantitative analyses of agonist concentration-responses, etomidate-induced left shift, and allosteric coagonist model fitting followed our approach described elsewhere (Stewart et al., 2008; Desai et al., 2009). Agonist (GABA or etomidate) concentration-response data were fitted with logistic functions using nonlinear least-squares (Origin 6.1; OriginLab, Northampton, MA; and GraphPad Prism 5.0; GraphPad Software, La Jolla, CA):
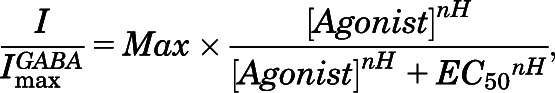 |
(1) |
where nH is Hill slope.
Etomidate left-shift values were calculated as the ratio of the GABA EC50 values in the absence of drug to that in the presence of 3.2 μM drug. Large EC50 ratios indicate strong modulation, whereas a ratio of 1.0 or less indicates no positive modulation.
Etomidate-dependent effects on pCMBS modification (protection analysis) were also analyzed by nonlinear least-squares fits to a logistic function with nH fixed at 1.0 and the minimum modification rate fixed at zero (based on results with high etomidate). We defined the PC50 as the etomidate concentration resulting in 50% reduction in the maximum modification rate.
PTX-sensitive leak currents (IPTX) normalized to 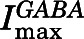 were measured to help estimate basal open probability, which was assessed in both α1β2M286Cγ2L receptors and in double-mutant α1L264Tβ2M286Cγ2L receptors. The α1L264T mutation confers spontaneous gating to the double mutant for comparison with α1L264Tβ2γ2L, which we have described previously (Rüsch et al., 2004). Maximal GABA efficacy was estimated using a single-sweep protocol with 2 μM alphaxalone as allosteric enhancer (Desai et al., 2009). If the addition of alphaxalone increases current beyond that elicited with maximal GABA alone, then GABA is not opening all activatable channels.
were measured to help estimate basal open probability, which was assessed in both α1β2M286Cγ2L receptors and in double-mutant α1L264Tβ2M286Cγ2L receptors. The α1L264T mutation confers spontaneous gating to the double mutant for comparison with α1L264Tβ2γ2L, which we have described previously (Rüsch et al., 2004). Maximal GABA efficacy was estimated using a single-sweep protocol with 2 μM alphaxalone as allosteric enhancer (Desai et al., 2009). If the addition of alphaxalone increases current beyond that elicited with maximal GABA alone, then GABA is not opening all activatable channels.
Estimated open probability ( ) was calculated by explicitly adding average spontaneous activity
) was calculated by explicitly adding average spontaneous activity  to normalized activated current
to normalized activated current  and renormalizing to the full range of open probability, bracketed by alphaxalone enhanced current
and renormalizing to the full range of open probability, bracketed by alphaxalone enhanced current 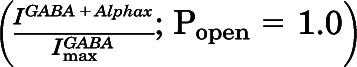 and PTX-blocked basal current (
and PTX-blocked basal current ( ):
):
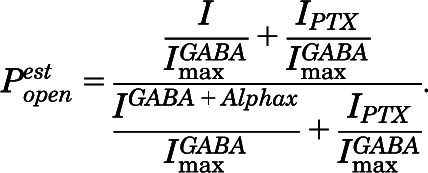 |
(2) |
Nonlinear least-squares fits to a MWC two-state coagonist mechanism (eq. 3) used  data (eq. 2) from GABA concentration-responses (with and without etomidate) and etomidate-dependent activation (with and without EC5 GABA). Both GABA and etomidate [ETO] were independent variables:
data (eq. 2) from GABA concentration-responses (with and without etomidate) and etomidate-dependent activation (with and without EC5 GABA). Both GABA and etomidate [ETO] were independent variables:
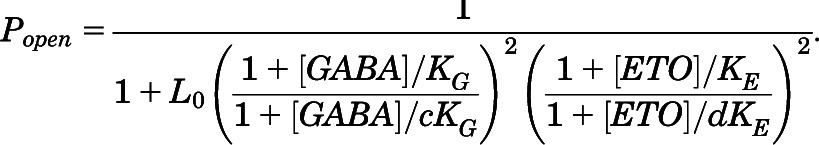 |
(3) |
L0 in eq. 3 is a dimensionless basal equilibrium gating variable, approximately P0-1. Spontaneous activity was undetectable in α1β2M286Cγ2L receptors. Therefore, we constrained L0 to a value based on previous estimates for wild-type L0 (25,000), corrected for the relative spontaneous activity of receptors with the α1L264T mutations in wild-type versus β2M286C backgrounds. The ratio of spontaneous open probabilities in these receptors is approximately 3 (see Results), so we constrained L0 to a value of 75,000 for α1β2M286Cγ2L receptors. KG and KE are equilibrium dissociation constants for GABA and etomidate binding to inactive receptors, and c and d are dimensionless parameters representing the respective ratios of dissociation constants in active versus inactive states. The intrinsic agonist efficacies of GABA and etomidate are inversely related to, respectively, c and d.
Analysis of patch macrocurrents for activation, desensitization, and deactivation kinetics was performed as described in Scheller and Forman (2002) using nonlinear least-squares fits to eq. 4 and F-tests at P = 0.99 (Clampfit8.0; Molecular Devices) to choose the best number of exponents:
| (4) |
Activation traces were fitted best with a single exponent; desensitization and deactivation were fitted best with two exponents.
Statistical Analysis.
Results are reported as mean ± S.D. Nonlinear regression errors are those from fits in Origin 6.1 (OriginLab), GraphPad Prism 5.02 (GraphPad Software), or Clampfit 8.0 (Molecular Devices). Statistical comparison of fitted parameters was performed using Prism 5.02. Statistical significance was inferred at P < 0.05.
Results
Functional Characterization of α1β2M286Cγ2L GABAA Receptors.
Voltage-clamped oocytes expressing α1β2M286Cγ2L receptors produced inward currents in response to GABA (Fig. 1A). Etomidate (3.2 μM) enhanced responses at all GABA concentrations (Fig. 1B). A logistic fit to pooled concentration-response data (Fig. 1C, solid squares; n ≥ 6) resulted in a GABA EC50 for this mutant of 180 μM; the average EC50 from independent analyses of six separate oocytes with complete data sets was 210 μM (Table 1). These values are about 6-fold higher than the wild-type GABA EC50 (Table 1). In the presence of 3.2 μM etomidate, GABA concentration responses shift leftward (Fig. 1C, open squares), reducing GABA EC50 2- to 3-fold. This is a significantly smaller leftward-shift ratio than that observed in α1β2γ2L receptors in the presence of 3.2 μM etomidate (Table 1).
Fig. 1.

GABA activation and etomidate modulation of α1β2M286Cγ2L GABAA receptors. (A) Current traces are from a single Xenopus oocyte expressing α1β2M286Cγ2L receptors, activated with different GABA concentrations. GABA application (labeled in micromolar) is indicated by horizontal bars above traces. (B) Current traces are from the same oocyte in (A), stimulated with variable GABA (labeled in micromolar above trave in A) plus 3.2 μM etomidate. GABA and etomidate application is indicated by horizontal bars above traces. (C) Averaged (± S.D.; n ≥ 6) GABA responses in the absence (solid squares) and presence of etomidate (3.2 μM; open squares) are shown. Current responses were normalized to the 3 mM GABA response in the same oocyte before averaging data from all cells. Lines represent logistic fits (eq. 1, in Materials and Methods) to data. Fitted parameters for GABA alone (solid line) are EC50 = 180 ± 19 μM and Hill slope = 1.3 + 0.16. Fitted parameters for GABA plus etomidate (dashed line) are maximum = 1.44 + 0.04, EC50 = 87 ± 11, and Hill slope = 1.5 ± 0.24. Restricting analyses to data below 3 mM GABA does not alter the relative maxima or GABA EC50 values. (D) Average (± S.D.; n ≥ 3) responses to etomidate alone (solid circles and line) or to etomidate combined with 20 μM GABA (EC3; open circles). Currents were normalized to 3 mM GABA responses in the same oocyte before averaging. Etomidate direct activation was never greater than 1.5% of maximal GABA response and was not further analyzed. The dashed line through GABA EC3 enhancement data represents a logistic fit: maximum = 67 ± 5.0%, EC50 = 24 ± 4.9 μM, Hill slope = 2.0 ± 0.75. (E) A voltage-clamp current trace recorded from an outside-out patch excised from an HEK293 cell expressing α1β2M286Cγ2L receptors and activated with 3 mM GABA for 1 second (bar above trace) delivered with sub-millisecond solution switching. The trace displays rapid activation, biphasic desensitization, and biphasic deactivation. Average rates for each phase are reported in Table 1.
TABLE 1.
Pharmacological, kinetic, and Monod-Wyman-Changeux (MWC) coagonist model parameters for α1β2γ2L and α1β2M286Cγ2L GABAA receptors
| Parameter | α1β2γ2L | α1β2M286Cγ2L |
|---|---|---|
| Mean ± S.D. (n)a | Mean ± S.D. (n) | |
| GABA EC50 (μM) | 37 ± 17 (7) | 210 ± 35 (6)** |
| nH | 1.6 ± 0.39 (7) | 1.5 ± 0.33 (6) |
| GABA efficacy (%)b | 88 ± 5 (4) | 64 ± 4.2 (4)** |
| GABA EC50 shift with 3.2 μM etomidatec | 19 ± 7.8 (7) | 2.7 ± 0.75 (4)** |
| Etomidate EC50 (μM) | 30 ± 11 (6) | Na |
| Etomidate efficacy (%) | 45 ± 12 (6) | < 1 (4)** |
| Maximal activation | ||
| Rate (s−1) | 3000 ± 1200 (4) | 2500 ± 940 (4) |
| Fast desensitization | ||
| Rate (s−1) | 20 ± 13 | 16 ± 9.4 |
| Amplitude (%) | 30 ± 11 (4) | 30 ± 18 (4) |
| Slow desensitization | ||
| Rate (s−1) | 1.0 ± 0.45 | 0.8 ± 0.29 |
| Amplitude (%)) | 70 ± 11 (4) | 70 ± 17 (4) |
| Fast deactivation | ||
| Rate (s−1) | 50 ± 18 | 57 ± 27 |
| Amplitude (%) | 60 ± 17 (4) | 70 ± 17 (4) |
| Slow deactivation | ||
| Rate (s−1) | 6 ± 2.5 | 4 ± 2.7 |
| Amplitude (%) | 40 ± 17 (4) | 30 ± 17 (4) |
| L0d | 25,000 | 75,000 |
| KG (μM) | 70 ± 22 | 270 ± 46** |
| c | (1.9 ± 0.38) × 10−3 | (2.4 ± 0.15) × 10−3 |
| KE (μM) | 40 ± 14 | 12 ± 2.4* |
| d | (7.6 ± 1.0) × 10−3 | 0.12 ± 0.018** |
n is the number of separately analyzed experiments in individual oocytes, patches or cells used to calculate averages and errors. Average values for wild-type include previously published data from Stewart et al. (2008) and Desai et al. (2009).
GABA efficacy was determined as the ratio of maximal GABA current to current elicited with maximal GABA + 2 μM alphaxalone.
EC50 shift is the ratio of GABA EC50 in the absence versus presence of 3.2 μM etomidate. Large values indicate significant positive modulation. A value of 1.0 represents no modulation.
MWC model parameters (L0, KG, KE, c, d) were derived from fits of eq. 3 to estimated Popen values (see Materials and Methods and Fig. 2).
P < 0.05; ** P < 0.01.
Etomidate at up to 1 mM elicited no significant direct activation of α1β2M286Cγ2L receptors (<1.5% of maximal GABA currents; Fig. 1D). We also studied etomidate-dependent modulation of low GABA (20 μM ≈ EC3) currents. Modulation of low GABA responses was characterized by an etomidate EC50 of 24 μM, and maximal enhancement at an etomidate concentration of 100 μM resulted in current responses averaging about 70% of maximal GABA currents (Fig. 1D).
Rapid superfusion and patch-clamp electrophysiology were used to study macrocurrent kinetics of α1β2M286Cγ2L receptors. Rapid application of 10 mM GABA to outside-out membrane patches from HEK293 cells expressing α1β2M286Cγ2L receptors resulted in currents with kinetic properties similar to those recorded with wild-type receptors (Fig. 1E; Table 1). The maximal current activation rate was approximately 3000 seconds−1, desensitization proceeds with two rates, and deactivation also displayed two distinct rates.
Allosteric Modeling of α1β2M286Cγ2L GABAA Receptor Function.
Quantitative analysis with the allosteric coagonist model (Fig. 2A) requires transformation of concentration-response data into estimates of open probability, which in turn is dependent on estimates of maximal GABA efficacy and spontaneous receptor activity. Alphaxalone, which does not interact with etomidate sites (Li et al., 2010), enhances maximal (3 mM) GABA currents by more than 50%, resulting in an estimated GABA efficacy of 0.64 (Fig. 2B). PTX-sensitive spontaneous activity was not detected in cells expressing α1β2M286Cγ2L receptors (Fig. 2C), so no Popen correction was needed for spontaneous activity. Estimated Popen values for averaged GABA-concentration responses (with and without etomidate) and etomidate-dependent modulation of GABA EC5 responses were calculated using eq. 2 (in Materials and Methods).
Fig. 2.

Allosteric coagonist modeling of GABA and etomidate-dependent activity in α1β2M286Cγ2L GABAA receptors. (A) Schematic of the MWC equilibrium coagonist mechanism for GABA and etomidate gating (eq. 3 in Materials and Methods). The mechanism is defined by five parameters: L0 is a dimensionless basal equilibrium gating variable for inactive/active (R/O) receptors, which was constrained as described in the text. KG and KE are equilibrium dissociation constants for GABA and etomidate binding to inactive (R) receptors, and c and d are dimensionless parameters representing the ratios of dissociation constants in active (O) versus inactive (R) states. The agonist efficacies of GABA and etomidate are inversely related to, respectively, c and d. (B) The trace depicts current recorded from a single oocyte expressing α1β2M286Cγ2L receptors, activated initially with 10 mM GABA, then with 10 mM GABA plus 2 μM alphaxalone, an allosteric modulator that does not interact directly with the etomidate site. The addition of alphaxalone increases the current elicited with GABA alone by about 50%, indicating that maximal GABA activates about 65% of receptors. (C) Two current traces are shown; these traces were recorded from the same oocyte expressing α1β2M286Cγ2L receptors; 3 mM GABA activates a large inward current. Picrotoxin (PTX, 2 mM) does not produce any discernable outward current, indicating that the spontaneous open probability of receptors is below the threshold of detection (0.1%) using this method. (D) Two current traces are shown, recorded from the same oocyte expressing α1L264Tβ2M286Cγ2L receptors. The α1L264T mutation confers spontaneous activity, evident from the apparent outward current with PTX application. The spontaneous activity is about 3% of the GABA-activated inward current. (E and F) Symbols represent estimated Popen values calculated from averaged data in Fig. 1, C and D. Lines through symbols represent fits to the MWC coagonist model (eq. 3, Materials and Methods) with parameters reported in Table 1. (E) GABA-dependent activation in the absence (solid squares) and presence of 3.2 μM etomidate (open squares). (F) Etomidate-dependent activation in the absence (solid circles) and presence of 20 μM GABA (open circles).
Based on previously published experiments, the basal gating parameter, L0, for wild-type α1β2γ2L is estimated to be around 25,000 (Rüsch et al., 2004; Desai et al., 2009). In double-mutant α1L264Tβ2M286Cγ2L receptors, PTX-sensitive spontaneous activity averaged 3% of the maximal GABA-activated currents (Fig. 2D). In comparison, oocyte-expressed α1L264Tβ2γ2L receptors display about 10% spontaneous activation (Rüsch et al., 2004; Desai et al., 2009). Assuming that the basal gating effects of α1L264T and β2M286C are independent, the β2M286C mutation reduces spontaneous activation around 3-fold, so we set the value of L0 for α1β2M286Cγ2L receptors in global analysis at 75,000, 3 times that of wild-type models.
With L0 thus constrained, the averaged values for α1β2M286Cγ2L resulted in a good overall fit to the MWC coagonist model (eq. 3 in Materials and Methods; R2 = 0.98), which is illustrated in Fig. 2, E and F. The fitted parameters are reported in Table 1. The fitted estimate for GABA affinity in inactive α1β2M286Cγ2L receptors (KG ≈ 270 μM) is higher than those in wild-type models (60–90 μM), whereas fitted GABA efficacy (c−1 ≈ 420) is not far from previous estimates for wild-type models (c−1 ≈ 530 to 590). The fitted etomidate dissociation constant (KE ≈ 12 μM) is lower than prior estimates in wild-type (20–40 μM); the largest difference between wild-type and α1β2M286Cγ2L models is the etomidate efficacy parameter (d−1 ≈ 8.5 in the β2M286C mutant model versus d−1 ≈ 104 to 130 in wild-type models).
pCMBS Modification of α1β2M286Cγ2L Receptors.
PCMBS exposure resulted in irreversible functional effects on α1β2M286Cγ2L receptors, both in the absence and presence of GABA (Fig. 3). Receptors exposed to pCMBS developed larger current responses to low GABA concentrations (30 μM, approx EC5) relative to high GABA concentrations (1–2 mM). Exposure to 50 μM pCMBS in the absence of GABA (inactive receptors) resulted in I30 μM/I2 mM increasing to about double its baseline value (Fig. 3, A and B) in 60–80 seconds. Exponential fits to normalized I30 μM/I2 mM as a function of pCMBS exposure time indicated modification rates averaging 1000 ± 470 M-1s−1 (mean ± S.D., n = 4). When receptors were exposed to pCMBS coapplied with 2 mM GABA, the apparent rate of receptor modification increased (Fig. 3, C and D). Exponential fits indicated rates of 1800 ± 580 M-1s−1 (mean ± S.D., n = 4). In addition, the amplitude of the pCMBS-induced gating effect was consistently larger when GABA was present (averaging 6-fold enhancement above baseline).
Fig. 3.

p-Chloromercuribenzene sulfonate modification of α1β2M286Cγ2L GABAA receptors is GABA dependent. (A) Traces represent currents recorded from a single oocyte expressing α1β2M286Cγ2L receptors, activated intermittently with 30 μM GABA (horizontal bars above each trace), each scaled to normalizing 2 mM GABA responses (not shown). Arrows indicate oocyte exposure to 50 μM pCMBS for 10 seconds (20 seconds for double arrows) followed by wash. The I30 μM approximately doubles relative to I2 mM after 60 seconds of pCMBS exposure. (B) Symbols represent the I30μM/I2mM ratios from (A) normalized to the initial value (t = 0), plotted against cumulative pCMBS exposure time. The solid line represents a single exponential fit to the data, characterized by an exponential time constant of 27 ± 4.5 seconds (apparent second-order reaction rate = 750 ± 130 M-1S−1). The dashed line is a linear fit to the first three data points. Its slope is 0.0255 second−1, resulting in an apparent initial reaction rate of 510 M-1s−1. (C) Current traces were recorded from another oocyte expressing α1β2M286Cγ2L receptors, activated intermittently with 30 μM and 2 mM GABA, each scaled to normalizing 2 mM GABA responses (not shown). Starred arrows represent coexposure to 50 μM pCMBS plus 2 mM GABA for 5 seconds (10 seconds for double-starred arrows) followed by wash. (D) Symbols represent I30 μM/I2 mM ratios from (C) normalized to the initial value (t = 0), plotted against cumulative exposure time. The I30 μM/I2 mM ratio increases more than 6-fold after 20 seconds of pCMBS/GABA exposure. A single exponential fit results in a time constant of 8.3 ± 1.2 seconds (apparent second-order reaction rate = 2400 ± 340 M-1s−1). A linear fit to the first three data points results in a slope of 0.454 second−1 (apparent initial rate = 9080 M-1s−1). (E) Symbols represent peak currents recorded from an oocyte expressing α1β2M286Cγ2L receptors activated with either 30 μM (triangles) or 2 mM (diamonds) GABA. The x-axis indicates cumulative exposure time to 50 μM pCMBS in the absence of GABA (open symbols, up to 120 seconds) and then in the presence of 50 μM pCMBS plus 2 mM GABA (solid symbols). (F) Symbols represent the I30 μM/I2 mM ratios from data in panel E, normalized to the initial value at t = 0 (0.08).
To examine the impact of GABA in more detail, we performed experiments where α1β2M286Cγ2L receptors were first modified with 50 μM pCMBS in the absence of GABA and then exposed to GABA + pCMBS. These studies reveal that the addition of GABA to pCMBS increases the response to low GABA (I30 μM; Fig. 3E, triangles) beyond that observed after pCMBS alone. Moreover, the pCMBS-induced response to high GABA (I2 mM; Fig. 3E, diamonds) drops far more after GABA + pCMBS exposure than with pCMBS alone. Together, these effects combine to increase the I30 μM/I2 mM ratio when GABA is present beyond the level achieved with pCMBS alone (Fig. 3F). Exposure to higher (250–500 μM) pCMBS also resulted in higher (3- to 4-fold) I30 μM/I2 mM ratios (unpublished data). These results suggest that partial modification of β2M286C was achieved with 50 μM pCMBS alone and that modification in the presence of GABA results in accumulation of deeply desensitized receptors that did not reactivate on the time scale of our experiments.
Because GABA affected the change in amplitude of I30 μM/I2 mM after pCMBS more than the apparent exponential rate change, we adopted an alternative approach to estimating initial rates of pCMBS modification using linear-slope analysis. Slope analyses resulted in average initial modification rates of around 400 M-1s−1 in the absence of GABA and 11,000 M-1s−1 in the presence of GABA. This finding suggests that GABA activation increases pCMBS modification rates over 20-fold, a much larger ratio than that based on exponential rate analysis (1.8-fold). Control experiments in α1β2γ2L receptors exposed to pCMBS (up to 2 mM for 60 seconds) failed to demonstrate significant (>10%) functional change in electrophysiological responses to zero, low or high GABA (unpublished data).
Etomidate Protection at α1β2M286Cγ2L Receptors.
For etomidate protection experiments, receptors were equilibrated with etomidate before and during pCMBS modification and postmodification functional testing. Because etomidate enhances responses to low GABA more than to high GABA, we reduced the low GABA concentration used for postmodification testing (1–10 μM) to produce about 5% maximal response in the presence of etomidate. Initial modification rates were quantified from linear fits to normalized Ilo GABA/I1-2 mM ratios after two or three pCMBS exposures.
In the absence of GABA, initial slope analysis resulted in an apparent β2M286C modification rate of 400 ± 96 M-1s−1 (Fig. 4A, red squares). The addition of 300 μM etomidate completely inhibited pCMBS modification (initial slope ≤0; Fig. 4A, brown diamonds). We also measured the initial rate of pCMBS modification at β2M286C in the presence of etomidate concentrations ranging from 3 to 100 μM. Data for 3 μM and 30 μM etomidate are shown in Fig. 4A. The modification rate versus (etomidate) data were fit by nonlinear least-squares to a logistic function with nH = 1, resulting in an estimate of 14 μM for the 50% protecting concentration (PC50; Fig. 4B).
Fig. 4.

Etomidate inhibition of the β2M286C-pCMBS reaction is enhanced by GABA. (A) Symbols represent data from a subset of oocytes expressing α1β2M286Cγ2L receptors used to study etomidate’s effects on pCMBS modification in the absence of GABA. Normalized IloGABA/I2 mM is plotted against cumulative pCMBS exposure (concentration × time). Lines represent linear fits to all points in each data set: pCMBS alone = red squares (370 ± 14 M-1s−1; n = 6); pCMBS + 3 μM etomidate = orange triangles (260 ± 14 M-1s−1; n = 7); pCMBS + 30 μM etomidate = blue circles (54 ± 11 M-1s−1; n = 7); pCMBS + 300 μM etomidate = brown diamonds (6 ± 8.5 M-1s−1; n = 4). (B) Symbols represent the average (± S.D.) of initial rates of pCMBS modification in the absence of GABA, from individual oocyte results, plotted against etomidate. The line through data represents a logistic fit. Max = 400 ± 26 M-1s−1 and PC50 = 14.3 μM (95% CI = 7.5 to 27.2 μM). (C) Symbols represent data from a subset of oocytes expressing α1β2M286Cγ2L receptors used to study etomidate effects on pCMBS modification in the presence of 2 mM GABA or GABA plus 2 μM alphaxalone. Normalized IloGABA/I2 mM is plotted against cumulative pCMBS exposure (concentration × time). Lines represent linear fits to all points in each data set: GABA + pCMBS= red squares (11,300 ± 580 M-1s−1; n = 5); GABA + pCMBS + 3 μM etomidate = orange triangles (3500 ± 270 M-1s−1; n = 4); GABA + pCMBS + 30 μM etomidate = blue circles (600 ± 76 M-1s−1; n = 6); GABA + pCMBS + 300 μM etomidate = brown diamonds (−40 ± 11 M-1s−1; n = 3); GABA + alphaxalone = green half-filled hexagons (16,600 ± 960 M-1s−1; n = 8). (D) Open circles represent the average (± S.D.) of initial rates of pCMBS modification in the presence of GABA, from individual oocyte results, plotted against etomidate. The solid line through data represents a logistic fit: max = 11,300 ± 490 M-1s−1 and PC50 = 2.2 μM (95% CI = 1.5 to 3.1 μM). The dashed line represents a logistic fit using the GABA + alphaxalone control: max = 16,600 ± 890 M-1s−1 and PC50 = 1.1 μM (95% CI = 0.65 to 1.9 μM). The red solid line represents a theoretical protection profile calculated using the fitted MWC model parameters for α1β2M286Cγ2L (Table 1). Open probability was calculated as a function of etomidate at GABA = 2 mM, protection was calculated using the predicted open-state etomidate dissociation constant (KE × d ≈ 1.4 μM), and a Hill slope of 1.0; control modification rate was set at the experimentally observed value (11,300 M-1s−1).
In the presence of GABA (2 mM), initial slope analysis revealed an apparent β2M286C modification rate of 11,300 ± 2000 M-1s−1 (Fig. 4C, red squares). The addition of alphaxalone (2 μM) to GABA further increased the apparent initial rate of β2M286C modification to 16,600 ± 4000 M-1s−1 (Fig. 4C, green half-filled hexagons). In contrast, addition of etomidate (range, 1–300 μM) reduced initial rates of GABA-activated α1β2M286Cγ2L receptor modification by pCMBS. Figure 4C displays data in the presence of 3, 30, and 300 μM etomidate. Logistic analysis of modification rates using the control with GABA alone results in a fitted etomidate PC50 value of 2.2 μM for GABA-activated receptors (Fig. 4D, solid black line). Substituting the control modification rate measured with alphaxalone plus GABA (closer to 100% open receptors) results in a leftward shift of the logistic fit, with PC50 = 1.1 μM (Fig. 4D, dashed black line).
Discussion
The major finding of this study is that GABAA receptor activation results in increased affinity for the allosteric agonist etomidate. Whereas state-dependent binding is inherent in theoretical models of receptor agonism and resting-state affinity may sometimes be inferred from functional analysis, direct measurement of agonist occupation in inactive receptors represents an experimental challenge. In this study, we assessed rates of pCMBS modification at cysteine-substituted β2M286 residues to estimate etomidate occupancy at its GABAA receptor binding sites.
The substituted cysteine accessibility method (SCAM) provides tools for probing the proximity of ligands and specific receptor residues. SCAM coupled with ligand protection has been used in heterologously expressed GABAA receptors to map residues in orthosteric agonist (GABA) sites (Wagner and Czajkowski, 2001) and transmembrane sites for propofol (Bali and Akabas, 2004). Our approach assumed that etomidate sterically hinders the formation of a covalent bond between pCMBS and the β2M286C sulfhydryl. Thus, as etomidate occupies an increasing fraction of its sites, the rate of covalent modification should drop from the control value. Etomidate site occupancy depends on its concentration and binding affinity. Other factors that could influence sulfhydryl modification rates include altering local pCMBS concentration, sulfhydryl ionization, and availability of water, which participates in covalent bond formation (Karlin and Akabas, 1998). Such factors may also change with the receptor’s functional state. Thus, to isolate the steric effects of ligand binding, other investigators using SCAM protection have established roughly equivalent mixes of receptor states in control modification and protection experiments (Bali and Akabas, 2004). Because of the unique features of the α1β2M286Cγ2L receptor, we were able to study etomidate protection under two conditions in which essentially all receptors were in either 1) the inactive state or 2) the GABA-activated state.
Functional characterization of α1β2M286Cγ2L receptors revealed a number of important changes from wild-type α1β2γ2L (Table 1). The α1β2M286Cγ2L receptors are characterized by a high GABA EC50 and reduced apparent GABA efficacy. In addition, direct etomidate agonism was absent in α1β2M286Cγ2L receptors. The reduced efficacies of both GABA and etomidate are partially explained by reduced spontaneous gating in the mutant receptors, which was evident in mutant-cycle experiments combining β2M286C with a spontaneous activation mutation, α1L264T (Fig. 2D). Etomidate modulation was also significantly diminished in α1β2M286Cγ2L receptors. Indeed, MWC coagonist model analysis (Fig. 2; Table 1) suggests that the mutation reduces the agonist efficacy of etomidate, but not of GABA. The etomidate efficacy parameter (d−1) for the mutant receptors is 8.5; estimates for wild-type receptors are between 100 and 130 (Rüsch et al., 2004; Stewart et al., 2008; Desai et al., 2009), a 12- to 15-fold difference in gating effect per etomidate site. Etomidate’s maximal agonist efficacy can be calculated from eq. 3 (in Materials and Methods) as (1 + L0d2)-1. Using values from Table 1, etomidate activates 41% of wild-type and 0.09% of α1β2M286Cγ2L receptors. Nonetheless, etomidate efficacy is sufficient to produce positive modulation of GABA-induced activation in the mutant channels (Fig. 1D).
Interestingly, some functional effects of the β2M286C mutation are opposite those seen with the β2M286W mutation, which increases spontaneous activation and sensitizes receptors to agonism by GABA (Stewart et al., 2008). However, both tryptophan and cysteine mutations at β2M286 reduce etomidate modulation and etomidate direct activation in comparison with the wild-type. Investigation of various βM286 mutations demonstrated that side-chain volume may be an important determinant of propofol effects (Krasowski et al., 2001). Our results show that the βM286 side-chain influences both anesthetic binding and basal channel gating and that the magnitude and directions of these effects are independent.
As previously reported for rat GABAA receptors (Bali and Akabas, 2004; Bali et al., 2009), we found that in human receptors, β2M286C is accessible to the water-soluble reagent pCMBS. Thus, the β2M286 side chain is located, at least part of the time, in a position where water permeates into the transmembrane domains of the receptor and an aqueous pathway from the extracellular space probably exists. Acceleration by GABA of β2M286C modification also indicates that the configuration of the receptor structures near β2M286 changes with GABA binding.
The highest etomidate concentration tested (300 μM) fully blocked irreversible gating effects associated with pCMBS modification. Complete etomidate protection most likely indicates an intimate steric relationship between the β2-286 residue and receptor-bound etomidate. The native β2M286 residue was photolabeled by both azi-etomidate and TDBzl-etomidate, supporting this inference. Nonetheless, because an ester linkage separates the photoreactive moieties of the photolabels from the hydrophobic core structures of etomidate, we cannot definitively conclude that the β2M286 side chain is within the etomidate binding pocket rather than just near it. An alternative mechanism is that etomidate blocks the aqueous pathway for pCMBS access. Bali and Akabas (2004) found that propofol also protects βM286C from modification in GABA-activated receptors. Propofol also inhibits azi-etomidate photolabeling (Li et al., 2010), although not completely. The most parsimonious interpretation is that both propofol and etomidate have partially overlapping binding sites that abut β2M286.
Etomidate occupancy of its sites, inferred from protection of β2M286C from pCMBS modification, is dependent on both the concentration of etomidate and on the receptor state. The PC50 for etomidate protection in the resting state is 14 μM (Fig. 4B), close to the α1β2M286Cγ2L resting state KE of 12 μM estimated from MWC allosteric model fitting. In the allosteric model, the estimated dissociation constant for etomidate binding to active α1β2M286Cγ2L receptors is KE × d = 1.4 μM. This is within the range of PC50 values (1.1–2.2 μM) derived from active-state protection experiments. Thus, both allosteric model analysis and protection experiments converge on similar affinity estimates, indicating that etomidate binds about 10-fold more tightly to active than to inactive α1β2M286Cγ2L receptors. The active-state PC50 value closest to KE × d was obtained using the control modification rate in the presence of alphaxalone, which increased the fraction of activated receptors without blocking pCMBS access to the etomidate site. Indeed, taking into account both etomidate’s positive gating modulation and its protective action at β2M286C, the MWC coagonist model closely predicts open-state protection results (Fig. 4D, solid red line). These results validate the MWC allosteric model and suggest that estimates for etomidate affinity and efficacy in wild-type GABAA receptors (Table 1) are reasonably accurate. More generally, this result is in accord with theories of agonist-receptor interactions (Buxton, 2006).
The close match between etomidate PC50 values and MWC model estimates for microscopic dissociation constants is notable because etomidate binding is reversible, whereas pCMBS modification is irreversible. In protection experiments, we reduced systematic bias from varying etomidate association rates at different concentrations by pre-equilibrating receptors with etomidate. As long as etomidate binding and dissociation rates are fast compared with the pCMBS reaction at β2M286C, a reasonable assumption under the conditions we selected, then the observed rate of covalent bond formation should reflect the initial fraction of unoccupied etomidate sites. Another potential source of divergence between our functional modeling and protection results is receptor desensitization, which is not factored into the two-state MWC model. Figure 1E shows that substantial desensitization of α1β2M286Cγ2L receptors occurs within seconds after exposure to high GABA. The fact that open state etomidate affinity estimates from equilibrium MWC modeling matches estimates from protection in a dynamic mixture of open and desensitized receptors suggests that affinities for both open and desensitized states are similar. This is consistent with observations that etomidate does not alter GABAA receptor desensitization kinetics (Zhong et al., 2008). To test this idea, studies comparing etomidate protection at β2M286C in open versus desensitized receptors are needed. Moreover, the fraction of desensitized receptors when GABA is absent is unknown and cannot be determined from functional studies, but the close match between KE from functional modeling and PC50 in inactive receptors suggests that at most a small fraction of channels are in high-affinity states.
Although a thorough characterization of GABA and etomidate-dependent receptor activity provided us with a mechanistic framework to interpret multiple effects of the β2M286C mutation, the major factor influencing the design and interpretation of SCAM protection experiments was the absence of etomidate agonism. Indeed, allosteric principles imply generally that in SCAM protection studies using ligands with agonist activity, the presence or absence of significant agonism in cysteine-substituted mutant channels determines the conditions under which protection may be tested. In mutant channels that, like wild-type GABAA receptors, are efficaciously activated by etomidate, the GABA-activated open state is the only viable control condition for protection studies, and protection may be observed using low ligand concentrations predicted to occupy most high-affinity active-state receptors. With mutations like β2M286C, in which etomidate efficacy is significantly weakened but not absent, open-state affinity is predicted to be only modestly higher than in inactive receptors, indicating the likely need for higher concentrations of the protective ligand and also establishing the potential for state-dependent protection experiments. More specifically, absent etomidate agonism coupled with retained GABA agonism in α1β2M286Cγ2L channels enabled protection studies in two different states. Finally if a cysteine mutation eliminates both etomidate agonism and modulation of GABA-activated responses, then both active and resting state receptors are expected to display low affinities, requiring high etomidate concentrations to test for protection.
Acknowledgments
The authors thank Aiping Liu and Youssef Jounaidi (both at Massachusetts General Hospital, Boston) for their expert technical support for this research. Keith Miller (Department of Anesthesia Critical Care & Pain Medicine, Massachusetts General Hospital, Boston) and Jonathan Cohen and David Chiara (Department of Neurobiology, Harvard Medical School, Boston) provided constructive suggestions on experiments and the manuscript.
Abbreviations
- GABAA
γ-aminobutyric acid type A
- HEK
human embryonic kidney
- MWC
Monod-Wyman-Changeux (model)
- pCMBS
para-chloromercuribenzene sulfonate
- PC50
50% protective ligand concentration
- PTX
picrotoxin
- SCAM
substituted cysteine accessibility method
Authorship Contributions
Participated in research design: Stewart, Forman.
Conducted experiments: Stewart, Hotta, Desai.
Performed data analysis: Stewart, Hotta, Forman, Desai.
Wrote or contributed to writing of manuscript: Stewart, Forman.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grants GM58448 and GM089745].
Part of this work was previously presented at the following meeting: Stewart D, Hotta M, and Forman SA (2013) State-dependent etomidate binding in GABAA receptors probed with cysteine substitution and protection from modification. 57th Biophysical Society Annual Meeting; 2013 Feb 2–6; Philadelphia, PA. Poster 228.20.
References
- Auerbach A. (2012) Thinking in cycles: MWC is a good model for acetylcholine receptor-channels. J Physiol 590:93–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali M, Akabas MH. (2004) Defining the propofol binding site location on the GABAA receptor. Mol Pharmacol 65:68–76 [DOI] [PubMed] [Google Scholar]
- Bali M, Akabas MH. (2012) Gating-induced conformational rearrangement of the γ-aminobutyric acid type A receptor β-α subunit interface in the membrane-spanning domain. J Biol Chem 287:27762–27770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali M, Jansen M, Akabas MH. (2009) GABA-induced intersubunit conformational movement in the GABAA receptor alpha 1M1-beta 2M3 transmembrane subunit interface: experimental basis for homology modeling of an intravenous anesthetic binding site. J Neurosci 29:3083–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxton ILO. (2006) Pharmacokinetics and pharmacodynamics, in Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th ed (Brunton LL, Lazo JS, Parker KL. eds) pp 1–39, McGraw-Hill, New York NY [Google Scholar]
- Chang Y, Weiss DS. (1999) Allosteric activation mechanism of the alpha 1 beta 2 gamma 2 gamma-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys J 77:2542–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux JP. (2012) Allostery and the Monod-Wyman-Changeux model after 50 years. Annu Rev Biophys 41:103–133 [DOI] [PubMed] [Google Scholar]
- Chiara DC, Dostalova Z, Jayakar SS, Zhou X, Miller KW, Cohen JB. (2012) Mapping general anesthetic binding site(s) in human α1β3 γ-aminobutyric acid type A receptors with [³H]TDBzl-etomidate, a photoreactive etomidate analogue. Biochemistry 51:836–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D. (1998) Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol 125:924–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai R, Ruesch D, Forman SA. (2009) Gamma-amino butyric acid type A receptor mutations at beta2N265 alter etomidate efficacy while preserving basal and agonist-dependent activity. Anesthesiology 111:774–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galzi JL, Edelstein SJ, Changeux J. (1996) The multiple phenotypes of allosteric receptor mutants. Proc Natl Acad Sci USA 93:1853–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitchounts G, Stewart DS, Forman SA. (2012) Two etomidate sites in α1β2γ2 γ-aminobutyric acid type A receptors contribute equally and noncooperatively to modulation of channel gating. Anesthesiology 116:1235–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A, Akabas MH. (1998) Substituted-cysteine accessibility method. Methods Enzymol 293:123–145 [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2004) Principles: receptor theory in pharmacology. Trends Pharmacol Sci 25:186–192 [DOI] [PubMed] [Google Scholar]
- Krasowski MD, Nishikawa K, Nikolaeva N, Lin A, Harrison NL. (2001) Methionine 286 in transmembrane domain 3 of the GABAA receptor beta subunit controls a binding cavity for propofol and other alkylphenol general anesthetics. Neuropharmacology 41:952–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GD, Chiara DC, Cohen JB, Olsen RW. (2010) Numerous classes of general anesthetics inhibit etomidate binding to gamma-aminobutyric acid type A (GABAA) receptors. J Biol Chem 285:8615–8620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. (2006) Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci 26:11599–11605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W. (2008) International Union of Pharmacology. LXX: Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev 60:243–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines DE, Krishnan NS. (1998) Transient low-affinity agonist binding to Torpedo postsynaptic membranes resolved by using sequential mixing stopped-flow fluorescence spectroscopy. Biochemistry 37:956–964 [DOI] [PubMed] [Google Scholar]
- Ruesch D, Neumann E, Wulf H, Forman SA. (2012) An allosteric coagonist model for propofol effects on α1β2γ2L γ-aminobutyric acid type A receptors. Anesthesiology 116:47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüsch D, Zhong H, Forman SA. (2004) Gating allosterism at a single class of etomidate sites on alpha1beta2gamma2L GABA A receptors accounts for both direct activation and agonist modulation. J Biol Chem 279:20982–20992 [DOI] [PubMed] [Google Scholar]
- Scheller M, Forman SA. (2002) Coupled and uncoupled gating and desensitization effects by pore domain mutations in GABA(A) receptors. J Neurosci 22:8411–8421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart DS, Desai R, Cheng Q, Liu A, Forman SA. (2008) Tryptophan mutations at azi-etomidate photo-incorporation sites on α1 or β2 subunits enhance GABAA receptor gating and reduce etomidate modulation. Mol Pharmacol 74:1687–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DA, Czajkowski C. (2001) Structure and dynamics of the GABA binding pocket: a narrowing cleft that constricts during activation. J Neurosci 21:67–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Rüsch D, Forman SA. (2008) Photo-activated azi-etomidate, a general anesthetic photolabel, irreversibly enhances gating and desensitization of gamma-aminobutyric acid type A receptors. Anesthesiology 108:103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]