

Abstract
Self-assembly is a fundamental process that drives structural organization in both inanimate and living systems. It is in the course of self-assembly of cells and tissues in early development that the organism and its parts eventually acquire their final shape. Even though developmental patterning through self-assembly is under strict genetic control it is clear that ultimately it is physical mechanisms that bring about the complex structures. Here we show, both experimentally and by using computer simulations, how tissue liquidity can be used to build tissue constructs of prescribed geometry in vitro. Spherical aggregates containing many thousands of cells, which form because of tissue liquidity, were implanted contiguously into biocompatible hydrogels in circular geometry. Depending on the properties of the gel, upon incubation, the aggregates either fused into a toroidal 3D structure or their constituent cells dispersed into the surrounding matrix. The model simulations, which reproduced the experimentally observed shapes, indicate that the control parameter of structure evolution is the aggregate–gel interfacial tension. The model-based analysis also revealed that the observed toroidal structure represents a metastable state of the cellular system, whose lifetime depends on the magnitude of cell–cell and cell–matrix interactions. Thus, these constructs can be made long-lived. We suggest that spherical aggregates composed of organ-specific cells may be used as “bio-ink” in the evolving technology of organ printing.
Self-assembly is the fundamental process, which generates structural organization across scales (1). Histogenesis and organogenesis are examples of self-assembly processes, in which, through cell–cell and cell–extracellular matrix interactions, the developing organism and its parts gradually acquire their final shape. In the present work we use both experimental and computational approaches to demonstrate how the self-organizing properties of cells and tissues, the basis for morphogenesis, can be exploited to build 3D biological structures of prescribed geometry.
Tissue engineering (2–7) offers the opportunity to study self-assembly processes during histo- and organogenesis in vitro, under controlled conditions. Tissue engineering aims not only to create desirable organs, but also to better understand the fundamental mechanisms and principles of biological organization in general and morphogenesis in particular. Classical tissue engineering is based on seeding cells into biodegradable polymer scaffolds or gels, culturing and expanding them in bioreactors for several weeks, and finally implanting the resulting tissue into the recipient organism, where the maturation of the new organ takes place.
It has recently been suggested to use cell aggregates, instead of individual cells, as building blocks in tissue engineering (8, 9). Cell aggregates have traditionally been used as a powerful tool to understand the principles of cell–cell (10) and cell–matrix adhesion (11), as well as cell sorting (12). In addition, rapid prototyping technology has successfully been applied for computer-aided deposition of cells in gels to create 3D tissue constructs (13, 14). We suggest that cell aggregates may be used as drops of “bio-ink,” which, upon implantation or “printing” into a scaffold (“bio-paper”), have the ability to fuse into 3D organ structures (15–17).
The ability of cell aggregates to fuse is based on the concept of tissue fluidity (18, 19), according to which embryonic tissues in many respects can be considered as liquids. In particular, in suspension or on nonadhesive surfaces, various multicellular aggregates round up into spherical shape similarly to liquid droplets (12). We hypothesize that closely placed aggregates in appropriately chosen 3D gels can fuse to form tissue constructs of desired geometry.
To demonstrate the feasibility of such a proposition, we used aggregates of genetically transformed cells with controlled adhesive properties, and arranged them to form a ring in gels of different chemical and mechanical properties. Our results demonstrate that contiguous aggregates under appropriate conditions, defined by the composition of the embedding gel, indeed can fuse into structures of specified morphology. We have also constructed and experimentally validated a mathematical model of cell aggregate fusion.
Taken together, these results suggest that cell aggregates can successfully be used as building blocks in tissue engineering technologies. They also provide support for the concept of self-assembling bio-ink, and thus justification for the use of cell aggregates in the evolving organ printing tools.
Materials and Methods
Cell Aggregate Preparation. Chinese Hamster Ovary (CHO) cells, transfected with N-cadherin (courtesy of A. Bershadsky, Weizmann Institute, Rehovot, Israel), were infected with histone binding H2B-YFP retrovirus (kindly provided by R. D. Lansford, Beckman Institute at California Institute of Technology). Confluent cell cultures (3–4 × 106 cells per 75-cm2 TC dish) grown in DMEM (GIBCO/BRL) supplemented with 10% FBS (U.S. Bio-Technologies, Pottstown, PA), 10 μg/ml penicillin, streptomycin, gentamicin, and kanamycin, 400 μg/ml geneticin), were washed twice with Hanks' balanced salt solution (HBSS) containing 2 mM CaCl2, then treated for 10 min with trypsin 0.1% (diluted from 2.5% stock, GIBCO/BRL). Depleted cells were centrifuged at 2,500 rpm for 4 min (Fisher Centrific model 225). The resulting pellet was transferred into capillary micropipettes of 500-μm diameter and incubated at 37°C with 5% CO2 for 10 min. The firm cylinders of cells removed from the pipettes were cut into fragments (500-μm height), then incubated in 10-ml tissue culture flasks (Bellco Glass, Vineland, NJ) with 3 ml of DMEM on a gyratory shaker at 120 rpm with 5% CO2 at 37°C for 24–36 h. This procedure reproducibly provides spherical aggregates of similar size (≈500-μm diameter).
Cell Aggregate–Gel Structures. NeuroGel (a biocompatible porous poly[N-(2-hydroxypropyl)methacrylamide] hydrogel) disks of 10-mm diameter and 2-mm thickness, containing Arg-Gly-Asp (RGD) fragments (kindly provided by Stephane Woerly, Organogel Canada, Quebec) were washed three times with DMEM to eliminate the storage medium. This gel has been shown to provide favorable conditions for spinal cord repair (20, 21). A 0.5-mm wide, 0.5-mm deep circular groove was cut into a disk, then filled with 10 aggregates, placed contiguously to form a closed circle. The groove was refilled with the gel to completely embed the aggregates. This structure was incubated at 37°C, 5% CO2 for 72 h in a tissue culture dish containing 10 ml of DMEM, washed with PBS, and finally embedded in Tissue-Tek OCT Compound (Electron Microscopy Sciences, Fort Washington, PA). The structure was slowly cooled (1°C per min) to -20°C in a Nalgene freezing container (Nalgene Labware, Rochester, NY). To visualize aggregate fusion at the end of the experiment, cryosectioning was performed with a Reichert 2800N Frigocut cryotome (Reichert Jung, Arnsberg, Germany), yielding 10- to 16-μm-thin slices mounted on microscope slides. Slices were examined on an Olympus IX-70 inverted microscope with fluorescent attachment at ×4 magnification.
To tune the strength of cell–gel interaction, further fusion experiments were conducted in rat-tail collagen type I (Sigma). Collagen was dissolved in 1 M acetic acid, then treated with Ham's F12 medium with sodium bicarbonate. At room temperature, this mixture gels in a few minutes depending on concentration. The gel–aggregate structure was achieved by creating a ring of 10 aggregates on the top of a previously (almost) solidified collagen layer, then covering with liquid collagen that embedded the aggregates after gelation. These samples were incubated under the same conditions as described above. Working with 1.0, 1.2, and 1.7 mg/ml collagen, the samples were transparent; thus, it was possible to follow pattern evolution in time by phase contrast and fluorescent microscopy.
Cell survivability was checked with Trypan Blue (GIBCO/BRL) at the end of each fusion experiment. A minimal number of uniformly distributed dead cells were found.
Modeling Structure Formation by Means of Cell Aggregate Fusion. To investigate shape changes of the evolving pattern, we constructed a simple 3D model, in which the sites of a cubic lattice are occupied either by point-like cells or gel volume elements. The total interaction energy, E of the system is written as
 |
[1] |
where r and r′ label lattice sites, and 〈r, r′〉 signifies summation over neighboring sites, each pair counted once. First, second, and third nearest neighbors are included, and we assume that a cell interacts with the same strength with all of the 26 cells it comes into contact with. To specify occupancy, we assign a spin value, σ, to each lattice site with values 0 for a “gel particle” and 1 for a cell. The interaction energy of two neighbors, J(σr, σr′), may take either of the values J(0, 0) = -εgg, J(1, 1) = -εcc, or J(0, 1) = J(1, 0) = -εcg. Here the positive parameters εgg, εcc, and εcg account for contact interaction strengths for cell–cell, gel–gel, and cell–gel pairs, respectively. More specifically, these are mechanical works needed to disrupt the corresponding bonds. (Note that εcc and εgg are works of cohesion, whereas εcg is work of adhesion per bond; ref. 22.) The strength of cell–cell interaction may be determined experimentally either directly (23) or by measuring the tissue surface tension (12, 24). The cell–gel interaction is tunable via the concentration of RGD groups in the gel (6, 25) or by the concentration of collagen. The gel–gel “bond energy” is an effective measure of gel filament density, interaction, and stiffness. It is determined by the specific chemistry of the gel.
The energy in Eq. 1 may be rewritten by separating interfacial and bulk terms in the sum. As a result we obtain
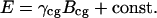 |
[2] |
Here, Bcg is the total number of cell–gel bonds, and γcg = (εcc + εgg)/2 - εcg is proportional to the cell–gel interfacial tension (22, 26, 27). The remaining terms in E do not change as the cellular pattern evolves. Our model is inspired by earlier efforts aiming at computer simulations of cell sorting (26–28), the morphogenetic phenomenon in which one of two, initially randomly intermixed cell populations sorts out and becomes surrounded by the other.
The evolution of the system is followed by using Monte Carlo simulations (29), relying on a random number generator of L'Ecuyer with Baym–Durham shuffle (30). The program identifies the cells on the aggregate–gel interface, picks one of them randomly, and exchanges it with an adjacent gel particle chosen by chance. The corresponding change in adhesive energy, ΔE, is calculated, and the new configuration is accepted with a probability P = 1 if ΔE ≤ 0 or P = exp(-βΔE) if ΔE > 0. β = 1/ET is the inverse of the average biological fluctuation energy ET, analogous to the thermal fluctuation energy (31), kBT (where kB is Boltzmann's constant and T is the absolute temperature). In statistical mechanics, this energy characterizes thermal agitation in a system of atoms or molecules. In the case of cells, it is a measure of the spontaneous, cytoskeleton-driven motion of cells, able to break adhesive bonds between neighbors via membrane ruffling (32), or more generally, via membrane protrusive activity (e.g., filopodial extensions). By definition, a Monte Carlo step (MCS) or “unit of time,” is completed when each cell in contact with the gel has been given the chance to move once. During each MCS, the interfacial sites are selected in random order. The gel boundary is treated as a fixed physical limit of the system, and cells are constrained to move within the gel.
Results
Structure Formation: Simulations. The result stated in Eq. 2 indicates that the evolution of the cellular pattern is governed by a single parameter, γcg/ET, which, for cells with specific adhesion apparatus, is controlled by the chemistry of the gel. The theoretical analysis shows that, once gel properties are appropriately tuned, efficient fusion of adjacent aggregates takes place. This is illustrated in the simulation shown in Fig. 1. For small γcg/ET (= 0.25 in Fig. 1 K and L), cells can spread in the bulk of the gel (i.e., permissive gel) and the pattern evolves toward its lowest energy state, being a sphere. (Because the interfacial energy is small in comparison to the fluctuation energy, the spheroidal structure in Fig. 1L is rather disperse.) Under optimal cell–gel interface properties, expressed in our model by a certain range in γcg/ET, fusion of aggregates results in a 3D toroidal structure. An example of this kind is depicted in Fig. 1 A and B (γcg/ET = 0.9, nonpermissive gel).
Fig. 1.

Initial (Upper) and final (Lower) cell aggregate configurations in the simulations and in experiments using various biocompatible gels. Panels A and B correspond to simulations with γcg/ET = 0.9, and K and L correspond to simulations with γcg/ET = 0.25. The 10 aggregates, each containing 925 cells, are one cell diameter from each other in the starting configurations. The final configurations are reached after 25,000 and 50,000 MCS, respectively. C–J correspond to CHO cell aggregates embedded in a neurogel with RGD fragments (C and D) and collagen gels of concentration 1.0 (E and F), 1.2 (G and H), and 1.7 (I and J)mg/ml. The nuclei of the cells are fluorescently labeled (see Materials and Methods), and the images of the cellular patterns were acquired with a ×4 objective. The average diameter of the aggregates is 500 μm.
Energetics During Structure Formation. Some shapes correspond to local minima of the interaction energy. These represent metastable configurations. They are identified from plateaus in the plot of the total interaction energy vs. MCS, and are important for tissue engineering, for they can be made long-lived. This is illustrated in more detail in the simulation shown in Fig. 2, where the initial state progresses toward a metastable toroidal configuration, whose energy is essentially unchanged in the entire interval between 104 and 6 × 104 MCS (Fig. 2 A). Eventually the toroid becomes unstable, and at ≈105 MCS it ruptures (Fig. 2B). Subsequent massive rearrangements lead to a pronounced energy decrease while the system evolves into three rounded aggregates. These remain stable for a long time because large spatial separations hinder their fusion into a single spheroid. Similar simulations showed that metastability depends on both system size and interaction strengths. Because the evolution of the cellular pattern is driven exclusively by energy minimization, and “time” is measured in MCS, in its present form, the model cannot provide information on the true dynamical behavior of the system.
Fig. 2.

(A) Metastable configuration at γcg/ET = 1.1. Fusion of 16 contiguous aggregates (123 cells each) takes place within the first 5,000 MCS. The plateau of the energy vs. MCS corresponds to a torus. (B) The evolution of the energy during 5 × 105 MCS and representative shapes. The final, rounded aggregates remained stable, and the energy was practically constant up to 106 MCS (not shown).
Once the structure reaches the metastable state, it can be stabilized by dissolving the supporting gel. In the simulations, this corresponds to increasing the value of γcg/ET. Indeed, if in the simulation shown in Fig. 2 A this quantity is changed to γcg/ET = 2 anywhere in the plateau region, the energy remains constant as long as the simulation is run (results not shown).
Experimental Realization of 3D Structure. To study the feasibility of engineering 3D tissue constructs of prescribed geometry, we have “manually printed” (i.e., embedded) aggregates of living cells into biocompatible gels. As indicated by our model, the ability of aggregates to fuse depends on the mutual properties of the cells and gel. The results of our experiments in Fig. 1 support this prediction. The interfacial tension, γcg contains three terms, which, in principle, can all be controlled. We performed experiments with fixed cell–cell adhesion and varying gel properties. For the purposes of this study, we used N-cadherin transfected CHO cells. Their adhesive properties were quantitatively assessed by measuring aggregate surface tension (for details on the method, see refs. 12, 24, 33, and 34). We used gels with differing chemical composition. The relative importance of cell–cell and cell–matrix interactions has been investigated quantitatively (11).
Results in Fig. 1 I and J show that collagen at concentration of 1.7 mg/ml is analogous to a permissive scaffold with small γcg/ET. Collagen at concentration of 1.0 and 1.2 mg/ml and the RGD containing neurogel match more the definition of the nonpermissive gel with high γcg/ET. These gels favor much less (collagen), or not at all (neurogel), the dispersion of the cells into the scaffold, thus facilitating fusion. During our measurements, we did not observe the collapse of the fused rings that, according to the model predictions, should eventually take place (Fig. 2).
The Influence of Scaffold on 3D Structure Formation. The above results demonstrate the well known fact that scaffold properties affect cellular structure (6, 11). The specific mechanism of how the gel influences pattern evolution depends on its detailed chemistry, and in general is not easy to discern.
Cells exert traction forces on their substrates and surrounding 3D matrices (35–38). In the case of collagen, numerous studies of this phenomenon have been performed (39–43). The evolution process in this case has some striking features. As the fusion of aggregates takes place, the pattern noticeably contracts, at least until ≈60 h (Figs. 3 and 4). For higher collagen concentration, contraction is more dramatic, whereas in the case of the neurogel no similar effects were observed (results not shown). Contraction results from the CHO cells pulling on collagen fibers. Furthermore, as aggregates fuse, at some point the pattern assumes a starburst appearance (see spikes at ≈60 h, but not before, in Fig. 5 for the 1.0 mg/ml collagen gel). Cells extend outward from the ring suggesting that by this time a network of radially aligned collagen fibers has developed in the vicinity of the aggregates. (For an explicit visualization of such an array, see ref. 42.) It is remarkable that no similar effect is observed inside the ring even at 144 h (Fig. 5). The probable reason collagen fibers do not align inside the ring is that the vectorial sum of the isotropically acting traction forces is zero. (An analogous cancellation makes the electric field in the interior of a conducting spherical shell to be zero.)
Fig. 3.

Time evolution of the cellular pattern in Fig. 1E. Note the initial strong contraction (see also Fig. 4). Also note that there are many more cells migrating into the gel than this image might suggest. Because the fluorescent signal of individual cells is much weaker than that of the fused aggregates, they are hard to spot (however, see Fig. 5). The collagen layers above and under the ring are too thin to observe any appreciable cell migration out of the plane of the ring.
Fig. 4.

Cell–matrix interaction induced contraction in collagen gels. The figure shows the change of the entire area inside the outer perimeter of the rings in Fig. 3. Squares, circles, and diamonds stand for collagen concentration 1.0, 1.2, and 1.7 mg/ml, respectively. Curves are exponential fits to the data.
Fig. 5.

Time course of aggregate fusion for collagen concentration 1.0 mg/ml. Bright field images of the cellular pattern were acquired with a ×4 objective. (Top) Evolution of the cellular boundary between adjacent aggregates. Note the radial, exclusively outward (top) directed migration of cells at around 60 h. (Middle) The entire pattern at selected times (Left, 36 h; Right, 144 h). (Bottom) variation of the angle between aggregate boundaries in Top as function of time. The curve is an exponential fit to the data (see text for details).
In Fig. 4, we quantified contraction in terms of the total area defined by the outer perimeter of the ring. The curves are exponential fits to the data in the form Aexp(-t/τcg) + B (A and B are constants). The quantity τcg defines a characteristic time scale of contraction. (For the 1.0 mg/ml collagen gel, the one we will use in later analysis, τcg ≈ 57 h.)
Kinetics of Aggregate Fusion. The transparency of collagen allows optically following structure evolution. Fig. 5 shows the time variation of the boundary between two adjacent aggregates in the 1.0 mg/ml collagen gel. A measure of fusion is the instantaneous value of the angle formed by the two aggregates. As aggregates coalesce, the angle between the tangents to their boundaries (drawn from the point where they join) approaches 180°. The curve in Fig. 5 is an exponential fit to the data in the form C[1 -exp(-t/τcc)] (C is constant), with τcc ≈ 23 h. Here, the quantity τcc defines a time scale of aggregate fusion.
Discussion
We have manually printed a simple, but nontrivial structure, a ring of spherical aggregates, each containing thousands of cells with specific adhesive properties. We have shown, both experimentally and in computer simulations, that under appropriate conditions the initially contiguous aggregates, positioned along a circle, fuse into a toroidal construct. If aggregates were printed in multiple layers, they would presumably fuse in both the horizontal and vertical directions, thus forming a lumenous organ-like module.
The major outcome of this work is the demonstration that spherical cell aggregates can be used as building blocks in tissue engineering applications. In particular, with the development of automated, computer-aided dispensers or bioprinters (which already exist for the delivery of cells; refs. 13 and 14) they could be used in the capacity of bio-ink.
The biophysical basis for bio-ink is tissue liquidity, a concept proposed by Steinberg (18). The differential adhesion hypothesis (DAH) provides the molecular foundation for tissue liquidity (18, 19). DAH explains the liquid-like behavior of cell populations in terms of tissue surface and interfacial tensions, generated by adhesive and cohesive interactions between the component subunits. In vitro experiments have demonstrated that tissue surface tension is a well defined physical parameter, which characterizes the equilibrium shape of multicellular aggregates. Measured values of the tensions in many cases account for the observed mutual envelopment behavior of tissues (12, 34). In vivo experiments give further support to DAH (44, 45).
According to DAH, the lowest energy configuration of any tissue fragment (containing motile cells, uniformly adhesive over their entire surfaces) embedded in a medium to which it adheres weakly, is a sphere. Thus, the final pattern in the 1.0 and 1.2 mg/ml collagen gels and the neurogel should also be a single spherical aggregate. However, as the simulations demonstrate, the system may be trapped in long-lived metastable intermediate states (Fig. 2), which correspond to particular fused conformations. This may provide sufficient time to dissolve the gel, thus freezing the desirable configuration and transferring the resulting tissue construct into a bioreactor for maintenance.
The cell aggregates we used in this work contain cells of only one type. Complex organs contain several cell types. There are indications that using aggregates comprised of heterocellular populations would lead to nontrivial structures. It has been shown, both experimentally (12, 34) and in computer simulations (26, 27), that pattern evolution in sorting is consistent with the predictions of DAH, and is indeed controlled and driven by interfacial tension. Moreover, when the mixture is composed of cells of tissues that are neighbors in normal development, in the course of sorting they recover their physiological configuration (46, 48).
Liquidity has been emphasized for embryonic tissues because it is primarily at this stage of development that tissues and cells must actively move to eventually give rise to organs. Here we used aggregates of N-cadherin transfected CHO cells, whose liquidity has been established earlier (49). It is quite likely that when structure formation is induced by the methods described here, aggregates of other cell types (in particular stem cells) will behave similarly to embryonic tissues, as far as their liquidity and ability to fuse is concerned.
Spherical cell aggregates can be made only of adhering cells. Fibroblasts, for example, normally do not adhere to each other directly, only through the extracellular matrix. Such cells can either be temporarily genetically manipulated to express cell adhesion molecules (50, 51) or embedded in a population of adhering cells.
Delivering or printing cell aggregates (instead of individual cells) into 3D scaffolds offers several advantages. Aggregates are prebuilt small tissue blocks, thus their fusion immediately results in 3D structures. Because they contain many thousands of cells, printing time could be dramatically reduced and cell survival greatly improved. Properly designed composite aggregates (with more than one cell type and potentially containing extracellular matrix; i.e., “multicolor bio-ink”) can enhance the creation of desired complex tissue constructs. Finally, the inescapable harsh mechanical conditions encountered in presently available printers are more critical for cells than cell aggregates. [Jet-based cell printers (15), due to their small orifice, are presently not appropriate for dispensing aggregates. Devices with the potential to print aggregates of several hundred microns (14) use micropipettes as cartridges with pressure-operated extruders, and thus provide more gentle conditions.]
Our work is based on the analysis of cell–hydrogel interactions in 3D. The computer simulations suggest that for the fusion of the aggregates to take place strict conditions on the embedding gel must be imposed. Our experiments support this prediction. In the case of collagen, because of the tendency of cells to reorganize the matrix, the situation is complex, and the interpretation of the experiments in terms of only gel–aggregate interfacial tension ignores important details. Figs. 4 and 5 imply that pattern evolution involves two distinct time scales, τcg ≈ 57 h and τcc ≈ 23 h for the 1.0 mg/ml collagen matrix, which characterize respectively the dynamical aspects of cell–gel interaction (i.e., how long it takes for the cells to reorganize the matrix) and cell–cell interactions (i.e., how long it takes for the aggregates to fuse). It is the competition between these interactions that determines the final pattern. Because for 1.0 mg/ml collagen τcg/τcc ≈2.5, contraction is considerably slower than fusion, and the ring has time to stabilize. For 1.7 mg/ml collagen concentration, contraction dominates pattern evolution and the fused ring does not have time to stabilize.
The ideal hydrogel for cell aggregate printing must allow cells to survive and provide favorable conditions for postprinting self-assembly. There already exist several candidates for such gels: thermo-reversible gels (52, 53), photo-sensitive gels (54–56), pH-sensitive gels (57), and gels sensitive to specific molecular entities (58, 59). Detailed studies must be performed to identify the optimal candidate for specific cells or their mixtures.
Our model to describe the observed pattern evolution clearly represents a strong oversimplification of the complex nature of both the cellular and scaffold system. Its major limitation is that it assumes that cells are identical point particles. Thus, in particular, it ignores the change in their shape, a factor known to be important in cell–matrix interactions. Similarly, it treats the embedding gel as a system without internal structure, which is not an adequate characterization of a network of fibers, in particular the one spanning a collagen matrix. Furthermore, cellular rearrangement within the model is exclusively driven by energy minimization (as implemented by the Monte Carlo method), thus, no information can be gained on the true time evolution of the pattern. The entire process is controlled by a single parameter, the aggregate–gel interfacial tension. It is intriguing that, despite these limitations, the similarity between the stimulated and experimentally obtained shapes is so strong. This suggests that our model may incorporate the most salient features of the observed phenomenon. Possible improvement of the model includes the treatment of cells as extended objects, capable of changing their shape. This can be accomplished for example within the large state Potts model (26, 27). Following the true time evolution of the pattern would require the detailed characterization of forces between cells and gel, and necessitate molecular dynamics based simulations instead of the Monte Carlo approach.
In conclusion, we have demonstrated that closely placed cell aggregates in 3D gels can self-organize into metastable tissue constructs of desired shape. It was also shown that adhesive and mechanical properties of the embedding hydrogels are critical for cell aggregate fusion. We have developed a mathematical model that accurately describes our experimental findings. When extended to incorporate more realistic cell and hydrogel properties, it may be used for the design and optimization of scaffold properties. Finally, on the basis of our findings, we have proposed that cell aggregates could be used as self-assembling bio-ink in automated delivery or printing devices.
Acknowledgments
This work was supported by research grants from the National Science Foundation (to G.F.) and the National Aeronautics and Space Administration (to G.F., V.M., and R.R.M.).
Abbreviations: CHO, Chinese hamster ovary; MCS, Monte Carlo step; DAH, differential adhesion hypothesis.
References
- 1.Whitesides, G. M. & Grzyboski, B. (2002) Science 295, 2418-2421. [DOI] [PubMed] [Google Scholar]
- 2.Langer, R. & Vacanti, J. P. (1993) Science 260, 920-926. [DOI] [PubMed] [Google Scholar]
- 3.Bonassar, L. J. & Vacanti, C. A. (1998) J. Cell Biochem. Suppl. 30–31, 297-303. [PubMed] [Google Scholar]
- 4.Lysaght, M. J., Nguy, N. A. & Sullivan, K. (1998) Tissue Eng. 4, 231-238. [DOI] [PubMed] [Google Scholar]
- 5.Marler, J., Upton, J., Langer, R. & Vacanti, J. (1998) Adv. Drug Del. Rev. 33, 165-182. [DOI] [PubMed] [Google Scholar]
- 6.Griffith, L. G. & Naughton, G. (2002) Science 295, 1009-1014. [DOI] [PubMed] [Google Scholar]
- 7.Wang, Y., Ameer, G. A., Sheppard, B. J. & Langer, R. (2002) Nat. Biotechnol. 20, 602-606. [DOI] [PubMed] [Google Scholar]
- 8.Martin, I., Dozin, B., Quarto, R., Cancedda, R. & Beltrame, F. (1997) Cytometry 28, 2141-2146. [DOI] [PubMed] [Google Scholar]
- 9.Layer, P. G., Robitzki, A., Rothermel, A. & Willbold, E. (2002) Trends Neurosci. 3, 131-134. [DOI] [PubMed] [Google Scholar]
- 10.Steinberg, M. S. (1996) Dev. Biol. 187, 377-388. [DOI] [PubMed] [Google Scholar]
- 11.Ryan, P. L., Foty, R. A., Kohn, J. & Steinberg, M. S. (2001) Proc. Natl. Acad. Sci. USA 98, 4323-4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foty, R. A., Pfleger, C. M., Forgacs, G. & Steinberg, M. S. (1996) Development (Cambridge, U.K.) 122, 1611-1620. [DOI] [PubMed] [Google Scholar]
- 13.Liu, V. A. & Bhatia, S. N. (2002) Biomed. Microdev. 4, 257-266. [Google Scholar]
- 14.Kachurin, A. M., Stewart, R. L., Church, K. H., Warren, W. L., Fisher, J. P., Mikos, A. G., Kraeft, S. K. & Chen, L. B. (2001) Proc. Mat. Res. Soc. 689, 651-656. [Google Scholar]
- 15.Wilson, W. C. & Boland, T. (2003) Anat Rec. 272A, 491-496. [DOI] [PubMed] [Google Scholar]
- 16.Boland, T., Mironov, V., Gutowska, A., Roth, E. A. & Markwald, R. R. (2003) Anat Rec. 272A, 497-502. [DOI] [PubMed] [Google Scholar]
- 17.Mironov, V., Boland, T., Trusk, T., Forgacs, G. & Markwald, R. R. (2003) Trends Biotechnol. 21, 157-161. [DOI] [PubMed] [Google Scholar]
- 18.Steinberg, M. S. (1963) Science 137, 762-763. [DOI] [PubMed] [Google Scholar]
- 19.Steinberg, M. S. and Poole, T. J. (1982) in Cell Behaviour, eds. Bellains, R., Curtis, A. S. G. & Dunn, G. (Cambridge Univ. Press, Cambridge, U.K.), pp. 583-607.
- 20.Woerly, S., Pinet, E., de Robertis, L., Van Diep, D. & Bousmina, M. (2001) Biomaterials 22, 1095-1111. [DOI] [PubMed] [Google Scholar]
- 21.Woerly, S., Van Diep, D., Sosa, N., de Vellis, J. & Espinosa, A. (2001) Int. J. Dev. Neurosci. 19, 63-83. [DOI] [PubMed] [Google Scholar]
- 22.Israelachvili, J. (1997) Intermolecular & Surface Forces (Academic, New York).
- 23.Benoit, M., Gabriel, D., Gerisch, G. & Gaub, H. E. (2000) Nat. Cell Biol. 2, 313-317. [DOI] [PubMed] [Google Scholar]
- 24.Forgacs, G., Foty, R. A., Shafrir, Y. & Steinberg, M. S. (1998) Biophys. J. 74, 2227-2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maheshwari, G., Brown, G., Lauffenburger, D. A., Wells, A. & Griffith, L. G. (2000) J. Cell Sci. 113, 1677-1686. [DOI] [PubMed] [Google Scholar]
- 26.Graner, F. & Glazier, J. A. (1992) Phys. Rev. Lett. 69, 2013-2016. [DOI] [PubMed] [Google Scholar]
- 27.Glazier, J. A. & Graner, F. (1993) Phys. Rev. E47, 2128-2154. [DOI] [PubMed] [Google Scholar]
- 28.Steinberg, M. S. (1975) J. Theor. Biol. 55, 431-443. [DOI] [PubMed] [Google Scholar]
- 29.Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., Teller, A. H. & Teller, E. (1953) J. Chem. Phys. 21, 1087-1092. [Google Scholar]
- 30.Press, W. H., Teukolsky, S. A., Vetterling, W. T. & Flannery, B. P. (2002) Numerical Recipes in C++: The Art of Scientific Computing (Cambridge Univ. Press, Cambridge, U.K.), 2nd Ed., pp. 275-286.
- 31.Beysens, D. A., Forgacs, G. & Glazier, J. A. (2000) Proc. Natl. Acad. Sci. USA 97, 9467-9471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mombach, J. C. M., Glazier, J. A. Raphael, R. C. & Zajac, M. (1995) Phys. Rev. Lett. 75, 2244-2247. [DOI] [PubMed] [Google Scholar]
- 33.Foty, R. & Steinberg, M. S. (1997) Cancer Res. 57, 5033-5036. [PubMed] [Google Scholar]
- 34.Davis, G. S., Phillips, H. M. & Steinberg, M. S. (1997) Dev. Biol. 192, 630-644. [DOI] [PubMed] [Google Scholar]
- 35.Harris, A. K., Wild, P. & Stopak, D. (1980) Science 208, 177-179. [DOI] [PubMed] [Google Scholar]
- 36.Harris, A. K., Stopak, D. & Wild, P. (1981) Nature 290, 249-251. [DOI] [PubMed] [Google Scholar]
- 37.Vernon, R. B. & Sage, E. H. (1992) Lab. Invest. 67, 807-808. [Google Scholar]
- 38.Vernon, R. B. & Sage, E. H. (1996) J. Cell. Biochem. 60, 185-197. [DOI] [PubMed] [Google Scholar]
- 39.Fray, T. R., Molloy, J. E., Armitage, M. P. & Sparrow, J. C. (1998) Tissue Eng. 4, 281-291. [DOI] [PubMed] [Google Scholar]
- 40.Vernon, R. B. & Sage, E. H. (1999) Microvasc. Res. 57, 118-133. [DOI] [PubMed] [Google Scholar]
- 41.Korff, T. & Augustin, H. G. (1999) J. Cell Sci. 112, 3249-3258. [DOI] [PubMed] [Google Scholar]
- 42.Sawhney, R. K. & Howard, J. (2002) J. Cell Biol. 157, 1083-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schreiber, D. I., Barocas, V. H. & Tranquillo, R. T. (2003) Biophys. J. 84, 4102-4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Godt, D. & Tepass, U. (1998) Nature 395, 387-391. [DOI] [PubMed] [Google Scholar]
- 45.González-Reyes, A. & St. Johnston, D. (1998) Development (Cambridge, U.K.) 125, 2837-2846. [DOI] [PubMed] [Google Scholar]
- 46.Townes, P. L. & Holtfreter, J. (1955) J. Exp Zool. 128, 53-120. [Google Scholar]
- 47.Techau, U. & Holstein, T. W. (1992) Dev. Biol. 151, 117-127. [DOI] [PubMed] [Google Scholar]
- 48.Rieu, J. P., Kataoka, N. & Sawada, Y. (1998) Phys. Rev. E 57, 924-931. [Google Scholar]
- 49.Robinson, E. E., Zazzali, K. M., Corbett, S. A. & Foty, R. A. (2003) J. Cell Sci. 116, 377-386. [DOI] [PubMed] [Google Scholar]
- 50.Steinberg, M. S. & Takeichi, M. (1994) Proc. Natl. Acad. Sci. USA 91, 206-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duguay, D., Foty, R. A. & Steinberg, M. S. (2003) Dev. Biol. 253, 309-323. [DOI] [PubMed] [Google Scholar]
- 52.Geong, B., Lee, K. M., Gutowska, A. & An, Y. H. (2002) Biomacromolecules 3, 865-868. [DOI] [PubMed] [Google Scholar]
- 53.Ohya, S., Nakayama, Y. & Matsuda, T. (2001) Biomacromolecules 2, 856-863. [DOI] [PubMed] [Google Scholar]
- 54.Elisseeff, J., Anseth, K., Sims, D., McIntosh, W., Randolph, M., Yaremchuk, M. & Langer, R. (1999) Plast. Reconstr. Surg. 104, 1014-1022. [DOI] [PubMed] [Google Scholar]
- 55.Baier-Leach, J., Bivens, K. A., Patrick, C. W., Jr., & Schmidt, C. E. (2003) Biotechnol. Bioeng. 82, 578-589. [DOI] [PubMed] [Google Scholar]
- 56.Park, Y. D., Tirelli, N. & Hubbell, J. A. (2003) Biomaterials 24, 893-900. [DOI] [PubMed] [Google Scholar]
- 57.Kulling, D., Woo, S., Demcheva, M. V., Hawes, R. H. & Vournakis, J. N. (1998) Endoscopy 30, S41-S42. [DOI] [PubMed] [Google Scholar]
- 58.Hoffman, A. S. (2002) Adv. Drug. Deliv. Rev. 54, 3-12. [DOI] [PubMed] [Google Scholar]
- 59.Lutolf, M. P., Lauer-Fields, J. L., Schmoekel, H. G., Metters, A. T., Weber, F. E., Fields, G. B. & Hubbell, J. A. (2003) Proc. Natl. Acad. Sci. USA 100, 5413-5418. [DOI] [PMC free article] [PubMed] [Google Scholar]