

Abstract
IL-17-producing T helper (Th17) cells comprise a distinct Th subset involved in epithelial cell- and neutrophil-mediated immune responses against extracellular microbes. At the same time, Th17 cells play significant roles in the development of autoimmune diseases including rheumatoid arthritis and multiple sclerosis. Since the identification of Th17 cells approximately a decade ago, the molecular mechanisms of their differentiation have been intensively studied and a number of signaling cascades and transcription factors have been shown to be involved. Here, we review the current knowledge regarding the function of Th17 cells in vivo as well as several key concepts for the molecular mechanisms of Th17 differentiation. We also discuss the emerging roles of phosphoinositide 3-kinase (PI3K), mammalian target of rapamycin complex 1 (mTORC1) and hypoxia-inducible factor 1 (HIF-1) in the differentiation of Th17 cells.
Introduction
Immune systems are generally divided into the innate and adaptive arms, and CD4+ T helper (Th) cells are indispensable for initiating the latter reaction. Th cells are subdivided into several subsets with distinct functions: T helper type 1 (Th1), T helper type 2 (Th2), IL-17-producing T helper (Th17), IL-9-producing T helper (Th9), or follicular T helper (Tfh) cells (Mosmann & Coffman 1989; Ouyang et al. 2008; Veldhoen et al. 2008b; Fazilleau et al. 2009). Th1 cells produce IFN-γ, activate macrophages and support granuloma formation as observed in Mycobacterium tuberculosis infection, whereas Th2 cells produce IL-4, IL-5 and IL-13, assist in the generation of IgE-producing plasma cells from naïve B cells, activate mast cells and eosinophils and support antihelminth immunity as well as allergic reactions. Th9 cells were recently identified as an IL-9-producing subtype possibly contributing to the induction of intestinal mucosal mast cells. Tfh cells produce IL-21 and provide B cell help in the lymph node germinal centers. There are also other CD4+ T-cell subsets with regulatory roles such as thymus-derived naturally occurring regulatory T cells (nTregs), inducible regulatory T cells (iTregs) and regulatory type 1 cells (Tr1) (Roncarolo et al. 2006; Sakaguchi et al. 2008).
Th17 cells are characterized by the production of IL-17A, IL-17F and IL-22. These Th17 cytokines induce the expression of numerous chemokines and antimicrobial peptides in epithelial cells and fibroblasts, which are important for the neutrophil-mediated immune reactions against extracellular microbes (Ouyang et al. 2008). Th17 cells are also a focus of attention because of their roles in the pathogenesis of various autoimmune diseases including rheumatoid arthritis (RA) and multiple sclerosis (MS). Since their identification, the molecular mechanisms of Th17 differentiation have been intensively studied, and numerous intracellular signaling cascades and transcriptional factors have now been identified. Recently, phosphoinositide 3-kinase (PI3K), mammalian target of rapamycin complex 1 (mTORC1) and hypoxia-inducible factor-1 (HIF-1) have been shown to regulate Th17 differentiation positively as well (Dang et al. 2011; Delgoffe et al. 2011; Ikejiri et al. 2011; Shi et al. 2011; Kurebayashi et al. 2012). Herein, we summarize the molecular mechanisms governing the Th17 differentiation in light of recent important findings. Although some of these findings still require further confirmation in vivo, understanding of these molecular complexities in Th17 differentiation may provide us the basis to define more clearly how these cells are generated and contribute to the host defense and to the development of autoimmune diseases.
Role of Th17 cells in bacterial and fungal infection
IL-17A was first identified as murine cytotoxic T lymphocyte–associated antigen-8 (mCTLA8) (Rouvier et al. 1993), which shows 57% homology with the amino acid sequence of the open reading frame 13 (ORF13) of T lymphotrophic virus Herpesvirus saimiri. Subsequently, IL-17A receptor (IL-17R) was cloned (Yao et al. 1995). Further studies expanded the IL-17 protein family from IL-17A to IL-17F (Kolls & Linden 2004), and Th17 cells were characterized as one of the major sources of IL-17A and IL-17F (Aggarwal et al. 2003; Bettelli & Kuchroo 2005; Langrish et al. 2005).
Early studies in murine infection models have established IL-17A derived from Th17 cells and IL-17-producing CD8+ T (Tc17) cells as important regulators of host defense against extracellular bacteria such as Klebsiella pneumoniae (Ye et al. 2001; Happel et al. 2005). In these models, IL-17A stimulates epithelial cells and fibroblasts to produce inflammatory mediators such as IL-6, macrophage inflammatory protein-2 (MIP-2), granulocyte colony-stimulating factor (G-CSF), prostaglandin E2 (PGE2) and several CXC chemokines, thus promoting granulopoiesis and neutrophil recruitment required for host defense against extracellular bacteria (Fossiez et al. 1996; Khader et al. 2009). IL-17A, IL-17F and IL-22 also induce the production of antimicrobial peptides such as β-defensin-2, S100 proteins and lipocalin-2 from mucosal (e.g. pulmonary and intestinal) epithelial cells (Khader et al. 2009). Later, studies indicate γδ T cells and invariant natural killer T (iNKT) cells as other major sources of IL-17A during K. pneumoniae infection (Price et al. 2012). In the intestinal mucosa as well, CD8+ T cells, γδ T cells, natural killer (NK) cells and innate lymphoid cells are also important producers of Th17 cytokines (Maynard et al. 2012); therefore, not only Th17 cells, but also other constellation of innate and adaptive sources of IL-17A, IL-17F and IL-22 are required for the effective host defense against extracellular bacterial infections. Host defense mechanisms against Citrobacter rodentium and Staphylococcus aureus also depend on Th17 cytokines (Ishigame et al. 2009; Khader et al. 2009).
Th17 cells are also required for the host defense against fungal infections depending on the species and the sites of infections. αβ T cells and their production of IL-17A are pivotal in the host defense against oral infection with Candida albicans, whereas Th1-related cytokine IL-12 prevents its systemic dissemination (Conti et al. 2009). In Aspergillus fumigatus infection, the host defense mainly relies on Th1 responses rather than Th17 responses (Romani 2011). In humans, patients with autosomal dominant hyper IgE syndrome (HIES) carry mutations in Stat3, presenting impaired Th17 differentiation and increased susceptibility to candidal and staphylococcal infections (Milner et al. 2008). Autosomal recessive IL-17RA deficiency and autosomal dominant IL-17F deficiency also lead to chronic mucocutaneous candidiasis (CMC) with S. aureus dermatitis (Puel et al. 2011). Similarly, those who develop autoantibodies against IL-17A, IL-17F and IL-22 suffer CMC (Kisand et al. 2010; Puel et al. 2010), which strikingly contrasts to the patients with anti-IFNγ autoantibody, who present disseminated nontuberous mycobacterial infections (Browne et al. 2012).
Additional studies revealed diverse roles of IL-17A in Th1-mediated immunity and B-cell biology. For instance, IL-17A directly activates macrophages and dendritic cells to produce various cytokines including IL-12, which enhances Th1 immunity and host defense against infection by the intracellular bacteria, Francisella tularensis (Lin et al. 2009). IL-17R is also expressed on B cells, and IL-17A promotes germinal center formation and B-cell survival and proliferation by activating NF-κB pathways (Hsu et al. 2008; Doreau et al. 2009; Xie et al. 2010). Recent findings show that ectopic lymph node formation in the lung upon infections requires IL-17A derived from CD4+ T cells (Rangel-Moreno et al. 2011).
Role of Th17 cells in the pathogenesis of autoimmunity
IL-17A also promotes the development of autoimmune diseases (Ouyang et al. 2008). In both murine collagen-induced arthritis (CIA) model and IL-1RA-/- arthritis model, deletion of IL-17A or p19 subunit of Th17-related cytokine IL-23 significantly attenuates the severity of disease (Murphy et al. 2003; Nakae et al. 2003a,b). IL-17A activates osteoblasts and synovial fibroblasts to express RANKL, which is required for osteoclast differentiation (Kotake et al. 1999; Sato et al. 2006), and thus, IL-17A contributes to bone destruction in RA. Experimental autoimmune encephalomyelitis (EAE) is an animal disease model of human MS, an autoimmune disease in central nervous system (CNS), and the deletion of IL-23 p19 subunit confers resistance to murine EAE (Cua et al. 2003). Th17 cell-derived IL-17A and IL-22 have been shown to act on blood–brain barrier (BBB) endothelial cells and disrupt the structure of BBB, enabling the infiltration of pathogenic Th17 cells into CNS lesion, suggesting the pathogenic role of Th17 cells. However, accumulating evidence shows that the deletion of IL-17A, IL-17F and IL-22 leads to only limited effects on the severity of EAE (Komiyama et al. 2006; Haak et al. 2009). Instead, RORγt-dependent expression of GM-CSF from Th cells activated by IL-23 has a pivotal role in the pathogenesis; Th cell-derived GM-CSF augments the infiltration of CD45hiCD11b+ myeloid cells into CNS and contributes to the development of EAE (Codarri et al. 2011). In murine colitis models, IL-17A production from CD4+ T cells is protective because IL-17A directly suppresses the development of colitogenic Th1 cells via IL-17R expressed on activated CD4+ T cells (O'Connor et al. 2009). In contrast, as in other autoimmune disease models, IL-23 accelerates the severity of murine colitis (Ahern et al. 2010). It is generally believed that IL-17-producing cells are protective but that IL-17/IFN-γ double producers are pathogenic and IL-23 accelerates the generation of double producers (Ahern et al. 2010; Hirota et al. 2011).
IL-17A is also required for Th2-mediated OVA-induced asthmatic reactions in mice (Nakae et al. 2002). Clinically, two different processes coexist in human asthma: corticosteroid-sensitive Th2 inflammatory mechanisms and corticosteroid-resistant airway remodeling characterized by subepithelial fibrosis and increased smooth muscle volume (Hackett 2012). Th17 cells are shown to contribute to the latter process in murine OVA-induced asthma model (Zhao et al. 2013). Bronchoalveolar lavage fluid (BALF) from asthma patients contains high amounts of IL-4/IL-17A double-positive CD4+ T cells (Wang et al. 2010), and moderate-to-severe human asthma patients have more IL-17A-positive cells in bronchial submucosa than mild asthma patients (Chakir et al. 2003). These observations indicate the role of Th17 cells in airway remodeling in asthma patients.
Developmental regulation of Th17 cells by cytokines and environmental factors
Upon antigen presentation by antigen-presenting cells (APCs), naïve CD4+ T cells differentiate into any Th subset based on the cytokine milieu produced by the presenting APCs and surrounding mesenchymal cells (Dong 2006; also see Fig. 1). For example, development of Th1 cells requires IL-12 and IFN-γ whereas that of Th2 cells requires IL-4. Each cytokine required for the differentiation of one Th subset negatively regulates the differentiation of the other, for example, IFN-γ and IL-12 inhibit Th2 differentiation and IL-4 inhibits Th1 differentiation.
Figure 1.
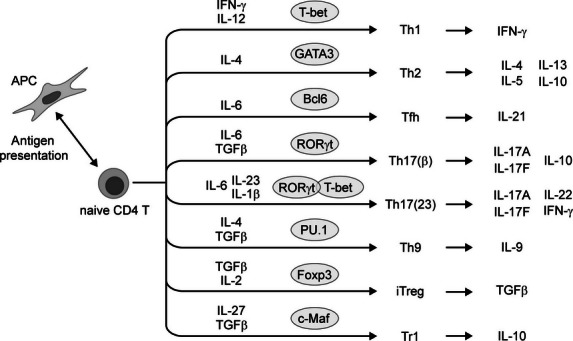
Cytokines and transcription factors required for Th differentiation. Upon antigen presentation, IL-12 and IFN-γ are required for Th1 differentiation, IL-4 for Th2 differentiation, IL-6 for Tfh differentiation, IL-6 and TGF-β for Th17(β) differentiation, IL-6, IL-23 and IL-1β for Th17(23) differentiation, IL-4 and TGF-β for Th9 differentiation, TGF-β and IL-2 for iTreg differentiation and IL-27 and TGF-β for Tr1 differentiation. These cytokines enable CD4+ T cells to express critical transcription factors that direct for the differentiation of each Th subset: T-bet for Th1 differentiation, GATA3 for Th2 differentiation, Bcl-6 for Tfh differentiation, RORγt for Th17(β) and Th17(23) differentiation, PU.1 for Th9 differentiation, Foxp3 for iTreg differentiation, and c-Maf for Tr1 differentiation. Th17(23) differentiation is characterized by the co-expression of RORγt and T-bet. Cytokines produced by each Th cell lineage are also indicated.
The cytokines initially linked to Th17 differentiation were IL-6 and IL-23 (Infante-Duarte et al. 2000; Aggarwal et al. 2003; Langrish et al. 2005). Later, TGF-β together with IL-6 was shown to initiate Th17 differentiation both in vitro and in vivo (Mangan et al. 2006; Veldhoen et al. 2006). It is now widely accepted that Th17 cells can be divided into two different subsets according to cytokine requirements for their differentiation and the expression profiles of cytokines and chemokines (Fig. 1). One is conventional Th17 (Th17(β)) cells differentiated by IL-6 and TGF-β, which express higher IL-10, CCL20 and CXCR6 in addition to IL-17A and IL-17F. The other is Th17(23) cells differentiated by IL-6, IL-23 and IL-1β without exogenous TGF-β, which are characterized by the expression of higher IL-22, CCL9 and CXCR3 (Ghoreschi et al. 2010). IL-21 is also indispensable for the development and expansion of both Th17(β) and Th17(23) cells in vivo both in humans and mice (Korn et al. 2007; Yang et al. 2008a), and IL-1β augments the differentiation of not only Th17(23) cells but also Th17(β) cells both in vitro and in vivo (Sutton et al. 2006; Gulen et al. 2010; Shaw et al. 2012). Accumulating evidence shows that Th17(23) cells possess higher pathogenic ability in autoimmune models (Ghoreschi et al. 2010), and IL-23 also has a pivotal role in the conversion of IL-17A single-positive Th17 cells into IL-17A/IFN-γ double producers in vivo (Hirota et al. 2011). Similar demarcation is observed in human memory Th17 cells, in which C. albicans-specific Th17 cells co-express higher IFN-γ than S. aureus-specific Th17 cells. The increased co-expression of IFN-γ is dependent on IL-1β, and only S. aureus-specific Th17 cells exhibit the ability for IL-10 expression upon restimulation (Zielinski et al. 2012).
In terms of cellular sources of these Th17-inducing cytokines, dendritic cells (DCs) produce IL-1β, IL-6 and IL-23 during antigen presentation, and it has recently been shown that Th17 cells support their own differentiation by producing TGF-β in an autocrine manner (Gutcher et al. 2011); however, the exact source of TGF-β in the initial differentiation of Th17 cells is still unclear. As with the opposing regulation between Th1 and Th2 differentiation, cytokines required for Th1 and Th2 differentiation (IFN-γ, IL-12 and IL-4) inhibit Th17 differentiation (Infante-Duarte et al. 2000; Harrington et al. 2005). Th1 and Th2 cells also inhibit Th17 differentiation through IFN-γ and IL-4, respectively. Th1 and Th2 cells expand with the help of IL-2 in an autocrine manner, but IL-2 severely dampens Th17 differentiation (Laurence et al. 2007). IL-27, which is also produced by APCs and induces the development of IL-10-producing Tr1 cells, inhibits Th17 differentiation and GM-CSF production, thus negatively regulating the severity of EAE (Stumhofer et al. 2006, 2007; Codarri et al. 2011). Notably, IL-6 in the absence of TGF-β initiates Tfh differentiation (Fazilleau et al. 2009) and TGF-β without IL-6 results in iTreg differentiation (Sakaguchi et al. 2008). It was in a way a surprise that TGF-β is required for the differentiation of Th17 cells in the presence of inflammatory cytokine IL-6 because TGF-β, an indispensable cytokine for the generation of iTreg cells, had been recognized as an anti-inflammatory cytokine with a regulatory nature. Hence, the balance between the production of pro- and anti-inflammatory cytokines from APCs is a key modulator of the development of each CD4+ T-cell lineage including Th17(β), Th17(23), Tfh and iTreg cells, which is determined as a result of the complex intracellular signaling interactions in APCs generated by the recognition of various antigens exposed on pathogens as described in detail elsewhere (Kawai & Akira 2011).
In a steady state, major population of Th17 cells harbor in the intestinal mucosa, whose development is largely dependent on the colonization of commensal microbiota (Ivanov et al. 2009). There is a complex regulation to balance effector immune reactions and tolerance acquisition against commensal microbiota, and the dysregulated activation of immune system results in the development of colitis. The steady-state intestinal Th17 cells are induced and maintained by TGF-β, which is abundant in intestinal mucosa, and microbiota-induced IL-1β and IL-23 (Ghoreschi et al. 2010; Maynard et al. 2012; Shaw et al. 2012). Microbiota-derived ATP even activates intestinal DCs to promote Th17 differentiation (Atarashi et al. 2008). The negative regulation of Th17 differentiation and inflammation in the intestine is mainly achieved by all-trans-retinoic acid, which is synthesized by CD103+ DCs and intestinal epithelial cells from food-derived vitamin A (Maynard et al. 2012). Microbiota-induced expression of indoleamine-pyrrole 2,3-dioxygenase (IDO) in epithelial cells and DCs catabolizes essential amino acid tryptophan and suppresses excessive Th cell differentiation (Romani 2011).
Th17 vs iTreg differentiation: RORγt vs Foxp3 and the role of hypoxia and HIF-1
The differentiation of each Th cell subset defined by the local cytokine milieu is achieved by the expression of specific transcription factors (Dong 2006; also see Fig. 1): T-bet in Th1 differentiation, GATA3 in Th2 differentiation, PU.1 in Th9 differentiation (Chang et al. 2010), or Bcl6 in Tfh differentiation (Fazilleau et al. 2009). Tregs are characterized by their expression of Foxp3 (Sakaguchi et al. 2008), and IL-10 production from Tr1 cells depends on c-Maf (Pot et al. 2009; Apetoh et al. 2010). In Th17 cells, RORγt, encoded by Rorc gene, is a pivotal transcription factor (Fig. 2A). In fact, transduction of RORγt is sufficient to convert unpolarized CD4+ T cells into Th17 cells (Ivanov et al. 2006). A related orphan receptor RORα has also been shown to initiate Th17 differentiation together with RORγt (Yang et al. 2008c). It has recently been shown that the anti-arrhythmic drug digoxin and its derivatives bind to RORγt and severely impair its transcriptional activity of RORγ and Th17 differentiation (Huh et al. 2011). This further underscores the critical role of RORγt in Th17 differentiation.
Figure 2.
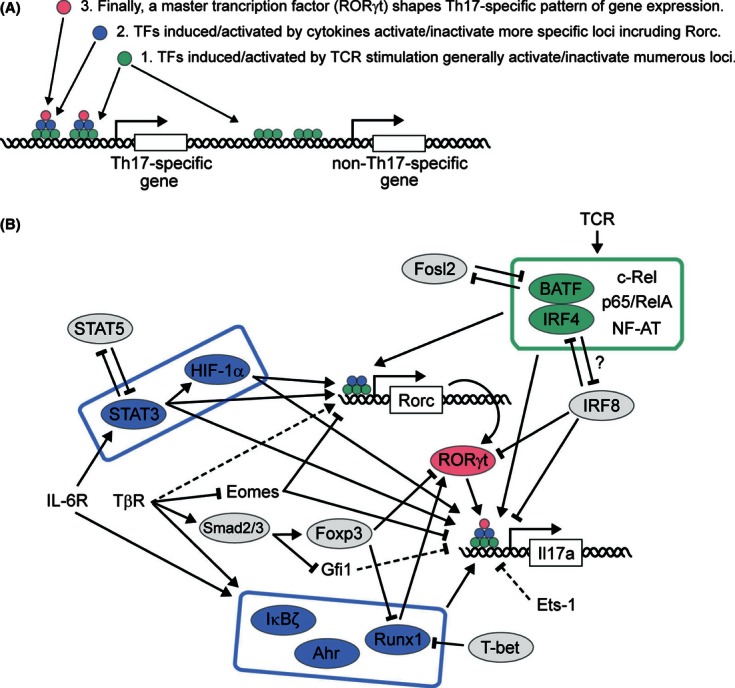
Transcription factors regulating Rorc and Il17a loci expression. (A) Schematic overview of the stepwise regulation of Th17-related loci expression. TCR-induced/TCR-activated transcription factors (TFs, green) bind to and activate/inactivate numerous Th17-specific and non-Th17-specific loci. Next, cytokine-induced/cytokine-activated TFs (blue) activate/inactivate more limited numbers of loci including a critical transcription factor RORγt (red), outlining the Th17-specific pattern of gene expression. Finally, a master transcription factor RORγt determines Th17-specific pattern of gene expression. (B) Schematic description of transcription factors regulating Th17 differentiation. BATF, IRF4, c-Rel, p65/RelA and NF-AT are TCR-induced/TCR-activated TFs generally activating/inactivating numerous loci (green box). Fosl2 and IRF8 compete with BATF and IRF4 for their target loci, respectively, and negatively regulate Th17 differentiation. Next, cytokine-induced/cytokine-activated TFs such as STAT3, HIF-1α, Runx1, IκBζ and Ahr outline the Th17-specific pattern of gene expression (blue box). STAT5 competes with STAT3 for their target loci and decreases Th17 differentiation. TGF-β-induced activation of Smad2/3 induces Foxp3 expression, which directly interacts with and inhibits the function of RORγt. Foxp3 also interacts with Runx1 and abrogates the positive interaction of Runx1 with RORγt. T-bet also directly interacts with Runx1 and interrupts its positive interaction with RORγt. TGF-β signaling decreases the expression of Eomes, a negative regulator of Rorc and Il17a expression. Gfi-1 and Ets-1 are negative regulators of Th17 differentiation without known functional mechanisms. The expression of Gfi-1 is also down-regulated by TGF-β signaling (see also Table 1).
As noted above, both pro-inflammatory Th17 and anti-inflammatory iTreg cells require TGF-β for their differentiation, and the molecular mechanism balancing Th17 versus iTreg differentiation has been intensively studied (Fig. 2B). During Th17(β) differentiation, RORγt expression is mainly induced by TGF-β (Ichiyama et al. 2008; Zhou et al. 2008) in a Smad2-/Smad3-independent manner (Takimoto et al. 2010) although the precise mechanisms of RORγt induction are still poorly understood. The induction of RORγt in Th17(23) cells without TGF-β signaling is more confusing, and the mechanism is largely unknown. Recently, it has been shown that TGF-β3 but not TGF-β1 (referred simply as TGF-β in this review unless otherwise indicated) is induced by IL-23 in addition to IL-6 and IL-1β, enabling RORγt expression and the development of more inflammatory Th17(23) cells (Lee et al. 2012). Of note, TGF-β is critical for RORγt induction but itself does not generate Th17(β) cells and instead initiates iTreg differentiation. This is because TGF-β also induces Foxp3 in a Smad2-/Smad3-dependent manner; Foxp3 interacts with RORγt and directly suppresses the transcriptional activity of RORγt, consequently blocking Th17(β) differentiation and initiating iTreg development (Ichiyama et al. 2008; Yang et al. 2008b; Zhou et al. 2008; also see Fig. 2B). The transcription factor Runx1 is a positive regulator of Th17 differentiation, which directly interacts with RORγt and increases its transcriptional activity (Zhang et al. 2008). Such Runx1-mediated increase in RORγt activity is also abrogated by the direct interaction of Foxp3 with Runx1; hence, Foxp3 directly inhibits Th17 differentiation through interaction with RORγt and indirectly through interaction with Runx1 (Zhang et al. 2008). The interaction of Runx1 with RORγt is also interrupted by tyrosine-phosphorylated T-bet, an important regulator of Th1 differentiation (Lazarevic et al. 2011), which is also induced in Th17(23) cells and actually limits IL-17A expression (Ghoreschi et al. 2010). The expression of Foxp3 is suppressed by addition of IL-6 and IL-21 in a STAT3-dependent manner, which enables RORγt to initiate Th17 differentiation (Ichiyama et al. 2008; Yang et al. 2008b; Zhou et al. 2008). Therefore, IL-6 and IL-21 serve as key cytokines in initiating Th17-mediated inflammatory reactions in the presence of TGF-β.
The mechanism how STAT3 down-regulates Foxp3 expression in Th17 differentiation had remained unclear, but it has recently been shown that STAT3-induced HIF-1α binds to Foxp3 and leads to the proteosomal degradation of Foxp3 during Th17 differentiation (Dang et al. 2011). HIF-1 is a well-characterized transcription factor induced under hypoxic conditions and consists of a heterodimer composed of an oxygen-sensitive HIF-1α subunit and a constitutively expressed HIF-1β subunit (Wang & Semenza 1993; Majmundar et al. 2010). Recent studies have shown that both hypoxia and HIF-1 positively and negatively regulate Th17 and iTreg differentiation, respectively, without apparent effect on Th1 or Th2 differentiation, and not only hypoxia but also IL-6 induces HIF-1α expression during Th17 differentiation via STAT3 activation (Dang et al. 2011; Ikejiri et al. 2011; Shi et al. 2011). Oxygen-dependent degradation of HIF-1α is mediated by E3 ubiquitin ligases, which contain von Hippel–Lindau tumor suppressor protein (pVHL), and the deletion of pVHL results in increased Th17 differentiation (Ikejiri et al. 2011). In addition to the proteosomal degradation of Foxp3 by HIF-1α, it also promotes Th17 differentiation via several other mechanisms. First, HIF-1 binds to hypoxia response elements (HREs) located in the proximal region of the Rorc locus and enhances its expression. HIF-1 also forms a complex with RORγt and recruits p300 to the Il17a, Il17f and Il23r loci. In addition, Shi et al. (2011) have shown that during Th17 differentiation, HIF-1 positively controls the glycolysis required for the rapid T-cell expansion after TCR stimulation.
In addition to inducing HIF-1α and down-regulating Foxp3 expression, IL-6 and STAT3 signaling have further important roles during Th17 differentiation. For example, STAT3 directly binds to and activates the expression of loci encoding Th17-related molecules and cytokines (Chen et al. 2006; Yang et al. 2011b; Ciofani et al. 2012), and interaction of STAT3 to Il17a and Il17f loci is directly competed by STAT5 (Yang et al. 2011b), which explains the negative regulatory role of IL-2 on Th17 differentiation. Intriguingly, even a combination of TGF-β treatment and the expression of a constitutively active form of STAT3 is insufficient for the full differentiation of Th17 cells (Zhou et al. 2007), indicating the presence of a yet to be identified factor for Th17 differentiation.
Regulation of Th17 differentiation by other transcription factors
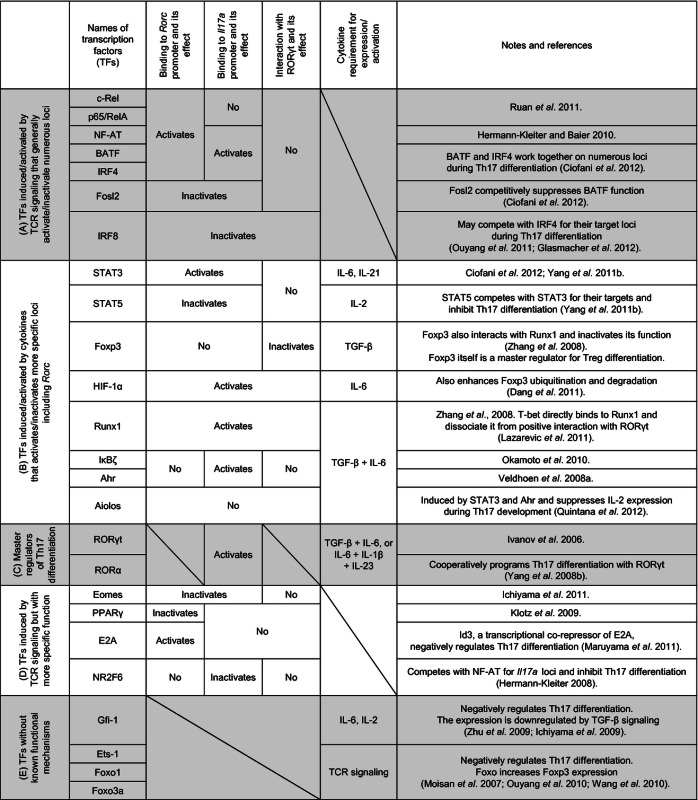
In addition to Foxp3, Runx1, STAT3 and HIF-1, the expression of RORγt and Th17 cytokines is also regulated by numerous other transcription factors. These transcription factors are induced in a stepwise fashion and can be classified into following five categories according to the order of their expression and function (Fig. 2A and Table 1): (A) those induced/activated by TCR stimulation, which contribute to general activation/inactivation of numerous loci including both Th17-specific and non-Th17-specific ones, (B) those induced/activated by cytokines, which activate/inactivate more specific loci including Rorc, (C) RORγt and RORα, which finally form a Th17-specific expression pattern, (D) those induced by TCR stimulation but with more specific function in Th17 differentiation, and (E) those lacking any established functional mechanisms but important in Th17 differentiation.
Table 1.
Classification of transcription factors regulating Th17 differentiation. Transcription factors (TFs) are classified into five categories (from A to E) based on the order of expression and their functions. Groups A, B and C correspond to TFs colored in green, blue and red in Fig. 2, respectively
 |
Initial signalings from TCR and costimulatory molecules induce and activate numerous transcription factors (group A in Table 1) that enable basal activation of CD4+ T cells required for further differentiation into each Th subset. Among the group A transcription factors, NF-κB and NF-AT family transcription factors generally activate many loci upon the activation of CD4+ T cells. During Th17 differentiation, RelA/p65 and c-Rel are two NF-κB family transcription factors required for the initiation of Rorc expression (Ruan et al. 2011; also see Fig. 2B). RelA/p65 and c-Rel directly bind to two putative Rel-binding site on the Rorc promoter and enhance RORγt expression, whereas none of NF-κB family transcription factors bind to Il17a promotor. RelA/p65 and c-Rel are also required for Foxp3 expression, and it forms a unique c-Rel enhanceome at Foxp3 promotor (Ruan et al. 2009). T-bet expression and Th1 differentiation are c-Rel-dependent as well (Hilliard et al. 2002). Therefore, RelA/p65 and c-Rel require other sets of transcription factors with limited functions to achieve a Th17-specific gene expression pattern. In addition to NF-κB activation, TCR stimulation also leads to the influx of Ca2+ into the cytoplasm and activates the calcineurin/NF-AT pathway, which also targets loci encoding various transcription factors and cytokines (Rao et al. 1997). Among the Th17-related gene loci, NF-AT binds to both Rorc and Il17a promoters and activates their expression (Hermann-Kleiter & Baier 2010). A nuclear orphan receptor NR2F6 competes with NF-AT for their targets in Th17-related genes and specifically inhibits Th17 differentiation (Hermann-Kleiter et al. 2008).
Recent studies have highlighted BATF and IRF4 as initial activators of Th17 differentiation (Ciofani et al. 2012; Glasmacher et al. 2012). Both BATF and IRF4 are induced by TCR stimulation and indispensable for proper Th17 differentiation (Brustle et al. 2007; Schraml et al. 2009). Ciofani et al. (2012) showed that BATF and IRF4 have common putative cis-regulatory modules (pCRMs) and function together in many loci, which include the most pCRMs for STAT3 and RORγt found in Il17a, Il17f, Il12b1, Il1r1 and Rorc loci. The binding of BATF and IRF4 to those loci increases chromatin accessibility for other transcription factors, and it is prerequisite for Th17 differentiation. Ciofani et al. (2012) also showed that another AP-1 family transcription factor Fosl2 competes with BATF for their target loci and works as a negative regulator of Th17 differentiation. Similarly, IRF8, a negative regulator of Th17 differentiation induced by TCR signaling (Ouyang et al. 2011), also shares many common pCRMs with IRF4 (Glasmacher et al. 2012); hence, IRF8 likely competes with IRF4 for their target loci and negatively regulates Th17 differentiation. IRF8 also directly binds to RORγt and suppresses its transcriptional activity (Ouyang et al. 2011). Notably, the activity of IRF4 is regulated through phosphorylation by ROCK2, a serine–threonine kinase induced during Th17 differentiation. Hence, the function of IRF4 is also partly under the control of Th17-inducing cytokines (Biswas et al. 2011).
Given the increased accessibility to numerous loci achieved by TCR-induced/TCR-activated transcription factors cytokine-induced/cytokine-activated transcription factors (group B of Table 1, see also Fig. 2B) form a more specific gene expression pattern for Th17 differentiation. These transcription factors include Runx1, STAT3, HIF-1, IκBζ, Ahr and Aiolos. The roles of Runx1, STAT3 and HIF-1 were mentioned above. During Th17 differentiation, the induction of IκBζ, Ahr and Aiolos requires both IL-6 and TGF-β. The induction and function of these transcription factors are mainly studied in Th17(β) cells; hence, it is still unclear whether these transcription factors are differently expressed in between Th17(β) and Th17(23) cells. IκBζ does not form complexes with RORγt but directly binds to and activates Il17a promoter. Among the three alternative splicing variants of IκBζ (IκBζ(L), IκBζ(S) and IκBζ(D)), IκBζ(L) and IκBζ(S) are expressed in and enhance the differentiation of Th17 cells (Okamoto et al. 2010). Ahr is also induced during Th17 differentiation, directly binds to Il17a promoter and activates the expression of IL-17A. One of the Ahr agonists 6-formylindolo(3,2-b)carbazole (FICZ) increases Th17 differentiation and exacerbates EAE, whereas Ahr antagonist resveratrol decreases the differentiation of Th17 cells (Quintana et al. 2008; Veldhoen et al. 2008a; Cui et al. 2011). Intriguingly, Ahr is also required for iTreg development and another Ahr agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) accelerates iTreg development and attenuates EAE severity; hence, Ahr regulates the differentiation of Th17 and iTreg cells in a ligand-specific manner (Quintana et al. 2008). Ahr also binds to Il10 promoter and enhances the expression of IL-10 together with c-Maf during Tr1 differentiation (Apetoh et al. 2010; Gandhi et al. 2010), which may also contribute to the production of IL-10 from Th17(β) cells. Aiolos is induced by STAT3 and Ahr downstream of IL-6 and TGF-β and shuts down the expression of IL-2 during Th17 differentiation (Quintana et al. 2012). These group A and B transcription factors briefly outline the Th17-specific gene expression patterns and induce the expression of a transcription factor RORγt (Ivanov et al. 2006; Ciofani et al. 2012). The function of RORγt is accomplished with the help of related nuclear orphan receptor RORα, which works together with RORγt and accelerates Th17 differentiation (Yang et al. 2008c). These group C molecules finally complete Th17-specific gene expression patterns (Fig. 2A,B).
Other transcription factors are induced by TCR stimulation but exert more specific functions in Th17 differentiation. These include Eomesodermin (Eomes), PPARγ and E2A (group D in Table 1). For instance, Eomes is induced by TCR signaling and works as a negative regulator of Rorc and Il17a expression (Ichiyama et al. 2011). The expression of Eomes is down-regulated by TGF-β-induced TAK1-JNK-c-Jun pathway, which explains one mechanism how Smad-independent upregulation of Rorc expression (Takimoto et al. 2010) is achieved by TGF-β during Th17 differentiation. PPARγ (Klotz et al. 2009) and E2A (Maruyama et al. 2011) are induced by TCR signaling, bind to Rorc promotor and inhibit and activate RORγt expression, respectively. Accordingly, antidiabetic thiazolidinediones pioglitazone, a PPARγ agonist, decreases Th17 differentiation and the severity of EAE, and Id3, a corepressor of E2A, also inhibits Th17 differentiation. Gfi1 (Ichiyama et al. 2009; Zhu et al. 2009), Ets-1 (Moisan et al. 2007), Foxo1 and Foxo3a (Ouyang et al. 2010; Wan et al. 2010) are also suggested as negative regulators of Th17 differentiation. However, the mechanisms by which these transcription factors work are still unclear (group E in Table 1). Regarding of iTreg differentiation, Foxo1 and Foxo3a directly bind to Foxp3 promoter and enhance its expression (Ouyang et al. 2010). The expression of Gfi-1 is induced by TCR stimulation and decreased by TGF-β during Th17 differentiation (Ichiyama et al. 2009; Zhu et al. 2009).
mTORC2-Akt-Foxo1/3a signaling in Th17 and Treg differentiation
Antigen presentation also leads to the activation of PI3K and mTORC2 in CD4+ T cells (Kurebayashi et al. 2012; Okkenhaug et al. 2006; also see Fig. 3). PI3K phosphorylates the third position of the hydroxyl group in the inositol ring of phosphatidylinositol and generates PIP3, which recruits a constitutively active kinase PDK1 and its substrate Akt to the cell membrane through interaction with their PH domains. PDK1 then phosphorylates Akt at Thr308 (Koyasu 2003). The expression of p110δ, a catalytic subunit of the class IA PI3K family, is restricted to lymphocytes. Replacement of wild-type p110δ with a kinase-defective p110δD910A, inhibition of p110δ by a specific inhibitor (IC87114), or deletion of p85α, a regulatory subunit that forms a heterodimer with p110δ, severely impair the phosphorylation of Akt at Thr308 upon TCR stimulation (Okkenhaug et al. 2006; Shiroki et al. 2007; Kurebayashi et al. 2012). There are two mTOR complexes: one is the rapamycin-sensitive mTORC1 composed of mTOR, mLST8 and Raptor, and the other is the rapamycin-insensitive mTORC2 composed of mTOR, mLST8 and Rictor (Wullschleger et al. 2006; also see Fig. 3). Among these, mTORC2 is able to phosphorylate Akt at Ser473 in TCR-stimulated CD4+ T cells (Lee et al. 2010). Although it is still poorly understood how mTORC2 is activated and phosphorylates Akt, studies have shown that this reaction requires the trafficking of Akt to the cell membrane upon PI3K activation (Andjelkovic et al. 1997). Therefore, the pan-PI3K inhibitor LY294002 also inhibits Ser473 phosphorylation of Akt. Interestingly, Thr308-phosphorylated Akt and Ser473-phosphorylated Akt phosphorylate distinct substrates; Thr308-phosphorylated Akt activates mTORC1 and enhances S6K1 phosphorylation, whereas Ser473-phosphorylated Akt preferentially phosphorylates Foxo1 and Foxo3a (Jacinto et al. 2006). Therefore, there are two major pathways crossing at Akt: one is the PI3K-Akt(pThr308)-mTORC1 signaling and the other is the mTORC2-Akt(pSer473)-Foxo1/3a signaling, and these demarcations are used in this review for convenience.
Figure 3.
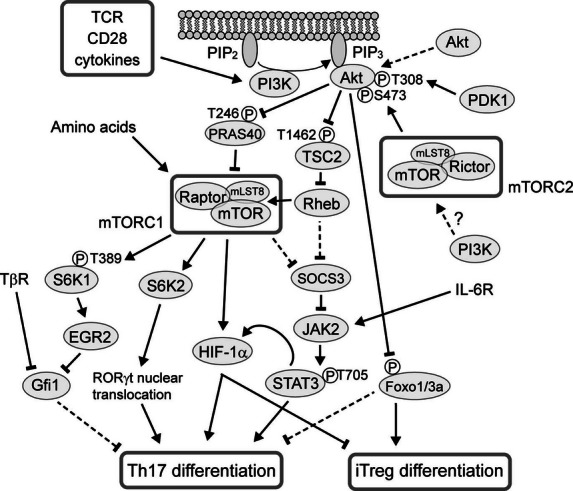
PI3K, Akt and mTOR complexes regulate Th17 differentiation. Stimulation of TCR, CD28 and cytokine receptors activates PI3K and mTORC2 in CD4+ T cells. PI3K activation enables Thr308 phosphorylation of Akt by PDK1 and Ser473 phosphorylation of Akt by mTORC2. TSC2 and PRAS40 are negative regulators of mTORC1 activity, and Thr308-phosphorylated Akt phosphorylates and inactivates these molecules. Activated mTORC1 enhances HIF-1α expression and RORγt nuclear translocation, whereas it negatively regulates the expression of Gfi1 and SOCS3, both of which are negative regulators of Th17 differentiation. Ser473-phosphorylated Akt also phosphorylates and inactivates Foxo1 and Foxo3a, both of which limit CD4+ T-cell activation and are suggested as negative regulators of Th17 differentiation.
The role of mTORC2-Akt-Foxo1/3a signaling is well characterized in the differentiation of both nTreg and iTreg cells. Foxo1 and Foxo3a directly bind to Foxp3 promoter and increase the expression of Foxp3, and the deletion of Foxo results in the collapse of T-cell homeostasis and in the severe autoimmunity (Harada et al. 2010; Ouyang et al. 2010). Foxo proteins are active in a dephosphorylated state, and phosphorylation by Akt results in the retention of Foxo proteins in the cytoplasm by 14-3-3 proteins; hence, constitutively active Akt impairs Treg development (Haxhinasto et al. 2008). In contrast, inhibition of PI3K by LY294002 increases iTreg differentiation in a Foxo1/3a-dependent manner (Sauer et al. 2008; Harada et al. 2010), and the deletion of Rictor, encoding a central component of mTORC2, also increases iTreg differentiation (Lee et al. 2010).
However, the contribution of mTORC2-Akt-Foxo1/3a signaling in Th17 differentiation is still controversial. The expression of a constitutively active form of Akt enhances the differentiation of all Th subsets including Th17 cells (Arimura et al. 2004; Kurebayashi et al. 2012), and T-cell-specific deletion of Foxo1 and Foxo3a results in the autoimmunity with an increased Th17 differentiation in vivo (Ouyang et al. 2010). It is also shown that the ectopic expression of Foxo inhibits IL-17A production by human CCR6+CD4+ T cells (Wan et al. 2010). All of these observations seem to indicate the contribution of mTORC2-Akt-Foxo1/3a signaling to Th17 differentiation; however, the deletion of Rictor does not influence on the differentiation of Th17 cells (Lee et al. 2010; Delgoffe et al. 2011).
PI3K-Akt-mTORC1 signaling in Th17 and Treg differentiation
The activation of PI3K and phosphorylation of Akt at Thr308 lead to the phosphorylation and inhibition of TSC1/TSC2 complex and PRAS40 (Jacinto et al. 2006; Wullschleger et al. 2006; Laplante & Sabatini 2012). TSC1/TSC2 complex negatively regulates mTORC1 activity by impairing Rheb GTPase activity, which is required for mTORC1 activation. In addition, PRAS40 directly associates with mTORC1 and down-regulates its activity. Extracellular amino acids also activate mTORC1 via Ragulator–Rag complexes. Rapamycin inhibits mTORC1 with high specificity, and mTORC1 inhibitors are applied to the chemotherapies against several types of cancers and for immune suppression in organ transplantation.
In CD4+ T cells, the inhibition of PI3K and mTORC1 increases iTreg differentiation (Sauer et al. 2008). In contrast, activation of mTORC1 by IL-1β-IRAK1/4 signaling, which degrades TSC1/TSC2 complex, promotes Th17 differentiation (Gulen et al. 2010), and malfunction of mTORC1 by deletion of Rheb and Raptor, encoding central components of mTORC1, impairs Th17 differentiation (Delgoffe et al. 2009, 2011; Kurebayashi et al. 2012). There are several independent mechanisms supposed to regulate Th17 differentiation via mTORC1. One is the positive regulation of HIF-1α expression downstream of mTORC1 pathway (Ikejiri et al. 2011; Shi et al. 2011). As reviewed above, HIF-1α expression positively regulates Th17 differentiation by directly promoting Rorc and Th17-related gene expression (Dang et al. 2011), and it also increases glycolytic activity required for rapid T-cell expansion (Shi et al. 2011). We showed that mild hypoxia (5% oxygen) during Th17 differentiation induces the activation of the mTORC1 pathway independently of PI3K, implying the existence of a positive feedback loop between mTORC1 and HIF-1 for Th17 differentiation (Ikejiri et al. 2011).
Second, mTOR complexes differently regulate the phosphorylation of STAT proteins during Th differentiation. Delgoffe et al. (2009) showed that the deletion of Flap, which encodes mTOR, deprives cells of both mTORC1 and mTORC2 and reduces tyrosine phosphorylation of STAT transcription factors and the differentiation of all Th subsets with increased iTreg development albeit without TGF-β. They also reported that the deletion of Rheb, which severely impairs mTORC1 function, selectively reduces the tyrosine phosphorylation of STAT3 and STAT4 by inducing SOCS3 expression and impairs the differentiation of Th1 and Th17 cells (Delgoffe et al. 2011). Although the deletion of Flap or Rheb impairs tyrosine phosphorylation of STAT3, mTORC1 inhibition by rapamycin does not interfere with the serine and tyrosine phosphorylation of STAT3 (Lee et al. 2010; Kurebayashi et al. 2012). Although the deletion of Raptor, encoding an essential component of mTORC1, results in embryonic lethality (Guertin et al. 2006), tamoxifen-induced deletion of Raptor has enabled us to examine the role of mTORC1 in cells of interest (Hoshii et al. 2012). Deletion of Raptor in T cells impairs Th17 differentiation without affecting tyrosine phosphorylation of STAT3 or Th1 differentiation (Kurebayashi et al. 2012). These observed differences between Flap, Rheb and Raptor deficiencies in the regulation of STAT phosphorylation and Th1 differentiation indicate some unknown mTORC1-independent roles of mTOR and Rheb in STAT phosphorylation.
We have also shown that Th17 differentiation is impaired by mTORC1 inhibition via decreased RORγt nuclear translocation and increased Gfi1 expression, whereas Th1 differentiation is maintained both in vivo and in vitro (Kurebayashi et al. 2012). Gfi1 expression is suppressed by EGR1 and EGR2 transcription factors, which directly bind to Gfi1 promoter (Laslo et al. 2006). Expression of EGR1 (Sarker & Lee 2004) and EGR2 (Carnevalli et al. 2011) is regulated by S6K1 downstream of mTORC1. Accordingly, we showed that forced expression of a constitutively active form of S6K1 in CD4+ T cells induced Egr2 expression, suppressed Gfi1 expression and accelerated Th17 differentiation (Kurebayashi et al. 2012), indicating that the PI3K-Akt-mTORC1-S6K1 pathway positively regulates IL-17 expression through the suppression of Gfi1. In addition to the suppression of Gfi1, mTORC1 accelerates the nuclear translocation of RORγt (Kurebayashi et al. 2012). RORγt does not have a nuclear localization signal (NLS), yet is localized in the nucleus in Th17 cells. We have shown that S6K2, a nuclear counterpart of S6K1, possesses a NLS, binds to RORγt and transports RORγt to the nucleus in a piggyback fashion. The expression of S6K2 is increased after TCR stimulation partly in a mTORC1-dependent fashion. Thus, the PI3K-Akt-mTORC1-S6K2 pathway also positively controls Th17 differentiation by nuclear translocation of RORγt (Kurebayashi et al. 2012).
In contrast to iTreg differentiation, nTreg development in the thymus is independent of mTORC1 activity. For example, the T-cell-specific deletion of TSC1, an inhibitory molecule of mTORC1, does not alter the size of the nTreg population in vivo (Yang et al. 2011a). Similarly, development of nTreg cells in vivo was little affected by the deletion of Raptor in T-cell lineage (S.M. & M.O. personal communication). Such difference in the susceptibility of nTreg and iTreg differentiation to the changes in mTORC1 activity is an intriguing subject for future studies.
Recently, a patient with a premature stop codon in PIK3R1, resulting in the absence of p85α but normal expression of p55α and p50α, has been reported (Conley et al. 2012). The patient shows agammaglobulinemia because of a severe defect in B-cell development in agreement with previous studies with mice lacking p85α (Suzuki et al. 1999, 2003). Further studies would reveal the role of PI3K in Th17 differentiation in humans.
Role of PI3K-Akt-mTORC1 signaling and HIF-1 in autoimmunity and host defense
Recent studies have also established the roles of PI3K-Akt-mTORC1 signaling and transcription factor HIF-1 in the development of autoimmunity. Mice expressing inactive form of p110δ (p110δD910A) show mild symptoms in EAE associated with decreased Th17 differentiation (Haylock-Jacobs et al. 2011). Although this study cannot exclude the possible contribution of PI3K in non-T cells to Th17 differentiation, adoptive transfer of p85α-deficient naive CD4+ T cells in murine T-cell transfer model of colitis shows decreased Th17 differentiation compared to wild-type CD4+ T cells, with Th1 differentiation maintained (our unpublished observations), indicating the pivotal roles of PI3K in CD4+ T cells in in vivo Th17 differentiation. Delgoffe et al. (2011) have shown that impaired mTORC1 function by deletion of Rheb in T cells also decreases in vivo generation of Th17 cells and attenuates the severity of EAE. Intriguingly, however, these mice presented an increased rate of ataxia because of mononuclear infiltration into the cerebellum instead of spinal cord. Similarly, the depletion of HIF-1α significantly decreases EAE severity by decreasing Th17 differentiation and increasing iTreg cells in vivo (Dang et al. 2011). Because RORγt controls GM-CSF expression in Th17 cells (Codarri et al. 2011), these data also implicate the role of PI3K, mTORC1 and HIF-1 in GM-CSF production from CD4+ T cells, a pivotal cytokine in the pathogenicity of myelin-reactive CD4+ T cells.
Treatment with mTORC1-specific inhibitor rapamycin in murine CD4+ T-cell transfer model of colitis also decreases Th17 differentiation and attenuates the decrease in body weights (Kurebayashi et al. 2012). This may partly be because mTORC1 inhibition decreases the expression of IL-23R on CD4+ T cells, a receptor for cytokine (IL-23) that is required for the induction of IL-17A/IFN-γ double-positive T cells and exacerbates the clinical course in murine colitis (Ahern et al. 2010). Additionally, mTORC1 function is required for T-cell proliferation and trafficking (Sinclair et al. 2008), which may also explain the regulatory effect of rapamycin in murine colitis. Especially, the importance of lymphopenia-driven proliferation in the development of CD4+ T-cell transfer model of colitis is well documented (Zhang & Bevan 2012). Notably, rapamycin treatment in CD4+ T-cell transfer model of colitis increases the differentiation of Th1 cells in mesenteric lymph nodes (Kurebayashi et al. 2012), possibly due to both T-cell intrinsic deviation to Th1 development and the increased production of IL-12 from APCs in the absence of PI3K-Akt-mTORC1 pathway (Fukao et al. 2002; Ohtani et al. 2008; Weichhart et al. 2008).
Compared to their function in the development of autoimmunity, the roles of PI3K-Akt-mTORC1 signaling and HIF-1 of CD4+ T cells in the host defense are still largely unknown, despite the major roles of PI3K-Akt-mTORC1 pathway in APCs to regulate Th1- and Th17-type immune reactions (Fukao et al. 2002; Ohtani et al. 2008; Weichhart et al. 2008). It has been shown that both Rheb and HIF-1α deficiency lead to the impaired Th17 differentiation in intestinal mucosa (Dang et al. 2011; Delgoffe et al. 2011). However, the contribution of PI3K and Akt in the generation of intestinal Th17 cells has not been reported. Cytokines and environmental factors that up-regulate mTORC1 activity and HIF-1 expression in the intestinal T cells are also largely unknown although inflammatory cytokines represented by IL-1β seem to be pivotal. The homeostatic differentiation and maintenance of Th17 cells in intestinal tissue mainly require IL-1β induced by microbiota (Shaw et al. 2012), and IL-1β is known to induce mTORC1 activation via IRAK1/4-mediated degradation of TSC1/2 complex in CD4+ T cells (Gulen et al. 2010). IL-1β is also known to induce HIF-1α expression even in a normoxic condition via PI3K and mTORC1 activation (Stiehl et al. 2002). TGF-β is another cytokine required for the de novo differentiation of intestinal Th17 cells (Ghoreschi et al. 2010), and TGF-β is a well-known activator of PI3K-Akt signaling (Zavadil & Bottinger 2005); hence, it is possible that TGF-β-induced activation of PI3K-Akt signaling also contributes to the differentiation and maintenance of Th17 cells in the intestinal mucosa.
Regarding the intestinal environment, the partial pressure of oxygen (pO2) in the capillary beds and mucosal interstitium of intestine is expected to be lower than those in pulmonary vein and alveoli, and this mildly hypoxic condition possibly supports the differentiation of Th17 cells in peripheral tissues including intestine as shown in vitro (Dang et al. 2011; Ikejiri et al. 2011; Shi et al. 2011). Immune homeostasis in the intestine is maintained in part by deprivation of essential amino acid tryptophan by IDO, and deprivation of essential amino acid potentially suppresses the activity of mTORC1 in CD4+ T cells as shown in several cell lines (Laplante & Sabatini 2012), which may result in the preferential suppression of Th17 differentiation. Actually, a small-molecule halofuginone selectively inhibits Th17 differentiation by activating amino acid starvation response and cytoprotective signaling pathway (Sundrud et al. 2009) and lower essential amino acid concentration preferentially decreases Th17 differentiation (our unpublished observations). Hence, PI3K-Akt-mTORC1 signaling and HIF-1 in CD4+ T cells may also contribute to the development of Th17 cells in mucosal tissues and to the maintenance of immune homeostasis with commensal microbiota.
Conclusion
Th17 cells have established a unique position among Th subsets by regulating neutrophil-mediated immune responses, and their differentiation and function is controlled by a number of intracellular signaling pathways and a complex transcription factor network as reviewed here. Recent findings further identified mTORC1 as another positive regulator of Th17 differentiation, acting via the cooperative regulation of STAT3 phosphorylation, RORγt nuclear translocation and Gfi1 and HIF-1α expression. This accumulating evidence now provides us more precise understandings of Th17 differentiation and clues to the pharmacological manipulation. However, it is also true that there are many issues that remain unresolved. For instance, although studies indicate the existence of both conventional Th17(β) cells and more pro-inflammatory Th17(23) cells in vivo, the spatiotemporal regulation and its molecular mechanism in the generation of these different Th17 subsets are still largely unknown. Studies on these matters would provide us more knowledge about T cell–mediated immunity and opportunities for manipulating immune systems in many inflammatory disorders and infections.
Acknowledgments
This work was in part supported by a Grant-in-Aid for Young Scientist (B) (21790476 to S.N.), from the Japan Society for the Promotion of Science, a Grant-in-Aid from the Takeda Science Foundation (S.N.), a Grant-in-Aid from the Mochida Memorial Foundation for Medical and Pharmaceutical Research (S.N.), a Health Sciences Research Grant for Research on Specific Diseases from the Ministry of Health, Labour and Welfare, Japan, a Grant-in-Aid for Scientific Research on Priority Areas (20060021 to S.N.), a National Grant-in-Aid for the Establishment of a High-Tech Research Center in a private University, a grant for the Promotion of the Advancement of Education and Research in Graduate Schools, and a Scientific Frontier Research Grant from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Disclosure
S.K. is a consultant for Medical and Biological Laboratories, Co. Ltd. The authors otherwise have no financial conflicts of interest.
References
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, Powrie F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279–288. doi: 10.1016/j.immuni.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, Burns EJ, Sherr DH, Weiner HL, Kuchroo VK. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010;11:854–861. doi: 10.1038/ni.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura Y, Shiroki F, Kuwahara S, Kato H, Dianzani U, Uchiyama T, Yagi J. Akt is a neutral amplifier for Th cell differentiation. J. Biol. Chem. 2004;279:11408–11416. doi: 10.1074/jbc.M309063200. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, Yagita H, Ishii N, Evans R, Honda K, Takeda K. ATP drives lamina propria TH17 cell differentiation. Nature. 2008;455:808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Kuchroo VK. IL-12- and IL-23-induced T helper cell subsets: birds of the same feather flock together. J. Exp. Med. 2005;201:169–171. doi: 10.1084/jem.20042279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas PS, Gupta S, Chang E, Song L, Stirzaker RA, Liao JK, Bhagat G, Pernis AB. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J. Clin. Invest. 2011;120:3280–3295. doi: 10.1172/JCI42856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne SK, Burbelo PD, Chetchotisakd P, et al. Adult-onset immunodeficiency in Thailand and Taiwan. N. Engl. J. Med. 2012;367:725–734. doi: 10.1056/NEJMoa1111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, Arpaia E, Mak TW, Kamradt T, Lohoff M. The development of inflammatory TH-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 2007;8:958–966. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- Carnevalli LS, Masuda K, Frigerio F, Le Bacquer O, Um SH, Gandin V, Topisirovic I, Sonenberg N, Thomas G, Kozma SC. S6K1 plays a critical role in early adipocyte differentiation. Dev. Cell. 2011;18:763–774. doi: 10.1016/j.devcel.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, Boulet LP, Hamid Q. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J. Allergy Clin. Immunol. 2003;111:1293–1298. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- Chang HC, Sehra S, Goswami R, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat. Immunol. 2010;11:527–534. doi: 10.1038/ni.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O'Shea JJ. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl Acad. Sci. USA. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, et al. A Validated Regulatory Network for Th17 Cell Specification. Cell. 2012;151:289–303. doi: 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- Conley ME, Dobbs AK, Quintana AM, Bosompem A, Wang YD, Coustan-Smith E, Smith AM, Perez EE, Murray PJ. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J. Exp. Med. 2012;209:463–470. doi: 10.1084/jem.20112533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, Filler SG, Masso-Welch P, Edgerton M, Gaffen SL. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q, Sheng H, Xi B, Zhang JZ, Zang YQ. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J. Clin. Invest. 2011;121:658–670. doi: 10.1172/JCI42974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, et al. Control of TH17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat. Rev. Immunol. 2006;6:329–333. doi: 10.1038/nri1807. [DOI] [PubMed] [Google Scholar]
- Doreau A, Belot A, Bastid J, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat. Immunol. 2009;10:778–785. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG. Follicular helper T cells: lineage and location. Immunity. 2009;30:324–335. doi: 10.1016/j.immuni.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossiez F, Djossou O, Chomarat P, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukao T, Tanabe M, Terauchi Y, Ota T, Matsuda S, Asano T, Kadowaki T, Takeuchi T, Koyasu S. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 2002;3:875–881. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, Kozoriz D, Weiner HL, Quintana FJ. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3+ regulatory T cells. Nat. Immunol. 2010;11:846–853. doi: 10.1038/ni.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang XP, et al. Generation of pathogenic TH17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasmacher E, Agrawal S, Chang AB, et al. A Genomic Regulatory Element That Directs Assembly and Function of Immune-Specific AP-1-IRF Complexes. Science. 2012;338:975–980. doi: 10.1126/science.1228309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Gulen MF, Kang Z, Bulek K, et al. The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity. 2010;32:54–66. doi: 10.1016/j.immuni.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity. 2011;34:396–408. doi: 10.1016/j.immuni.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, Waisman A. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J. Clin. Invest. 2009;119:61–69. doi: 10.1172/JCI35997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett TL. Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr. Opin. Allergy Clin. Immunol. 2012;12:53–59. doi: 10.1097/ACI.0b013e32834ec6eb. [DOI] [PubMed] [Google Scholar]
- Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, Odden AR, Shellito JE, Bagby GJ, Nelson S, Kolls JK. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J. Exp. Med. 2005;202:761–769. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada Y, Elly C, Ying G, Paik JH, DePinho RA, Liu YC. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J. Exp. Med. 2010;207:1381–1391. doi: 10.1084/jem.20100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J. Exp. Med. 2008;205:565–574. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haylock-Jacobs S, Comerford I, Bunting M, Kara E, Townley S, Klingler-Hoffmann M, Vanhaesebroeck B, Puri KD, McColl SR. PI3Kδ drives the pathogenesis of experimental autoimmune encephalomyelitis by inhibiting effector T cell apoptosis and promoting Th17 differentiation. J. Autoimmun. 2011;36:278–287. doi: 10.1016/j.jaut.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Hermann-Kleiter N, Baier G. NFAT pulls the strings during CD4+ T helper cell effector functions. Blood. 2010;115:2989–2997. doi: 10.1182/blood-2009-10-233585. [DOI] [PubMed] [Google Scholar]
- Hermann-Kleiter N, Gruber T, Lutz-Nicoladoni C, Thuille N, Fresser F, Labi V, Schiefermeier N, Warnecke M, Huber L, Villunger A, Eichele G, Kaminski S, Baier G. The nuclear orphan receptor NR2F6 suppresses lymphocyte activation and T helper 17-dependent autoimmunity. Immunity. 2008;29:205–216. doi: 10.1016/j.immuni.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard BA, Mason N, Xu L, Sun J, Lamhamedi-Cherradi SE, Liou HC, Hunter C, Chen YH. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J. Clin. Invest. 2002;110:843–850. doi: 10.1172/JCI15254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshii T, Tadokoro Y, Naka K, Ooshio T, Muraguchi T, Sugiyama N, Soga T, Araki K, Yamamura K, Hirao A. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J. Clin. Invest. 2012;122:2114–2129. doi: 10.1172/JCI62279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HC, Yang P, Wang J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat. Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- Huh JR, Leung MW, Huang P, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature. 2011;472:486–490. doi: 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyama K, Hashimoto M, Sekiya T, Nakagawa R, Wakabayashi Y, Sugiyama Y, Komai K, Saba I, Moroy T, Yoshimura A. Gfi1 negatively regulates T(h)17 differentiation by inhibiting RORgammat activity. Int. Immunol. 2009;21:881–889. doi: 10.1093/intimm/dxp054. [DOI] [PubMed] [Google Scholar]
- Ichiyama K, Sekiya T, Inoue N, Tamiya T, Kashiwagi I, Kimura A, Morita R, Muto G, Shichita T, Takahashi R, Yoshimura A. Transcription factor Smad-independent T helper 17 cell induction by transforming-growth factor-beta is mediated by suppression of eomesodermin. Immunity. 2011;34:741–754. doi: 10.1016/j.immuni.2011.02.021. [DOI] [PubMed] [Google Scholar]
- Ichiyama K, Yoshida H, Wakabayashi Y, Chinen T, Saeki K, Nakaya M, Takaesu G, Hori S, Yoshimura A, Kobayashi T. Foxp3 inhibits RORγt-mediated IL-17A mRNA transcription through direct interaction with RORγt. J. Biol. Chem. 2008;283:17003–17008. doi: 10.1074/jbc.M801286200. [DOI] [PubMed] [Google Scholar]
- Ikejiri A, Nagai S, Goda N, Kurebayashi Y, Osada-Oka M, Takubo K, Suda T, Koyasu S. Dynamic regulation of Th17 differentiation by oxygen concentrations. Int. Immunol. 2011;24:137–146. doi: 10.1093/intimm/dxr111. [DOI] [PubMed] [Google Scholar]
- Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J. Immunol. 2000;165:6107–6115. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Khader SA, Gaffen SL, Kolls JK. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 2009;2:403–411. doi: 10.1038/mi.2009.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisand K, Boe Wolff AS, Podkrajsek KT, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 2010;207:299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz L, Burgdorf S, Dani I, et al. The nuclear receptor PPARγ selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med. 2009;206:2079–2089. doi: 10.1084/jem.20082771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Invest. 1999;103:1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyasu S. The role of PI3K in immune cells. Nat. Immunol. 2003;4:313–319. doi: 10.1038/ni0403-313. [DOI] [PubMed] [Google Scholar]
- Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y, Yamada T, Egami S, Hoshii T, Hirao A, Matsuda S, Koyasu S. PI3K-Akt-mTORC1-S6K1/2 Axis Controls Th17 Differentiation by Regulating Gfi1 Expression and Nuclear Translocation of RORγ. Cell Rep. 2012;1:360–373. doi: 10.1016/j.celrep.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR Signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee HJ, Sciammas R, Gantner BN, Dinner AR, Singh H. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–766. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O'shea JJ. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, Oukka M, Kuchroo VK, Glimcher LH. T-bet represses TH17 differentiation by preventing Runx1-mediated activation of the gene encoding RORγt. Nat. Immunol. 2011;12:96–104. doi: 10.1038/ni.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. 2010;32:743–753. doi: 10.1016/j.immuni.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012;13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Ritchea S, Logar A, et al. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity. 2009;31:799–810. doi: 10.1016/j.immuni.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-β induces development of the TH17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Li J, Vaque JP, Konkel JE, Wang W, Zhang B, Zhang P, Zamarron BF, Yu D, Wu Y, Zhuang Y, Gutkind JS, Chen W. Control of the differentiation of regulatory T cells and TH17 cells by the DNA-binding inhibitor Id3. Nat. Immunol. 2011;12:86–95. doi: 10.1038/ni.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489:231–241. doi: 10.1038/nature11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JD, Brenchley JM, Laurence A, et al. Impaired TH17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisan J, Grenningloh R, Bettelli E, Oukka M, Ho IC. Ets-1 is a negative regulator of Th17 differentiation. J. Exp. Med. 2007;204:2825–2835. doi: 10.1084/jem.20070994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J. Immunol. 2003a;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc. Natl Acad. Sci. USA. 2003b;100:5986–5990. doi: 10.1073/pnas.1035999100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor W, Jr, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, Kolls JK, Flavell RA. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani M, Nagai S, Kondo S, Mizuno S, Nakamura K, Tanabe M, Takeuchi T, Matsuda S, Koyasu S. Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide-induced interleukin-12 production in dendritic cells. Blood. 2008;112:635–643. doi: 10.1182/blood-2008-02-137430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Iwai Y, Oh-Hora M, Yamamoto M, Morio T, Aoki K, Ohya K, Jetten AM, Akira S, Muta T, Takayanagi H. IκBζ regulates TH17 development by cooperating with ROR nuclear receptors. Nature. 2010;464:1381–1385. doi: 10.1038/nature08922. [DOI] [PubMed] [Google Scholar]
- Okkenhaug K, Patton DT, Bilancio A, Garcon F, Rowan WC, Vanhaesebroeck B. The p110δ isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J. Immunol. 2006;177:5122–5128. doi: 10.4049/jimmunol.177.8.5122. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat. Immunol. 2010;11:618–627. doi: 10.1038/ni.1884. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Zhang R, Yang J, et al. Transcription factor IRF8 directs a silencing programme for TH17 cell differentiation. Nat. Commun. 2011;2:314. doi: 10.1038/ncomms1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pot C, Jin H, Awasthi A, Liu SM, Lai CY, Madan R, Sharpe AH, Karp CL, Miaw SC, Ho IC, Kuchroo VK. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J. Immunol. 2009;183:797–801. doi: 10.4049/jimmunol.0901233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AE, Reinhardt RL, Liang HE, Locksley RM. Marking and quantifying IL-17A-producing cells in vivo. PLoS ONE. 2012;7:e39750. doi: 10.1371/journal.pone.0039750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65–68. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Doffinger R, Natividad A, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 2010;207:291–297. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Jin H, Burns EJ, Nadeau M, Yeste A, Kumar D, Rangachari M, Zhu C, Xiao S, Seavitt J, Georgopoulos K, Kuchroo K. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat. Immunol. 2012;13:770–777. doi: 10.1038/ni.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, Kolls JK, Khader SA, Randall TD. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat. Immunol. 2011;12:639–646. doi: 10.1038/ni.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Romani L. Immunity to fungal infections. Nat. Rev. Immunol. 2011;11:275–288. doi: 10.1038/nri2939. [DOI] [PubMed] [Google Scholar]
- Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol. Rev. 2006;212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J. Immunol. 1993;150:5445–5456. [PubMed] [Google Scholar]
- Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, Tone M, Chen YH. Development of Foxp3+ regulatory T cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–940. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Q, Kameswaran V, Zhang Y, Zheng S, Sun J, Wang J, DeVirgiliis J, Liou HC, Beg AA, Chen YH. The Th17 immune response is controlled by the Rel-RORγ-RORγ T transcriptional axis. J. Exp. Med. 2011;208:2321–2333. doi: 10.1084/jem.20110462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Sarker KP, Lee KY. L6 myoblast differentiation is modulated by Cdk5 via the PI3K-AKT-p70S6K signaling pathway. Oncogene. 2004;23:6064–6070. doi: 10.1038/sj.onc.1207819. [DOI] [PubMed] [Google Scholar]
- Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006;203:2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O'Connor E, Shokat KM, Fisher AG, Merkenschlager M. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl Acad. Sci. USA. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraml BU, Hildner K, Ise W, et al. The AP-1 transcription factor Batf controls TH17 differentiation. Nature. 2009;460:405–409. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw MH, Kamada N, Kim YG, Nunez G. Microbiota-induced IL-1β, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J. Exp. Med. 2012;209:251–258. doi: 10.1084/jem.20111703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiroki F, Matsuda S, Doi T, Fujiwara M, Mochizuki Y, Kadowaki T, Suzuki H, Koyasu S. The p85α regulatory subunit of class IA phosphoinositide 3-kinase regulates β-selection in thymocyte development. J. Immunol. 2007;178:1349–1356. doi: 10.4049/jimmunol.178.3.1349. [DOI] [PubMed] [Google Scholar]
- Sinclair LV, Finlay D, Feijoo C, Cornish GH, Gray A, Ager A, Okkenhaug K, Hagenbeek TJ, Spits H, Cantrell DA. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat. Immunol. 2008;9:513–521. doi: 10.1038/ni.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiehl DP, Jelkmann W, Wenger RH, Hellwig-Burgel T. Normoxic induction of the hypoxia-inducible factor 1α by insulin and interleukin-1β involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002;512:157–162. doi: 10.1016/s0014-5793(02)02247-0. [DOI] [PubMed] [Google Scholar]
- Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, Villarino AV, Huang Q, Yoshimura A, Sehy D, Saris CJ, O'Shea JJ, Hennighausen L, Ernst M, Hunter CA. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat. Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, Ernst M, Saris CJ, O'Shea JJ, Hunter CA. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat. Immunol. 2007;8:1363–1371. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule-Smith A, Lefebvre RE, Unutmaz D, Mazitschek R, Waldner H, Whitman M, Keller T, Rao A. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324:1334–1338. doi: 10.1126/science.1172638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Matsuda S, Terauchi Y, Fujiwara M, Ohteki T, Asano T, Behrens TW, Kouro T, Takatsu K, Kadowaki T, Koyasu S. PI3K and Btk differentially regulate B cell antigen receptor mediated signal transduction. Nat. Immunol. 2003;4:280–286. doi: 10.1038/ni890. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Terauchi Y, Fujiwara M, Aizawa S, Yazaki Y, Kadowaki T, Koyasu S. Xid-like immunodeficiency in mice with disruption of the p85α subunit of phosphoinositide 3-kinase. Science. 1999;283:390–392. doi: 10.1126/science.283.5400.390. [DOI] [PubMed] [Google Scholar]
- Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, Takahashi R, Asakawa M, Muto G, Mori T, Hasegawa E, Saika S, Hara T, Nomura M, Yoshimura A. Smad2 and Smad3 are redundantly essential for the TGF-β-mediated regulation of regulatory T plasticity and Th1 development. J. Immunol. 2010;185:842–855. doi: 10.4049/jimmunol.0904100. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008a;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-β initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat. Immunol. 2006;7:1151–1156. doi: 10.1038/ni1391. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, Martin B, Wilhelm C, Stockinger B. Transforming growth factor-β ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 2008b;9:1341–1346. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- Wan Q, Kozhaya L, ElHed A, Ramesh R, Carlson TJ, Djuretic IM, Sundrud MS, Unutmaz D. Cytokine signals through PI-3 kinase pathway modulate Th17 cytokine production by CCR6+ human memory T cells. J. Exp. Med. 2010;208:1875–1887. doi: 10.1084/jem.20102516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J. Biol. Chem. 1993;268:21513–21518. [PubMed] [Google Scholar]
- Wang YH, Voo KS, Liu B, Chen CY, Uygungil B, Spoede W, Bernstein JA, Huston DP, Liu YJ. A novel subset of CD4+ TH2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J. Exp. Med. 2010;207:2479–2491. doi: 10.1084/jem.20101376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, Kolbe T, Stulnig TM, Horl WH, Hengstschlager M, Müller M, Säemann MD. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–577. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Xie S, Li J, Wang JH, Wu Q, Yang P, Hsu HC, Smythies LE, Mountz JD. IL-17 activates the canonical NF-kappaB signaling pathway in autoimmune B cells of BXD2 mice to upregulate the expression of regulators of G-protein signaling 16. J. Immunol. 2010;184:2289–2296. doi: 10.4049/jimmunol.0903133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat. Immunol. 2011a;12:888–897. doi: 10.1038/ni.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA. IL-21 and TGF-beta are required for differentiation of human TH17 cells. Nature. 2008a;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, Feng XH, Jetten AM, Dong C. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008b;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity. 2008c;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, Hirahara K, Sun HW, Wei L, Vahedi G, Kanno Y, O'Shea JJ, Laurence A. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat. Immunol. 2011b;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Ye P, Rodriguez FH, Kanaly S, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavadil J, Bottinger EP. TGF-β and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat. Immunol. 2008;9:1297–1306. doi: 10.1038/ni.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Bevan MJ. TGF-β signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat. Immunol. 2012;13:667–673. doi: 10.1038/ni.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Lloyd CM, Noble A. Th17 responses in chronic allergic airway inflammation abrogate regulatory T-cell-mediated tolerance and contribute to airway remodeling. Mucosal Immunol. 2013 doi: 10.1038/mi.2012.76. in press. doi: 10.1038/mi.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Davidson TS, Wei G, Jankovic D, Cui K, Schones DE, Guo L, Zhao K, Shevach EM, Paul WE. Down-regulation of Gfi-1 expression by TGF-β is important for differentiation of Th17 and CD103+ inducible regulatory T cells. J. Exp. Med. 2009;206:329–341. doi: 10.1084/jem.20081666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, Monticelli S, Lanzavecchia A, Sallusto F. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature. 2012;484:514–518. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]